

Abstract
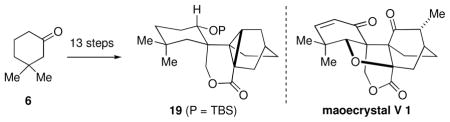
An approach towards the synthesis of the complex polycyclic diterpene maoecrystal V (1) is described. Construction of the advanced tetracyclic core structure (i.e., 19) was achieved in 13 steps from 3,3-dimethylcyclohexanone (6) by employing a stereoselective Nazarov cyclization followed by a Diels–Alder reaction to forge the two contiguous quaternary stereocenters.
In 2004, Sun and coworkers reported the structure of maoecrystal V (1), a topologically complex polycyclic diterpene that possesses a previously unknown ring system (Figure 1).1 Apart from its fascinating structure, which includes vincinal quaternary stereocenters, a rigid [2.2.2] bicyclooctane core, and a highly strained tetrahydrofuran ring, maoecrystal V (1) displayed potent and highly selective activity against HeLa cells (IC50 = 60 nM). Maoecrystal V (1) was nontoxic towards several other cancer cell lines (K562, 0.19 M; A549, 0.8 M).
Figure 1.

The structure of maoecrystal V 1.
As a consequence of maoecrystal V’s challenging structure and interesting biological profile, several research groups have developed approaches towards 1,2 but to date a completed total synthesis has not been reported. We now describe our ongoing efforts towards maoecrystal V (1), which have resulted in a clear strategy for constructing the carbogenic framework of the natural product.
The underlying logic of our approach towards maoecrystal V (1) is outlined retrosynthetically in Scheme 1A.3 A number of retrosynthetic functional group interconversions (FGI’s) within the natural product led back to pentacyclic compound 2. We chose to mask the six-membered lactone as a cyclopentanone, reasoning that the lactone could be revealed by a late-stage oxidative cleavage of the indicated carbon–carbon bond within 2. While it was tempting to install the functionality required to form the cyclic ether at an early stage in the synthesis, we elected to pursue a strategy that called for late-stage introduction to more rapidly simplify subtargets. Thus, we disconnected the left-hand carbon–oxygen bond within 1, leading to tetracyclic compound 2. Our plan to form this carbon–oxygen bond in the synthetic direction involves remote functionalization induced by the free alcohol within 2.4 Further retrosynthetic functional group manipulations led us to [2.2.2] bicyclic structure 3, which we further simplified to diene 4 by application of a ketene equivalent Diels–Alder cycloaddition transform.5 The stereoselectivity of the forward cycloaddition would be controlled by the protected alcohol within 4, which clearly blocks dienophile approach from the back face. This Diels–Alder reaction is an alternative construction to that employed by the groups of Yang, Danishefsky, Baran and Nicolaou.2 Recognition of the cyclopentenone embedded within 4 keyed application of a Nazarov cyclization transform,6 generating the simplified precursor 5. In a forward sense, the conversion of enone 5 into the tetracyclic compound 3 would proceed through maximum use of sequential carbon-skeleton bond-forming reactions and fashion the congested vicinal quaternary stereocenters required for the natural product efficiently and directly.
Scheme 1.

