

Abstract
Rationale
Gap junctions mediate cell-to-cell electrical coupling of cardiomyocytes. The primary gap junction protein in the working myocardium, connexin43 (Cx43), exhibits increased localization at the lateral membranes of cardiomyocytes in a variety of heart diseases, although the precise location and function of this population is unknown.
Objective
To define the subcellular location of lateralized gap junctions at the light and electron microscopic level, and further characterize the biochemical regulation of gap junction turnover.
Methods and Results
By electron microscopy we characterized gap junctions formed between cardiomyocyte lateral membranes in failing canine ventricular myocardium. These gap junctions were varied in structure and appeared to be extensively internalizing. Internalized gap junctions were incorporated into multi-lamellar membrane structures, with features characteristic of autophagosomes. Intracellular Cx43 extensively co-localized with the autophagosome marker GFP-LC3 when both proteins were exogenously expressed in HeLa cells, and endogenous Cx43 co-localized with GFP-LC3 in neonatal rat ventricular myocytes. Furthermore, a distinct phosphorylated form of Cx43, as well as the autophagosome-targeted form of LC3 targeted to lipid rafts in cardiac tissue, and both were increased in heart failure.
Conclusions
Our data demonstrate a previously unrecognized pathway of gap junction internalization and degradation in the heart, and identify a cellular pathway with potential therapeutic implications.
Keywords: gap junctions, connexin43, heart failure, autophagy, lipid rafts
INTRODUCTION
Gap junctions (GJs) allow ions and small molecules to be exchanged between the cytoplasm of adjacent cells. Intercellular communication through GJs mediates coordinated cell behavior in nearly every mammalian tissue, contributing to diverse physiological processes. Efficient electrical activation and action potential propagation in the heart requires current passage between cardiomyocytes, a function carried out by GJs. Thus, accurate targeting and maintenance of GJs in cardiomyocytes is essential for normal heart function. Consistent with this concept are studies that demonstrate impaired heart function upon genetic disruption of connexin43 (Cx43), the primary GJ channel protein in mammalian ventricular muscle1-3. Furthermore, a variety of structural heart diseases in both humans and animal models are associated with remodeling of GJ proteins, including decreased expression and altered subcellular distribution of Cx434-10. Despite the critical role GJs play in the heart, and extensive evidence for GJ remodeling in cardiac disease, a mechanistic understanding of the GJ life cycle in cardiomyocytes is limited.
GJ channels are assembled from two hexameric hemichannels, or connexons, (formed by connexins) which dock together at contacts between cells. GJ channels cluster into tightly packed two dimensional arrays consisting of a few to thousands of channels, known as GJ plaques. In cardiac tissue GJs are formed primarily at the intercalated disk (ID), which is the site of contact between the ends of cardiomyocytes. This arrangement accounts for anisotropic current flow, with conduction progressing rapidly in the direction of tissue fiber orientation. GJ proteins have been shown to have a remarkably rapid rate of turnover, with half lives on the order of 1.5-5 hours both in vitro and in vivo11-13. Such rapid turnover kinetics necessitates well coordinated and regulated trafficking. GJ plaques are internalized by a cellular process in which the plasma membranes of both coupled cells are internalized into one of the two cells, producing double membrane intracellular inclusions14. Intracellular circular GJ membranes, termed annular GJs, have been morphologically characterized in isolated cells and tissues by electron microscopy15, 16. Dynamic studies of GJs in cell systems have demonstrated internalization of entire plaques forming annular GJs consisting of membranes from both coupled cells17, 18. Since their earliest description, annular gap junctions have been implicated in degradative pathways involving lysosomes14, 15, 19-22 or the proteasome23. More recent studies have sought to characterize the cellular processes by which GJs are internalized and degraded18, 24, although many details remain unknown, especially in the physiological context. Cx43 phosphorylation is widely implicated in the assembly and turnover of GJs. Cx43 can be phosphorylated on multiple amino acids on its carboxyl-terminal tail, with different phosphorylation sites having distinct effects on GJ function. It has been reported that the phosphorylation state of Cx43 is altered with cardiac disease and upon pharmacological treatment of cardiac cells6, 7, 25-29; however, the precise phosphorylation changes that occur, and the effect they have on GJ function are unresolved.
In this paper we provide an ultrastructural characterization of GJs formed between cardiomyocyte lateral cell borders in failing canine cardiac tissue. Internalized GJs are incorporated into heterogeneous multi-lamellar membranes, reminiscent of autophagosomes. These data suggest that an autophagic pathway is enhanced in the failing heart, and involves a distinct phosphorylated form of Cx43 that targets to lipid rafts.
METHODS
A detailed methods section is available in the online supplement, and describes antibodies and plasmids, cell culture methods, biochemical fractionation and analysis of tissue, and both light and electron microscopic methods used in the study.
Canine Heart Failure Model
Dogs were rapidly paced into heart failure as previously described6.
Neonatal rat ventricular myocyte (NRVM) isolation and culture
NRVMs were enzymatically dissociated from the ventricles of 2-day-old rats as previously described30.
Statistical analysis
For the frequency of lateralized gap junctions (normal vs. failing hearts), statistical significance was determined by a proportion test of the mean frequencies. Statistical significance of Western blot data was determined using unpaired, two-tailed Student’s T-tests. Error bars represent standard deviation of the mean.
All procedures involving animals were approved by the Johns Hopkins Animal Care and Use Committee.
RESULTS
Lateralized Cx43 does not colocalize with ZO-1 or cadherin
Lateralized Cx43 was first characterized in canine ventricular tissue by immunofluorescent confocal microscopy. Figure 1A-B shows representative failing cardiac tissue sections stained with antibodies to Cx43, zonula occludens-1 (ZO-1) or a pan-specific cadherin antibody, revealing extensive targeting of both Cx43 and mechanical junction proteins to the ID (boxed regions). Extensive Cx43 staining was also observed along cardiomyocyte lateral borders, a pattern previously shown to be exaggerated in a variety of structural heart diseases (arrowheads). The lateralized population of Cx43 exhibited diminished co-localization with ZO-1 and cadherin. ZO-1 staining was also present in spaces between cardiomyocytes, representing staining of non-cardiomyocyte cell types including vascular endothelial cells within capillaries and fibroblasts.
FIGURE 1. Immunofluorescent localization of Cx43, ZO-1 and Cadherin in canine cardiac myocardium.
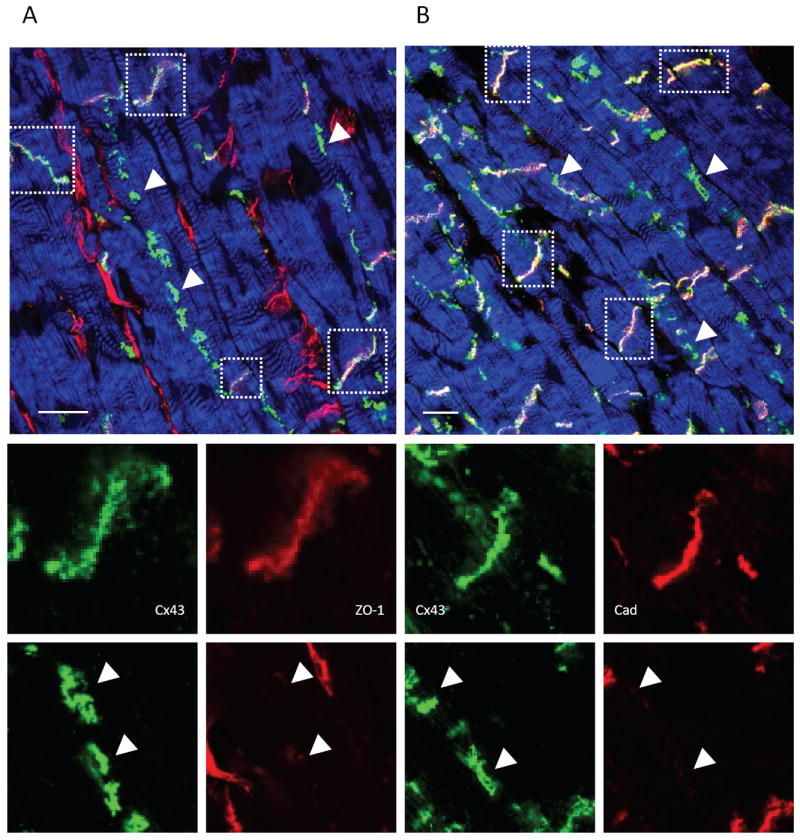
Failing canine left ventricular tissue sections were stained for Cx43 (green) and (A) ZO-1(red) or (B) cadherin (red) and imaged by confocal microscopy. Actin was stained using phalloidin to reveal tissue architecture (blue). (A) and (B) are optical sections showing intercalated disks (IDs) (boxed regions) and lateralized GJs (arrowheads). The IDs exhibit staining for Cx43 and ZO-1 and Cx43 and cadherin, whereas lateralized Cx43 signal exhibits diminished signal for ZO-1 and Cadherin.
