

Abstract
Statins are 3-hydroxy-3-methyglutaryl coenzyme A (HMG-CoA) reductase inhibitors, which are prescribed extensively for cholesterol lowering in the primary and secondary prevention of cardiovascular disease. Recent compelling evidence suggests that the beneficial effects of statins may not only be due to their cholesterol lowering effects, but also, to their cholesterol-independent or pleiotropic effects. Through these so-called pleiotropic effects, statins are directly involved in restoring or improving endothelial function, attenuating vascular remodeling, inhibiting vascular inflammatory response, and perhaps, stabilizing atherosclerotic plaques. These cholesterol-independent effects of statins are due predominantly to their ability to inhibit isoprenoid synthesis, the products of which are important lipid attachments for intracellular signaling molecules, such as Rho, Rac and Cdc42. In particular, inhibition of Rho and its downstream target, Rho-associated coiled-coil containing protein kinase (ROCK), has emerged as the principle mechanisms underlying the pleiotropic effects of statins. This review provides an update of statin-mediated vascular effects beyond cholesterol lowering and highlights recent findings from bench to bedside to support the concept of statin pleiotropy.
Keywords: Statin, vascular, cholesterol, inflammation, nitric oxide, rho kinase
Introduction
The 3-hydroxy-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, or statins, are potent inhibitors of cholesterol biosynthesis. They have emerged as the leading therapeutic class of lipid lowering agents and are established in the primary and secondary prevention of coronary artery disease. However, in contrast to the original rationale of the biological effect of statins, it has become increasingly apparent that the overall benefits observed with statins are not mediated solely by their lipid-lowering properties [1-3]. Indeed, several studies have demonstrated beneficial effects of statins in cardiovascular disease that appears to be independent of cholesterol lowering.
The term “pleiotropy” comes from the Greek words pleio, which means many, and trepein, which means influencing. Over the past decades the term “pleiotropic effects” has been ascribed to some of the effects of statins in the treatment and prevention of cardiovascular events. In particular, statins may play a key role in immunomodulation [4], neuroprotection [5] and cellular senescence [6].
Statin Pharmacology
Statins were initially identified as secondary metabolites of fungi [7]. One of the first natural inhibitors of HMG-CoA reductase, ML-236B, was isolated as a metabolite from cultures of Penicillium citrinum and was shown to be an extremely potent competitive inhibitor of HMG-CoA reductase [8]. Thus, statins inhibit HMG-CoA reductase through binding to the enzyme's active site and block the substrate-product transition state of the enzyme [9]. Each of the statins is unique in its tissue permeability and pharmacokinetics. Although all statins can enter hepatic cells through either active or passive transport, hydrophilic statins, such as pravastatin and rosuvastatin are less likely to enter non-hepatic cells, while lipophilic statins, e.g. atorvastatin and simvastatin are more likely to hepatic and non-hepatic cells through passive diffusion. This difference in tissue permeability and metabolism may account for some of the differential pleiotropic effects among the statins [10].
A second mechanism by which statins exert extrahepatic effects is their ability to prevent the synthesis of other important isoprenoid intermediates of the cholesterol biosynthetic pathway, such as farnesylpyrophosphate (FPP) and geranyl-geranylpyrophosphate (GGPP) that are downstream from L-mevalonic acid [11]. These intermediates serve as important lipid attachments for the post-translational modification of proteins, including nuclear lamins, Ras, Rho, Rac and Rap [12]. The inhibition of isoprenoid, therefore, may contribute to some of the pleiotropic effects of statins (Fig. 1). Interestingly, statins have also been shown to interact with the leukocyte function-associated antigen-1 (LFA-1), which is independent of mevalonate synthesis. LFA –1 belongs to the integrin family and plays an important role in leukocyte trafficking and T-cell activation. Lovastatin binds to an allosteric site within the beta2-integrin LFA-1 and inhibits the LFA-1 intercellular adhesion molecule-1 interaction [13].
Fig. (1). Biological actions of isoprenoids.
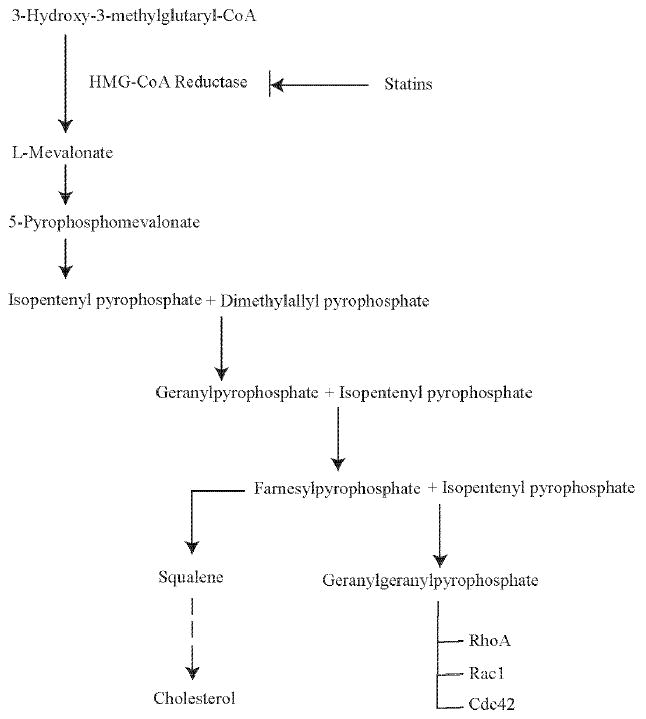
Statins inhibit HMG-CoA reductase activity leading to a decrease in isoprenylation of signaling molecules, such as RhoA, Rac1 and cdc42.
Statins and its Diverse Points of Action
Statins and Rho/ROCK
Rho kinases (ROCKs) are protein serine/threonine kinases of ∼ 160 kDa and are downstream effectors of the small GTPase Rho [14]. They were initially characterized by their ability to mediate the formation of RhoA-induced stress fibers and focal adhesions through increasing the phosphorylation of myosin light chain (MLC) [15]. Statins block the synthesis of isoprenoids, and therefore, the subsequent geranygeranylation of Rho GTPases [16]. Through post-translational modifications, isoprenylation is critical for intracellular trafficking and function of small GTP-binding proteins [17]. In particular, by inhibiting mevalonate synthesis, statins prevent membrane targeting of Rho and its subsequent activation of ROCK. Indeed, in vitro studies suggest that many of the pleiotropic effects of statins are due to alterations in the RhoA/ROCK signaling pathways [18-20]. For example, similar to the effects of statins, the administration of ROCK inhibitors has been shown to prevent cerebral vasospasm after subarachnoidal hemorrhage [21] and to prevent arterial remodeling after vascular injury [22].
Statins and Rac
Rac is a 20-30 kDa monomeric G protein and a member of the small GTPase subfamily. The Rac signaling pathway is involved in actin cytoskeletal remodeling and reactive oxygen species (ROS) generation. Within this context, Rac has received great attention for its involvement in myocardial signaling since the development of myocardial hypertrophy and heart failure is exhibited by ventricular remodeling and increased oxidative stress [23]. Rac1 influences multiple actin cytoskeletal remodeling proteins, such as Wiskott-Aldrich Syndrome protein, calmodulin-binding GTPase activation proteins and p21 activated kinase [24]. Rac1 also binds to p67phox and leads to activation of the NADPH oxides system and subsequent generation of ROS [25]. As demonstrated in fibroblasts, Rac activity is closely related to NADPH-dependent ROS production in response to growth factors and inflammatory cytokines [26].
Some of the pleiotropic effects of statins may be mediated through inhibition of Rac1. In animal models, simvastatin prevented angiotensin II (Ang II) or pressure overload induced hypertrophy through inhibition of Rac1-mediated NADPH oxidase activity in vascular smooth muscle and heart [27,28]. Indeed, inducible and selective deletion of Rac1 in the adult mouse heart protects against Ang II-induced cardiac hypertrophy [29]. This finding is further supported by analysis of failing human heart tissues, where upregulation of ROS release is associated with increase Rac1 activity, both of which are attenuated by statin treatment [30]. Atorvastatin decreases cardiac Rac1 and RhoA activities and downregulates atrial natriuretic peptide, as well as myosin light chain 2V expression in spontaneously hypertensive rats [16]. Interestingly, a recent study in a transgenic rabbit model demonstrated that atorvastatin prevented cardiac hypertrophy was associated with reduced levels of membrane Ras and extracellular regulated kinase (ERK) activation, but without decreases in GTP-bound or total RhoA or Rac1 [31]. Nevertheless, these findings support effects of statins that are not due to lipid lowering.
Statins and the Peroxisome Proliferators-Activated Receptor
Peroxisome proliferators-activated receptors (PPARs) are members of the nuclear steroid-hormone receptor superfamily and bind to specific DNA response elements as heterodimers with the retinoid X receptor [32]. Currently, three isoforms of PPAR have been identified. PPARs are major players in the energy homeostasis as well as in the regulation of inflammatory responses [33,34]. Recent studies reveal that PPARs may contribute to the pleiotropic effect of statins. For example, statins induce PPARγ transcriptional activity in macrophages. Statins inhibit lipopolysaccharide (LPS)-induced tumor necrosis factor α (TNFα) and monocyte chemoattractant protein-1 (MCP-1) expression as well as repress the transcriptional activity of nuclear transcription factor (NFκB) and activator protein (AP1) through PPARα and PPARγ [35]. These findings are supported by data demonstrating that statins stabilize atherosclerotic plaques through the activation of PPARγ and that combined administration of simvastatin and PPARγ agonists elicit additive effects on atherosclerotic plaque regression [36,37]. Indeed, acute treatment with statins inhibits LPS-induced expression of inflammatory genes via a PPARα-proteinkinase C (PKC)-dependent mechanism [38,39]. These findings support the anti-inflammatory properties of statins and suggest evidence for the existence of a cross-talk between the statin and PPAR pathways. Further studies are required to determine whether combination therapy of statins and PPAR agonist has synergistic effects in reducing cardiovascular disease.
Statin and its Pleiotropic Effects
Statins and the Endothelium
The endothelium forms the inner lining of blood vessels and represents a metabolically active system. In addition to its function as a barrier, the endothelium senses and responds to environmental factors and serves as an important autocrine and paracrine organ that regulates the contractile state of blood vessels, blood cell trafficking, hemostatic balance and other cellular compositions [40]. Endothelial dysfunction, as defined by decreased bioavailability of endothelium-derived nitric oxide (eNOS), is one of the earliest manifestations of atherosclerosis [41,42]. An important characteristic of endothelial dysfunction is the impaired synthesis, release, and activity of eNOS as well as a decrease in NO bioavailability through inactivation by increased ROS production [43,44]. The anti-atherogenic effects of eNOS include enhancing vascular relaxation [45] and inhibiting of platelet aggregation [46], vascular smooth muscle cell proliferation [47], and leukocyte-endothelial interactions [48].
Clinically, statins have been shown to improve endothelial function in patients with hypercholesterolemia and atherosclerosis. Interestingly, this benefit occurs through both cholesterol-dependent and –independent mechanisms. The cholesterol-dependent mechanism is evident in patients after LDL apheresis, which removes plasma LDL particles physically. These patients show rapid improvement in endothelium-dependent vasomotion through acute reduction in serum cholesterol levels [49]. On the other hand however, several studies have demonstrated that statins improve endothelial function even before significant reduction in serum cholesterol levels has occurred [50,51]. These effects have been linked to the upregulation of eNOS by statins [52,53] and is further supported by studies in normocholesterolemic animals, where statins protect against stroke, myocardial ischemia and reperfusion injury through non-cholesterol mechanisms [54,55].
