

Abstract
Atherosclerosis is a complex inflammatory process characterized by the cross-talk between excessive inflammation and lipid accumulation. In the past few years, compelling evidence suggests that statins can decrease vascular inflammation and attenuate the development of atherosclerosis through their so-called “pleiotropic effects”. These cholesterol-independent effects are predominantly due to their ability to inhibit isoprenoid synthesis. In particular, inhibition of geranylgeranylpyrophosphate synthesis leads to inhibition of Rho and its downstream target, Rho-kinase (ROCK). Thus, one of the beneficial effects of statin therapy could be due to inhibitory effects on ROCK. ROCK is involved in mediating diverse cellular functions such as smooth muscle contraction, cell migration and proliferation. While increased ROCK activity is associated with endothelial dysfunction, cerebral ischemia, coronary vasospasms and metabolic syndrome, the inhibition of ROCK by statins or selective ROCK inhibitors leads to up-regulation of endothelial nitric oxide synthase (eNOS), decreased vascular inflammation, and reduced atherosclerotic plaque formation. This review will focus on the impact of ROCK in cardiovascular disease and its contributory role to vascular inflammation and the atherosclerosis.
Keywords: Rho-kinase, inflammation, atherosclerosis, statin
Introduction
Atherosclerosis is a complex inflammatory process that is characterized by the cross-talk between excessive inflammation and lipid accumulation [1]. Its development is partly initiated by local endothelial cell dysfunction leading to the activation of endothelial cells and recruitment of proinflammatory cells. Local inflammation then promotes the adhesion of leukocytes and recruitment of activated platelets to the damaged endothelium, leading to increased permeability of blood vessels for lipid components in the plasma [2]. Subsequently, monocytes that are loaded with cell-activating lipids, accumulate in the arterial intima and acquire the morphological characteristics of macrophages leading to the transformation into foam cells [3,4]. Following the accumulation of additional inflammatory cell subsets and extracellular lipids, these early plaques, also known as fatty streaks progress into mature atherosclerotic plaques. Plaque cells promote their own growth by secreting cytokines and growth factors resulting in further deposition of extracellular matrix components and progression of plaques and stenosis. At the same time, matrix-degrading proteases and cytokines are secreted by plaque cells and result in thinning of the fibrous cap, eventually leading to disintegration of the cap and plaque erosion [5].
In the past few years, statins have been shown to prevent or reduce atherosclerosis. In particular, statins have been shown to modulate immune activation by decreasing the number of inflammatory cells in atherosclerotic plaques and to contribute to plaque stability by reducing plaque size or by modifying the molecular composition of the lipid core [6]. Statins exert their extrahepatic effects through its ability to prevent the synthesis of other important isoprenoid intermediates of the cholesterol biosynthetic pathway, such as farnesylpyrophosphate (FPP) and geranylgeranylpyrophosphate (GGPP). As a results, recent research have shifted the focus to isoprenoid intermediates, especially since they serve as important lipid attachments for the post-translational modification of intracellular signaling molecules, such as Rho, Rac and Cdc42 [7]. In particular, the inhibition of Rho and its downstream target, Rho-associated coiled-coil forming protein kinase (ROCK), has emerged as the principle mechanisms, which contributes to the pleiotropic effects of statins.
Rho KINASE
Rho kinases (ROCKs) are protein serine/threonine kinases of 160 kDa and are downstream effectors of the small GTPase Rho [8]. They were initially characterized by their ability to mediate the formation of RhoA-induced stress fibers and focal adhesions through increasing the phosphorylation of myosin light chain (MLC) [9]. ROCKs consist of an amino-terminal kinase domain, followed by a mid-coiled-coil-forming lesion containing a Rho-binding domain (RBD), and a carboxy-terminal cysteine-rich domain (CRD) located within the pleckstrin homology (PH) motif. The two isoforms ROCK1 and ROCK2 share a 65% homology in their amino acid sequence and 92% homology in their kinase domains [8]. The carboxy-terminal regions of ROCKs, which contain the PH domain and the RBD, serve as an autoregulatory inhibitor of the amino-terminal kinase domain [10]. The interaction of GTP-bound RhoA to the RBD of ROCKs increases ROCK activity through repression of the carboxy-terminal RBD-PH domains on the amino-terminal kinase domain, leading to an active “open” kinase conformation. This open conformation can also be formed by the binding of arachidonic acid to the PH domain or by cleavage of the carboxy terminus by caspase-3 [11-13]. Interestingly, ROCKs can also be activated independently of RhoA through amino-terminal transphosphorylation caused by protein oligomerization. Other small GTP-binding proteins such as Gem and Rad specifically regulate either ROCK1 or ROCK2-mediated cell rounding and neurite retraction [14]. Although further studies are needed to uncover the precise mechanism, these results indicate that ROCK1 and ROCK2 may have different physiological roles in cellular function.
ROCKs are important regulators of cellular apoptosis, growth, metabolism and migration via control of the actin cytoskeletal assembly and cell contraction. Stimulation of tyrosine kinase and G protein-coupled receptors recruits and activates Rho GEFs, leading to activation of RhoA. ROCKs are pivotal downstream effectors of RhoA in regulating the actin cytoskeleton by phosphorylation and inhibition of MLCP, which increases MLC phosphorylation and cellular contraction. By affecting tight and adherent junctions through actin cytoskeletal contractions, ROCKs can also regulate macrophage phagocytic activity and endothelial cell permeability.
Although ROCK1 and ROCK2 are ubiquitously expressed in mouse tissues from early embryonic development to adulthood, ROCK1 mRNA is preferentially expressed in lung, liver, spleen, kidney and testis, whereas ROCK2 mRNA is highly expressed in the heart, skeletal muscle, adipose tissue, and brain [15-17]. Growing evidence suggests a pivotal role for ROCK in the pathophysiology of cardiovascular diseases, such as hypertension, myocardial hypertrophy, cerebral ischemia, neointima formation and atherosclerosis (Fig. 1). The emergence of this linkage coincides with the growing acceptance of the pleiotropic effects of statins, as a therapeutic ROCK inhibitor. Indeed, it has become increasingly apparent that the overall benefits observed with statins are not mediated solely by their lipid-lowering properties, but by a cascade of cholesterol-independent or pleiotropic effects [18,19].
Fig. 1. Biological actions of ROCK in the vasculature.