Gap junction ultrastructure
We used transmission electron microscopy (TEM) of canine cardiac tissue to examine the ultrastructure of GJs. By TEM, GJs appear as pentalaminar membranes. Immunogold labeling of Cx43 revealed extensive staining on pentalaminar membranes within the ID, representing GJs formed between cells (not shown). In both normal and failing hearts Cx43-labeled pentalaminar membranes were also observed in circular structures emanating from the ID region, representing internalizing GJs destined to become annular GJs (AGJs) (Figure 2A).
FIGURE 2. Transmission electron microscopy (TEM) of cardiac tissue sections.
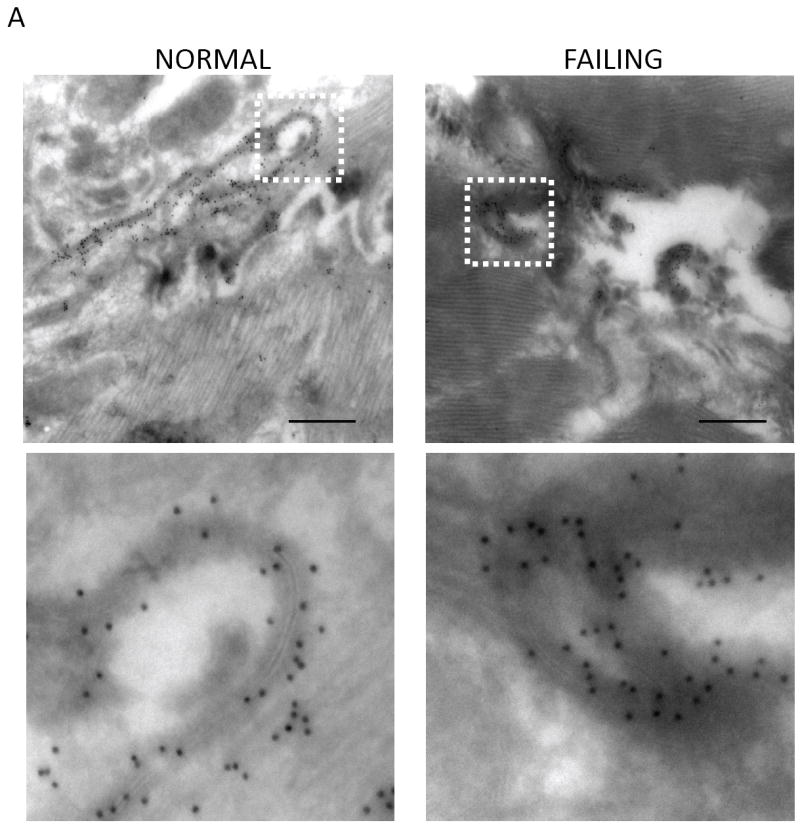
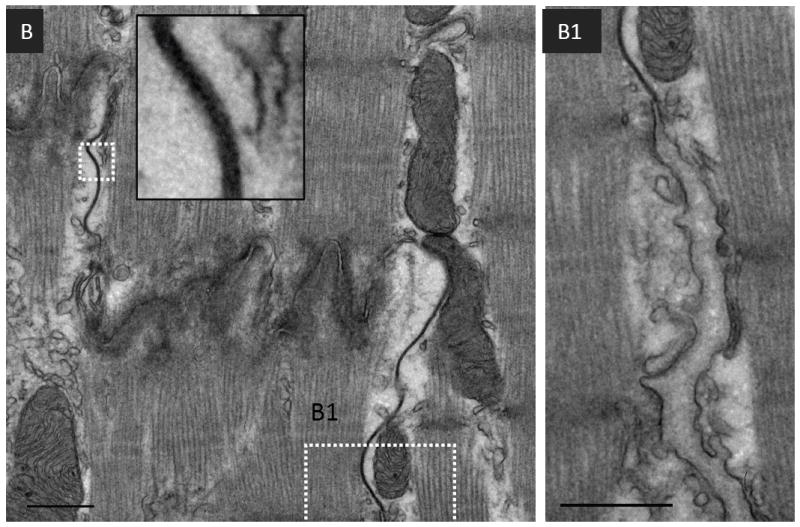
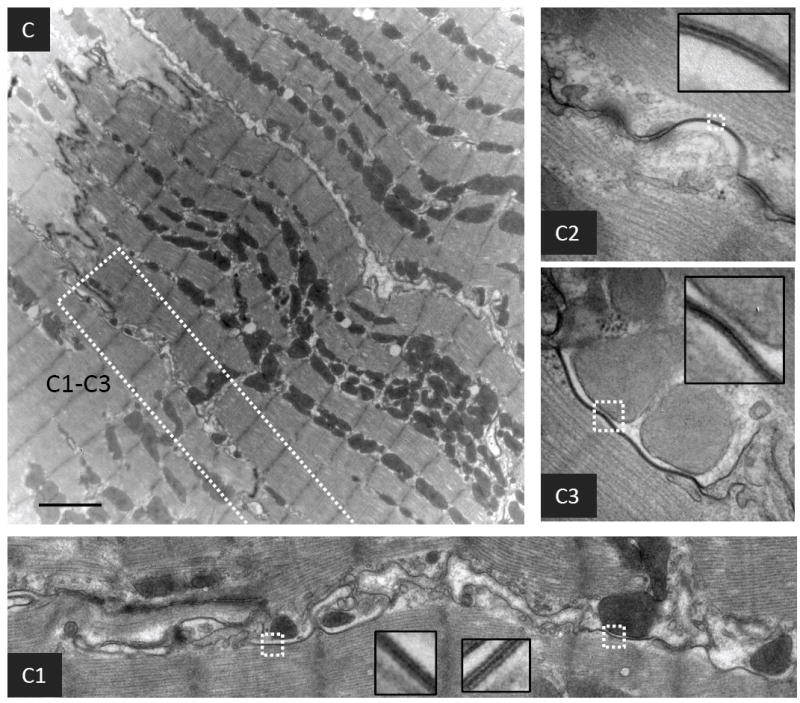
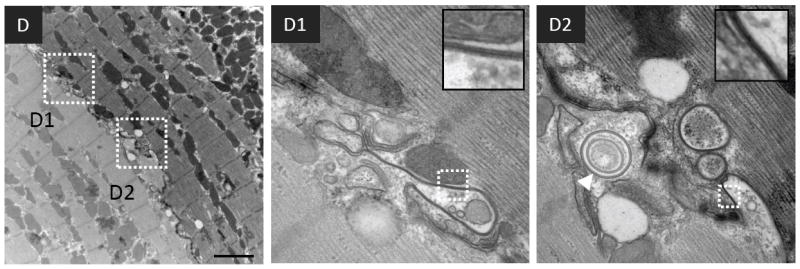
(A) Micrographs of normal and failing canine ventricular tissue sections labeled with anti-Cx43 primary antibody and a secondary antibody conjugated to12 nm colloidal gold then imaged by TEM. The bottom panels show magnified regions of the boxed areas. (B) Conventional TEM of non-failing canine left ventricular tissue sections. The intercalated disk is magnified in the inset. B1 is a magnified and extended view of the boxed region in panel B. (C) and (D) Micrographs of failing canine ventricular tissue sections, highlighting contacts between cardiomyocyte lateral cell borders. C1-C3 and D1-D2 are magnified images from within the boxed regions in panels C and D respectively. The insets in each of the panels are from boxed regions highlighting pentalaminar membranes. A concentric internalized GJ is indicated by a white arrowhead in D2. Scale bars A, B, = 500nm C, D = 2μm
Conventional TEM was used to study the ultrastructure of GJs in greater detail. Figure 2B shows an electron micrograph of normal cardiac tissue demonstrating the orientation of GJs within the ID. The mechanical junctions formed between cardiomyocytes are formed by adherens junctions and desmosomes, and run perpendicular to the direction of tissue fibers. GJs are located between intervening portions of mechanical junctions and run parallel to the fiber orientation (Figure 2B boxed region). GJs located at the outer edge of the ID are not typically flanked by mechanical junctions at the edge which is continuous with lateral membranes. (Figure 2B1).
