

Abstract
In prostate cancer (PCa) patients, initial responsiveness to androgen deprivation therapy is frequently followed by relapse due to development of treatment-resistant androgen-independent PCa. This is typically associated with acquisition of mutations in AR that allow activity as a transcription factor in the absence of ligand, indicating that androgen-independent PCa remains dependent on AR function. Our strategy to effectively target AR in androgen-independent PCa involved using a cell-based readout to isolate small molecules that inhibit AR transactivation function through mechanisms other than modulation of ligand binding. A number of the identified inhibitors were toxic to AR-expressing PCa cells regardless of their androgen dependence. Among these, some only suppressed PCa cell growth (ARTIS), while others induced cell death (ARTIK). ARTIK, but not ARTIS, compounds caused disappearance of AR protein from treated cells. siRNA against AR behaved like ARTIK compounds, while a dominant negative AR mutant that prevents AR-mediated transactivation but does not eliminate the protein showed only a growth suppressive effect. These observations reveal a transcription-independent function of AR that is essential for PCa cell viability and, therefore, is an ideal target for anti-PCa treatment. Indeed, several of the identified AR inhibitors demonstrated in vivo efficacy in mouse models of PCa and are candidates for pharmacologic optimization.
Introduction
Prostate cancer (PCa) tumors are composed primarily of prostate luminal epithelial cells. These cells express androgen receptor (AR), a transcription factor that controls differentiation of prostate luminal epithelial cells by driving expression of prostate-specific markers and controls survival of the cells through mechanisms that remain unclear 1–5.
More than 60 years ago, Huggins showed that depletion of androgens, ligands of AR, causes death of prostate luminal epithelial cells 6, thus demonstrating the critical role of the AR pathway in survival of prostate cells. Cancerous prostate cells continue to express AR and their survival also depends on the presence of androgens, which makes androgen deprivation the therapy of choice for patients with advanced PCa. Androgen withdrawal or anti-androgen therapy is applied in different forms, including castration (surgical removal of the testes, the major organ producing the androgen, testosterone), inhibition of testosterone synthesis or conversion to a more active form, dihydrotestosterone (DHT), AR antagonists (compounds binding to the AR LBD, but not causing AR activation) and AR inhibitors (see below and reviews 7, 8). There are steroid and non-steroid antagonists of AR (for review see 9). Steroid antagonists act by competing with androgens for binding sites in the AR LBD, while non-steroid antagonists alter the conformation of the LBD such that it cannot bind androgens 8. Thus, non-steroid antagonists are functionally similar to AR inhibitors, compounds with various effects on AR that lead to inhibition of AR function, but do not involve competition with androgen binding. The AR inhibitors or non-steroid AR antagonists currently used in the clinic are flutamide and bicalutamide. A new androgen antagonist that entered recently clinical trials is MDV3100 10. Importantly, the clinically available (flutamide, casodex) and newly discovered (MDV3100) AR inhibitors all bind to the LBD of AR 10. Anti-androgen therapies, including use of the inhibitors flutamide and bicalutamide, are usually effective initially, but rarely result in a complete cure. PCa relapse occurs in most of patients treated with such therapies, which leads to androgen-independent, chemotherapy-resistant tumors with poor prognosis 11. Thus, resistance to anti-androgen therapy is a major obstacle in successful treatment of PCa.
Analysis of the mechanisms of androgen-independence acquired by PCa during tumor progression indicates that loss of AR signalling is involved rarely 1. On the contrary, androgen-independent PCa is typically characterized by heightened AR activity due to expression of AR mutants that are ligand-independent (constitutively active) or responsive to non-androgen ligands 12–17. Increased rate of point in frame mutations localised predominantly in LBD of AR proposes role of AR in relapced castration resistant prostate cancer 12–14, 17.
Data confirming the critical role of AR in PCa consists of results of studies using various strategies to inhibit AR, such as microinjection of neutralizing antibodies or transfection of anti-sense or interfering RNAs targeting AR. Such strategies lead to inactivation of AR and suppression of growth or induction of death of PCa cells, including both androgen-dependent and -independent variants 1–5. These data experimentally confirmed dependence of androgen-independent PCa on the presence of functional AR.