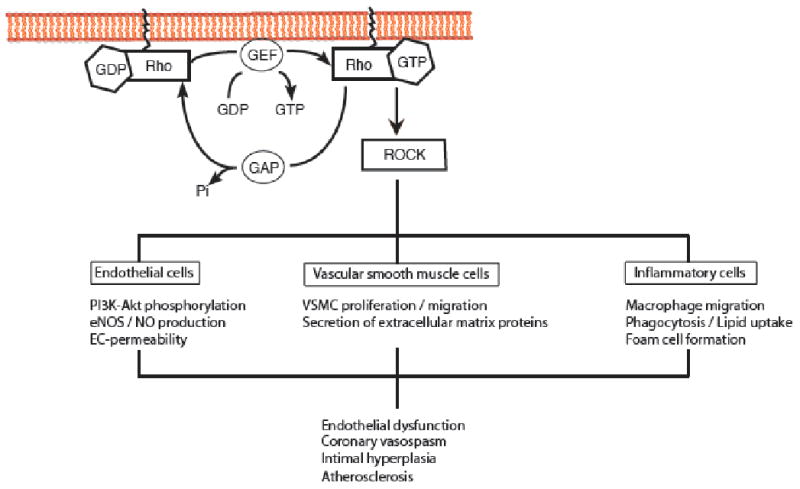
In endothelial cells the inhibition of ROCK leads to a rapid phosphorylation and activation of PI3K/Akt resulting in increased production of NO. In vascular smooth muscle cells ROCK inhibition regulates cell migration and proliferation and is involved in the pathomechanism of vascular inflammation and injury. Finally, the inhibition of ROCK either pharmacologically or genetically prevents the development of atherosclerosis by inhibition altered chemotaxis of macrophages and its transformation into foam cells. (Adapted from Wang et al. [97]).
Statins and ROCK
Statins have emerged as the leading therapeutic class of lipid lowering agents and are established therapy in the primary and secondary prevention of coronary artery diseases. As potent competitive inhibitors of the 3-hydroxy-methylglutaryl coenzyme A (HMG-CoA) reductase, statins bind to the enzyme's active site and block the substrate-product transition state of the enzyme [20,21]. However, in contrast to the original rationale of the biological effect of statins, it has become increasingly apparent that the overall benefits observed with statins are not mediated solely by their lipid-lowering properties, but by cholesterol independent or pleiotropic effects [18,19]. Indeed, statins prevent the synthesis of other important isoprenoid intermediates of the cholesterol biosynthetic pathway, such as farnesylpyrophosphate (FPP) and geranylgeranylpyrophosphate (GGPP) that are downstream from L-mevalonic acid [22]. These intermediates serve as important lipid attachments for the post-translational modification of proteins, including nuclear lamins, Ras, Rho, Rac and Rap [7]. Through posttranslational modifications, isoprenylation is critical for intracellular trafficking and function of small GTP-binding proteins [23]. In particular, by inhibiting mevalonate synthesis, statins prevent membrane targeting of Rho and its subsequent activation of ROCK. Indeed, in-vitro studies suggest that many of the pleiotropic effects of statins are due to alterations in the RhoA/ROCK signaling pathways [24-26]. For example, similar to the effects of statins, the administration of ROCK inhibitors has been shown to prevent cerebral vasospasm after subarachnoidal hemorrhage [27] and to prevent arterial remodeling after vascular injury [28].
The concept of statin pleiotropy is still controversial, because it has been difficult to separate the cholesterol-lowering effects of statins from their pleiotropic effects in humans. Previous data indicate that statin pleiotropy on endothelial function and inflammation appears to be dose related. Recently, a new cholesterol inhibitor ezetimibe, which inhibits intestinal cholesterol absorption, has been shown to reduce cholesterol by 15-20% when used alone [29]. If used in the so-called dual therapy, i.e. in conjunction with statins, it can enhance the cholesterol-lowering effect of statins up to 40% [30]. Comparing high-dose statin mono-therapy with equivalent cholesterol-lowering efficacy of the same statin at a lower dose plus ezetimibe, our group has recently shown that high-dose simvastatin alone improves endothelial function as assessed by decreased ROCK activity and increased flow-mediated dilation (FMD), more than dual therapy of ezetimibe and low-dose simvastatin [31]. This finding underlines the concept that inhibition of ROCK contributes to some of the pleiotropic effects of statins therapy and also supports the hypothesis that ROCK inhibition may elicit protective effects on the cardiovascular system.
ROCK in the Vasculature
ROCK and Endothelial Cells
The vascular endothelium forms the inner lining of blood vessels and serves as a physical barrier in the healthy vasculature. In addition, the endothelium is also a secreting organ, releasing vasoactive substances such as NO, prostacyclin, endothelium-derived hyperpolarizing factor and endothelins [32]. Endothelial dysfunction, defined by decreased bioavailability of endothelium-derived nitric oxide (eNOS), is one of the earliest manifestations of atherosclerosis [33,34]. In particular, eNOS plays an important role in the regulation of vascular tone, inhibition of platelet aggregation [35], suppression of smooth muscle cell proliferation [36] and prevention of leukocyte recruitment to the vessel wall [37]. Increased bioavailability of NO is partly dependent on increased expression and activity of eNOS as well as on decreased inactivation of NO by reactive oxygen species (ROS). Although various conditions and factors such as laminar shear stress, oxygen tension and TGFβ can regulate eNOS expression at the transcriptional level, eNOS expression also can be regulated at the posttranscriptional level. For example, chronic hypoxia, tumor necrosis factor (TNF), thrombin, oxLDL and cellular proliferation are known to decrease eNOS mRNA stability. Chronic hypoxia and cellular proliferation are known to activate RhoA and ROCK. In contrast, statins, which have been shown to increase eNOS mRNA stability, inhibit RhoA geranyl-geranylation and ROCK activity. Thus, RhoA/ROCK inversely regulates eNOS expression through alteration in eNOS mRNA stability [24,38].
The involvement of ROCK in the regulation of endothelial nitric oxide synthase (NOS) has been profoundly demonstrated in several studies. In human endothelial cells, ROCK negatively regulates phosphorylation of eNOS through inhibition of protein kinase B/Akt [39]. Moreover, inhibition of ROCK leads to a rapid phosphorylation and activation of Akt via the phosphatidylinositol 3-kinase (PI3K), leading to increased NO production [40]. These data suggest an important role of ROCK in the regulation of eNOS in the peripheral circulation of healthy subjects. Previous data in human umbilical vein endothelial cells (HUVEC) and human coronary artery endothelial cells (HCAE) have demonstrated that native lipoproteinA (Lp(a)) elicits re-arrangement of the actin cytoskeleton through its apo(a) component. This re-arrangement is characterized by increased central stress fiber formation, dispersion of vascular endothelial (VE)-cadherin, and increased cell permeability, whereas treatment with LDL or plasminogen had no effect [41]. Interestingly, this effect was mediated by increased MLC phosphorylation through a ROCK-dependent signaling pathway. In a follow-up study the same group has identified that the strong lysine binding sites (LBS) in apo(a) mutants has a key functional role in mediating a Rho/ROCK/MYPT1 signaling transduction pathway to enhance MLC phosphorylation via inactivation of MLCP, which thereby increases endothelial cell contraction and permeability [42].