Figures 2C and 2D show electron micrographs of failing cardiac tissue. GJs are extensively formed between the lateral membranes of adjacent myocytes, in regions distant from the ID (Figure 2C1-3 and 2D1-2). These lateralized GJs are not flanked by typical mechanical junctions. Lateralized GJs are atypical of GJs formed at IDs in that they exhibit complex membrane bending, and are often in close association with mitochondria, and other indistinct cellular material. Lateralized GJs were more frequently observed in failing compared with normal tissue. A series of images of similar tissue orientations were acquired from 4 normal and 3 failing dog hearts, and the number of cell-cell pairs possessing lateralized GJs was counted (Figure 3A). In tissue sections from 4 normal dogs 1/21, 1/22, 4/25, and 11/21 cell-cell pairs possessed lateralized GJs (average frequency = 0.19), whereas in tissue sections from 3 failing dogs 7/14, 14/21, and 9/12 cell-cell pairs possessed lateralized GJs (average frequency = 0.64) (P=0.013) (Figure 3B).
FIGURE 3. Frequency of lateralized GJs in normal and failing myocardium.
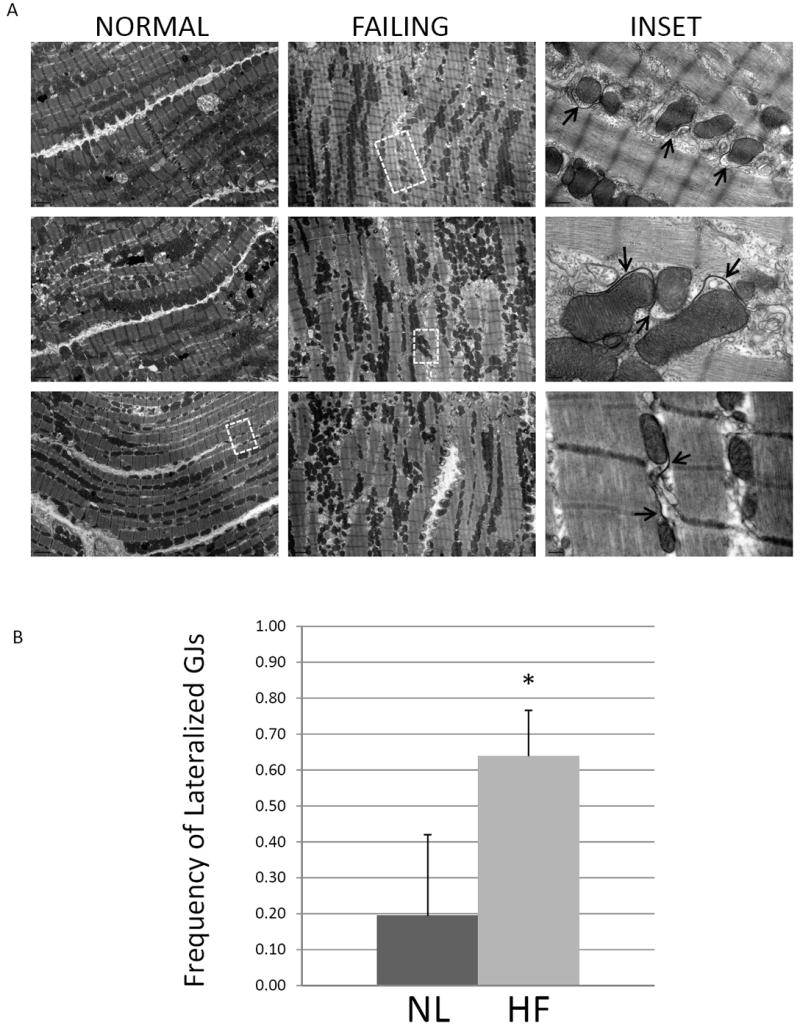
(A) Representative micrographs of apposing lateral cell borders in normal and failing canine myocardium. The inset panel images are from the boxed regions, highlighting GJs formed between myocyte lateral cell membranes. Pentalaminar GJ membranes are indicated by arrows. (B) The frequency with which apposing lateral cell borders exhibited GJ formation was quantified in normal and failing tissue sections. In normal hearts (n=4), 1/22, 1/22, 4/25, and 11/21 cell-cell borders possessed lateralized GJs whereas in failing hearts (n=3), 7/14, 14/21, and 9/12 cell-cell borders possessed lateralized GJs. *P=0.013
In failing cardiac tissue internalized GJs were often observed as concentric rings, suggesting extensive involution of GJ membranes during internalization. (Figure 2D2 arrowhead, Figures 4A - 4D). Internalized GJs are typically large (~0.1-1 micron in diameter), highly heterogeneous structures with undefined cellular debris in their lumens (Figure 4A). Internalized GJ membranes also formed multi-lamellar membrane structures (Figures 4 B and C) reminiscent of autophagosomes. Further morphological support for autophagic sequestration of internalized GJs were crescent-shaped, putative isolation membranes that appeared to envelope internalized GJs (Figure 4D arrowheads), as well as multiple putative isolation membranes in close proximity to the ID (Figure 4E).
FIGURE 4. Ultrastructure of internalized GJs from failing myocardium.