It was recently shown that unlike normal prostate stem cells, prostate “tumor initiating cells” or “cancer stem cells”, a minor cell population believed to be the major source of self-renewing tumor cells, express functional AR 18. This, together with the observed maintenance of AR activity in PCa tumors that have progressed to the stage of castration resistance, suggests that AR is a highly promising potential therapeutic target on all stages of PCa.
In this study, we sought to isolate small molecules able to inhibit AR function in androgen–independent PCa cells with the expectation that such inhibitors would likely be toxic to PCa cells and therefore useful for development of new anti-PCa drugs. We deliberately designed our search for AR inhibitors to identify those targeting aspects of AR function distinct from AR–ligand interaction. Isolation of several small molecule inhibitors of AR with different mechanism of actions allow demonstrating of efficacy of AR inhibition for the treatment of PCa and demonstrated a new transcription independent role of AR in the control of survival of Pca cells.
Results
Identification of novel inhibitors of AR-dependent transcription
To identify new inhibitors of AR, we chose to screen a library of small molecules. We selected a cell-based readout approach for a number of reasons, including (i) extending of the potential target from AR protein itself to the entire AR pathway, which is not possible with an in vitro biochemical assay; (ii) absence of knowledge of a mechanism of AR control of cell survival; and (iii) absence of the crystal structure of full length AR.
We used PCa CWR22R cells, which proliferation is not affected by the absence of androgens and AR transcriptional activity is decreased minimally (compared to other PCa cell lines) as a result of androgen ablation 4. These cells expresses a truncated mutant of AR lacking the LBD (ARΔLBD) 19. More than 50% of the AR protein present in CWR22R cells is the ARΔLBD form (19 and Fig. 1), while the remainder is full length with LBD mutations 19. We reasoned that using cells in which much of the AR activity is due to a mutant lacking the LBD would increase the chances of isolating small molecule inhibitors that act through mechanisms other than inhibition of AR–ligand interaction.
Figure 1. Choice of CWR22R cells expressing ARΔLBD for AR inhibitor screening: ARΔLBD has ligand-independent AR function.

A. ARΔLBD is transcriptionally active in serum-free media (SFM). HeLa cells were transfected in SFM with the AR-dependent reporter construct pARE-Luc and wild type (wt) AR, ARΔLBD or negative control (empty vector) expression constructs. Luciferase activity was measured 24 hrs post-transfection. Error bars – standard deviations from three replicates within experiment B. ARΔLBD has constant nuclear localization. HeLa cells transfected in SFM as in (A) were left untreated or treated with 1nM dehydrotestosterone (DHT) for 24 hrs followed by immunofluorescent staining with anti-AR antibody. C–E. Substitution of the endogenous AR in CWR22R cells with ectopic ARΔLBD by combined lentiviral transduction of shRNA targeting the untranslated region of the endogenous AR transcript and an ARΔLBD expression construct. C. Western blotting of cell lysates prepared with or without transduction. Blots were probed with anti-AR and anti-actin antibodies. D. AR-dependent reporter assay (pARE-Luc) in original and transduced cells grown in SFM with addition of different concentrations of DHT. E. Growth rate (cell counts) of original and transduced cells in regular serum-containing medium. The assays shown in D and E were performed in triplicate. Error bars indicate standard deviation.
To demonstrate that ARΔLBD has ligand-independent AR function and is a suitable target for identification of inhibitors, we experimentally generated CWR22R cells in which only ARΔLBD is present by co-transduction of shRNA targeting the untranslated region of the endogenous AR transcript and an expression construct directing ectopic expression of ARΔLBD (Fig. 1). In these cells, the ARΔLBD mutant is constitutively active as a transcription factor and is localized to the nucleus even in the absence of any ligands (i.e., in steroid-free medium (SFM) made with charcoal-stripped serum Fig. 1A and B). We also demonstrated that while elimination of all AR protein is toxic for CWR22R cells 4, ectopically expressed ARΔLBD can support proliferation of CWR22R cells in the absence of full length AR (Fig. 1C–E). These results indicate that the ARΔLBD mutant form of AR has the same transcriptional and pro-survival activity as the full length protein, but without the requirement for ligand.