Statins can upregulate eNOS by several different mechanisms (Fig. 2). First, statins increase the stability of eNOS mRNA through the Rho/ROCK pathway, thereby leading to increased eNOS expression [18,52]. For example, RhoA negatively regulates eNOS expression and activity as demonstrated by studies showing that direct inhibition of Rho by Clostridium botulinum C3 transferase or overexpression of a dominant-negative mutant of RhoA increases eNOS expression [56,57]. Although the effects of statins on Ras and Rho isoprenylation are reversed in the presence of FPP and GGP, respectively, the effects of statins on eNOS expression are reversed only by GGPP, but not by FPP or LDL cholesterol [56]. These findings indicate that the Rho/ROCK pathway rather than the Ras pathway predominantly regulates eNOS expression.
Fig. (2). Upregulation of eNOS by statins.
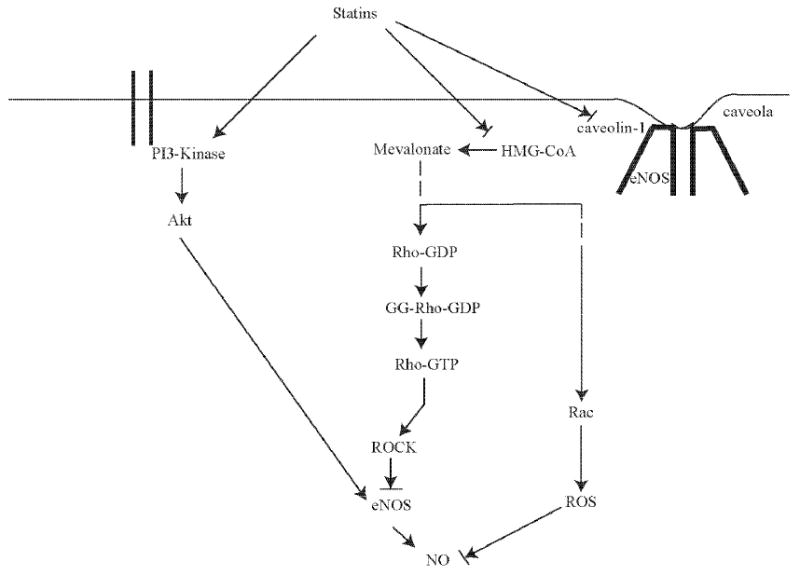
Statins modulate eNOS expression and activity through three major mechanisms: 1) Increased eNOS mRNA stability through inhibition of Rho isoprenylation. 2) Increased Akt-mediated eNOS phosphorylation through PI3K-dependent signaling. 3) Restoration of eNOS activity through reduction of caveolin-1 abundance.
A second important mechanism by which statins activate eNOS is mediated through the serine-threonine protein kinase Akt. Akt is activated through protein tyrosine kinase and G protein-coupled receptors and has been shown to be involved in mediating growth, metabolism, and survival through downstream targets such as glycogen synthase kinase (GSK)-β and Forkhead/FOXO transcription factors. Interestingly, eNOS is also a substrate of Akt and is phosphorylated at Ser 1177/1179 [58]. Thus, statins have been shown to rapidly promote the activation of Akt in endothelial cells leading to eNOS phosphorylation and increased NO production. Because this process is inhibited by the phosphatidylinositol-3 kinase (PI3K) inhibitors, wortmannin and LY294002, these findings indicate that statins activates Akt by upregulating phosphatidylinositol-3-kinase (PI3K) signaling [59]. The statin-Akt pathway appears to be specific for endothelial cells, as activation of Akt by statins is not observed in cardiac or smooth muscle cells [59]. Although higher doses of statins promote an increase in eNOS protein expression, recent reports show that statins at higher doses also exerts toxic effects on endothelial cells, presumably due to excessive inhibition of protein prenylation [18]. This finding is further supported in animal models where low statin concentrations enhances angiogenesis in ischemic hindlimbs of normochloesterolemic animals through an eNOS-dependent mechanism, while high doses inhibit angiogenesis [60,61]. Through the PI3K/Akt pathway, statins also enhance the mobilization of endothelial progenitor cells (EPC) from bone marrow to newly forming blood vessels [62,63]. Indeed, statins promote EPC mobilization in patients with stable coronary heart diseases [64].
A third mechanism by which statins may regulate eNOS activity is through their effects on caveolin-1. Caveolin-1 is an integral membrane protein that binds to eNOS in caveolae and thereby inhibit NO production directly [65]. Its allosteric competitor calmodulin (CaM), which promotes the calcium-dependent activation of eNOS through binding to the CaM-binding motif, is thought to displace an adjacent autoinhibitory loop on eNOS. [66,67]. Atorvastatin has been shown to reduce caveolin-1 abundance leading to restoration of eNOS activity in endothelial cells. This effect is completely reversed by the addition of mevalonate [68]. Furthermore, fluvastatin has been shown to prevent hypoxia-induced interaction between eNOS and caveolin-1, as well as the dissociation of heat shock proteins (HSP90) from eNOS in pulmonary endothelial cells [69]. Both of these effects of fluvastatin would lead to greater eNOS activity.
Finally, statins may have beneficial non-cholesterol effects through enhancing the moblization of EPCs. EPCs augment ischemia-induced neovascularization, accelerate reendothelialization after carotid balloon injury and improve postischemic cardiac function. Indeed, statins induce angiogenesis by promoting the mobilization, proliferation, migration, and survival of circulating EPCs. This effect is dependent upon the activation of the PI3K/Akt pathway [62]. Interestingly, as mentioned above, this effect is observed at lower concentrations of statins, while higher concentrations of statins elicit anti-angiogenic effects [61,63]. Similar effects were observed clinically. One-week treatment with 40 mg atorvastain leads to a 1.5-fold increase of EPCs in elderly patients with stable coronary artery diseases [64]. This is confirmed by a recent study demonstrating the efficiency of EPC mobilization in healthy, young male volunteers after short-term therapy with simvastatin or rosuvastatin [70].
Statins and VSMC
Vascular smooth muscle cells (VSMC) contibute to vascular proliferative diseases, including postangioplasty restenosis, transplant arteriosclerosis and venous graft occlusion [71]. Recent studies have shown that statins can attenuate cytokine-mediated VSMC proliferation in coronary artery smooth muscle cells and also inhibit pathological proliferation such as that observed in transplant-associated arteriopathy [72-74]. Because transplant-associated arteriopathy is more dependent on immunological mechanisms rather than lipid disorders, this finding further underscores the importance of statin pleiotropy.
Early studies have shown that statins can inhibit cell proliferation through an isoprenoid-dependent way [75]. In fibroblasts, G1 cell cycle arrest induced by lovastatin is reversed by the addition of mevalonate or GGPP [76]. Inhibition of platelet-derived growth factor (PDGF)-induced DNA synthesis in VSMCs is reversed by isoprenoid, but not cholesterol [19,77]. Because the small GTP-binding proteins, Ras and Rho, require posttranslational modification for membrane localization and activity and are implicated in cell-cycle regulation, they are likely targets for the direct antiproliferative vascular effects of statins. Ras can promote cell-cycle progression via activation of the mitogen-activated protein kinase pathway, whereas Rho causes cellular proliferation through destabilization of the cyclin-dependent kinase inhibitor, p27Kip1 [78,79]. It appears that inhibition of Rho may be the predominant effect of statins on VSMC proliferation as the inhibition is reversed by GGPP, but not by farnesyl pyrophosphate or LDL cholesterol [19]. Indeed, direct inhibition of Rho by Clostridium botulinum C3 transferase or by a dominant-negative Rho mutant increases p27Kip1 and inhibits SMC proliferation after PDGF stimulation. Another study showed that atorvastatin inhibits serotonin-induced mitogenesis and migration through inhibition of GTP-RhoA formation in pulmonary artery smooth muscle cells [80]. This effect is also reversed by GGPP, but not FPP and is accompanied by a reduction of nuclear phospho-ERK. Taken together, these findings indicate that Rho mediates SMC proliferation and that inhibition of Rho by statins is the predominant mechanism by which statins inhibit vascular SMC proliferation.
ROS are generally formed by one or two electron reduction of molecular oxygen resulting in the formation of superoxide anion or hydrogen peroxide, and are involved in cellular proliferation. They directly affect vascular homeostasis through scavenging and decreasing the bioavailability of NO and thus contribute to vascular dysfunction. Statins have been shown to exert antioxidant effects by increasing eNOS activity and decreasing ROS production [52,59]. Specifically, statins prevent lipid oxidation and decrease the Ang II receptor (AT-1)-dependent ROS generation [28,81,82]. Statins also affect ROS generation through the inhibition of NAD(P)H oxidase activity and assembly by preventing isoprenylation of the small p21 rac protein [83]. Recently, it has been reported that atorvastatin reduces vascular mRNA expression of essential NADPH oxidase subunits p22phox and nox1 (gp91phox) by a mechanism that involves the translocation of Rac1 from the cytosol to the cell membrane [28]. Because NO is scavenged by ROS, these findings indicate that the antioxidant properties of statins may also contribute to their ability to inhibit VSMC proliferation.
Statins and the Myocardium
Cardiac hypertrophy is an adaptive response of the heart to pressure overload. Ras was the first member of the small GTPase linked to cardiac remodeling [84]. Following this finding, the Rho/Rac/cdc42 family has been shown to participate in myocardial remodeling [23,85,86]. Indeed, recent animal studies suggest that NADPH oxidase-dependent ROS production in response to pressure overload, stretch and Ang II-infusion require the guanine nucleotide-binding proteins Rac1 and Rap for activation [87,88]. During activation, Rac1 binds GTP and migrates to the membrane with the core cytosolic complex, leading to NADPH oxidase-dependent ROS production [16,89]. Because Rac1 is required for NADPH oxidase activity and cardiac hypertrophy is mediated, in part, by myocardial oxidative stress, it is likely that statins could inhibit cardiac hypertrophy through an antioxidant mechanism involving inhibition of Rac1 geranylgeranylation. Indeed, statins inhibit angiotensin II-induced oxidative stress and cardiac hypertrophy in rodents [27]. Furthermore, a recent study demonstrated that treatment with lovastatin failed to protect AngII induced cardiac hypertrophy in p21 knockout mice. Molecular analysis demonstrated that lovastatin activated the FoxO3a transcription factor complex which in turn activates the growth-inhibitor gene p21 and thus mediated its protective role to prevent cardiac hypertrophy [90]. Oral statin treatment in patients with heart failure has also been shown to decrease Rac1 function in the human heart and thus reduce NADPH-oxidase mediated ROS activity [30]. Together, these findings strongly support the impact of statin in preventing the development of cardiac hypertrophy.
Statins also exert protective function against ischemic myocardial injury. For example, pretreatment of rats with simvastatin for 18 hours prior to the induction of ischemia-reperfusion significantly reduced cardiac dysfunction and improved coronary flow [91]. Several follow-up studies confirmed these findings in normocholesterolemic as well as hypercholesterolemic mice [92]. One of the main contributors to this protective effect of statins is the increase in NO availability. NO-mediated vasodilation facilitates regional myocardial blood flow under hypoxic conditions and also inhibits platelet activation which may aggregate at sites of vascular lesions [93,94]. Furthermore, statin-induced NO production has been shown to inhibit the upregulation of adhesion molecules involved in leukocyte-endothelial cell interactions, such as P-selectin, vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) [95,96]. Finally, simvastatin has been demonstrated to preserve mitochondrial membrane potential in response to oxidative stress in cardiac myocytes on a NO-dependent manner [97]. All of these mechanisms underscore the important role of statins in cardioprotection.