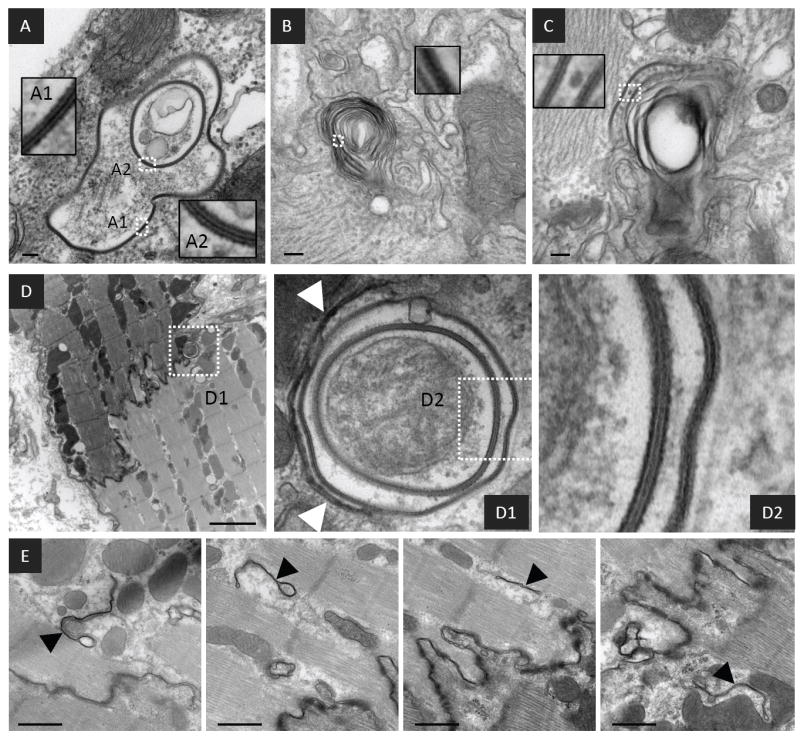
Representative micrographs of failing myocardium showing internalized GJs with concentric pentalaminar membranes and cellular debris in their lumens (A) as well as pentalminar membranes incorporated into multi-lamellar membrane structures (B) and (C). (D) An internalized and/or internalizing concentric GJ in close association with a putative isolation membrane shown at higher resolution in D1(arrowheads). (E) Multiple double membrane structures with the appearance of isolation membranes in close proximity to the ID. The insets show the detail of pentalaminar membranes from boxed regions. Scale bars A-C = 100nm D = 2μm E = 500nm
To test the hypothesis that internalized GJs associate with autphagosomes we transiently transfected HeLa cells with both Cx43 and a specific marker of autophagosomes, microtubule associated protein light chain 3 (LC3) fused to EGFP (GFP-LC3). Figure 5A shows two HeLa cells each expressing Cx43 and GFP-LC3. GJs are formed at sites of contact between the two cells (arrowhead Figure 5A merge) and exhibit strong Cx43 staining. A significant fraction of Cx43 staining is intracellular and punctate, representing trafficking intermediates of Cx43. GFP-LC3 is expressed as a cytoplasmic protein, resulting in diffuse green signal throughout the cell. However, GFP-LC3 is also concentrated on maturing autophagosomes by a covalent lipid modification, resulting in punctate green staining of autophagosomes. A proportion of intracellular Cx43 signal co-localized with GFP-LC3 (Figure 5A merge), suggesting an association of internalized GJs with autophagosomes. To determine if endogenous cardiac Cx43 associates with autophagosomes we cultured primary neonatal rat ventricular myocytes (NRVMs) that were transiently transfected with GFP-LC3. Under baseline culture conditions cytoplasmic Cx43 and GFP-LC3 only sporadically co-localized (data not shown). We hypothesized that internalized GJs may be rapidly cleared from the cytoplasm through lysosomal degradation. We therefore treated cells with a lysosomal inhibitor (10μM chloroquine) for two hours prior to fixing, immunostaining and imaging the cells. Inhibiting lysosomal activity resulted in significant intracellular co-localization between endogenous Cx43 and expressed GFP-LC3 (Figure 5B).
FIGURE 5. Association of internalized gap junctions with autophagosomes.
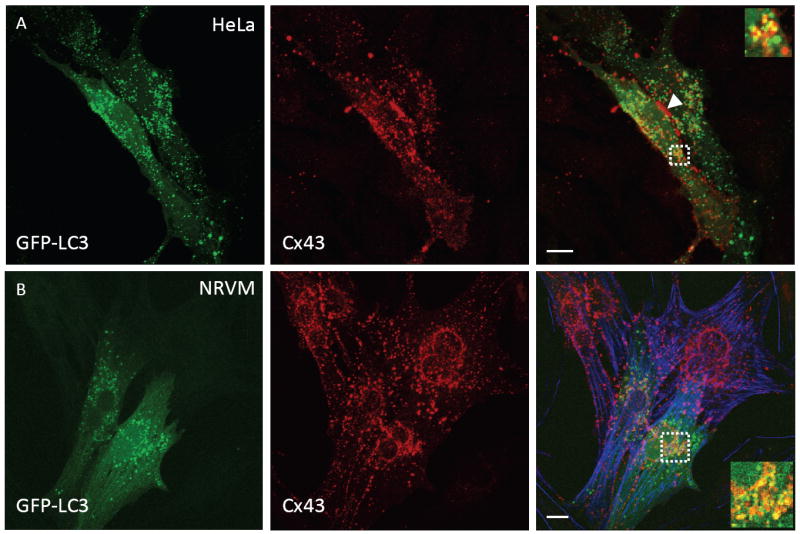
(A) HeLa cells were transfected with Cx43 and GFP-LC3. Fixed cells were immunostained and imaged by confocal microscopy (GFP-LC3 (green, left) Cx43 (red, middle) merged panel (right)). (B) Cultured neonatal rat ventricular myocytes (NRVMs) were transfected with GFP-LC3 and treated with a lysosomal inhibitor (10μM chloroquine for 2 hours prior to fixation). Fixed cells were then immunostained for endogenous Cx43 and imaged as in panel A. NRVMs were stained for actin using phalloidin (blue). All images are projections of 17 optical sections. Inset images in merged panels are single optical sections from within boxed regions. Scale bars = 2μm
Lipid raft targeting of Cx43 and LC3-II
Cx43 has been reported to target to buoyant, cholesterol and sphingolipid-rich membrane domains termed lipid rafts (LRs) 31-33, although the significance of LR targeting is not known. Our observation that internalized GJs associate with multi-lamellar membranes led us to hypothesize these structures might represent detergent resistant, buoyant populations of Cx43. We prepared LRs from cardiac tissue by cold Triton X-100 extraction and sucrose density centrifugation. Western blotting revealed two populations of Cx43 (Figure 6A): a buoyant population (fraction 5) which co-fractionated (but is not necessarily associated) with the muscle specific caveolar protein caveolin-3 and the glycosylated sphingolipid GM1 (markers of LR domains), and a dense population (fractions 9-11). Cx43 is well known to separate into multiple bands by SDS-PAGE, representing distinct phosphorylated species of the protein34. To better separate Cx43 into its distinct phosphorylated forms we analyzed the sucrose fractions by large format SDS-PAGE (Figure 6B upper panel). The slowly migrating form of Cx43 (designated P2) was uniquely targeted to the LR fraction, while intermediate and fast migrating bands (designated P1 and P0 respectively) remained predominantly in the dense non-LR fractions. To confirm that the slow migration of LR targeted Cx43 was due to phosphorylation and not an artifact of the LR isolation procedure, we treated proteins isolated from fraction 5 with AP (Figure 6C). AP treatment increased the mobility of Cx43, indicating that LR-targeted Cx43 constitutes a highly phosphorylated form of the protein.
FIGURE 6. Lipid raft targeting of Cx43 and LC3 in cardiac tissue.