AR activity in CWR22R cells was monitored by integrated AR-dependent luciferase reporter (ARE-Luc) 4. From a library of 34,000 small molecules applied on a readout cells we selected compounds that inhibited luciferase activity least two-fold after 24 hrs incubation. These compounds were tested for the absence of non-specific inhibition transcription, translation, luciferase activity and non-specific toxicity on non-PCa cells (HT1080), expressing luciferase under CMV promoter (data not shown).
To confirm the AR-inhibitory activity of compounds on an endogenous AR target gene, we assessed the effect of compounds on Prostate Specific Antigen (PSA) expression in a different androgen-independent PCa cell line, C4-2 20,21, to exclude compounds specific for one cell type. Compounds that inhibited PSA secretion by C4-2 cells by >60% were selected for further analysis (14 compounds, Table 1). The selected compounds inhibited AR transcriptional activity in both cell lines (CWR22R and C4-2) in the presence of steroids naturally present in FBS (Table 1) or in the presence of up to 10nM dihydrotestosterone (DHT) in SFM (data not shown). Therefore, the selected compounds are inhibitors of AR transcriptional activity in at least two models of androgen-independent PCa.
Table 1.
AR inhibitors described in the study
| Name | ID | Structure | Activity of ARE-Luc in CWR22R11 cellsa | PSA secretion in C4-2 cellsb | Survival of cells in the presence of 10μM of a compound (DMSO=100%)c | Inhibition of GR or MR | Effect on PCa cellse | Decrease of AR levelf | Type of inhibitor | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % of DMSO | % of DMSO | LNCaP | C4-2 | CWR22R | Non-PCa (average)d | |||||||
| C5 | 6099112 | 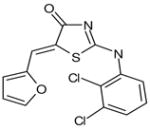 |
33 | 44 | 18 | 44 | 5 | 100 | no | A | no | ARTIS |
| C6 | 5652306 | 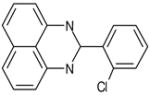 |
30 | 0 | 24 | 53 | 65 | 100 | no | A | no | ARTIS |
| C8 | 5771015 | 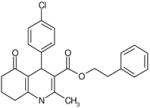 |
46 | 0 | 16 | 93 | 63 | Bic-like | ||||
| C11 | 6005978 | 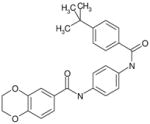 |
41 | 14 | 27 | 58 | 17 | 100 | no | A | no | ARTIS |
| C16 | 6605765 | 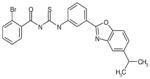 |
49 | 14 | 34 | 104 | 77 | Bic-like | ||||
| C17 | 6611064 | 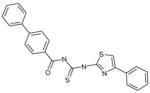 |
28 | 0 | 30 | 55 | 45 | 100 | no | A | no | ARTIS |
| C18 | 5935083 | 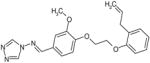 |
40 | 0 | 23 | 56 | 41 | 100 | no | A | no | ARTIS |
| C47 | 6463344 | 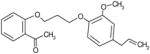 |
19 | 20 | 48 | 100 | 100 | Bic-like | ||||
| C52 | 5582367 |  |
27 | 11 | 53 | 36 | 10 | 92 | no | D | yes | ARTIK |
| C55 | 6104475 |  |
33 | 20 | 73 | 102 | 100 | Bic-like | ||||
| C61 | 5857595 |  |
30 | 22 | 68 | 97 | 94 | Bic-like | ||||
| C66 | 5872746 |  |
42 | 0 | 10 | 16 | 5 | 59 | yes | Toxic compound | ||
| C73 | 6443213 | 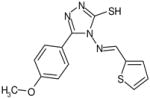 |
32 | 0 | 55 | 99 | 92 | Bic-like | ||||
| C85 | 6028717 | 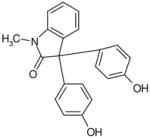 |
29 | 0 | 22 | 20 | 35 | 100 | no | D | yes | ARTIK |
| Bic | 43 | 0 | 19 | 91 | 100 | no | ||||||
| shAR6 | 9 | 3 | 13 | 26 | 39 | 100 | no | D | yes | |||
luciferase activity in CWR22R-ARE-Luc cells incubated for 24h in the presence of 10μM of compounds presented as a percentage of luciferase activity in cells includated with 0.1% DMSO Effect of shAR6 was measured at 72h.