ROCK and Vascular Smooth Muscle Cells
In vascular smooth muscle cells (VSMC), the Rho/Rho kinase system is involved in proliferation and migration [43,44]. Furthermore, ROCK mediates angiotensin II-induced expression of monocyte chemoattractant protein-1 (MCP-1) [45] and plasminogen activator inhibitor-1(PAI-1) [46]. In addition several in-vivo studies have identified ROCK activation in experimental models of vascular inflammation or injury [47-49]. In terms of atherogenesis, smooth muscle cells (SMC) from the vessel wall respond to growth factors and migrate and proliferate throughout the intima [50]. Besides the production of collagen and other extracellular matrix proteins, SMCs secrete vascular endothelial growth factor, TNF-α, IL-1 and other pro-inflammatory molecules [51]. Fibrous tissue and proliferating SMCs overlay the mature lipid core, which becomes rich in necrotic debris. The phenotype of SMC migration and proliferation is a critical component of plaque stability, and, interestingly, the lack of SMCs may paradoxically increase the occurrence of arterial thrombosis [52].
The involvement of ROCK in vascular inflammation and remodeling has been demonstrated in several studies. In L-NAME treated rats, ROCK inhibitors attenuate the inflammatory response and vascular remodeling [48,53]. In addition, ROCK activity is increased in the neointima following balloon-induced vascular injury, which is suppressed by ROCK inhibitors or gene transfer of a dominant-negative mutant of ROCK [28,49,54]. Most of these studies however, utilized pharmacological inhibitors of the Rho kinase: Fasudil or Y-27632. Both inhibitors function through inhibition of the ATP-dependent kinase domain, which is highly homologous between the two ROCK iso-forms [55,56]. Therefore, neither Fasudil, nor Y-27632 can distinguish between cellular processes mediated by ROCK1 and ROCK2. Furthermore, when applied in-vivo for prolonged periods and at higher concentrations, these pharmacological inhibitors could also inhibit other serine-threonine kinases such as PKA and PKC.
Recently, several groups have successfully generated mutant mice with deletion of the ROCK1 and ROCK2 allele. While mice harboring homozygous deletion of both ROCK1 or ROCK2 alleles are embryonic and postnatal lethal, haploinsufficient mice are fertile and phenotypically normal [57-60]. Investigating the effect of ROCK on neointima formation after vascular injury, we have found that VSMC proliferation was decreased in the neointima of ROCK1+/- mice following carotid artery ligation [61]. Interestingly, VSMC proliferation in response to serum or PDGF was not different between wildtype and ROCK1+/- mice, while the migration of VSMC in response to PDGF was substantially reduced in ROCK1+/- mice, suggesting that ROCK1 may contribute to increase VSMC migration and survival following vascular injury.
ROCK and Atherosclerosis
Pharmacological inhibition of ROCK activity has been demonstrated to protect against atherosclerosis. In a porcine model, long-term inhibition of Rho-kinase results in a regression of arteriosclerotic coronary lesions [62]. In LDLr-/- mice on high-fat diet, application of the Rho kinase inhibitor Y-27632 significantly reduced atherosclerotic lesion size by 35% compared to control mice fed with high-fat diet [63,64]. Interestingly, the expression of macrophage, smooth muscle cell and collagen in the plaque did not differ between the Y-27632 treated and saline treated animals. But instead, the number of CD3-positive T-lymphocytes per lesion was reduced by 40% in Y-27632 treated mice. The authors conclude that Rho kinase may have direct effects on T-lymphocytes and indeed, Y-27632 was found to exert a profound inhibitory and dose-dependent effect on conA-induced proliferation of spleen-derived T-lymphocytes. In accordance to this study, several other studies have previously demonstrated modulatory role of the Rho/Rho kinase system in T-lymphocyte homeostasis [64-66]. In another study with ApoE knockout mice treated with or without ROCK inhibitor, a cell type-selective distribution and phosphorylation of ROCK target proteins ERM and MLC could be demonstrated [67]. Y-27632 inhibited ERM phosphorylation in the plaque, but, however, failed to demonstrate dose dependence. MLC and phospho MLC were associated with SMC and did not respond to the Y-27632 treatment.
A recent study from our group showed that macrophage ROCK1 is involved in the pathogenesis for atherosclerosis [68]. Transplantation of bone marrow-derived macrophages from ROCK1-/- mice to LDLr-/- mice demonstrates that ROCK1 in bone marrow-derived macrophages is critical to the development of atherosclerosis. Specifically, lipid accumulation and atherosclerotic lesions were reduced in atherosclerosis-prone LDLr-/- mice, whose bone marrows have been replaced with bone marrows derived from ROCK1-/- mice. Indeed, bone marrow-derived ROCK1-deficient macrophages exhibited impaired chemotaxis to MCP-1, reduced ability in lipid uptake and subsequent transformation into foam cells. These findings indicate that bone marrow-derived ROCK1 may be an important therapeutic target for vascular inflammation and atherosclerosis. However, it remains to be determined whether and how ROCK2 is involved in this system. There is evidence that ROCK1 expression rather than ROCK2 is up-regulated upon macrophage adhesion [69]. At the same time, a study by Yoneda et al. showed that phagocytic uptake of fibronectin-coated beads was down-regulated in ROCK2-depleted cells, but not in ROCK1-depleted cells [70].
As mentioned above, endothelial permeability is controlled by a variety of chemical stimuli and abrogation of the integrity of the endothelial monolayer contribute to pathological conditions such as atherosclerosis. Several studies have demonstrated the importance of Rho family GTPases in the controlling and maintaining of endothelial barrier function [71,72]. Also, extravasation of leukocytes has been shown to induce stress fiber formation and increased MLC phosporylation and increased endothelial permeability [73,74].
Clinical Implication of ROCK and ROCK Inhibitors
ROCK has undoubtedly attracted significant interests as a potential target for the treatment of cardiovascular disease and numerous clinical studies have already demonstrated a linkage for ROCK and cardiovascular disease in human. In Japan, the isoquinoline derivative fasudil has been used in clinics since 1995 and proven to be successful in preventing vasospasm associated with subarachnoid hemorrhage (SAH) [75], acute ischemic stroke [76], angina pectoris [77,78], coronary artery spasm [79,80], pulmonary arterial hypertension [81] and atherosclerosis [82,83].