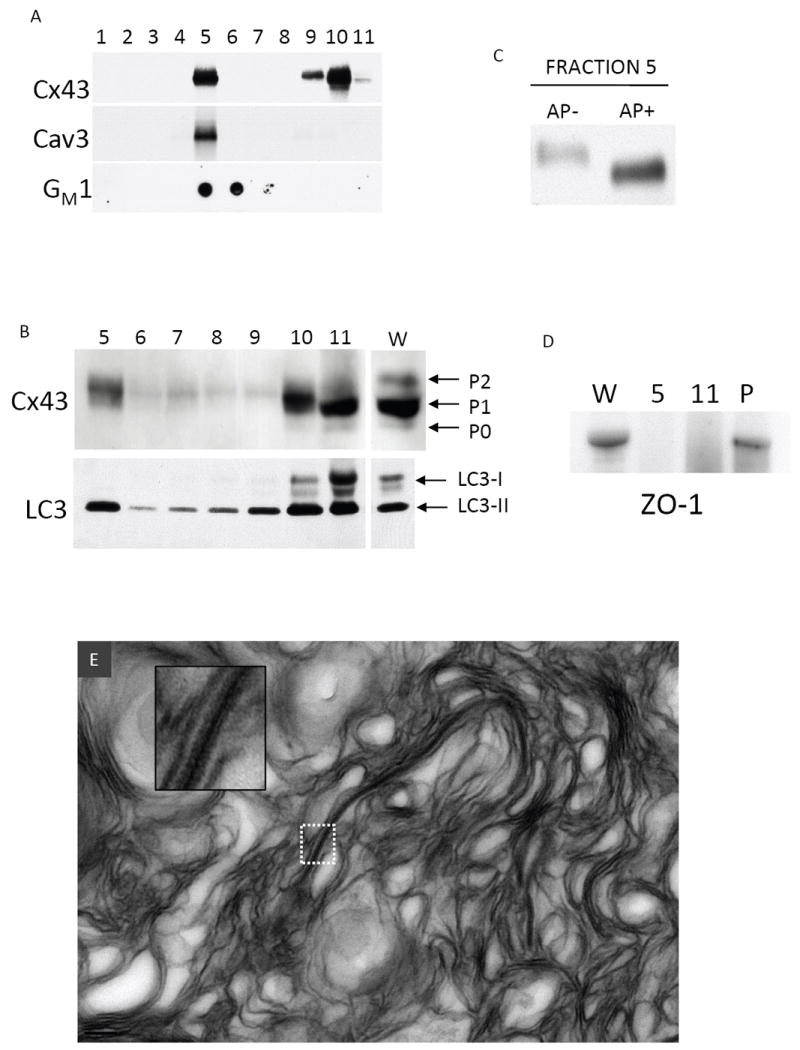
(A) Sucrose gradient fractionation of canine ventricular myocardium Western blotted for Cx43, caveolin-3 (Cav-3), and GM1 (dot blot probed with HRP conjugated cholera toxin B subunit). Fractions 1-4 correspond to 5% sucrose, 5-10 correspond to 38% sucrose, 11 corresponds to 40% sucrose. (B) Fractions 5-11 probed for Cx43 (separated by large format (18cm2) 10% acrylamide gel) and LC3. The lane marked W is a whole tissue lysate. Distinct phosphorylated species of Cx43 (P0, P1, P2) and LC3-I and LC3-II are indicated with arrows. (C) Cx43 Western blot of Fraction 5 that was untreated (AP-) or treated (AP+) with alkaline phosphatase and separated on a 10% acrylamide gel. (D) ZO-1 is absent from fraction 5 and 11, but was present in the pellet (P). (E) Material in fraction 5 was pelleted and prepared for TEM. A representative TEM of fraction 5 exhibits primarily multilamellar membranous material with an incorporated pentalaminar membrane (the inset is a higher power view of the boxed region).
We examined the distribution of the autophagosome marker LC3 in LR fractions (Figure 6B lower panel). LC3 migrates as two forms, LC3-I and LC3-II. LC3-I is cytoplasmic and migrates at ~18kDa. During autophagy LC3-I is partially cleaved and covalently linked to maturing autophagosomes, resulting in the LC3-II form which migrates at ~15kDa. The dense, non-LR fractions contained both LC3-I and LC3-II. The buoyant fraction 5 contained LC3-II, indicating that a subpopulation of autophagosomes possess the physicochemical properties of LRs. The LR fraction, as expected, is devoid of ZO-1, which was confined to the pellet (Figure 6D).
We then examined the ultrastructure of membranous material from fraction 5 (Figure 6E). By TEM fraction 5 consists primarily of multi-lamellar membrane structures with no discernable structure or morphology. However, interspersed in this material we observed pentalaminar membranes. The edges of these pentalaminar membranes were not associated with structures consistent with mechanical junctions (fascia adherens and desmosomes). This observation is consistent with a population of Cx43 possessing LR properties after incorporation into GJs, but not associated with mechanical junctions, which may include the internalized, multi-lamellar membranes associated with GJs we observed in cardiac tissue.
Cx43 and LC3 expression in heart failure
We examined LR fractions from both normal (NL) and pacing induced heart failure (HF) dog ventricles (Figure 7A)6, 7. LR domains isolated from failing hearts exhibited an approximately 3.5-fold greater Cx43 signal than LR domains from NL hearts (P=0.011). In these same samples the total Cx43 levels decreased by approximately 2-fold (P=0.01). We examined the expression levels of LC3-I and LC3-II in NL and HF hearts (Figure 7B). The levels of LC3-I were not statistically different, whereas the levels of LC3-II increased by approximately 2-fold in HF hearts as compared to NL hearts (P=0.016), indicative of increased autophagosome formation in failing hearts.
FIGURE 7. Lipid raft targeted Cx43 and LC3-II are increased in heart failure.
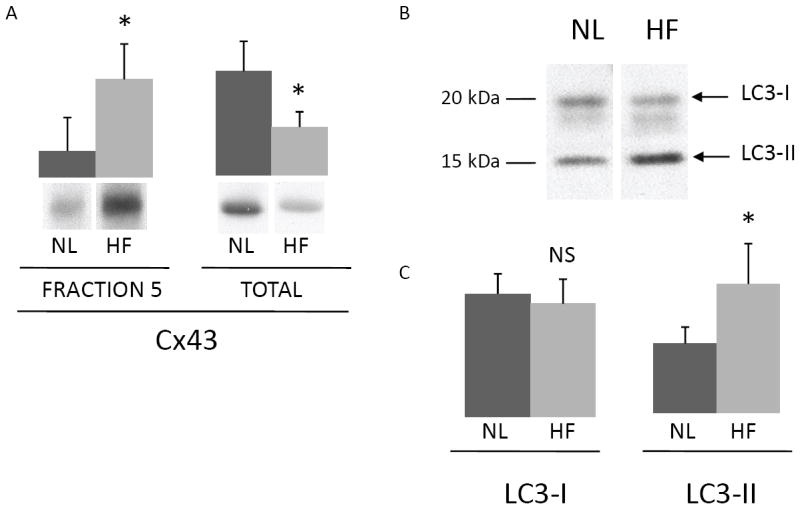
(A) Bar graphs and representative Western blots of fraction 5 (left) from normal (NL) and failing (HF) dog hearts (n=5 each) and whole tissue lysates (right) from the same hearts (separated by 4-12% acrylamide mini gels to collapse Cx43 bands) probed for Cx43 (arbitrary units, *P<0.02). (B) The whole tissue lysates used in panel A were also probed for LC3. The migration of LC3-I and LC3-II are indicated by arrows. (C) Bar graphs of LC3-I and LC3-II levels in normal and failing hearts (n=5 each, arbitrary units, NS=not significant, *P<0.02).
DISCUSSION
In cardiac tissue, Cx43 forms GJs primarily at the ID. A prominent feature of structural heart disease is a redistribution of Cx43 to the lateral cell borders and a decrease in Cx43 expression levels6, 7, 29. Despite the consistency of altered Cx43 expression and distribution in structural heart disease, the functional status and structural nature of lateralized Cx43 is incompletely understood. In this study we demonstrate that GJs are formed between the lateral membranes of cardiomyocytes with increased frequency in a canine model of pacing induced HF. However, the lack of co-localization with ZO-1 and cadherin, and the heterogeneous structure of lateralized GJs demonstrate that they are structurally and likely functionally distinct from those formed at the ID.
Cx43 has been shown to be directly bound by ZO-135 and it is thought that ZO-1 in part regulates the assembly of Cx43 into the periphery of GJ plaques36. Changes in the relationship between ZO-1 and Cx43 have been reported in cardiac disease37, 38 and is emerging as a potentially critical interaction for the maintenance of proper GJ mediated intercellular communication. Furthermore, Cx43-containing vesicles have been shown to be directly delivered to cadherin-containing adherens junctions via microtubules39. The formation of apparently atypical GJs at cardiomyocyte lateral membranes, devoid of ZO-1 and cadherin co-localization, suggests impairment in the mechanisms responsible for direct delivery of Cx43 to sites of mechanical junction formation.