level of PSA in the medium of C4-2 cells incubated for 72h in the presence of 10μM of compounds presented as a percentage of the PSA level in cells incubated with 0.1% DMSO. Effect of shAR6 was measured at 72h.
the number of colonies grown in the presence of 10μM of compounds presented as a percentage of colonly formation in the presence of 0.1% DMSO.
average of three cell lines: HeLa, HT1080 and DU145.
classification of compound effect on CWR22R cells: A – growth arrest, D – death
results of Western blotting and immunofluorescent staining with anti-AR antibodies
White cells contain acceptable parameters for further testing, light grey - borderline acceptable, dark grey cells – non-acceptable.
All numbers in the table are presented as rounded mean of two independent experiments, done in triplicates. Confident intervals were <10% and are omitted from the table.
Effect of the selected hits on the growth of PCa cells
To test the toxicity of 14 AR inhibitors towards cells of different origins, we performed colony formation assays using a single dose of each compound (10μM) on several PCa (LNCaP, C4-2, CWR22R and DU145) and two non-PCa (HT1080 and HeLa) cell lines (Fig. 2A). With the exception of a single compound (c66), the selected small molecules did not affect the growth of non-PCa cells or AR-negative PCa cells (DU145). Almost all of the molecules tested, as well as the positive control bicalutamide (an AR inhibitor currently used in the clinic), suppressed growth of AR-expressing androgen-sensitive LNCaP cells to different degrees (Fig. 2A). At the same time, bicalutamide did not suppress growth of either C4-2 or CWR22R cells (AR-expressing, androgen-independent cell lines), while some of the selected molecules did. Thus, we identified several compounds (c5, c6, c11, c17, c18, c52, c85) with different degrees of toxicity towards three AR-expressing PCa cell lines but no toxicity towards non-PCa cells or AR-negative cells (Table 1). However, several compounds (c8, c16, c47, c55, c61, c73) behaved similarly to bicalutamide in that they suppressed growth of LNCaP cells, but not CWR22R or C4-2 cells. These compounds were not weaker inhibitors of AR transcriptional activity in CWR22R or C4-2 cells than compounds that inhibited growth of the cells (Table 1). These findings demonstrate that inhibition of AR-dependent transcriptional activity is not, in itself, sufficient to suppress growth of androgen-independent PCa cells.
Figure 2. Toxicity of AR inhibitors to different cells.

A. Colony formation assay of indicated cells treated with different chemicals for 7 days. B. Comparison of EC50 (inhibition of AR-dependent reporter in CWR22R cells) and IC50 (toxicity to CWR22R cells) of the hits. C. Dose-dependent toxicity of several hits to a panel of cell lines of different origins. Error bars in A and C – standard deviation between triplicates within experiment; in B – confidence intervals of EC50 and IC50. See Material and Methods for details.