A common sequela of decompensated cardiac hypertrophy is the development of congestive heart failure (CHF). Emerging evidences suggest the importance of statins in the treatment for these patients. Several animal models of myocardial hypertrophy and heart failure, such as aortic banding, myocardial infarction have proven that statins can preserve cardiac functions under this conditions [16,98,99]. Furthermore, statins may improve endothelial dysfunction and increase vascular tone that characterize patients with heart failure [100]. Concurrently, retrospective analysis of large statin trials, such as the Scandinavian Simvastatin Survival Study (4S) and Treating to New Target (TNT) study, suggests that statins reduce the incidence and morbidity of heart failure [101,102]. In a recent prospective, double blind, placebo-controlled study, patients with symptomatic, nonischemic, dilated cardiomyopathy were randomly divided into two groups receiving statin or placebo for 14 weeks [103]. Although patients receiving statins exhibited a modest reduction in serum cholesterol level compared to patients receiving placebo, these patients demonstrated a significant improvement in exercise endurance, as exhibited by a lower NYHA functional class compared to patients receiving placebo. This corresponded to improved left ventricular ejection fraction in the statin group (33 ± 4 to 41 ± 4%, p < 0.01), but not in the placebo group. The improvements in their exercise endurance and heart function were in addition to the improvements already observed with two current treatments for heart failure, beta blockers and ACE inhibitors. Furthermore, plasma concentrations of TNFα, interleukin-6 (IL-6), and brain natriuretic peptide (BNP) were lower in the statin group compared to the placebo group. This study indicates that short-term statin therapy improves cardiac function, neurohormonal imbalance, and symptoms associated with idiopathic dilated cardiomyopathy and further strengthens the therapeutic benefits statins may have in patients with heart failure irrespective of serum cholesterol levels or atherosclerotic heart disease.
Statins and Atherosclerosis
Atherosclerosis is a complex inflammatory process that is characterized by the cross-talk between excessive inflammation and lipid accumulation [104,105]. In the presence of monocytes, macrophages and T-lymphocytes, inflammatory cytokines are secreted and subsequently modify endothelial function, SMC proliferation, collagen degradation, and thrombosis [106]. The early step in atherogenesis is characterized by an inflammatory response to injury, and involves the recruitment of monocytes to the artery wall, followed by penetration into the subendothelial space [107]. In the past few years, statins have been found to modulate immune activation and to exert anti-inflammatory effects on the vascular wall by decreasing the number of inflammatory cells in atherosclerotic plaques [108]. The mechanism is due, in part, to statins' ability to decrease the expression of endothelial adhesion molecules, such as ICAM-1, VCAM-1 and E-selectin [109-111]. Furthermore, statins attenuate P-selectin expression and leukocyte adhesion in normocholesterolemic animals by increasing endothelial NO production [55,112]. This cholesterol-independent effect of statins was absent in eNOS-deficient mice, suggesting that eNOS mediates the vascular protective effects of statins [113].
Once monocytes become resident in the arterial intima, they acquire the morphological characteristics of macrophages, express scavenger receptors that bind internalized lipoprotein particles leading to transformation into foam cells [107]. The direct cholesterol-lowering properties of statins represent the one side of anti-oxidative properties of statins. On the other hand, statins also inhibit the oxidation of LDL through lipid-independent properties, which is demonstrated by the upregulation of anti-oxidative enzyme catalase in the aorta and reduced vascular expression of NADPH oxidase subunits p22phox and nox1 in hypertensive rats after atorvastatin treatment [28]. During atherogenesis T-lymphocytes also play an important role in the pathomechanism. The activation of T-lymphocytes and the control of the immune response are mediated by the major histocompatibility complex class II (MHC-II) and CD40/CD40L. Under physiological conditions, antigen-presenting cells express MHC-II constitutively, whereas the induction of interferon-γ (INF-γ) leads to an increase of MHC-II expression in numerous cells, including human endothelial cells and monocytes. An important regulator in this pathway is the class II transactivator (CIITA). Statins inhibit MHC-II expression on endothelial cells and monocyte-macrophages via inhibition of the promoter IV of the transactivator CIITA and thereby repress MHC-II mediated T cell activation [114]. In addition, statins also have been shown to decrease INF-γ induced CD40 expression and CD40-related activation of vascular cells [115, 116].
The atherosclerotic lesion contains highly thrombogenic materials in the lipid core that are separated from the bloodstream by a fibrous cap [117]. Fissuring, erosion, and ulceration of the fibrous cap eventually lead to plaque rupture and thrombosis [118], which is the major cause of acute coronary syndromes [106,119]. Lipid lowering by statins contribute to plaque stability by reducing plaque size or by modifying the physiochemical properties of the lipid core [120,121]. However, changes in plaque size by lipid lowering tend to occur over extended time and are quite minimal as assessed by coronary angiography or intravascular ultrasound. Rather, the clinical benefits from lipid lowering are probably due to decreases in macrophage accumulation in atherosclerotic lesions and inhibition of matrix metalloproteinase (MMP) production by activated macrophages [122]. Indeed, statins inhibit the expression of MMPs and tissue factor by cholesterol-dependent and –independent mechanisms [120,123]. The cholesterol-independent mechanisms are mainly mediated through a combined reduction in lipids, macrophages, and MMPs [124]. These effects of statins may reduce the incidence of acute coronary syndromes by lessening the propensity for plaque to rupture and may explain the rapid time course of event reduction in patients at high risk for recurrent coronary ischemia in the MIRACL (Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering) and PROVE-IT (Pravastatin or Atorvastatin Evaluation and Infection Therapy and Thrombolysis in Myocardial Infarction) trials [125,126].
Finally, statins can also modulate LFA, a major counterreceptor for ICAM-1 on leukocytes [127]. By binding directly to its regulatory site in the beta-2 integrin, statins can inhibit T cell activation and suppress the inflammatory response independent of HMG-CoA reductase inhibition and small GTPases [13,128].
While inflammatory processes characterize the initiation of atherosclerosis, platelets play a critical role in the manifestation of acute coronary syndromes. Circulating platelets are associated with mural thrombus formation at the site of plaque rupture and vascular injury and acute thrombus formation is present in most episodes of acute coronary syndromes [129]. Hypercholesterolemia has been associated with increases in platelet reactivity [130,131]. These abnormalities are linked with impairment of platelet-derived NO release, an increase in the cholesterol-to-phospholipid ratio in platelets, and elevated thromboxane A2 biosynthesis [132-135]. Statins have been shown to modulate platelet function through multiple mechanisms [136,137]. One mechanism includes the statin-mediated upregulation of eNOS that is associated with downregulation of platelet reactivity [138]. This observation is further supported by a recent published work showing that the combination of subtherapeutic doses of simvastatin and dipyridamole increases cerebral blood flow and decreases stroke volume through an eNOS-dependent pathway [139]. Also fluvastatin has been shown to reduce platelet aggregation in association with greater platelet-derived NO release and less oxidative stress [140]. Interestingly, this study also supports previous assumptions that the reduced platelet activity under statins is due to its inhibitory effect on Rho [141]. For example, fluvastatin inhibited platelet nitrotyrosine expressions, which was reversed by supplementation of GGPP. Further possibilities include a reduction in the production of thromboxane A2 and modifications in the cholesterol content of platelet membranes [108,142]. Indeed, animal studies suggest that statin therapy inhibits platelet deposition on damaged vessels and reduces platelet thrombus formation. Finally, statins inhibit tissue factor expression by macrophages and thereby potentially reducing the thrombotic potential of the vascular wall [122].
Statins in Clinical Trials: Evidence for Pleiotropy
Because serum cholesterol level is strongly associated with coronary heart disease, it has been generally assumed that the beneficial effects underlying statin therapy are predominantly due to cholesterol reduction. Several meta-analyses of lipid-lowering trials have suggested lipid modification alone accounts for the clinical benefits associated with statin therapy [143]. However, when comparing statin with non-statin lipid-lowering trials – such as the Lipid Research Clinics – Coronary Primary Prevention Trial (LRC-CPPT) or the Program on the Surgical Control of the Hyperlipidemias (POSCH) trial, clinical studies with statin therapy demonstrate benefits within 5 years compared with greater than 7 years for non-statin trials [144]. The European WOSCOPS (West of Scotland Coronary Prevention Study), a double-blind, randomized placebo-controlled trial has evaluated the effects of 40mg pravastatin daily on preventing the coronary events in hyperlipidemic subjects during a time-frame of 4.9 years. Although no significant differences could be found in non-cardiac death, the onset of non-fatal myocardial infarction or death resulting from coronary artery disease was significantly lower compared to the placebo group [145]. Furthermore, subgroup analysis of WOSCOPS and CARE (The Cholesterol and Recurrent Events Trial) studies indicate that, despite comparable serum cholesterol levels among patients in the statin and placebo groups, patients who are treated with statin have 47% lower risk of developing subsequent events [146,147]. These findings suggest clinical benefits of statin therapy, which are independent of LDL cholesterol reduction.
Previous findings indicate that statin therapy lowers high-sensitivity C-reactive protein (hs-CRP), an independent marker shown to predict future vascular events [148,149]. This anti-inflammatory property of statins has been suggested to be one explanation for its beneficial effects on endothelial dysfunction. As demonstrated in the PROVE IT study, patients with acute coronary syndromes who were treated with statins and achieved a target level of CRP less than 2mg/l, showed a significant improvement in event-free survival [150]. In a second step, the REVERSAL trial (Reversal of atherosclerosis with aggressive lipid lowering) has demonstrated that intensive treatment with atorvastatin not only significantly reduced atherogenic lipoproteins, but also resulted in a 36.4% reduction of C-reactive protein (CRP) in the atorvastatin group compared with 5.2% reduction in the pravastatin group. This is further accompanied by a lower progression rate of atheroma volume in the atorvastatin group, underlining the relationship between CRP and atherosclerosis [151]. Finally, to evaluate whether long-term treatment with rosuvastatin (20mg/day) would decrease the rate of cardiovascular events among apparently healthy men and women with acceptable levels of LDL cholesterol, but increased levels of hsCRP, the JUPITER trial (Justification for the Use of statin in Primary prevention: an Intervention Trial Evaluating Rosuvastatin) was initiated in 2003 and stopped in March this year, because of significant reduction in cardiovascular morbidity and mortality in patients who received rosuvastatin [152]
Recently, two clinical trials with ezetimibe support the notion of statin pleiotropy in humans. Ezetimibe is a lipid-lowering agent, which acts by decreasing cholesterol absorption in the intestine [153]. It is used alone or in combination with statin therapy to enhance lipid lowering [154]. In patients with heart failure, 4 weeks of simvastatin but not ezetimibe treatment improved endothelial function and reduced oxidative stress, despite comparable reduction in serum cholesterol levels [155]. Similarly, 40 mg atorvastatin improved forearm blood flow to a greater extent than 20 mg simvastatin/10 mg ezetimibe, despite comparable reduction in LDL cholesterol [156]. Furthermore in the recently published Ezetimibe and and Simvastatin in Hypercholesterolemia Enhances Atherosclerosis Regression (ENHANCE) study – a double blind, randomized trial comparing ezetimibe/simvastatin (10mg/80mg) to the highest recommended dose of 80mg simvastatin, ezetimide/simvastatin combination did not reduce the intima-media thickness of the carotid-artery wall in patients with familial hypercholesterolemia, despite significant incremental reductions in levels of both LDL cholesterol and C-reactive protein [157]. One possible interpretation of this result could be that the lowering of LDL cholesterol levels by a drug other than a statin might not be as effective for slowing atherosclerosis as statin monotherapy. This explanation would emphasize the pleiotropic effect of statins. Indeed, direct comparison between ezetimibe and statins revealed differential effects on endothelial function favoring statin therapy despite similar reductions in LDL cholesterol [155]. Currently, the IMPROVE-IT study comparing Vytorin (ezetimibe/simvastatin) versus simvastatin alone in subjects with acute coronary syndromes is ongoing. The expected completion for this trial is awaited for 2012 and will hopefully help determine whether there are any beneficial effects of statins beyond LDL-lowering.