Annular GJs have been described in a number of cells and tissues14, 17, 18 including isolated cardiomyocytes15, 40 and cardiac tissue subjected to stress29, 41. Annular GJs have been suggested to be endocytosed GJ plaques destined for lysosomal or proteasomal degradation14, 15, 19, 21, 23. Direct evidence for the origin of annular GJs comes from the work of Jordan et al17 using time lapse studies of fluorescently tagged Cx43, and Piehl et al18 using dye injection studies to demonstrate internalization of intercellular GJ plaques into one of two coupled cells. We suggest that intracellular uni- and multi-lamellar GJ-containing membrane rings observed in cardiac tissue represent annular GJs in progressively advanced stages of processing for degradation.
The incorporation of internalized GJs into multi-lamellar membranes, their close association with cellular debris, and the co-localization of Cx43 with LC3 is suggestive of autophagy playing a role in GJ clearance. The association of internalized GJs with autophagosomes has been suggested in the literature15, 42 but is not an accepted mechanism of GJ degradation. Autophagosomes are dynamic organelles which sequester cell contents for delivery to lysosomes43. In post-mitotic cells, such as cardiomyocytes, autophagy is constitutively active to help maintain cell size and integrity. Under stressful conditions the rate of autophagy is increased, where it serves an adaptive function by providing a source of nutrients through catabolism of “unnecessary” cellular contents, as well as clearing damaged organelles44. Autophagy has been implicated in the pathogenesis of many diseases, including ischemia/reperfusion injury and heart failure45, 46, as a mechanism of cell survival. Internalized GJs are quite large and structurally distinct relative to typical endocytic organelles, and therefore connexins are relatively non-standard endocytic cargo. The cellular machinery involved in GJ internalization and degradation is therefore likely to be distinct from that involved in more conventional endoctyic processes. Thus, it is plausible that GJs would be sequestered by autophagic machinery for delivery to lysosomes.
The signaling mechanisms regulating the internalization and degradation of Cx43 GJs are poorly understood, but likely involve post-translational modification of Cx43. Cx43 is phosphorylated extensively on its carboxyl-terminus, regulating both the trafficking and permeability of Cx43 GJ channels47, 48. By SDS-PAGE the three major bands observed for Cx43 are typically referred to as P0 (fastest migrating, least phosphorylated), P1 and P2 (slowest migrating, most phosphorylated), and the P2 form only appears after Cx43 has reached the plasma membrane and formed gap junctions34. Changes in the phosphorylation state of cardiac Cx43 have been reported in heart disease7, 29; however, the precise changes that occur and the functional consequences of these changes are unclear. In cell-based models, pharmacological interventions which promote the phosphorylation of Cx43 to the P2 state have been shown to enhance internalization and degradation of Cx43 GJs, suggesting that hyperphosphorylation of Cx43 may be involved in GJ degradation24, 49-52. Other studies have reported enhanced expression and GJ formation upon pharmacological treatments also likely to result in Cx43 phosphorylation26, 28. It is likely that the specific amino acids which are phosphorylated dictate the effects that Cx43 phosphorylation has on GJs. Thus, increased Cx43 phosphorylation, or perhaps differences in the pattern of phosphorylation, may result in more rapid kinetics at multiple stages of the GJ life cycle enhancing the formation of GJs as well as their internalization and degradation, increasing the overall rate of GJ assembly and turnover. Further studies will be required to determine the effect that specific phosphorylation events have on GJ function, and to determine which of these events are altered in disease.
Cx43 has been reported to target to buoyant, cholesterol and sphingolipid-rich membrane domains termed lipid rafts (LRs)31-33, and there is evidence that the phosphorylation state may differ between LR and non-LR targeted populations32-34, 53. Our observation that the P2 form of Cx43 is uniquely targeted to LRs suggests that LR targeting of Cx43 is a late event in the GJ life cycle, and may be involved in GJ degradation. Consistent with this hypothesis are studies which demonstrate that connexins can target to LRs, but that intercellular GJs themselves are not LRs31. Musil et al34 have described the relationship between Cx43 Triton solubility, phosphorylation state, and assembly into GJs in vitro. The authors suggest that Cx43 begins as a Triton soluble, hypo-phosphorylated form during its progression towards the plasma membrane. Upon arrival at the plasma membrane Cx43 acquires Triton resistance and matures into a hyper-phosphorylated form. The authors further suggest that the acquisition of Triton resistance and maturation to the P2 phosphorylated form corresponds to the formation of mature GJ plaques. Based on our ultrastructural characterization of fraction 5 (Figure 6E), which contains P2 phosphorylated Cx43, we do not believe these buoyant pentalaminar membranes represent GJ plaques coupling two cells at intercalated disks, but rather internalized GJs incorporated into multi-lamellar membranes. We therefore hypothesize that maturation to the P2 form in fact corresponds to the degradation of GJs through autophagic processing, although further experiments will be required to confirm this hypothesis.
Our data demonstrate that GJs with highly variable morphology are formed between cardiomyocyte lateral membranes with increased frequency in HF, and that GJ turnover likely occurs via an autophagic pathway. The mechanisms by which Cx43 GJs are targeted for internalization and degradation by autophagy appears to involve changes in phosphorylation, as well as processing through membranes with physicochemical properties of lipid rafts. The regulated process of GJ turnover via autophagy suggests a novel pathway for the regulation of cardiac GJs, and therefore conduction in the mammalian ventricle. These findings offer insight into the mechanisms regulating electrical conduction in mammalian myocardium, and may in part contribute to the arrhythmogenic electrical remodeling associated with heart failure.
There are limitations to the present study. We did not perform serial sectioning and/or 3D reconstruction of TEM sections. It is therefore possible that apparently internalized GJs are in reality extensively convoluted GJs still attached to the plasma membrane. Regardless of this possibility, the presence of GJs at lateral membranes is nonetheless more frequent in failing hearts, and lateral GJs are more convoluted, if not more extensively internalized, than GJs at the ID.
These data were obtained in a canine model of heart failure that in many respects mimics human heart failure, but is clearly different from the most common forms of heart failure occurring in humans. The detailed regulation of Cx43 and GJ localization may not be identical with human disease. Reassuringly, many of the features of GJ remodeling characterized in this model are similar to those previously observed in diseased human hearts5, 9
Although we have provided ultrastructural, microscopic and biochemical evidence in support of GJ association with autophagosomes, we have not demonstrated connexin degradation via autophagy per se. The precise contribution of autophagy to connexin turnover will require a more quantitative examination.
Supplementary Material
Acknowledgments
We acknowledge Dr. David R. Graham for assistance with lipid raft isolation.
SOURCES OF FUNDING This work is supported by NIH grant PO1 HL077180. Dr. Tomaselli holds the Michel Mirowski MD Professorship in Cardiology.
NON-STANDARD ABBREVIATIONS AND ACRONYMS
- AGJ
annular gap junction
- AP
alkaline phosphatase
- Cx43
connexin43
- GJ
gap junction
- HF
heart failure
- ID
intercalated disk
- LC3
microtubule associated protein light chain 3
- LR
lipid raft
- NL
normal
- NRVM
neonatal rat ventricular myocytes
- SDS-PAGE
SDS polyacrylamide gel electrophoresis
- TEM
transmission electron microscopy
- ZO-1
zonula occludens-1
NOVELTY AND SIGNIFICANCE
“What is known?”
Gap junctions (GJs), formed by connexins, allow electrical current to flow from one cardiomyocyte to another, and are required for synchronous electrical activation and contraction of cardiac muscle.
In healthy hearts, GJs are formed primarily between the ends of cardiomyocytes (intercalated disks); however, in a variety of heart diseases the major ventricular GJ protein (Cx43) increases its localization at cardiomyocyte lateral membranes as well as decreases in overall expression.
The precise location, the functional status, and mechanisms of regulation of lateralized Cx43 are not known.
“What new information does this article contribute?”
This article demonstrates increased GJ formation between the lateral membranes of adjacent cardiomyocytes, which are not flanked by typical mechanical junctions, have highly variable morphology, and appear to undergo extensive internalization.