Retrosynthetic analysis of maoecrystal V (1) and construction of the core rings.
Our synthesis (Scheme 1B) commenced with the regioselective Rubottom oxidation of 3,3-dimethylcyclohexanone (6),7 followed by conversion of the intermediate secondary alcohol to its corresponding tert-butyldimethylsilyl (TBS) ether (7, 70% yield over two steps). A Horner–Wadsworth–Emmons reaction, using phosphonate 8,8 afforded the Weinreb amide in 79% yield and as a single stereoisomer. Formation of the desired dienone 10 proceeded smoothly in 93% yield following the addition of vinyl anion 9 (generated from the corresponding bromide) to the Weinreb amide.9 Upon exposure to stoichiometric Fe(III) chloride,10 dienone 10 was transformed into the desired spirocyclic ketone 4 in good yield and as a single stereoisomer. Reduction of the carbonyl group proved necessary to facilitate the subsequent Diels–Alder reaction with nitroethylene11 and reduce byproducts due to oxidation of the cyclohexadiene ring. Thus, the [2.2.2] bicyclic ring system (i.e., 11) was produced (44% over two steps) as a single regioisomer, with only the endo-adduct detected by NMR spectroscopic analysis. Confirmation of the structure of 11 was obtained by single X-ray structure analysis, which revealed that, as planned, the OTBS substituent contolled the facial selectivity of both the Nazarov and Diels–Alder cyclizations.
Attempts to conduct a Nef reaction12 on 11 were met with minimal success at this juncture, although treatment with KOH resulted in complete epimerization of the nitro group – an outcome that facilitated stereoselective installation of the requisite alcohol (i.e., 13), yet led to some unexpected chemistry (see below). The desired substrate (i.e., 13) for our planned remote functionalization to form the ether ring was prepared by formation of ketone 12 from alcohol 11 (86% yield over three steps), followed by a completely stereoselective Rubottom oxidation of the thermodynamically most stable enol silane.13 The structure of 13 was confirmed by X-ray structural analysis. We next tested a number of conditions to bring about the desired ring-to-ring functionalization. Under a variety of conditions,14 such as those reported by Suárez and coworkers for similar transformations (PhI(OAc)2/I2, hν),14a alcohol 13 was converted to a compound whose structure initially proved problematic to assign. Ultimately, X-ray analysis established the product to be the [2.2.5] bicyclic ring system 14. While this structure was unexpected, its formation may be rationalized by formation of the desired oxygen centered radical, which in turn may undergo a Grob fragmentation due to favorable alignment of the broken σC-C with σ*C-N of the leaving group.15
At this juncture we elected to install the lactone before attempting to form the bridging cyclic ether. Initially, we attempted to conduct a Baeyer–Villiger reaction16 directly on ketone 13 (Scheme 2), reasoning that the congested nature of the structure might favor a conformation of the Criegee intermediate that would lead to selective migration of the desired bond.17
Scheme 2.

Baeyer–Villiger Reaction of 13 (P = TBS).
Exposure of 13 to trifluoroperoxyacetic acid, however, afforded lactone 15 in 52% yield. X-ray structure analysis revealed insertion had occurred on the wrong side of the carbonyl group, and that under the acidic reaction conditions the TBS ether had been cleaved thus inducing ketalization.
Attempts to generate the enol silane of ketone 13, as a prelude to oxidative cleavage were unsuccessful due to steric congestion. Likewise, attempted formation of the less substituted enol silane derived from ketone 12 (see Scheme 1B) led only to formation of the more substituted alkene, presumably as a consequence of severe steric hindrance. To avoid these issues, we elected to conduct a Rubottom oxidation on enone 16 (Scheme 3), which was derived in two steps from the initial Diels–Alder cycloadduct 11 (see Scheme 1B). Enol silane formation could be achieved upon the addition of TMSI and HMDS,13 and thus, following the addition of m-CPBA, α-hydroxy ketone 17 was accessed in 88% yield.18 Attempted oxidative cleavage of 17 led to complex reaction mixtures, while reduction of the enone to generate the saturated ketone proved capricious. Curiously, under a variety of conditions examined, the major product was cyclopropane 18, which could be reproducably formed in 71% yield upon exposure to Pd/C and H2.
Scheme 3.