Relation of compounds toxicity to inhibition of AR activity was further demonstrated by comparison of 50% effective concentration (EC50, inhibition of AR-dependent luciferase activity in CWR22R cells measured 24h after compound addition) and 50% inhibitory concentration (IC50, inhibition of growth of CWR22R cells measured 6 days after compound addition (Fig. 2B). Importantly, none of the compounds demonstrated toxicity at 24h (data not shown). This is similar to the delayed toxic effect of AR-targeting siRNA on these cells, which becomes apparent 4–6 days after application of the siRNA (4). Figure 2B shows that the EC50 for inhibition of the AR-dependent reporter at 24h is within the same range as the IC50 for toxicity at 6 days. This demonstrates that inhibition of AR-dependent transcription occurs before toxicity and that there is a correlation between AR inhibition and toxicity to PCa cells for those compounds that are toxic to PCa cells. These results, together with the fact that some AR inhibitors are not toxic, suggests that there are different modes by which AR can be inactivated: (i) inhibition of only the transcriptional activity of AR is not toxic for androgen-independent PCa cells (similar to the effect of androgen withdrawal); (ii) inhibition of AR transcriptional activity together with some other transcription-independent function of AR that controls survival of PCa cells is toxic to both androgen-dependent and – independent PCa cells; and (iii) inhibition of only the postulated transcription-independent pro-survival function of AR without affecting AR-mediated transcription would be expected to be toxic to all PCa cells, although such inhibitors were not identified so far. Since our readout system was based on monitoring AR-dependent transactivation, compounds acting through the hypothetical third mode could not be isolated in our screen.
We also tested the effect of the selected compounds on a larger group (n=10) of non-target cell lines (non-PCa cell lines and PCa cells that do not express AR). Examples of these data are shown in Figure 2C. Selected hits were toxic only to target cells and non-toxic to AR-negative cells at all concentrations tested. Thus, we can conclude with a high level of confidence that the selected hits that display toxicity are toxic only to PCa cells expressing AR; and therefore, the toxicity of these compounds is likely to be related to their effect on AR.
Observation of compounds effect on PCa cells allowed separation of small molecules into two categories: compounds c52 and c85 kill practically all of the PCa cells in the culture by day 7; in contrast, other compounds caused reduced growth as compared to untreated control cells, but not obvious cell death This suggests that a subset of the compounds suppress proliferation of PCa cells, but do not kill them. To test this possibility, we assessed the cell cycle distribution of CWR22R cells treated with both categories of compounds. As expected, treatment with compounds of the first type (c52, c85) leads to the appearance of cells with sub-G1 DNA content characteristic of apoptotic cells (Fig. 3D). In contrast, treatment of the cells with representative compounds of the second type (c5, c6, c11) did not result in appearance of cells with sub-G1 DNA content, but did lead to changes in cell cycle distribution indicative of cell cycle arrest (an increase in the proportion of cells in the G1 phase of the cell cycle and a decrease in the proportion of cells in S phase). Therefore, different subsets of compounds inhibiting AR-dependent transcription in androgen-independent PCa cells cause either growth arrest or apoptosis in these cells. Whether a compound causes growth arrest or apoptosis is not determined by the potency of the compound as an inhibitor of AR-dependent transcription (Table 1 and Fig. 2B).
Figure 3. Reduction of AR protein levels is associated with toxicity of the selected AR inhibitor hits to PCa cells.

A. Western blotting of lysates from CWR22R cells treated with 10mM of the indicated hits for 36 hrs. AR antibody reveals the full length and DLBD forms of AR naturally present in these cells. GAPDH antibody was used to control for protein loading. B. Unlike parental CWR22R cells, a clone selected in the presence of c52 (c52R) is resistant to the effects of c52. AR protein expression in the c52R clone of CWR22R is not reduced by c52 treatment. Western blotting of parental and c52-resistant CWR22R cells with or without c52 treatment (10mM for 36 hrs). C. Cell viability measured by methylene blue staining 4 days after compound addition. Error bars – standard deviation between triplicates within experiment. D. Effect of the selected compounds on cell cycle distribution of CWR22R cells measured by propidium iodide staining of cellular DNA content by fluorescent activated cell sorting. Assay was done twice. Error bars – deviation between two measurements.