Internalized GJs are incorporated into multilamellar membrane structures characteristic of autophagosomes, and Cx43 colocalizes with an autophagosome marker (LC3-II) in both HeLa cells and primary cultured neonatal rat ventricular myocytes.
This article also shows that a distinct phosphorylated form of Cx43 targets to lipid rafts, and it is this population that we hypothesize is involved in the autophagic degradation of GJs.
We and others hypothesize that remodeling of GJ expression and localization contributes, at least in part, to the high incidence of lethal arrhythmias associated with cardiovascular disease. In this study we have demonstrated that GJs are formed between adjacent cardiomyocyte lateral membranes with increased frequency in failing hearts. Lateralized GJs have highly variable morphology and appear to be extensively internalized, and internalized GJs associate with autophagosomes. Furthermore, a distinct phosphorylated form of Cx43 targets to lipid rafts, and we hypothesize that it is this population that undergoes autophagic degradation. Of particular novelty in this study is the ultrastructural characterization of lateralized GJs. Until now, the precise location of lateralized Cx43 was unknown. Our observation suggests that Cx43 may be aberrantly targeted to lateral membranes with increased frequency in a disease setting, highlighting the importance of further study into the mechanisms of Cx43 targeting to the plasma membrane. Finally, our study suggests that GJs are degraded through autophagy, which until now has not been incorporated into models of GJ degradation. From this observation should stem further study into the role of autophagy in GJ turnover, both in the heart, and in general.
Footnotes
DISCLOSURES None
References
- 1.Reaume AG, de Sousa PA, Kulkarni S, Langille BL, Zhu D, Davies TC, Juneja SC, Kidder GM, Rossant J. Cardiac malformation in neonatal mice lacking connexin43. Science. 1995;267(5205):1831–1834. doi: 10.1126/science.7892609. [DOI] [PubMed] [Google Scholar]
- 2.Guerrero PA, Schuessler RB, Davis LM, Beyer EC, Johnson CM, Yamada KA, Saffitz JE. Slow ventricular conduction in mice heterozygous for a connexin43 null mutation. J Clin Invest. 1997;99(8):1991–1998. doi: 10.1172/JCI119367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gutstein DE, Morley GE, Tamaddon H, Vaidya D, Schneider MD, Chen J, Chien KR, Stuhlmann H, Fishman GI. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res. 2001;88(3):333–339. doi: 10.1161/01.res.88.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uzzaman M, Honjo H, Takagishi Y, Emdad L, Magee AI, Severs NJ, Kodama I. Remodeling of gap junctional coupling in hypertrophied right ventricles of rats with monocrotaline-induced pulmonary hypertension. Circ Res. 2000;86(8):871–878. doi: 10.1161/01.res.86.8.871. [DOI] [PubMed] [Google Scholar]
- 5.Smith JH, Green CR, Peters NS, Rothery S, Severs NJ. Altered patterns of gap junction distribution in ischemic heart disease. An immunohistochemical study of human myocardium using laser scanning confocal microscopy. Am J Pathol. 1991;139(4):801–821. [PMC free article] [PubMed] [Google Scholar]
- 6.Akar FG, Nass RD, Hahn S, Cingolani E, Shah M, Hesketh GG, DiSilvestre D, Tunin RS, Kass DA, Tomaselli GF. Dynamic changes in conduction velocity and gap junction properties during development of pacing-induced heart failure. Am J Physiol Heart Circ Physiol. 2007;293(2):H1223–1230. doi: 10.1152/ajpheart.00079.2007. [DOI] [PubMed] [Google Scholar]
- 7.Akar FG, Spragg DD, Tunin RS, Kass DA, Tomaselli GF. Mechanisms underlying conduction slowing and arrhythmogenesis in nonischemic dilated cardiomyopathy. Circ Res. 2004;95(7):717–725. doi: 10.1161/01.RES.0000144125.61927.1c. [DOI] [PubMed] [Google Scholar]
- 8.Gard JJ, Yamada K, Green KG, Eloff BC, Rosenbaum DS, Wang X, Robbins J, Schuessler RB, Yamada KA, Saffitz JE. Remodeling of gap junctions and slow conduction in a mouse model of desmin-related cardiomyopathy. Cardiovasc Res. 2005;567(3):539–547. doi: 10.1016/j.cardiores.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 9.Sepp R, Severs NJ, Gourdie RG. Altered patterns of cardiac intercellular junction distribution in hypertrophic cardiomyopathy. Heart. 1996;76(5):412–417. doi: 10.1136/hrt.76.5.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Severs NJ, Bruce AF, Dupont E, Rothery S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc Res. 2008;80(1):9–19. doi: 10.1093/cvr/cvn133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beardslee MA, Laing JG, Beyer EC, Saffitz JE. Rapid turnover of connexin43 in the adult rat heart. Circ Res. 1998;83(6):629–635. doi: 10.1161/01.res.83.6.629. [DOI] [PubMed] [Google Scholar]
- 12.Fallon RF, Goodenough DA. Five-hour half-life of mouse liver gap-junction protein. J Cell Biol. 1981;90(2):521–526. doi: 10.1083/jcb.90.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laird DW, Puranam KL, Revel JP. Turnover and phosphorylation dynamics of connexin43 gap junction protein in cultured cardiac myocytes. Biochem J. 1991;273(Pt 1):67–72. doi: 10.1042/bj2730067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Larsen WJ, Hai N. Origin and fate of cytoplasmic gap junctional vesicles in rabbit granulosa cells. Tissue Cell. 1978;10(3):585–598. doi: 10.1016/s0040-8166(16)30351-2. [DOI] [PubMed] [Google Scholar]
- 15.Severs NJ, Shovel KS, Slade AM, Powell T, Twist VW, Green CR. Fate of gap junctions in isolated adult mammalian cardiomyocytes. Circ Res. 1989;65(1):22–42. doi: 10.1161/01.res.65.1.22. [DOI] [PubMed] [Google Scholar]
- 16.Marquart KH. So-called annular gap junctions in bone cells of normal mice. Experientia. 1977;33(2):270–272. doi: 10.1007/BF02124106. [DOI] [PubMed] [Google Scholar]
- 17.Jordan K, Chodock R, Hand AR, Laird DW. The origin of annular junctions: a mechanism of gap junction internalization. J Cell Sci. 2001;114(Pt 4):763–773. doi: 10.1242/jcs.114.4.763. [DOI] [PubMed] [Google Scholar]
- 18.Piehl M, Lehmann C, Gumpert A, Denizot JP, Segretain D, Falk MM. Internalization of large double-membrane intercellular vesicles by a clathrin-dependent endocytic process. Mol Biol Cell. 2007;18(2):337–347. doi: 10.1091/mbc.E06-06-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ginzberg RD, Gilula NB. Modulation of cell junctions during differentiation of the chicken otocyst sensory epithelium. Dev Biol. 1979;68(1):110–129. doi: 10.1016/0012-1606(79)90247-1. [DOI] [PubMed] [Google Scholar]
- 20.Larsen WJ, Tung HN, Murray SA, Swenson CA. Evidence for the participation of actin microfilaments and bristle coats in the internalization of gap junction membrane. J Cell Biol. 1979;83(3):576–587. doi: 10.1083/jcb.83.3.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murray SA, Larsen WJ, Trout J, Donta ST. Gap junction assembly and endocytosis correlated with patterns of growth in a cultured adrenocortical tumor cell (SW-13) Cancer Res. 1981;41(10):4063–4074. [PubMed] [Google Scholar]
- 22.Vaughan DK, Lasater EM. Acid phosphatase localization in endocytosed horizontal cell gap junctions. Vis Neurosci. 1992;8(1):77–81. doi: 10.1017/s0952523800006519. [DOI] [PubMed] [Google Scholar]