There is a large pool of data supporting the pleiotropic effect of statins on the vasculature. But recent clinical studies have also focused on pleiotropic effects of statins beyond the vascular tree. As demonstrated by Horwich et al. statins also improve heart function and survival in patients with non-ischemic heart failure [158-160]. While ischemic heart failure is due to atherosclerosis and associated with high cholesterol level, non-ischemic heart failure is usually unrelated to atherosclerosis. Short-term statin therapy has been demonstrated to improve cardiac function, neurohormonal imbalance and symptoms associated with idiopathic dilated cardiomyopathy [103]. Furthermore, two prospective trials have recently investigated the effect of statin on ischemic and non-ischemic heart failure. Results of the CORONA (Controlled Rosuvastatin Multinational Study in Heart Failure) trial have demonstrated that daily treatment with 10mg of rosuvastatin in patients with ischemic, systolic heart failure (NYHA class III-IV, or EF < 35% in NYHA class II) significantly reduced hospitalization of the patients. However, rosuvastatin treatment did not show significant changes on the NYHA class compared to placebo treatment, nor did it reduce the composite outcome of death from cardiovascular causes or nonfatal myocardial infarction or stroke [161] The outcome of patients with non-ischemic heart failure and a preserved ejection fraction are enrolled in the GISSI-HF (Gruppo Italiano per lo Studio della Sopravvivenza nell'Infarto Miocardico-Insufficienza Cardiaca) trial. This trial compares rosuvastatin with placebo and n-3 polyunsaturated fatty acids with placebo in 6975 patients, with 4574 patients assigned to participate in the rosuvastsatin portion of the study [162]. The results are expected to be announced in August 2008 and will further help to determine whether statin therapy is effective in patients with non-ischemic heart failure.
Recently, a meta-analysis of randomized trials with statins on the end point of incidence or recurrence of atrial fibrillation demonstrated that statins significantly decreased the risk of incidence or recurrence of atrial fibrillation (AF) in patients with a history of previous AF or patients undergoing cardiac surgery or after acute coronary syndrome [163]. This analysis included six studies with a total of 3,557 patients. Interestingly, the beneficial effect in the prevention of AF recurrences is stronger than in primary prevention of AF. The mechanism could be due to the interaction of statins with the renin-angiotensin system. Ang II has been demonstrated to enhance cardiac myocyte growth and promote the proliferation of vascular smooth muscle cells and fibroblasts resulting in remodeling and fibrosis of the atria that could finally lead to the pro-arrhythmia [164]. At the same time statins has been shown to decrease oxidative stress and downregulate the renin-angiotensin system [165], which might in part explain a possible antiarrhythmic effect of statins against AF [166]. Statins have also been suggested to modulate the autonomic nervous system, which might explain the protective role of statins in the particular setting of postoperative patients with enhanced sympathetic activity [167].
Underlining the pleiotropic effect of statins outside the cardiovascular system, several clinical trials focusing on other organ system have been conducted over the past years, including the neurovascular, renal and immunological system. Especially the impact of statins in the prevention of stroke has been the spotlight recently [168]. Although myocardial infarction is closely associated with serum cholesterol levels, several observational studies looking at the relationship between elevated cholesterol level and cerebrovascular disease reported contradictory results [169,170]. However, many clinical trials primarily designed to examine the coronary benefits of statins also demonstrated reduction in risk of stroke [171]. While WOSCOPS and the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS) did not show any decrease in stroke events with statin therapy, the Anglo-Scandinavian Cardiac Out-cmomes Trial (ASCOT)-Lipid Lowering Arm (LLA) demonstrated a 27% reduction of fatal or nonfatal stroke in the atorvastatin group compared to placebo [147,172,173]. In the recently completed Collaborative Atorvastatin Diabetes Study (CARDS), atorvastatin treatment resulted in 48% reduction in the relative risk of stroke in patients with diabetes and at least one other cardiovascular risk factor, but no history of coronary artery disease [174]. Further non-cardiovascular clinical studies supporting the effects of statins beyond cholesterol reduction include the beneficial effects of statin in the treatment of chronic kidney disease [175,176], rheumatologic diseases, neurological disease like multiple sclerosis [177,178] or Alzheimer's disease [108]. These data all support the concept of stain pleiotropy and emphasizes the rationale for its clinical use beyond cholesterol lowering.
Conclusion
Statin pleiotropy – once regarded as being “too good to be true”, is still a work-in-progress. Most of the pleiotropic effects of statins are mediated through inhibition of the isoprenoid synthesis, with subsequent downstream effects on small GTPase signaling pathways. In particular statins can lead to increased expression of atheroprotective genes and inhibition of pro-inflammatory mediators. These benefits include endothelial protective effects, enhancing the stability of atherosclerotic plaques, and inhibiting vascular smooth muscle proliferation and platelet aggregation. It remains to be determined to what extend these pleiotropic effects account for the clinical benefits of statin therapy beyond cholesterol lowering.
Acknowledgments
This work is supported by grants from the National Institutes of Health (HL052233 and HL080187) and intramural funding from the Department of Cardiology, University of Freiburg, Germany (to Q.Z.).
References
- 1.Maron DJ, Fazio S, Linton MF. Current perspectives on statins. Circulation. 2000;101:207–13. doi: 10.1161/01.cir.101.2.207. [DOI] [PubMed] [Google Scholar]
- 2.Palinski W. New evidence for beneficial effects of statins unrelated to lipid lowering. Arterioscler Thromb Vasc Biol. 2001;21:3–5. doi: 10.1161/01.atv.21.1.3. [DOI] [PubMed] [Google Scholar]
- 3.Wang CY, Liu PY, Liao JK. Pleiotropic effects of statin therapy: molecular mechanisms and clinical results. Trends Mol Med. 2008;14:37–44. doi: 10.1016/j.molmed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greenwood J, Steinman L, Zamvil SS. Statin therapy and autoimmune disease: from protein prenylation to immunomodulation. Nat Rev Immunol. 2006;6:358–70. doi: 10.1038/nri1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kivipelto M, Solomon A, Winblad B. Statin therapy in Alzheimer's disease. Lancet Neurol. 2005;4:521–2. doi: 10.1016/S1474-4422(05)70150-2. [DOI] [PubMed] [Google Scholar]
- 6.Brouilette SW, Moore JS, McMahon AD, Thompson JR, Ford I, Shepherd J, et al. Telomere length, risk of coronary heart disease, and statin treatment in the West of Scotland Primary Prevention Study: a nested case-control study. Lancet. 2007;369:107–14. doi: 10.1016/S0140-6736(07)60071-3. [DOI] [PubMed] [Google Scholar]
- 7.Alberts AW. Discovery, biochemistry and biology of lovastatin. Am J Cardiol. 1988;62:10J–5J. doi: 10.1016/0002-9149(88)90002-1. [DOI] [PubMed] [Google Scholar]
- 8.Alberts AW, Chen J, Kuron G, Hunt V, Huff J, Hoffman C, et al. Mevinolin: a highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc Natl Acad Sci U S A. 1980;77:3957–61. doi: 10.1073/pnas.77.7.3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Istvan ES, Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science. 2001;292:1160–4. doi: 10.1126/science.1059344. [DOI] [PubMed] [Google Scholar]
- 10.Germershausen JI, Hunt VM, Bostedor RG, Bailey PJ, Karkas JD, Alberts AW. Tissue selectivity of the cholesterol-lowering agents lovastatin, simvastatin and pravastatin in rats in vivo. Biochem Biophys Res Commun. 1989;158:667–75. doi: 10.1016/0006-291x(89)92773-3. [DOI] [PubMed] [Google Scholar]
- 11.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–30. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 12.Van Aelst L, D'Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 1997;11:2295–322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- 13.Weitz-Schmidt G, Welzenbach K, Brinkmann V, Kamata T, Kallen J, Bruns C, et al. Statins selectively inhibit leukocyte function antigen-1 by binding to a novel regulatory integrin site. Nat Med. 2001;7:687–92. doi: 10.1038/89058. [DOI] [PubMed] [Google Scholar]
- 14.Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4:446–56. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
- 15.Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–79. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 16.Laufs U, Kilter H, Konkol C, Wassmann S, Bohm M, Nickenig G. Impact of HMG CoA reductase inhibition on small GTPases in the heart. Cardiovasc Res. 2002;53:911–20. doi: 10.1016/s0008-6363(01)00540-5. [DOI] [PubMed] [Google Scholar]
- 17.Hall A. G proteins and small GTPases: distant relatives keep in touch. Science. 1998;280:2074–5. doi: 10.1126/science.280.5372.2074. [DOI] [PubMed] [Google Scholar]
- 18.Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97:1129–35. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- 19.Laufs U, Marra D, Node K, Liao JK. 3-Hydroxy-3-methylglutaryl-CoA reductase inhibitors attenuate vascular smooth muscle proliferation by preventing rho GTPase-induced down-regulation of p27(Kip1) J Biol Chem. 1999;274:21926–31. doi: 10.1074/jbc.274.31.21926. [DOI] [PubMed] [Google Scholar]
- 20.Mundy G, Garrett R, Harris S, Chan J, Chen D, Rossini G, et al. Stimulation of bone formation in vitro and in rodents by statins. Science. 1999;286:1946–9. doi: 10.1126/science.286.5446.1946. [DOI] [PubMed] [Google Scholar]