Generation of the tetracyclic core 19 (P = TBS).
The formation of cyclopropane species 18 was completely unexpected and at this juncture we do not have firm evidence for a mechanistic rationale of the transformation. We speculate, however, that the close proximity and alignment of σ*C-N of the nitro group allows for an intramolecular alkylation of a presumed palladium enolate intermediate.19 We observed similar reactivity when Pd(OAc)2 was used as the catalyst, but Pd/C provided the greatest yield. Oxidative cleavage of 18 proceeded smoothly upon treatment with methanolic periodic acid, to afford the intermediate enal-acid, which was reduced with NaBH4. Lactonization occurred spontaneously under these conditions, providing 19 in 75% yield from ketone 18 and thus established the key carbocyclic elements of maoecrystal V (1).
In summary, we have devised a route to access the challenging core structure (i.e., lactone 19) of maoecrystal V in 13 steps from 3,3-dimethylcyclohexanone. Systematic application of powerful carbon–carbon bond forming reactions (i.e., Nazarov and Diels–Alder cyclizations) enabled rapid assembly of the central quaternary carbon stereocenters and the complex carbon framework. Continued work towards maoecrystal V (1) and preliminary biological studies are underway in our laboratories.
Supplementary Material
Acknowledgments
We gratefully acknowledge support from Northwestern University, the NIH (training grant T32AG000260) and the American Cancer Society by way of an institutional grant (ACS-IRG 93-037-15) to the Robert H. Lurie Comprehensive Cancer Center at NU. DXH was the recipient of a NU Undergraduate Research Grant. We thank Amy A. Sarjeant (NU) for help with X-ray analysis.
Footnotes
Supporting Information Available Experimental procedures, spectral data and crystallographic data (PDF, CIF). This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Li SH, Wang J, Niu XM, Shen YH, Zhang HJ, Sun HD, Li ML, Tian QE, Lu Y, Cao P, Zheng QT. Org Lett. 2004;6:4327–4330. doi: 10.1021/ol0481535. [DOI] [PubMed] [Google Scholar]
- 2.(a) Gong J, Lin G, Li CC, Yang Z. Org Lett. 2009;11:4770–4773. doi: 10.1021/ol9014392. [DOI] [PubMed] [Google Scholar]; (b) Krawczuk PJ, Schöne N, Baran PS. Org Lett. 2009;11:4774–4776. doi: 10.1021/ol901963v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Peng F, Yu ML, Danishefsky SJ. Tetrahedron Lett. 2009;50:6586–6587. doi: 10.1016/j.tetlet.2009.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Nicolaou KC, Dong L, Deng LJ, Talbot AC, Chen DYK. Chem Commun. 2010;46:70–72. doi: 10.1039/b917045f. [DOI] [PubMed] [Google Scholar]
- 3.Corey EJ. Angew Chem Int Ed. 1991;30:455–465. [Google Scholar]
- 4.For a review, see: Heusler K, Kalvoda J. Angew Chem Int Ed. 1964;3:525–596.
- 5.For a review on ketene equivalents for Diels–Alder cycloadditions, see: Aggarwal VK, Ali A, Coogan MP. Tetrahedron. 1999;55:293–312.
- 6.For recent reviews of the Nazarov cyclization, see: Pellissier H. Tetrahedron. 2005;61:6479–6517.Frontier AJ, Collison C. Tetrahedron. 2005;61:7577–7606.
- 7.(a) Rubottom GM, Vazquez MA, Pelegrina DR. Tetrahedron Lett. 1974;15:4319–4322. [Google Scholar]; (b) Hassner A, Reuss RH, Pinnick HW. J Org Chem. 1975;40:3427–3429. [Google Scholar]
- 8.Nuzillard JM, Boumendjel A, Massiot G. Tetrahedron Lett. 1989;30:3779–3780. [Google Scholar]
- 9.Nahm S, Weinreb SM. Tetrahedron Lett. 1981;22:3815–3818. [Google Scholar]
- 10.Denmark SE, Jones TK. J Am Chem Soc. 1982;104:2642–2645. [Google Scholar]
- 11.For a relevant example of a closely related Diels–Alder reaction using nitroethylene, see: Corey EJ, Myers AG. J Am Chem Soc. 1985;107:5574–5576.
- 12.For a review on the Nef reaction see: Ballini R, Petrini M. Tetrahedron. 2004;60:1017–1047.
- 13.Miller RD, McKean DR. Synthesis. 1979:730–732. [Google Scholar]
- 14.(a) Concepción JI, Francisco CG, Hernández R, Salazar JA, Suárez E. Tetrahedron Lett. 1984;25:1953–1956. [Google Scholar]; (b) Dorta RL, Francisco CG, Freire R, Suárez E. Tetrahedron Lett. 1988;29:5429–5432. [Google Scholar]; (c) Ceccherelli P, Curini M, Marcotullio MC, Mlari BL, Wenkert E. J Org Chem. 1986;51:1505–1509. [Google Scholar]
- 15.For a recent review of the Grob fragmentation, see: Prantz K, Mulzer J. Chem Rev. ASAP; 2010. http://pubs.acs.org/doi/pdf/10.1021/cr900386h.. See also: Ho T-L. Heterolytic Fragmentation of Organic Molecules. Wiley; New York: 1993.
- 16.Baeyer A, Villiger V. Chem Ber. 1899;32:3625–3622. [Google Scholar]
- 17.For an experimental study of the reactive conformations involved in Baeyer–Villiger reactions, see: Goodman RM, Kishi Y. J Am Chem Soc. 1998;120:9392–9393.
- 18.Rubottom GM, Gruber JM. J Org Chem. 1978;43:1599–1602. [Google Scholar]
- 19.For a paper regarding the structure of palladium enolates, see: Veya P, Floriani C, Chiesi-Villa A, Rizzoli C. Organometallics. 1993;12:4899–4907.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.