- 23.Laing JG, Beyer EC. The gap junction protein connexin43 is degraded via the ubiquitin proteasome pathway. J Biol Chem. 1995;270(44):26399–26403. doi: 10.1074/jbc.270.44.26399. [DOI] [PubMed] [Google Scholar]
- 24.Leithe E, Brech A, Rivedal E. Endocytic processing of connexin43 gap junctions: a morphological study. Biochem J. 2006;393(Pt 1):59–67. doi: 10.1042/BJ20050674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ai X, Pogwizd SM. Connexin 43 downregulation and dephosphorylation in nonischemic heart failure is associated with enhanced colocalized protein phosphatase type 2A. Circ Res. 2005;96(1):54–63. doi: 10.1161/01.RES.0000152325.07495.5a. [DOI] [PubMed] [Google Scholar]
- 26.Dodge SM, Beardslee MA, Darrow BJ, Green KG, Beyer EC, Saffitz JE. Effects of angiotensin II on expression of the gap junction channel protein connexin43 in neonatal rat ventricular myocytes. J Am Coll Cardiol. 1998;32(3):800–807. doi: 10.1016/s0735-1097(98)00317-9. [DOI] [PubMed] [Google Scholar]
- 27.Salameh A, Frenzel C, Boldt A, Rassler B, Glawe I, Schulte J, Muhlberg K, Zimmer HG, Pfeiffer D, Dhein S. Subchronic alpha- and beta-adrenergic regulation of cardiac gap junction protein expression. Faseb J. 2006;20(2):365–367. doi: 10.1096/fj.05-4871fje. [DOI] [PubMed] [Google Scholar]
- 28.Salameh A, Krautblatter S, Baessler S, Karl S, Rojas Gomez D, Dhein S, Pfeiffer D. Signal transduction and transcriptional control of cardiac connexin43 up-regulation after alpha 1-adrenoceptor stimulation. J Pharmacol Exp Ther. 2008;326(1):315–322. doi: 10.1124/jpet.108.136663. [DOI] [PubMed] [Google Scholar]
- 29.Sasano C, Honjo H, Takagishi Y, Uzzaman M, Emdad L, Shimizu A, Murata Y, Kamiya K, Kodama I. Internalization and dephosphorylation of connexin43 in hypertrophied right ventricles of rats with pulmonary hypertension. Circ J. 2007;71(3):382–389. doi: 10.1253/circj.71.382. [DOI] [PubMed] [Google Scholar]
- 30.Iravanian S, Nabutovsky Y, Kong CR, Saha S, Bursac N, Tung L. Functional reentry in cultured monolayers of neonatal rat cardiac cells. Am J Physiol Heart Circ Physiol. 2003;285(1):H449–456. doi: 10.1152/ajpheart.00896.2002. [DOI] [PubMed] [Google Scholar]
- 31.Locke D, Liu J, Harris AL. Lipid rafts prepared by different methods contain different connexin channels, but gap junctions are not lipid rafts. Biochemistry. 2005;44(39):13027–13042. doi: 10.1021/bi050495a. [DOI] [PubMed] [Google Scholar]
- 32.Schubert AL, Schubert W, Spray DC, Lisanti MP. Connexin family members target to lipid raft domains and interact with caveolin-1. Biochemistry. 2002;41(18):5754–5764. doi: 10.1021/bi0121656. [DOI] [PubMed] [Google Scholar]
- 33.Langlois S, Cowan KN, Shao Q, Cowan BJ, Laird DW. Caveolin-1 and -2 Interact with Connexin43 and Regulate Gap Junctional Intercellular Communication in Keratinocytes. Mol Biol Cell. 2007 doi: 10.1091/mbc.E07-06-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Musil LS, Goodenough DA. Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J Cell Biol. 1991;115(5):1357–1374. doi: 10.1083/jcb.115.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giepmans BN, Moolenaar WH. The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr Biol. 1998;8(16):931–934. doi: 10.1016/s0960-9822(07)00375-2. [DOI] [PubMed] [Google Scholar]
- 36.Hunter AW, Barker RJ, Zhu C, Gourdie RG. Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol Biol Cell. 2005;16(12):5686–5698. doi: 10.1091/mbc.E05-08-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kostin S. Zonula occludens-1 and connexin 43 expression in the failing human heart. J Cell Mol Med. 2007;11(4):892–895. doi: 10.1111/j.1582-4934.2007.00063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bruce AF, Rothery S, Dupont E, Severs NJ. Gap junction remodeling in human heart failure is associated with increased interaction of connexin43 with ZO-1. Cardiovasc Res. 2007 doi: 10.1093/cvr/cvm083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shaw RM, Fay AJ, Puthenveedu MA, von Zastrow M, Jan YN, Jan LY. Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell. 2007;128(3):547–560. doi: 10.1016/j.cell.2006.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mazet F, Wittenberg BA, Spray DC. Fate of intercellular junctions in isolated adult rat cardiac cells. Circ Res. 1985;56(2):195–204. doi: 10.1161/01.res.56.2.195. [DOI] [PubMed] [Google Scholar]
- 41.Emdad L, Uzzaman M, Takagishi Y, Honjo H, Uchida T, Severs NJ, Kodama I, Murata Y. Gap junction remodeling in hypertrophied left ventricles of aortic-banded rats: prevention by angiotensin II type 1 receptor blockade. J Mol Cell Cardiol. 2001;33(2):219–231. doi: 10.1006/jmcc.2000.1293. [DOI] [PubMed] [Google Scholar]
- 42.Pfeifer U. Autophagic sequestration of internalized gap junctions in rat liver. Eur J Cell Biol. 1980;21(3):244–246. [PubMed] [Google Scholar]
- 43.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8(11):931–937. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 44.Levine B, Kroemer G. Autophagy in the Pathogenesis of Disease. Cell. 2008;132(1):27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13(5):619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 46.Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117(7):1782–1793. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laird DW. Connexin phosphorylation as a regulatory event linked to gap junction internalization and degradation. Biochim Biophys Acta. 2005;1711(2):172–182. doi: 10.1016/j.bbamem.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 48.Solan JL, Lampe PD. Connexin phosphorylation as a regulatory event linked to gap junction channel assembly. Biochim Biophys Acta. 2005;1711(2):154–163. doi: 10.1016/j.bbamem.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 49.Guan X, Ruch RJ. Gap junction endocytosis and lysosomal degradation of connexin43-P2 in WB-F344 rat liver epithelial cells treated with DDT and lindane. Carcinogenesis. 1996;17(9):1791–1798. doi: 10.1093/carcin/17.9.1791. [DOI] [PubMed] [Google Scholar]
- 50.Leithe E, Rivedal E. Ubiquitination and down-regulation of gap junction protein connexin-43 in response to 12-O-tetradecanoylphorbol 13-acetate treatment. J Biol Chem. 2004;279(48):50089–50096. doi: 10.1074/jbc.M402006200. [DOI] [PubMed] [Google Scholar]
- 51.Ren P, Mehta PP, Ruch RJ. Inhibition of gap junctional intercellular communication by tumor promoters in connexin43 and connexin32-expressing liver cells: cell specificity and role of protein kinase C. Carcinogenesis. 1998;19(1):169–175. doi: 10.1093/carcin/19.1.169. [DOI] [PubMed] [Google Scholar]
- 52.Spinella F, Rosano L, Di Castro V, Nicotra MR, Natali PG, Bagnato A. Endothelin-1 decreases gap junctional intercellular communication by inducing phosphorylation of connexin 43 in human ovarian carcinoma cells. J Biol Chem. 2003;278(42):41294–41301. doi: 10.1074/jbc.M304785200. [DOI] [PubMed] [Google Scholar]
- 53.Mograbi B, Corcelle E, Defamie N, Samson M, Nebout M, Segretain D, Fenichel P, Pointis G. Aberrant Connexin 43 endocytosis by the carcinogen lindane involves activation of the ERK/mitogen-activated protein kinase pathway. Carcinogenesis. 2003;24(8):1415–1423. doi: 10.1093/carcin/bgg093. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.