- 21.Shibuya M, Suzuki Y, Sugita K, Saito I, Sasaki T, Takakura K, et al. Effect of AT877 on cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Results of a prospective placebo-controlled double-blind trial. J Neurosurg. 1992;76:571–7. doi: 10.3171/jns.1992.76.4.0571. [DOI] [PubMed] [Google Scholar]
- 22.Sawada N, Itoh H, Ueyama K, Yamashita J, Doi K, Chun TH, et al. Inhibition of rho-associated kinase results in suppression of neointimal formation of balloon-injured arteries. Circulation. 2000;101:2030–3. doi: 10.1161/01.cir.101.17.2030. [DOI] [PubMed] [Google Scholar]
- 23.Brown JH, Del Re DP, Sussman MA. The Rac and Rho hall of fame: a decade of hypertrophic signaling hits. Circ Res. 2006;98:730–42. doi: 10.1161/01.RES.0000216039.75913.9e. [DOI] [PubMed] [Google Scholar]
- 24.Nakagawa H, Miki H, Ito M, Ohashi K, Takenawa T, Miyamoto S. N-WASP, WAVE and Mena play different roles in the organization of actin cytoskeleton in lamellipodia. J Cell Sci. 2001;114:1555–65. doi: 10.1242/jcs.114.8.1555. [DOI] [PubMed] [Google Scholar]
- 25.Gregg D, Rauscher FM, Goldschmidt-Clermont PJ. Rac regulates cardiovascular superoxide through diverse molecular interactions: more than a binary GTP switch. Am J Physiol Cell Physiol. 2003;285:C723–34. doi: 10.1152/ajpcell.00230.2003. [DOI] [PubMed] [Google Scholar]
- 26.Sundaresan M, Yu ZX, Ferrans VJ, Sulciner DJ, Gutkind JS, Irani K, et al. Regulation of reactive-oxygen-species generation in fibroblasts by Rac1. Biochem J. 1996;318(Pt 2):379–82. doi: 10.1042/bj3180379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takemoto M, Node K, Nakagami H, Liao Y, Grimm M, Takemoto Y, et al. Statins as antioxidant therapy for preventing cardiac myocyte hypertrophy. J Clin Invest. 2001;108:1429–37. doi: 10.1172/JCI13350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wassmann S, Laufs U, Baumer AT, Muller K, Konkol C, Sauer H, et al. Inhibition of geranylgeranylation reduces angiotensin II-mediated free radical production in vascular smooth muscle cells: involvement of angiotensin AT1 receptor expression and Rac1 GTPase. Mol Pharmacol. 2001;59:646–54. doi: 10.1124/mol.59.3.646. [DOI] [PubMed] [Google Scholar]
- 29.Satoh M, Ogita H, Takeshita K, Mukai Y, Kwiatkowski DJ, Liao JK. Requirement of Rac1 in the development of cardiac hypertrophy. Proc Natl Acad Sci U S A. 2006;103:7432–7. doi: 10.1073/pnas.0510444103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maack C, Kartes T, Kilter H, Schafers HJ, Nickenig G, Bohm M, et al. Oxygen free radical release in human failing myocardium is associated with increased activity of rac1-GTPase and represents a target for statin treatment. Circulation. 2003;108:1567–74. doi: 10.1161/01.CIR.0000091084.46500.BB. [DOI] [PubMed] [Google Scholar]
- 31.Senthil V, Chen SN, Tsybouleva N, Halder T, Nagueh SF, Willer-son JT, et al. Prevention of cardiac hypertrophy by atorvastatin in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circ Res. 2005;97:285–92. doi: 10.1161/01.RES.0000177090.07296.ac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–50. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 33.Vidal-Puig AJ, Considine RV, Jimenez-Linan M, Werman A, Pories WJ, Caro JF, et al. Peroxisome proliferator-activated receptor gene expression in human tissues. Effects of obesity, weight loss, and regulation by insulin and glucocorticoids. J Clin Invest. 1997;99:2416–22. doi: 10.1172/JCI119424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cunard R, Eto Y, Muljadi JT, Glass CK, Kelly CJ, Ricote M. Repression of IFN-gamma expression by peroxisome proliferator-activated receptor gamma. J Immunol. 2004;172:7530–6. doi: 10.4049/jimmunol.172.12.7530. [DOI] [PubMed] [Google Scholar]
- 35.Yano M, Matsumura T, Senokuchi T, Ishii N, Murata Y, Taketa K, et al. Statins activate peroxisome proliferator-activated receptor gamma through extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase-dependent cyclooxygenase-2 expression in macrophages. Circ Res. 2007;100:1442–51. doi: 10.1161/01.RES.0000268411.49545.9c. [DOI] [PubMed] [Google Scholar]
- 36.Zelvyte I, Dominaitiene R, Crisby M, Janciauskiene S. Modulation of inflammatory mediators and PPARgamma and NFkappaB expression by pravastatin in response to lipoproteins in human monocytes in vitro. Pharmacol Res. 2002;45:147–54. doi: 10.1006/phrs.2001.0922. [DOI] [PubMed] [Google Scholar]
- 37.Corti R, Osende JI, Fallon JT, Fuster V, Mizsei G, Jneid H, et al. The selective peroxisomal proliferator-activated receptor-gamma agonist has an additive effect on plaque regression in combination with simvastatin in experimental atherosclerosis: in vivo study by high-resolution magnetic resonance imaging. J Am Coll Cardiol. 2004;43:464–73. doi: 10.1016/j.jacc.2003.08.048. [DOI] [PubMed] [Google Scholar]
- 38.Paumelle R, Blanquart C, Briand O, Barbier O, Duhem C, Woerly G, et al. Acute antiinflammatory properties of statins involve peroxisome proliferator-activated receptor-alpha via inhibition of the protein kinase C signaling pathway. Circ Res. 2006;98:361–9. doi: 10.1161/01.RES.0000202706.70992.95. [DOI] [PubMed] [Google Scholar]
- 39.Blanquart C, Mansouri R, Paumelle R, Fruchart JC, Staels B, Glineur C. The protein kinase C signaling pathway regulates a molecular switch between transactivation and transrepression activity of the peroxisome proliferator-activated receptor alpha. Mol Endocrinol. 2004;18:1906–18. doi: 10.1210/me.2003-0327. [DOI] [PubMed] [Google Scholar]
- 40.Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. 2007;100:158–73. doi: 10.1161/01.RES.0000255691.76142.4a. [DOI] [PubMed] [Google Scholar]
- 41.Werns SW, Walton JA, Hsia HH, Nabel EG, Sanz ML, Pitt B. Evidence of endothelial dysfunction in angiographically normal coronary arteries of patients with coronary artery disease. Circulation. 1989;79:287–91. doi: 10.1161/01.cir.79.2.287. [DOI] [PubMed] [Google Scholar]
- 42.Steinberg D, Parthasarathy S, Carew TE, Khoo JC, Witztum JL. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N Engl J Med. 1989;320:915–24. doi: 10.1056/NEJM198904063201407. [DOI] [PubMed] [Google Scholar]
- 43.Alderson LM, Endemann G, Lindsey S, Pronczuk A, Hoover RL, Hayes KC. LDL enhances monocyte adhesion to endothelial cells in vitro. Am J Pathol. 1986;123:334–42. [PMC free article] [PubMed] [Google Scholar]
- 44.Liao JK. Inhibition of Gi proteins by low density lipoprotein attenuates bradykinin-stimulated release of endothelial-derived nitric oxide. J Biol Chem. 1994;269:12987–92. [PubMed] [Google Scholar]
- 45.Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A. 1987;84:9265–9. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Radomski MW, Rees DD, Dutra A, Moncada S. S-nitroso-glutathione inhibits platelet activation in vitro and in vivo. Br J Pharmacol. 1992;107:745–9. doi: 10.1111/j.1476-5381.1992.tb14517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garg UC, Hassid A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J Clin Invest. 1989;83:1774–7. doi: 10.1172/JCI114081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gauthier TW, Scalia R, Murohara T, Guo JP, Lefer AM. Nitric oxide protects against leukocyte-endothelium interactions in the early stages of hypercholesterolemia. Arterioscler Thromb Vasc Biol. 1995;15:1652–9. doi: 10.1161/01.atv.15.10.1652. [DOI] [PubMed] [Google Scholar]
- 49.Tamai O, Matsuoka H, Itabe H, Wada Y, Kohno K, Imaizumi T. Single LDL apheresis improves endothelium-dependent vasodilatation in hypercholesterolemic humans. Circulation. 1997;95:76–82. doi: 10.1161/01.cir.95.1.76. [DOI] [PubMed] [Google Scholar]
- 50.Anderson TJ, Meredith IT, Yeung AC, Frei B, Selwyn AP, Ganz P. The effect of cholesterol-lowering and antioxidant therapy on endothelium-dependent coronary vasomotion. N Engl J Med. 1995;332:488–93. doi: 10.1056/NEJM199502233320802. [DOI] [PubMed] [Google Scholar]
- 51.O'Driscoll G, Green D, Taylor RR. Simvastatin, an HMG-coenzyme A reductase inhibitor, improves endothelial function within 1 month. Circulation. 1997;95:1126–31. doi: 10.1161/01.cir.95.5.1126. [DOI] [PubMed] [Google Scholar]
- 52.Laufs U, Fata VL, Liao JK. Inhibition of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase blocks hypoxia-mediated down-regulation of endothelial nitric oxide synthase. J Biol Chem. 1997;272:31725–9. doi: 10.1074/jbc.272.50.31725. [DOI] [PubMed] [Google Scholar]
- 53.Essig M, Nguyen G, Prie D, Escoubet B, Sraer JD, Friedlander G. 3-Hydroxy-3-methylglutaryl coenzyme A reductase inhibitors increase fibrinolytic activity in rat aortic endothelial cells. Role of geranylgeranylation and Rho proteins. Circ Res. 1998;83:683–90. doi: 10.1161/01.res.83.7.683. [DOI] [PubMed] [Google Scholar]
- 54.Endres M, Laufs U, Huang Z, Nakamura T, Huang P, Moskowitz MA, et al. Stroke protection by 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 1998;95:8880–5. doi: 10.1073/pnas.95.15.8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lefer AM, Campbell B, Shin YK, Scalia R, Hayward R, Lefer DJ. Simvastatin preserves the ischemic-reperfused myocardium in normocholesterolemic rat hearts. Circulation. 1999;100:178–84. doi: 10.1161/01.cir.100.2.178. [DOI] [PubMed] [Google Scholar]
- 56.Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem. 1998;273:24266–71. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- 57.Takemoto M, Sun J, Hiroki J, Shimokawa H, Liao JK. Rho-kinase mediates hypoxia-induced downregulation of endothelial nitric oxide synthase. Circulation. 2002;106:57–62. doi: 10.1161/01.cir.0000020682.73694.ab. [DOI] [PubMed] [Google Scholar]
- 58.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–5. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 59.Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, et al. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6:1004–10. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sata M, Nishimatsu H, Suzuki E, Sugiura S, Yoshizumi M, Ouchi Y, et al. Endothelial nitric oxide synthase is essential for the HMG-CoA reductase inhibitor cerivastatin to promote collateral growth in response to ischemia. Faseb J. 2001;15:2530–2. doi: 10.1096/fj.01-0415fje. [DOI] [PubMed] [Google Scholar]