ROCK in Endothelial Dysfunction
A significant relationship between endothelial dysfunction and increased ROCK activity in young, current smokers has been previously demonstrated, showing a significant relationship between ROCK activity with age, systolic blood pressure, serum concentration of total cholesterol, and number of pack-years smoked in healthy male subjects [84,85]. In a multivariate analysis the authors demonstrated that age and number of pack-years smoked were independent predictors of ROCK activity among these candidates. In addition, the concentration of serum malondialdehyde-modified (MDA)-LDL, a marker for oxidative stress and impaired aortic stiffness as assessed by pulse wave velocity (PWV) also correlated with ROCK activity [83]. Indeed, oxidative stress has been linked with impaired aortic stiffness and has been shown to be an independent marker to estimate subjects with cardiovascular disease [86-89]. Furthermore, our group has previously demonstrated a correlation between elevated ROCK activity and impaired endothelial function in coronary artery disease (CAD) patients [82]. Treatment with the ROCK inhibitor fasudil reduced the over-activity of ROCK in patients with atherosclerosis and improved endothelium-dependent vasodilation as well as flow-mediated, endothelium-dependent dilation. Of note, this finding was only present in patients with CAD, but not in healthy individuals, where ROCK is presumably not “overactive”. Most interestingly, endothelium-dependent vasodilation in healthy subjects tended to worsen with fasudil therapy compared with placebo.
In animal models, Rho expression is regulated by a negative feedback mechanism mediated by the actin cytoskeleton [90]. Under physiological conditions, such as in healthy individuals in whom basal ROCK activity is not up-regulated, inhibition of ROCK may lead to decreased negative feedback and thus to increased transcription of Rho, which in turn may lead to a subsequent compensatory increase in the downstream effects of Rho including suppression of endothelial NO production. On the other hand, ROCK inhibition in healthy individuals may also lead to an excess of NO production, resulting in the formation of peroxynitrite, which might lead to eNOS uncoupling and worsening endothelial function [91]. From our clinical study, it appears that some basal ROCK activity is probably required for the maintenance of vascular homeostasis.
ROCK in Metabolic Syndrome
In a previous paper by Liu et al., a correlation between ROCK activity and metabolic syndrome in the Taiwanese population has been demonstrated [92]. The authors showed that increased levels of ROCK activity independently predicted the diagnosis of metabolic syndrome, and that in addition, higher ROCK activity was also associated with greater body mass index (BMI), waist circumference, greater hs-CRP as well as lower adiponectin levels. This clinical finding is supported by several in vitro studies, where increased ROCK is involved in the phosphorylation of insulin receptor substrate (IRS)-1, which prevents insulin signaling and glucose transporter-4 translocation leading to altered glucose metabolism [93-95]. Interestingly, the insulin sensitivity is ameliorated by ROCK inhibition in obese, but not in lean rats, supporting the clinical finding of elevated ROCK activity in obese patients [93]. Also, ROCK has been demonstrated to mediate high glucose-induced NF-κB activation, resulting in PAI-1 expression in endothelial cells [96].
Conclusions
Growing evidence from animal and clinical studies suggest the importance of ROCK in the pathogenesis of cardiovascular disease. Indeed, many of the pleiotropic or cholesterol-independent effects of statins may be due to their ability to block isoprenoid synthesis and inhibit the Rho/ROCK pathway. Thus, targeting ROCK with specific inhibitors may be an important therapeutic strategy in the prevention and treatment of cardiovascular disease in patients with normal or low lipid levels. As ROCKs are involved in various aspects of vascular function and disease, understanding their role in the vascular wall may provide key insights into how the vasculature as a whole is regulated under normal and pathophysiological conditions. However, despite a growing number of reports demonstrating that ROCK activity is increased under a variety of pathologic conditions, little is known regarding the molecular mechanisms that contribute to increased ROCK activity or what the downstream targets for ROCK are. Also, determining the precise role of ROCK in the vascular wall for each isoform will certainly provide more insights into the molecular function of ROCK.
Acknowledgments
This work is supported by grants from the National Institutes of Health (HL052233 and HL080187) to JK. Liao and Deutsche Forschungsgemeinschaft (ZH 231/1-1) to Q. Zhou.
References
- 1.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–9. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 2.Langer HF, Gawaz M. Platelet-vessel wall interactions in atherosclerotic disease. Thromb Haemost. 2008;99:480–6. doi: 10.1160/TH07-11-0685. [DOI] [PubMed] [Google Scholar]
- 3.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–74. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 4.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–43. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 5.Aukrust P, Halvorsen B, Yndestad A, Ueland T, Oie E, Otterdal K, et al. Chemokines and cardiovascular risk. Arterioscler Thromb Vasc Biol. 2008;28:1909–19. doi: 10.1161/ATVBAHA.107.161240. [DOI] [PubMed] [Google Scholar]
- 6.Fukumoto Y, Libby P, Rabkin E, Hill CC, Enomoto M, Hirouchi Y, et al. Statins alter smooth muscle cell accumulation and collagen content in established atheroma of watanabe heritable hyperlipidemic rabbits. Circulation. 2001;103:993–9. doi: 10.1161/01.cir.103.7.993. [DOI] [PubMed] [Google Scholar]