- 61.Weis M, Heeschen C, Glassford AJ, Cooke JP. Statins have biphasic effects on angiogenesis. Circulation. 2002;105:739–45. doi: 10.1161/hc0602.103393. [DOI] [PubMed] [Google Scholar]
- 62.Dimmeler S, Aicher A, Vasa M, Mildner-Rihm C, Adler K, Tiemann M, et al. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. J Clin Invest. 2001;108:391–7. doi: 10.1172/JCI13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Llevadot J, Murasawa S, Kureishi Y, Uchida S, Masuda H, Kawamoto A, et al. HMG-CoA reductase inhibitor mobilizes bone marrow -- derived endothelial progenitor cells. J Clin Invest. 2001;108:399–405. doi: 10.1172/JCI13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vasa M, Fichtlscherer S, Adler K, Aicher A, Martin H, Zeiher AM, et al. Increase in circulating endothelial progenitor cells by statin therapy in patients with stable coronary artery disease. Circulation. 2001;103:2885–90. doi: 10.1161/hc2401.092816. [DOI] [PubMed] [Google Scholar]
- 65.Plenz GA, Hofnagel O, Robenek H. Differential modulation of caveolin-1 expression in cells of the vasculature by statins. Circulation. 2004;109:e7–8. doi: 10.1161/01.CIR.0000111128.83347.7A. author reply e7-8. [DOI] [PubMed] [Google Scholar]
- 66.Michel JB, Feron O, Sase K, Prabhakar P, Michel T. Caveolin versus calmodulin. Counterbalancing allosteric modulators of endothelial nitric oxide synthase. J Biol Chem. 1997;272:25907–12. doi: 10.1074/jbc.272.41.25907. [DOI] [PubMed] [Google Scholar]
- 67.Michel JB, Feron O, Sacks D, Michel T. Reciprocal regulation of endothelial nitric-oxide synthase by Ca2+-calmodulin and caveolin. J Biol Chem. 1997;272:15583–6. doi: 10.1074/jbc.272.25.15583. [DOI] [PubMed] [Google Scholar]
- 68.Feron O, Dessy C, Desager JP, Balligand JL. Hydroxymethylglutaryl-coenzyme A reductase inhibition promotes endothelial nitric oxide synthase activation through a decrease in caveolin abundance. Circulation. 2001;103:113–8. doi: 10.1161/01.cir.103.1.113. [DOI] [PubMed] [Google Scholar]
- 69.Murata T, Kinoshita K, Hori M, Kuwahara M, Tsubone H, Karaki H, et al. Statin protects endothelial nitric oxide synthase activity in hypoxia-induced pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2005;25:2335–42. doi: 10.1161/01.ATV.0000186184.33537.48. [DOI] [PubMed] [Google Scholar]
- 70.Spiel AO, Mayr FB, Leitner JM, Firbas C, Sieghart W, Jilma B. Simvastatin and rosuvastatin mobilize Endothelial Progenitor Cells but do not prevent their acute decrease during systemic inflammation. Thromb Res. 2008 doi: 10.1016/j.thromres.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 71.Braun-Dullaeus RC, Mann MJ, Dzau VJ. Cell cycle progression: new therapeutic target for vascular proliferative disease. Circulation. 1998;98:82–9. doi: 10.1161/01.cir.98.1.82. [DOI] [PubMed] [Google Scholar]
- 72.Chandrasekar B, Mummidi S, Mahimainathan L, Patel DN, Bailey SR, Imam SZ, et al. Interleukin-18-induced human coronary artery smooth muscle cell migration is dependent on NF-kappaB- and AP-1-mediated matrix metalloproteinase-9 expression and is inhibited by atorvastatin. J Biol Chem. 2006;281:15099–109. doi: 10.1074/jbc.M600200200. [DOI] [PubMed] [Google Scholar]
- 73.Shimizu K, Aikawa M, Takayama K, Libby P, Mitchell RN. Direct anti-inflammatory mechanisms contribute to attenuation of experimental allograft arteriosclerosis by statins. Circulation. 2003;108:2113–20. doi: 10.1161/01.CIR.0000092949.67153.74. [DOI] [PubMed] [Google Scholar]
- 74.Kobashigawa JA, Katznelson S, Laks H, Johnson JA, Yeatman L, Wang XM, et al. Effect of pravastatin on outcomes after cardiac transplantation. N Engl J Med. 1995;333:621–7. doi: 10.1056/NEJM199509073331003. [DOI] [PubMed] [Google Scholar]
- 75.Jakobisiak M, Bruno S, Skierski JS, Darzynkiewicz Z. Cell cycle-specific effects of lovastatin. Proc Natl Acad Sci U S A. 1991;88:3628–32. doi: 10.1073/pnas.88.9.3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vogt A, Qian Y, McGuire TF, Hamilton AD, Sebti SM. Protein geranylgeranylation, not farnesylation, is required for the G1 to S phase transition in mouse fibroblasts. Oncogene. 1996;13:1991–9. [PubMed] [Google Scholar]
- 77.Yang Z, Kozai T, van der Loo B, Viswambharan H, Lachat M, Turina MI, et al. HMG-CoA reductase inhibition improves endothelial cell function and inhibits smooth muscle cell proliferation in human saphenous veins. J Am Coll Cardiol. 2000;36:1691–7. doi: 10.1016/s0735-1097(00)00924-4. [DOI] [PubMed] [Google Scholar]
- 78.Hughes DA. Control of signal transduction and morphogenesis by Ras. Semin Cell Biol. 1995;6:89–94. doi: 10.1016/1043-4682(95)90005-5. [DOI] [PubMed] [Google Scholar]
- 79.Hengst L, Reed SI. Translational control of p27Kip1 accumulation during the cell cycle. Science. 1996;271:1861–4. doi: 10.1126/science.271.5257.1861. [DOI] [PubMed] [Google Scholar]
- 80.Li M, Liu Y, Dutt P, Fanburg BL, Toksoz D. Inhibition of serotonin-induced mitogenesis, migration, and ERK MAPK nuclear translocation in vascular smooth muscle cells by atorvastatin. Am J Physiol Lung Cell Mol Physiol. 2007;293:L463–71. doi: 10.1152/ajplung.00133.2007. [DOI] [PubMed] [Google Scholar]
- 81.Suzumura K, Odawara A, Yasuhara M, Tanaka K, Narita H, Suzuki T. In vitro inhibitory effects of the optical isomers and metabolites of fluvastatin on copper ion-induced LDL oxidation. Biol Pharm Bull. 1999;22:971–4. doi: 10.1248/bpb.22.971. [DOI] [PubMed] [Google Scholar]
- 82.Aviram M, Rosenblat M, Bisgaier CL, Newton RS. Atorvastatin and gemfibrozil metabolites, but not the parent drugs, are potent antioxidants against lipoprotein oxidation. Atherosclerosis. 1998;138:271–80. doi: 10.1016/s0021-9150(98)00032-x. [DOI] [PubMed] [Google Scholar]
- 83.Wagner AH, Kohler T, Ruckschloss U, Just I, Hecker M. Improvement of nitric oxide-dependent vasodilatation by HMG-CoA reductase inhibitors through attenuation of endothelial superoxide anion formation. Arterioscler Thromb Vasc Biol. 2000;20:61–9. doi: 10.1161/01.atv.20.1.61. [DOI] [PubMed] [Google Scholar]
- 84.Thorburn A, Thorburn J, Chen SY, Powers S, Shubeita HE, Feramisco JR, et al. HRas-dependent pathways can activate morphological and genetic markers of cardiac muscle cell hypertrophy. J Biol Chem. 1993;268:2244–9. [PubMed] [Google Scholar]
- 85.Clerk A, Cullingford TE, Fuller SJ, Giraldo A, Markou T, Pikkarainen S, et al. Signaling pathways mediating cardiac myocyte gene expression in physiological and stress responses. J Cell Physiol. 2007;212:311–22. doi: 10.1002/jcp.21094. [DOI] [PubMed] [Google Scholar]
- 86.Lezoualc'h F, Metrich M, Hmitou I, Duquesnes N, Morel E. Small GTP-binding proteins and their regulators in cardiac hypertrophy. J Mol Cell Cardiol. 2008;44:623–32. doi: 10.1016/j.yjmcc.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 87.Li JM, Gall NP, Grieve DJ, Chen M, Shah AM. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension. 2002;40:477–84. doi: 10.1161/01.hyp.0000032031.30374.32. [DOI] [PubMed] [Google Scholar]
- 88.Aikawa R, Komuro I, Yamazaki T, Zou Y, Kudoh S, Zhu W, et al. Rho family small G proteins play critical roles in mechanical stress-induced hypertrophic responses in cardiac myocytes. Circ Res. 1999;84:458–66. doi: 10.1161/01.res.84.4.458. [DOI] [PubMed] [Google Scholar]
- 89.Higuchi Y, Otsu K, Nishida K, Hirotani S, Nakayama H, Yamaguchi O, et al. The small GTP-binding protein Rac1 induces cardiac myocyte hypertrophy through the activation of apoptosis signal-regulating kinase 1 and nuclear factor-kappa B. J Biol Chem. 2003;278:20770–7. doi: 10.1074/jbc.M213203200. [DOI] [PubMed] [Google Scholar]
- 90.Hauck L, Harms C, Grothe D, An J, Gertz K, Kronenberg G, et al. Critical role for FoxO3a-dependent regulation of p21CIP1/WAF1 in response to statin signaling in cardiac myocytes. Circ Res. 2007;100:50–60. doi: 10.1161/01.RES.0000254704.92532.b9. [DOI] [PubMed] [Google Scholar]
- 91.Lefer AM, Campbell B, Scalia R, Lefer DJ. Synergism between platelets and neutrophils in provoking cardiac dysfunction after ischemia and reperfusion: role of selectins. Circulation. 1998;98:1322–8. doi: 10.1161/01.cir.98.13.1322. [DOI] [PubMed] [Google Scholar]
- 92.Scalia R, Kochilas L, Campbell B, Lefer AM. Effects of defibrotide on leukocyte-endothelial cell interaction in the rat mesenteric vascular bed: role of P-selectin. Methods Find Exp Clin Pharmacol. 1996;18:669–76. [PubMed] [Google Scholar]
- 93.Schafer A, Wiesmann F, Neubauer S, Eigenthaler M, Bauersachs J, Channon KM. Rapid regulation of platelet activation in vivo by nitric oxide. Circulation. 2004;109:1819–22. doi: 10.1161/01.CIR.0000126837.88743.DD. [DOI] [PubMed] [Google Scholar]
- 94.Hwang YS, Tsai WC, Lu YH, Lin CC, Chen YF. Effect of atorvastatin on the expression of CD40 ligand and P-selectin on platelets in patients with hypercholesterolemia. Am J Cardiol. 2004;94:364–6. doi: 10.1016/j.amjcard.2004.04.037. [DOI] [PubMed] [Google Scholar]
- 95.Davenpeck KL, Gauthier TW, Lefer AM. Inhibition of endothelial-derived nitric oxide promotes P-selectin expression and actions in the rat microcirculation. Gastroenterology. 1994;107:1050–8. doi: 10.1016/0016-5085(94)90229-1. [DOI] [PubMed] [Google Scholar]
- 96.De Caterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA, Jr, et al. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest. 1995;96:60–8. doi: 10.1172/JCI118074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jones SP, Teshima Y, Akao M, Marban E. Simvastatin attenuates oxidant-induced mitochondrial dysfunction in cardiac myocytes. Circ Res. 2003;93:697–9. doi: 10.1161/01.RES.0000097262.21507.DF. [DOI] [PubMed] [Google Scholar]
- 98.Bauersachs J, Galuppo P, Fraccarollo D, Christ M, Ertl G. Improvement of left ventricular remodeling and function by hydroxymethylglutaryl coenzyme a reductase inhibition with cerivastatin in rats with heart failure after myocardial infarction. Circulation. 2001;104:982–5. doi: 10.1161/hc3401.095946. [DOI] [PubMed] [Google Scholar]