- 7.Van Aelst L, D'Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 1997;11:2295–322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- 8.Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4:446–56. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
- 9.Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–79. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 10.Amano M, Chihara K, Nakamura N, Kaneko T, Matsuura Y, Kaibuchi K. The COOH terminus of Rho-kinase negatively regulates rho-kinase activity. J Biol Chem. 1999;274:32418–24. doi: 10.1074/jbc.274.45.32418. [DOI] [PubMed] [Google Scholar]
- 11.Feng J, Ito M, Ichikawa K, Isaka N, Nishikawa M, Hartshorne DJ, et al. Inhibitory phosphorylation site for Rho-associated kinase on smooth muscle myosin phosphatase. J Biol Chem. 1999;274:37385–90. doi: 10.1074/jbc.274.52.37385. [DOI] [PubMed] [Google Scholar]
- 12.Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol. 2001;3:339–45. doi: 10.1038/35070009. [DOI] [PubMed] [Google Scholar]
- 13.Sebbagh M, Renvoize C, Hamelin J, Riche N, Bertoglio J, Breard J. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat Cell Biol. 2001;3:346–52. doi: 10.1038/35070019. [DOI] [PubMed] [Google Scholar]
- 14.Ward Y, Yap SF, Ravichandran V, Matsumura F, Ito M, Spinelli B, et al. The GTP binding proteins Gem and Rad are negative regulators of the Rho-Rho kinase pathway. J Cell Biol. 2002;157:291–302. doi: 10.1083/jcb.200111026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Cunto F, Imarisio S, Hirsch E, Broccoli V, Bulfone A, Migheli A, et al. Defective neurogenesis in citron kinase knockout mice by altered cytokinesis and massive apoptosis. Neuron. 2000;28:115–27. doi: 10.1016/s0896-6273(00)00090-8. [DOI] [PubMed] [Google Scholar]
- 16.Nakagawa O, Fujisawa K, Ishizaki T, Saito Y, Nakao K, Narumiya S. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. 1996;392:189–93. doi: 10.1016/0014-5793(96)00811-3. [DOI] [PubMed] [Google Scholar]
- 17.Wei L, Roberts W, Wang L, Yamada M, Zhang S, Zhao Z, et al. Rho kinases play an obligatory role in vertebrate embryonic organogenesis. Development. 2001;128:2953–62. doi: 10.1242/dev.128.15.2953. [DOI] [PubMed] [Google Scholar]
- 18.Maron DJ, Fazio S, Linton MF. Current perspectives on statins. Circulation. 2000;101:207–13. doi: 10.1161/01.cir.101.2.207. [DOI] [PubMed] [Google Scholar]
- 19.Palinski W. New evidence for beneficial effects of statins unrelated to lipid lowering. Arterioscler Thromb Vasc Biol. 2001;21:3–5. doi: 10.1161/01.atv.21.1.3. [DOI] [PubMed] [Google Scholar]
- 20.Alberts AW, Chen J, Kuron G, Hunt V, Huff J, Hoffman C, et al. Mevinolin: a highly potent competitive inhibitor of hydroxymethyl-glutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc Natl Acad Sci USA. 1980;77:3957–61. doi: 10.1073/pnas.77.7.3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Istvan ES, Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science. 2001;292:1160–4. doi: 10.1126/science.1059344. [DOI] [PubMed] [Google Scholar]
- 22.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–30. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 23.Hall A. G proteins and small GTPases: distant relatives keep in touch. Science. 1998;280:2074–5. doi: 10.1126/science.280.5372.2074. [DOI] [PubMed] [Google Scholar]
- 24.Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97:1129–35. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- 25.Laufs U, Marra D, Node K, Liao JK. 3-Hydroxy-3-methylglutaryl-CoA reductase inhibitors attenuate vascular smooth muscle proliferation by preventing rho GTPase-induced down-regulation of p27(Kip1) J Biol Chem. 1999;274:21926–31. doi: 10.1074/jbc.274.31.21926. [DOI] [PubMed] [Google Scholar]
- 26.Mundy G, Garrett R, Harris S, Chan J, Chen D, Rossini G, et al. Stimulation of bone formation in vitro and in rodents by statins. Science. 1999;286:1946–9. doi: 10.1126/science.286.5446.1946. [DOI] [PubMed] [Google Scholar]
- 27.Shibuya M, Suzuki Y, Sugita K, Saito I, Sasaki T, Takakura K, et al. Effect of AT877 on cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Results of a prospective placebo-controlled double-blind trial. J Neurosurg. 1992;76:571–7. doi: 10.3171/jns.1992.76.4.0571. [DOI] [PubMed] [Google Scholar]
- 28.Sawada N, Itoh H, Ueyama K, Yamashita J, Doi K, Chun TH, et al. Inhibition of rho-associated kinase results in suppression of neointimal formation of balloon-injured arteries. Circulation. 2000;101:2030–3. doi: 10.1161/01.cir.101.17.2030. [DOI] [PubMed] [Google Scholar]
- 29.Sudhop T, Lutjohann D, Kodal A, Igel M, Tribble DL, Shah S, et al. Inhibition of intestinal cholesterol absorption by ezetimibe in humans. Circulation. 2002;106:1943–8. doi: 10.1161/01.cir.0000034044.95911.dc. [DOI] [PubMed] [Google Scholar]
- 30.Gagne C, Gaudet D, Bruckert E. Efficacy and safety of ezetimibe coadministered with atorvastatin or simvastatin in patients with homozygous familial hypercholesterolemia. Circulation. 2002;105:2469–75. doi: 10.1161/01.cir.0000018744.58460.62. [DOI] [PubMed] [Google Scholar]
- 31.Liu PY, Liu YW, Lin LJ, Chen JH, Liao JK. Evidence for Statin Pleiotropy in Humans. Differential Effects of Statins and Ezetimibe on Rho-Associated Coiled-Coil Containing Protein Kinase Activity, Endothelial Function, and Inflammation. Circulation. 2008 doi: 10.1161/CIRCULATIONAHA.108.813311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. 2007;100:158–73. doi: 10.1161/01.RES.0000255691.76142.4a. [DOI] [PubMed] [Google Scholar]
- 33.Werns SW, Walton JA, Hsia HH, Nabel EG, Sanz ML, Pitt B. Evidence of endothelial dysfunction in angiographically normal coronary arteries of patients with coronary artery disease. Circulation. 1989;79:287–91. doi: 10.1161/01.cir.79.2.287. [DOI] [PubMed] [Google Scholar]
- 34.Steinberg D, Parthasarathy S, Carew TE, Khoo JC, Witztum JL. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N Engl J Med. 1989;320:915–24. doi: 10.1056/NEJM198904063201407. [DOI] [PubMed] [Google Scholar]
- 35.Yao SK, Ober JC, Krishnaswami A, Ferguson JJ, Anderson HV, Golino P, et al. Endogenous nitric oxide protects against platelet aggregation and cyclic flow variations in stenosed and endothelium-injured arteries. Circulation. 1992;86:1302–9. doi: 10.1161/01.cir.86.4.1302. [DOI] [PubMed] [Google Scholar]
- 36.Ignarro LJ, Buga GM, Wei LH, Bauer PM, Wu G, del Soldato P. Role of the arginine-nitric oxide pathway in the regulation of vascular smooth muscle cell proliferation. Proc Natl Acad Sci USA. 2001;98:4202–8. doi: 10.1073/pnas.071054698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zeiher AM, Fisslthaler B, Schray-Utz B, Busse R. Nitric oxide modulates the expression of monocyte chemoattractant protein 1 in cultured human endothelial cells. Circ Res. 1995;76:980–6. doi: 10.1161/01.res.76.6.980. [DOI] [PubMed] [Google Scholar]
- 38.Laufs U, Fata VL, Liao JK. Inhibition of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase blocks hypoxia-mediated down-regulation of endothelial nitric oxide synthase. J Biol Chem. 1997;272:31725–9. doi: 10.1074/jbc.272.50.31725. [DOI] [PubMed] [Google Scholar]
- 39.Ming XF, Viswambharan H, Barandier C, Ruffieux J, Kaibuchi K, Rusconi S, et al. Rho GTPase/Rho kinase negatively regulates endothelial nitric oxide synthase phosphorylation through the inhibition of protein kinase B/Akt in human endothelial cells. Mol Cell Biol. 2002;22:8467–77. doi: 10.1128/MCB.22.24.8467-8477.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wolfrum S, Dendorfer A, Rikitake Y, Stalker TJ, Gong Y, Scalia R, et al. Inhibition of Rho-kinase leads to rapid activation of phosphatidylinositol 3-kinase/protein kinase Akt and cardiovascular protection. Arterioscler Thromb Vasc Biol. 2004;24:1842–7. doi: 10.1161/01.ATV.0000142813.33538.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pellegrino M, Furmaniak-Kazmierczak E, LeBlanc JC, Cho T, Cao K, Marcovina SM, et al. The apolipoprotein(a) component of lipoprotein(a) stimulates actin stress fiber formation and loss of cell-cell contact in cultured endothelial cells. J Biol Chem. 2004;279:6526–33. doi: 10.1074/jbc.M309705200. [DOI] [PubMed] [Google Scholar]
- 42.Cho T, Jung Y, Koschinsky ML. Apolipoprotein(a), through its strong lysine-binding site in KIV(10′), mediates increased endothelial cell contraction and permeability via a Rho/Rho kinase/MYPT1-dependent pathway. J Biol Chem. 2008;283:30503–12. doi: 10.1074/jbc.M802648200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seasholtz TM, Majumdar M, Kaplan DD, Brown JH. Rho and Rho kinase mediate thrombin-stimulated vascular smooth muscle cell DNA synthesis and migration. Circ Res. 1999;84:1186–93. doi: 10.1161/01.res.84.10.1186. [DOI] [PubMed] [Google Scholar]
- 44.Sauzeau V, Le Mellionnec E, Bertoglio J, Scalbert E, Pacaud P, Loirand G. Human urotensin II-induced contraction and arterial smooth muscle cell proliferation are mediated by RhoA and Rho-kinase. Circ Res. 2001;88:1102–4. doi: 10.1161/hh1101.092034. [DOI] [PubMed] [Google Scholar]
- 45.Funakoshi Y, Ichiki T, Shimokawa H, Egashira K, Takeda K, Kaibuchi K, et al. Rho-kinase mediates angiotensin II-induced monocyte chemoattractant protein-1 expression in rat vascular smooth muscle cells. Hypertension. 2001;38:100–4. doi: 10.1161/01.hyp.38.1.100. [DOI] [PubMed] [Google Scholar]
- 46.Takeda K, Ichiki T, Tokunou T, Iino N, Fujii S, Kitabatake A, et al. Critical role of Rho-kinase and MEK/ERK pathways for angiotensin II-induced plasminogen activator inhibitor type-1 gene expression. Arterioscler Thromb Vasc Biol. 2001;21:868–73. doi: 10.1161/01.atv.21.5.868. [DOI] [PubMed] [Google Scholar]
- 47.Morishige K, Shimokawa H, Eto Y, Hoshijima M, Kaibuchi K, Takeshita A. In vivo gene transfer of dominant-negative rho-kinase induces regression of coronary arteriosclerosis in pigs. Ann N Y Acad Sci. 2001;947:407–11. doi: 10.1111/j.1749-6632.2001.tb03974.x. [DOI] [PubMed] [Google Scholar]
- 48.Kataoka C, Egashira K, Inoue S, Takemoto M, Ni W, Koyanagi M, et al. Important role of Rho-kinase in the pathogenesis of cardiovascular inflammation and remodeling induced by long-term blockade of nitric oxide synthesis in rats. Hypertension. 2002;39:245–50. doi: 10.1161/hy0202.103271. [DOI] [PubMed] [Google Scholar]
- 49.Shibata R, Kai H, Seki Y, Kato S, Morimatsu M, Kaibuchi K, et al. Role of Rho-associated kinase in neointima formation after vascular injury. Circulation. 2001;103:284–9. doi: 10.1161/01.cir.103.2.284. [DOI] [PubMed] [Google Scholar]
- 50.Bentzon JF, Weile C, Sondergaard CS, Hindkjaer J, Kassem M, Falk E. Smooth muscle cells in atherosclerosis originate from the local vessel wall and not circulating progenitor cells in ApoE knockout mice. Arterioscler Thromb Vasc Biol. 2006;26:2696–702. doi: 10.1161/01.ATV.0000247243.48542.9d. [DOI] [PubMed] [Google Scholar]
- 51.Doran AC, Meller N, McNamara CA. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:812–9. doi: 10.1161/ATVBAHA.107.159327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schwartz SM, Virmani R, Rosenfeld ME. The good smooth muscle cells in atherosclerosis. Curr Atheroscler Rep. 2000;2:422–9. doi: 10.1007/s11883-000-0081-5. [DOI] [PubMed] [Google Scholar]
- 53.Ikegaki I, Hattori T, Yamaguchi T, Sasaki Y, Satoh SI, Asano T, et al. Involvement of Rho-kinase in vascular remodeling caused by long-term inhibition of nitric oxide synthesis in rats. Eur J Pharmacol. 2001;427:69–75. doi: 10.1016/s0014-2999(01)01181-5. [DOI] [PubMed] [Google Scholar]