- 99.Dechend R, Fiebeler A, Park JK, Muller DN, Theuer J, Mervaala E, et al. Amelioration of angiotensin II-induced cardiac injury by a 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitor. Circulation. 2001;104:576–81. doi: 10.1161/hc3001.092039. [DOI] [PubMed] [Google Scholar]
- 100.Drexler H. Endothelium as a therapeutic target in heart failure. Circulation. 1998;98:2652–5. doi: 10.1161/01.cir.98.24.2652. [DOI] [PubMed] [Google Scholar]
- 101.Kjekshus J, Pedersen TR, Olsson AG, Faergeman O, Pyorala K. The effects of simvastatin on the incidence of heart failure in patients with coronary heart disease. J Card Fail. 1997;3:249–54. doi: 10.1016/s1071-9164(97)90022-1. [DOI] [PubMed] [Google Scholar]
- 102.LaRosa JC, Grundy SM, Waters DD, Shear C, Barter P, Fruchart JC, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med. 2005;352:1425–35. doi: 10.1056/NEJMoa050461. [DOI] [PubMed] [Google Scholar]
- 103.Node K, Fujita M, Kitakaze M, Hori M, Liao JK. Short-term statin therapy improves cardiac function and symptoms in patients with idiopathic dilated cardiomyopathy. Circulation. 2003;108:839–43. doi: 10.1161/01.CIR.0000084539.58092.DE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ross GW, Petrovitch H, White LR, Masaki KH, Li CY, Curb JD, et al. Characterization of risk factors for vascular dementia: the Honolulu-Asia Aging Study. Neurology. 1999;53:337–43. doi: 10.1212/wnl.53.2.337. [DOI] [PubMed] [Google Scholar]
- 105.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–9. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 106.Libby P. Molecular bases of the acute coronary syndromes. Circulation. 1995;91:2844–50. doi: 10.1161/01.cir.91.11.2844. [DOI] [PubMed] [Google Scholar]
- 107.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–74. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 108.Vaughan CJ, Gotto AM, Jr, Basson CT. The evolving role of statins in the management of atherosclerosis. J Am Coll Cardiol. 2000;35:1–10. doi: 10.1016/s0735-1097(99)00525-2. [DOI] [PubMed] [Google Scholar]
- 109.Chung HK, Lee IK, Kang H, Suh JM, Kim H, Park KC, et al. Statin inhibits interferon-gamma-induced expression of intercellular adhesion molecule-1 (ICAM-1) in vascular endothelial and smooth muscle cells. Exp Mol Med. 2002;34:451–61. doi: 10.1038/emm.2002.63. [DOI] [PubMed] [Google Scholar]
- 110.Rezaie-Majd A, Prager GW, Bucek RA, Schernthaner GH, Maca T, Kress HG, et al. Simvastatin reduces the expression of adhesion molecules in circulating monocytes from hypercholesterolemic patients. Arterioscler Thromb Vasc Biol. 2003;23:397–403. doi: 10.1161/01.ATV.0000059384.34874.F0. [DOI] [PubMed] [Google Scholar]
- 111.Rasmussen LM, Hansen PR, Nabipour MT, Olesen P, Kristiansen MT, Ledet T. Diverse effects of inhibition of 3-hydroxy-3-methylglutaryl-CoA reductase on the expression of VCAM-1 and E-selectin in endothelial cells. Biochem J. 2001;360:363–70. doi: 10.1042/0264-6021:3600363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Scalia R, Gooszen ME, Jones SP, Hoffmeyer M, Rimmer DM, 3rd, Trocha SD, et al. Simvastatin exerts both anti-inflammatory and cardioprotective effects in apolipoprotein E-deficient mice. Circulation. 2001;103:2598–603. doi: 10.1161/01.cir.103.21.2598. [DOI] [PubMed] [Google Scholar]
- 113.Stalker TJ, Lefer AM, Scalia R. A new HMG-CoA reductase inhibitor, rosuvastatin, exerts anti-inflammatory effects on the microvascular endothelium: the role of mevalonic acid. Br J Pharmacol. 2001;133:406–12. doi: 10.1038/sj.bjp.0704070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kwak B, Mulhaupt F, Myit S, Mach F. Statins as a newly recognized type of immunomodulator. Nat Med. 2000;6:1399–402. doi: 10.1038/82219. [DOI] [PubMed] [Google Scholar]
- 115.Mulhaupt F, Matter CM, Kwak BR, Pelli G, Veillard NR, Burger F, et al. Statins (HMG-CoA reductase inhibitors) reduce CD40 expression in human vascular cells. Cardiovasc Res. 2003;59:755–66. doi: 10.1016/s0008-6363(03)00515-7. [DOI] [PubMed] [Google Scholar]
- 116.Wagner AH, Gebauer M, Guldenzoph B, Hecker M. 3-hydroxy-3-methylglutaryl coenzyme A reductase-independent inhibition of CD40 expression by atorvastatin in human endothelial cells. Arte-rioscler Thromb Vasc Biol. 2002;22:1784–9. doi: 10.1161/01.atv.0000037098.20829.31. [DOI] [PubMed] [Google Scholar]
- 117.Fernandez-Ortiz A, Badimon JJ, Falk E, Fuster V, Meyer B, Mailhac A, et al. Characterization of the relative thrombogenicity of atherosclerotic plaque components: implications for consequences of plaque rupture. J Am Coll Cardiol. 1994;23:1562–9. doi: 10.1016/0735-1097(94)90657-2. [DOI] [PubMed] [Google Scholar]
- 118.Fuster V, Stein B, Ambrose JA, Badimon L, Badimon JJ, Chesebro JH. Atherosclerotic plaque rupture and thrombosis. Evolving concepts. Circulation. 1990;82:II47–59. [PubMed] [Google Scholar]
- 119.Fuster V. Elucidation of the role of plaque instability and rupture in acute coronary events. Am J Cardiol. 1995;76:24C–33C. doi: 10.1016/s0002-9149(99)80467-6. [DOI] [PubMed] [Google Scholar]
- 120.Fukumoto Y, Libby P, Rabkin E, Hill CC, Enomoto M, Hirouchi Y, et al. Statins alter smooth muscle cell accumulation and collagen content in established atheroma of watanabe heritable hyperlipidemic rabbits. Circulation. 2001;103:993–9. doi: 10.1161/01.cir.103.7.993. [DOI] [PubMed] [Google Scholar]
- 121.Koh KK. Effects of statins on vascular wall: vasomotor function, inflammation, and plaque stability. Cardiovasc Res. 2000;47:648–57. doi: 10.1016/s0008-6363(00)00146-2. [DOI] [PubMed] [Google Scholar]
- 122.Aikawa M, Rabkin E, Sugiyama S, Voglic SJ, Fukumoto Y, Furukawa Y, et al. An HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation. 2001;103:276–83. doi: 10.1161/01.cir.103.2.276. [DOI] [PubMed] [Google Scholar]
- 123.Bourcier T, Libby P. HMG CoA reductase inhibitors reduce plasminogen activator inhibitor-1 expression by human vascular smooth muscle and endothelial cells. Arterioscler Thromb Vasc Biol. 2000;20:556–62. doi: 10.1161/01.atv.20.2.556. [DOI] [PubMed] [Google Scholar]
- 124.Crisby M, Nordin-Fredriksson G, Shah PK, Yano J, Zhu J, Nilsson J. Pravastatin treatment increases collagen content and decreases lipid content, inflammation, metalloproteinases, and cell death in human carotid plaques: implications for plaque stabilization. Circulation. 2001;103:926–33. doi: 10.1161/01.cir.103.7.926. [DOI] [PubMed] [Google Scholar]
- 125.Schwartz GG, Olsson AG, Ezekowitz MD, Ganz P, Oliver MF, Waters D, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. Jama. 2001;285:1711–8. doi: 10.1001/jama.285.13.1711. [DOI] [PubMed] [Google Scholar]
- 126.Cannon CP, Braunwald E, McCabe CH, Rader DJ, Rouleau JL, Belder R, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495–504. doi: 10.1056/NEJMoa040583. [DOI] [PubMed] [Google Scholar]
- 127.Yamada A, Hara A, Inoue M, Kamizono S, Higuchi T, Itoh K. Beta 2-integrin-mediated signal up-regulates counterreceptor ICAM-1 expression on human monocytic cell line THP-1 through tyrosine phosphorylation. Cell Immunol. 1997;178:9–16. doi: 10.1006/cimm.1997.1117. [DOI] [PubMed] [Google Scholar]
- 128.Weitz-Schmidt G, Welzenbach K, Dawson J, Kallen J. Improved lymphocyte function-associated antigen-1 (LFA-1) inhibition by statin derivatives: molecular basis determined by x-ray analysis and monitoring of LFA-1 conformational changes in vitro and ex vivo. J Biol Chem. 2004;279:46764–71. doi: 10.1074/jbc.M407951200. [DOI] [PubMed] [Google Scholar]
- 129.Lacoste L, Lam JY, Hung J, Letchacovski G, Solymoss CB, Waters D. Hyperlipidemia and coronary disease. Correction of the increased thrombogenic potential with cholesterol reduction. Circulation. 1995;92:3172–7. doi: 10.1161/01.cir.92.11.3172. [DOI] [PubMed] [Google Scholar]
- 130.Opper C, Clement C, Schwarz H, Krappe J, Steinmetz A, Schneider J, et al. Increased number of high sensitive platelets in hypercholesterolemia, cardiovascular diseases, and after incubation with cholesterol. Atherosclerosis. 1995;113:211–7. doi: 10.1016/0021-9150(94)05448-r. [DOI] [PubMed] [Google Scholar]
- 131.Carvalho AC, Colman RW, Lees RS. Platelet function in hyperlipoproteinemia. N Engl J Med. 1974;290:434–8. doi: 10.1056/NEJM197402212900805. [DOI] [PubMed] [Google Scholar]
- 132.Ikeda H, Takajo Y, Murohara T, Ichiki K, Adachi H, Haramaki N, et al. Platelet-derived nitric oxide and coronary risk factors. Hypertension. 2000;35:904–7. doi: 10.1161/01.hyp.35.4.904. [DOI] [PubMed] [Google Scholar]
- 133.Takajo Y, Ikeda H, Haramaki N, Murohara T, Imaizumi T. Augmented oxidative stress of platelets in chronic smokers. Mechanisms of impaired platelet-derived nitric oxide bioactivity and augmented platelet aggregability. J Am Coll Cardiol. 2001;38:1320–7. doi: 10.1016/s0735-1097(01)01583-2. [DOI] [PubMed] [Google Scholar]
- 134.Belton O, Byrne D, Kearney D, Leahy A, Fitzgerald DJ. Cyclooxygenase-1 and -2-dependent prostacyclin formation in patients with atherosclerosis. Circulation. 2000;102:840–5. doi: 10.1161/01.cir.102.8.840. [DOI] [PubMed] [Google Scholar]
- 135.Montrucchio G, Alloatti G, Camussi G. Role of platelet-activating factor in cardiovascular pathophysiology. Physiol Rev. 2000;80:1669–99. doi: 10.1152/physrev.2000.80.4.1669. [DOI] [PubMed] [Google Scholar]
- 136.Huhle G, Abletshauser C, Mayer N, Weidinger G, Harenberg J, Heene DL. Reduction of platelet activity markers in type II hypercholesterolemic patients by a HMG-CoA-reductase inhibitor. Thromb Res. 1999;95:229–34. doi: 10.1016/s0049-3848(99)00037-7. [DOI] [PubMed] [Google Scholar]
- 137.Hale LP, Craver KT, Berrier AM, Sheffield MV, Case LD, Owen J. Combination of fosinopril and pravastatin decreases platelet response to thrombin receptor agonist in monkeys. Arterioscler Thromb Vasc Biol. 1998;18:1643–6. doi: 10.1161/01.atv.18.10.1643. [DOI] [PubMed] [Google Scholar]
- 138.Laufs U, Gertz K, Huang P, Nickenig G, Bohm M, Dirnagl U, et al. Atorvastatin upregulates type III nitric oxide synthase in thrombocytes, decreases platelet activation, and protects from cerebral ischemia in normocholesterolemic mice. Stroke. 2000;31:2442–9. doi: 10.1161/01.str.31.10.2442. [DOI] [PubMed] [Google Scholar]