- 54.Eto Y, Shimokawa H, Hiroki J, Morishige K, Kandabashi T, Matsumoto Y, et al. Gene transfer of dominant negative Rho kinase suppresses neointimal formation after balloon injury in pigs. Am J Physiol Heart Circ Physiol. 2000;278:H1744–50. doi: 10.1152/ajpheart.2000.278.6.H1744. [DOI] [PubMed] [Google Scholar]
- 55.Sasaki Y, Suzuki M, Hidaka H. The novel and specific Rho-kinase inhibitor (S)-(+)-2-methyl-1-[(4-methyl-5-isoquinoline)sulfonyl]-homopiperazine as a probing molecule for Rho-kinase-involved pathway. Pharmacol Ther. 2002;93:225–32. doi: 10.1016/s0163-7258(02)00191-2. [DOI] [PubMed] [Google Scholar]
- 56.Ishizaki T, Uehata M, Tamechika I, Keel J, Nonomura K, Maekawa M, et al. Pharmacological properties of Y-27632, a specific inhibitor of rho-associated kinases. Mol Pharmacol. 2000;57:976–83. [PubMed] [Google Scholar]
- 57.Rikitake Y, Oyama N, Wang CY, Noma K, Satoh M, Kim HH, et al. Decreased perivascular fibrosis but not cardiac hypertrophy in ROCK1+/- haploinsufficient mice. Circulation. 2005;112:2959–65. doi: 10.1161/CIRCULATIONAHA.105.584623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rikitake Y, Kim HH, Huang Z, Seto M, Yano K, Asano T, et al. Inhibition of Rho kinase (ROCK) leads to increased cerebral blood flow and stroke protection. Stroke. 2005;36:2251–7. doi: 10.1161/01.STR.0000181077.84981.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shimizu Y, Thumkeo D, Keel J, Ishizaki T, Oshima H, Oshima M, et al. ROCK-I regulates closure of the eyelids and ventral body wall by inducing assembly of actomyosin bundles. J Cell Biol. 2005;168:941–53. doi: 10.1083/jcb.200411179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mukai Y, Rikitake Y, Shiojima I, Wolfrum S, Satoh M, Takeshita K, et al. Decreased vascular lesion formation in mice with inducible endothelial-specific expression of protein kinase Akt. J Clin Invest. 2006;116:334–43. doi: 10.1172/JCI26223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Noma K, Rikitake Y, Oyama N, Yan G, Alcaide P, Liu PY, et al. ROCK1 mediates leukocyte recruitment and neointima formation following vascular injury. J Clin Invest. 2008;118:1632–44. doi: 10.1172/JCI29226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shimokawa H, Morishige K, Miyata K, Kandabashi T, Eto Y, Ikegaki I, et al. Long-term inhibition of Rho-kinase induces a regression of arteriosclerotic coronary lesions in a porcine model in vivo. Cardiovasc Res. 2001;51:169–77. doi: 10.1016/s0008-6363(01)00291-7. [DOI] [PubMed] [Google Scholar]
- 63.Mallat Z, Gojova A, Sauzeau V, Brun V, Silvestre JS, Esposito B, et al. Rho-associated protein kinase contributes to early atherosclerotic lesion formation in mice. Circ Res. 2003;93:884–8. doi: 10.1161/01.RES.0000099062.55042.9A. [DOI] [PubMed] [Google Scholar]
- 64.Woodside DG, Wooten DK, McIntyre BW. Adenosine diphosphate (ADP)-ribosylation of the guanosine triphosphatase (GTPase) rho in resting peripheral blood human T lymphocytes results in pseudopodial extension and the inhibition of T cell activation. J Exp Med. 1998;188:1211–21. doi: 10.1084/jem.188.7.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vicente-Manzanares M, Rey M, Perez-Martinez M, Yanez-Mo M, Sancho D, Cabrero JR, et al. The RhoA effector mDia is induced during T cell activation and regulates actin polymerization and cell migration in T lymphocytes. J Immunol. 2003;171:1023–34. doi: 10.4049/jimmunol.171.2.1023. [DOI] [PubMed] [Google Scholar]
- 66.Tharaux PL, Bukoski RC, Rocha PN, Crowley SD, Ruiz P, Nataraj C, et al. Rho kinase promotes alloimmune responses by regulating the proliferation and structure of T cells. J Immunol. 2003;171:96–105. doi: 10.4049/jimmunol.171.1.96. [DOI] [PubMed] [Google Scholar]
- 67.Rekhter M, Chandrasekhar K, Gifford-Moore D, Huang XD, Rutherford P, Hanson J, et al. Immunohistochemical analysis of target proteins of Rho-kinase in a mouse model of accelerated atherosclerosis. Exp Clin Cardiol. 2007;12:169–74. [PMC free article] [PubMed] [Google Scholar]
- 68.Wang HW, Liu PY, Oyama N, Rikitake Y, Kitamoto S, Gitlin J, et al. Deficiency of ROCK1 in bone marrow-derived cells protects against atherosclerosis in LDLR-/- mice. Faseb J. 2008;22:3561–70. doi: 10.1096/fj.08-108829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fox R, Nhan TQ, Law GL, Morris DR, Liles WC, Schwartz SM. PSGL-1 and mTOR regulate translation of ROCK-1 and physiological functions of macrophages. Embo J. 2007;26:505–15. doi: 10.1038/sj.emboj.7601522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yoneda A, Multhaupt HA, Couchman JR. The Rho kinases I and II regulate different aspects of myosin II activity. J Cell Biol. 2005;170:443–53. doi: 10.1083/jcb.200412043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ridley AJ. Rho family proteins: coordinating cell responses. Trends Cell Biol. 2001;11:471–7. doi: 10.1016/s0962-8924(01)02153-5. [DOI] [PubMed] [Google Scholar]
- 72.van Nieuw Amerongen GP, Vermeer MA, van Hinsbergh VW. Role of RhoA and Rho kinase in lysophosphatidic acid-induced endothelial barrier dysfunction. Arterioscler Thromb Vasc Biol. 2000;20:E127–33. doi: 10.1161/01.atv.20.12.e127. [DOI] [PubMed] [Google Scholar]
- 73.Garcia JG, Verin AD, Schaphorst K, Siddiqui R, Patterson CE, Csortos C, et al. Regulation of endothelial cell myosin light chain kinase by Rho, cortactin, and p60(src) Am J Physiol. 1999;276:L989–98. doi: 10.1152/ajplung.1999.276.6.L989. [DOI] [PubMed] [Google Scholar]
- 74.Del Maschio A, Zanetti A, Corada M, Rival Y, Ruco L, Lampugnani MG, et al. Polymorphonuclear leukocyte adhesion triggers the disorganization of endothelial cell-to-cell adherens junctions. J Cell Biol. 1996;135:497–510. doi: 10.1083/jcb.135.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhao J, Zhou D, Guo J, Ren Z, Zhou L, Wang S, et al. Effect of fasudil hydrochloride, a protein kinase inhibitor, on cerebral vasospasm and delayed cerebral ischemic symptoms after aneurysmal subarachnoid hemorrhage. Neurol Med Chir (Tokyo) 2006;46:421–8. doi: 10.2176/nmc.46.421. [DOI] [PubMed] [Google Scholar]
- 76.Shibuya M, Hirai S, Seto M, Satoh S, Ohtomo E. Effects of fasudil in acute ischemic stroke: results of a prospective placebo-controlled double-blind trial. J Neurol Sci. 2005;238:31–9. doi: 10.1016/j.jns.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 77.Vicari RM, Chaitman B, Keefe D, Smith WB, Chrysant SG, Tonkon MJ, et al. Efficacy and safety of fasudil in patients with stable angina: a double-blind, placebo-controlled, phase 2 trial. J Am Coll Cardiol. 2005;46:1803–11. doi: 10.1016/j.jacc.2005.07.047. [DOI] [PubMed] [Google Scholar]
- 78.Fukumoto Y, Mohri M, Inokuchi K, Ito A, Hirakawa Y, Masumoto A, et al. Anti-ischemic effects of fasudil, a specific Rho-kinase inhibitor, in patients with stable effort angina. J Cardiovasc Pharmacol. 2007;49:117–21. doi: 10.1097/FJC.0b013e31802ef532. [DOI] [PubMed] [Google Scholar]
- 79.Masumoto A, Mohri M, Shimokawa H, Urakami L, Usui M, Takeshita A. Suppression of coronary artery spasm by the Rho-kinase inhibitor fasudil in patients with vasospastic angina. Circulation. 2002;105:1545–7. doi: 10.1161/hc1002.105938. [DOI] [PubMed] [Google Scholar]
- 80.Mohri M, Shimokawa H, Hirakawa Y, Masumoto A, Takeshita A. Rho-kinase inhibition with intracoronary fasudil prevents myocardial ischemia in patients with coronary microvascular spasm. J Am Coll Cardiol. 2003;41:15–9. doi: 10.1016/s0735-1097(02)02632-3. [DOI] [PubMed] [Google Scholar]
- 81.Fukumoto Y, Matoba T, Ito A, Tanaka H, Kishi T, Hayashidani S, et al. Acute vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients with severe pulmonary hypertension. Heart. 2005;91:391–2. doi: 10.1136/hrt.2003.029470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nohria A, Grunert ME, Rikitake Y, Noma K, Prsic A, Ganz P, et al. Rho kinase inhibition improves endothelial function in human subjects with coronary artery disease. Circ Res. 2006;99:1426–32. doi: 10.1161/01.RES.0000251668.39526.c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Noma K, Goto C, Nishioka K, Jitsuiki D, Umemura T, Ueda K, et al. Roles of rho-associated kinase and oxidative stress in the pathogenesis of aortic stiffness. J Am Coll Cardiol. 2007;49:698–705. doi: 10.1016/j.jacc.2006.06.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Noma K, Higashi Y, Jitsuiki D, Hara K, Kimura M, Nakagawa K, et al. Smoking activates rho-kinase in smooth muscle cells of forearm vasculature in humans. Hypertension. 2003;41:1102–5. doi: 10.1161/01.HYP.0000067062.92836.9E. [DOI] [PubMed] [Google Scholar]
- 85.Noma K, Goto C, Nishioka K, Hara K, Kimura M, Umemura T, et al. Smoking, endothelial function, and Rho-kinase in humans. Arterioscler Thromb Vasc Biol. 2005;25:2630–5. doi: 10.1161/01.ATV.0000189304.32725.bd. [DOI] [PubMed] [Google Scholar]
- 86.Laurent S, Katsahian S, Fassot C, Tropeano AI, Gautier I, Laloux B, et al. Aortic stiffness is an independent predictor of fatal stroke in essential hypertension. Stroke. 2003;34:1203–6. doi: 10.1161/01.STR.0000065428.03209.64. [DOI] [PubMed] [Google Scholar]
- 87.Mattace-Raso FU, van der Cammen TJ, Hofman A, van Popele NM, Bos ML, Schalekamp MA, et al. Arterial stiffness and risk of coronary heart disease and stroke: the Rotterdam Study. Circulation. 2006;113:657–63. doi: 10.1161/CIRCULATIONAHA.105.555235. [DOI] [PubMed] [Google Scholar]
- 88.Blacher J, Guerin AP, Pannier B, Marchais SJ, Safar ME, London GM. Impact of aortic stiffness on survival in end-stage renal disease. Circulation. 1999;99:2434–9. doi: 10.1161/01.cir.99.18.2434. [DOI] [PubMed] [Google Scholar]
- 89.de Simone G, Roman MJ, Koren MJ, Mensah GA, Ganau A, Devereux RB. Stroke volume/pulse pressure ratio and cardiovascular risk in arterial hypertension. Hypertension. 1999;33:800–5. doi: 10.1161/01.hyp.33.3.800. [DOI] [PubMed] [Google Scholar]
- 90.Laufs U, Endres M, Custodis F, Gertz K, Nickenig G, Liao JK, et al. Suppression of endothelial nitric oxide production after withdrawal of statin treatment is mediated by negative feedback regulation of rho GTPase gene transcription. Circulation. 2000;102:3104–10. doi: 10.1161/01.cir.102.25.3104. [DOI] [PubMed] [Google Scholar]
- 91.Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708–14. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- 92.Liu PY, Chen JH, Lin LJ, Liao JK. Increased Rho kinase activity in a Taiwanese population with metabolic syndrome. J Am Coll Cardiol. 2007;49:1619–24. doi: 10.1016/j.jacc.2006.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kanda T, Wakino S, Homma K, Yoshioka K, Tatematsu S, Hasegawa K, et al. Rho-kinase as a molecular target for insulin resistance and hypertension. Faseb J. 2006;20:169–71. doi: 10.1096/fj.05-4197fje. [DOI] [PubMed] [Google Scholar]
- 94.Sordella R, Jiang W, Chen GC, Curto M, Settleman J. Modulation of Rho GTPase signaling regulates a switch between adipogenesis and myogenesis. Cell. 2003;113:147–58. doi: 10.1016/s0092-8674(03)00271-x. [DOI] [PubMed] [Google Scholar]
- 95.Begum N, Sandu OA, Ito M, Lohmann SM, Smolenski A. Active Rho kinase (ROK-alpha) associates with insulin receptor substrate-1 and inhibits insulin signaling in vascular smooth muscle cells. J Biol Chem. 2002;277:6214–22. doi: 10.1074/jbc.M110508200. [DOI] [PubMed] [Google Scholar]
- 96.Iwasaki H, Okamoto R, Kato S, Konishi K, Mizutani H, Yamada N, et al. High glucose induces plasminogen activator inhibitor-1 expression through Rho/Rho-kinase-mediated NF-kappaB activation in bovine aortic endothelial cells. Atherosclerosis. 2008;196:22–8. doi: 10.1016/j.atherosclerosis.2006.12.025. [DOI] [PubMed] [Google Scholar]
- 97.Wang CY, Liu PY, Liao JK. Pleiotropic effects of statin therapy: molecular mechanisms and clinical results. Trends Mol Med. 2008;14:37–44. doi: 10.1016/j.molmed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]