- 139.Kim HH, Sawada N, Soydan G, Lee HS, Zhou Z, Hwang SK, et al. Additive effects of statin and dipyridamole on cerebral blood flow and stroke protection. J Cereb Blood Flow Metab. 2008 doi: 10.1038/jcbfm.2008.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Haramaki N, Ikeda H, Takenaka K, Katoh A, Sugano R, Yamagishi S, et al. Fluvastatin alters platelet aggregability in patients with hypercholesterolemia: possible improvement of intraplatelet redox imbalance via HMG-CoA reductase. Arterioscler Thromb Vasc Biol. 2007;27:1471–7. doi: 10.1161/ATVBAHA.106.128793. [DOI] [PubMed] [Google Scholar]
- 141.Kaneider NC, Egger P, Dunzendorfer S, Wiedermann CJ. Rho-GTPase-dependent platelet-neutrophil interaction affected by HMG-CoA reductase inhibition with altered adenosine nucleotide release and function. Arterioscler Thromb Vasc Biol. 2002;22:1029–35. doi: 10.1161/01.atv.0000018306.68268.86. [DOI] [PubMed] [Google Scholar]
- 142.Lijnen P, Echevaria-Vazquez D, Petrov V. Influence of cholesterol-lowering on plasma membrane lipids and function. Methods Find Exp Clin Pharmacol. 1996;18:123–36. [PubMed] [Google Scholar]
- 143.Robinson JG, Smith B, Maheshwari N, Schrott H. Pleiotropic effects of statins: benefit beyond cholesterol reduction? A meta-regression analysis. J Am Coll Cardiol. 2005;46:1855–62. doi: 10.1016/j.jacc.2005.05.085. [DOI] [PubMed] [Google Scholar]
- 144.Buchwald H, Varco RL, Matts JP, Long JM, Fitch LL, Campbell GS, et al. Effect of partial ileal bypass surgery on mortality and morbidity from coronary heart disease in patients with hypercholesterolemia. Report of the Program on the Surgical Control of the Hyperlipidemias (POSCH) N Engl J Med. 1990;323:946–55. doi: 10.1056/NEJM199010043231404. [DOI] [PubMed] [Google Scholar]
- 145.Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med. 1998;339:1349–57. doi: 10.1056/NEJM199811053391902. [DOI] [PubMed] [Google Scholar]
- 146.Sacks FM, Pfeffer MA, Moye LA, Rouleau JL, Rutherford JD, Cole TG, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N Engl J Med. 1996;335:1001–9. doi: 10.1056/NEJM199610033351401. [DOI] [PubMed] [Google Scholar]
- 147.Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFarlane PW, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med. 1995;333:1301–7. doi: 10.1056/NEJM199511163332001. [DOI] [PubMed] [Google Scholar]
- 148.Ridker PM, Rifai N, Rose L, Buring JE, Cook NR. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med. 2002;347:1557–65. doi: 10.1056/NEJMoa021993. [DOI] [PubMed] [Google Scholar]
- 149.Ridker PM. C-reactive protein and the prediction of cardiovascular events among those at intermediate risk: moving an inflammatory hypothesis toward consensus. J Am Coll Cardiol. 2007;49:2129–38. doi: 10.1016/j.jacc.2007.02.052. [DOI] [PubMed] [Google Scholar]
- 150.Ridker PM, Cannon CP, Morrow D, Rifai N, Rose LM, McCabe CH, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med. 2005;352:20–8. doi: 10.1056/NEJMoa042378. [DOI] [PubMed] [Google Scholar]
- 151.Nissen SE, Tuzcu EM, Schoenhagen P, Brown BG, Ganz P, Vogel RA, et al. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. Jama. 2004;291:1071–80. doi: 10.1001/jama.291.9.1071. [DOI] [PubMed] [Google Scholar]
- 152.Ridker PM. Rosuvastatin in the primary prevention of cardiovascular disease among patients with low levels of low-density lipoprotein cholesterol and elevated high-sensitivity C-reactive protein: rationale and design of the JUPITER trial. Circulation. 2003;108:2292–7. doi: 10.1161/01.CIR.0000100688.17280.E6. [DOI] [PubMed] [Google Scholar]
- 153.Sudhop T, Lutjohann D, Kodal A, Igel M, Tribble DL, Shah S, et al. Inhibition of intestinal cholesterol absorption by ezetimibe in humans. Circulation. 2002;106:1943–8. doi: 10.1161/01.cir.0000034044.95911.dc. [DOI] [PubMed] [Google Scholar]
- 154.Bruckert E, Giral P, Tellier P. Perspectives in cholesterol-lowering therapy: the role of ezetimibe, a new selective inhibitor of intestinal cholesterol absorption. Circulation. 2003;107:3124–8. doi: 10.1161/01.CIR.0000072345.98581.24. [DOI] [PubMed] [Google Scholar]
- 155.Landmesser U, Bahlmann F, Mueller M, Spiekermann S, Kirchhoff N, Schulz S, et al. Simvastatin versus ezetimibe: pleiotropic and lipid-lowering effects on endothelial function in humans. Circulation. 2005;111:2356–63. doi: 10.1161/01.CIR.0000164260.82417.3F. [DOI] [PubMed] [Google Scholar]
- 156.Fichtlscherer S, Schmidt-Lucke C, Bojunga S, Rossig L, Heeschen C, Dimmeler S, et al. Differential effects of short-term lipid lowering with ezetimibe and statins on endothelial function in patients with CAD: clinical evidence for ‘pleiotropic’ functions of statin therapy. Eur Heart J. 2006;27:1182–90. doi: 10.1093/eurheartj/ehi881. [DOI] [PubMed] [Google Scholar]
- 157.Kastelein JJ, Akdim F, Stroes ES, Zwinderman AH, Bots ML, Stalenhoef AF, et al. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med. 2008;358:1431–43. doi: 10.1056/NEJMoa0800742. [DOI] [PubMed] [Google Scholar]
- 158.Horwich TB, MacLellan WR, Fonarow GC. Statin therapy is associated with improved survival in ischemic and non-ischemic heart failure. J Am Coll Cardiol. 2004;43:642–8. doi: 10.1016/j.jacc.2003.07.049. [DOI] [PubMed] [Google Scholar]
- 159.Foody JM, Shah R, Galusha D, Masoudi FA, Havranek EP, Krumholz HM. Statins and mortality among elderly patients hospitalized with heart failure. Circulation. 2006;113:1086–92. doi: 10.1161/CIRCULATIONAHA.105.591446. [DOI] [PubMed] [Google Scholar]
- 160.Go AS, Lee WY, Yang J, Lo JC, Gurwitz JH. Statin therapy and risks for death and hospitalization in chronic heart failure. Jama. 2006;296:2105–11. doi: 10.1001/jama.296.17.2105. [DOI] [PubMed] [Google Scholar]
- 161.Kjekshus J, Apetrei E, Barrios V, Bohm M, Cleland JG, Cornel JH, et al. Rosuvastatin in older patients with systolic heart failure. N Engl J Med. 2007;357:2248–61. doi: 10.1056/NEJMoa0706201. [DOI] [PubMed] [Google Scholar]
- 162.Tavazzi L, Tognoni G, Franzosi MG, Latini R, Maggioni AP, Marchioli R, et al. Rationale and design of the GISSI heart failure trial: a large trial to assess the effects of n-3 polyunsaturated fatty acids and rosuvastatin in symptomatic congestive heart failure. Eur J Heart Fail. 2004;6:635–41. doi: 10.1016/j.ejheart.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 163.Fauchier L, Pierre B, de Labriolle A, Grimard C, Zannad N, Babuty D. Antiarrhythmic effect of statin therapy and atrial fibrillation a meta-analysis of randomized controlled trials. J Am Coll Cardiol. 2008;51:828–35. doi: 10.1016/j.jacc.2007.09.063. [DOI] [PubMed] [Google Scholar]
- 164.Healey JS, Baranchuk A, Crystal E, Morillo CA, Garfinkle M, Yusuf S, et al. Prevention of atrial fibrillation with angiotensin-converting enzyme inhibitors and angiotensin receptor blockers: a meta-analysis. J Am Coll Cardiol. 2005;45:1832–9. doi: 10.1016/j.jacc.2004.11.070. [DOI] [PubMed] [Google Scholar]
- 165.Rosenson RS. Statins in atherosclerosis: lipid-lowering agents with antioxidant capabilities. Atherosclerosis. 2004;173:1–12. doi: 10.1016/S0021-9150(03)00239-9. [DOI] [PubMed] [Google Scholar]
- 166.Lozano HF, Conde CA, Florin T, Lamas GA. Treatment and prevention of atrial fibrillation with non-antiarrhythmic pharmacologic therapy. Heart Rhythm. 2005;2:1000–7. doi: 10.1016/j.hrthm.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 167.Liu T, Li GP. Statins may prevent postoperative atrial fibrillation through autonomic modulation. Am J Cardiol. 2006;97:1266. doi: 10.1016/j.amjcard.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 168.Nassief A, Marsh JD. Statin therapy for stroke prevention. Stroke. 2008;39:1042–8. doi: 10.1161/STROKEAHA.107.501361. [DOI] [PubMed] [Google Scholar]
- 169.Cholesterol, diastolic blood pressure, and stroke: 13,000 strokes in 450,000 people in 45 prospective cohorts. Prospective studies collaboration. Lancet. 1995;346:1647–53. [PubMed] [Google Scholar]
- 170.Iso H, Jacobs DR, Jr, Wentworth D, Neaton JD, Cohen JD. Serum cholesterol levels and six-year mortality from stroke in 350,977 men screened for the multiple risk factor intervention trial. N Engl J Med. 1989;320:904–10. doi: 10.1056/NEJM198904063201405. [DOI] [PubMed] [Google Scholar]
- 171.Amarenco P, Labreuche J, Lavallee P, Touboul PJ. Statins in stroke prevention and carotid atherosclerosis: systematic review and up-to-date meta-analysis. Stroke. 2004;35:2902–9. doi: 10.1161/01.STR.0000147965.52712.fa. [DOI] [PubMed] [Google Scholar]
- 172.Downs JR, Clearfield M, Weis S, Whitney E, Shapiro DR, Beere PA, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. Jama. 1998;279:1615–22. doi: 10.1001/jama.279.20.1615. [DOI] [PubMed] [Google Scholar]
- 173.Sever PS, Dahlof B, Poulter NR, Wedel H, Beevers G, Caulfield M, et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial--Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet. 2003;361:1149–58. doi: 10.1016/S0140-6736(03)12948-0. [DOI] [PubMed] [Google Scholar]
- 174.Colhoun HM, Betteridge DJ, Durrington PN, Hitman GA, Neil HA, Livingstone SJ, et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet. 2004;364:685–96. doi: 10.1016/S0140-6736(04)16895-5. [DOI] [PubMed] [Google Scholar]
- 175.Strippoli GF, Navaneethan SD, Johnson DW, Perkovic V, Pellegrini F, Nicolucci A, et al. Effects of statins in patients with chronic kidney disease: meta-analysis and meta-regression of randomised controlled trials. Bmj. 2008;336:645–51. doi: 10.1136/bmj.39472.580984.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sandhu S, Wiebe N, Fried LF, Tonelli M. Statins for improving renal outcomes: a meta-analysis. J Am Soc Nephrol. 2006;17:2006–16. doi: 10.1681/ASN.2006010012. [DOI] [PubMed] [Google Scholar]
- 177.Sena A, Pedrosa R, Graca Morais M. Therapeutic potential of lovastatin in multiple sclerosis. J Neurol. 2003;250:754–5. doi: 10.1007/s00415-003-1070-8. [DOI] [PubMed] [Google Scholar]
- 178.Vollmer T, Key L, Durkalski V, Tyor W, Corboy J, Markovic-Plese S, et al. Oral simvastatin treatment in relapsing-remitting multiple sclerosis. Lancet. 2004;363:1607–8. doi: 10.1016/S0140-6736(04)16205-3. [DOI] [PubMed] [Google Scholar]