Correlation between the toxicity of compounds towards PCa cells and their effect on AR protein levels
Based on the results described above, we can distinguish three categories of compounds among the inhibitors of AR-dependent transcription identified in our screen (Table 1): (i) bicalutamide-like compounds that suppress growth of androgen-sensitive LNCaP cells but do not affect growth of androgen-independent C4-2 or CWR22R cells (c8, c16, c47, c55, c61, and c73); (ii) compounds that suppress growth of all (androgen-dependent and – independent) PCa cells expressing AR (c5, c6, c11, c17, and c18); and (iii) compounds that kill all PCa cells expressing AR (c52 and c85). The AR inhibitors in the first category likely target ligand binding and, therefore, were not within the focus of our study. The other two categories were of interest as they appeared to represent new types of AR inhibitors for potential treatment of androgen-independent PCa and we named them ARTIS (AR Transcription Inhibitors – Suppressive, including compounds c5, c6, c11, c17, c18) and ARTIK (AR Transcription Inhibitors – Killing, including compounds c52 and c85).
In an effort to understand why ARTIS and ARTIK compounds have different effects on the growth of PCa cells, we compared AR protein levels in CWR22R cells treated with both categories of compounds as well as several hits of the first category that are not toxic to PCa cells. This analysis showed that treatment with ARTIKs (c52 and c85) led to complete disappearance of AR protein from the cells. In contrast, treatment with ARTIS compounds or non-toxic AR inhibitors did not affect the level of AR protein in the cells (Fig. 3A, Table 1). This supports our hypothesis that AR controls survival of androgen-independent PCa cells through a transcription-independent mechanism.
The critical importance of AR elimination for the toxic effect of one ARTIK compound was demonstrated by in vitro selection of a CWR22R clone resistant to ARTIK toxicity (Fig. 3B, see Material and Methods for details). A single clone was obtained after growth of 107 cells in the presence of c52 and this clone retained expression of AR even when grown in the constant presence of c52 (Fig. 3C). No clones resistant to compound c85 were obtained.
As an additional test of our hypothesis that a transcription-independent function of AR controls survival of PCa cells, we compared the effects of two gene constructs on the survival of CWR22R cells. ShRNA targeting AR causes suppression of AR expression leading to elimination of AR protein similar to ARTIK compounds 4. On the other hand, the AR mutant, HD1-KRAB-AR122, in which part of the AR transactivation domain (aa 1–122) is substituted with the transrepression domains of KRAB and HDAC1, does not affect endogenous AR protein levels, but completely abolishes AR-dependent transcription by competing with endogenous AR protein for binding to ARE DNA sequences 22. These two constructs were tested for their effect on AR-dependent transcription over a range of doses in CWR22R cotransfection assays with the ARE-Luc reporter (Fig. 4A) and for their effect on CWR22R survival in colony formation assays (Fig. 4B). Both constructs almost completely suppressed AR-dependent luciferase expression in CWR22R cells, with the HD1-KRAB-AR122 mutant being slightly more potent than shRNA constructs upon transfection of an equal amount of DNA. However, as shown previously, shRNAs against AR suppressed growth of CWR22R colonies 4, while the HD1-KRAB-AR122 construct did not. This situation mimics that observed with our panel of AR inhibitory small molecules and provides additional support for the hypothesis that AR controls survival of PCa cells through a transactivation-independent function that has not been characterized previously.
Figure 4. Comparison of the effects of shRNAs targeting AR and the AR mutant HD1-KRAB-AR122 on AR-dependent transcription and survival of AR-expressing PCa cells.

A. Reporter assay on CWR22R cells cotransfected with pARE-Luc and different amounts of shRNA or AR mutant HD1-KRAB-AR122 constructs as shown on the x-axis. The total amount of DNA in the transfection mixture was kept equal by adding shControl vector DNA. Transfection efficiency was normalized by cotransfection of pCMV-b-gal construct. Assays were run in duplicate. B. Colony formation assay on CWR22R cells transfected with the indicated constructs and selected for puromycin resistance. Assay was done in duplicate. Error bars – deviation of the results between two measurements.
Analysis of potential effects of AR inhibitors on other steroid receptors
Several steroid receptors (SR) are highly homologous, including AR, and the estrogen (ER), progesterone (PR), glucocorticoid (GR) and mineralcorticoid (MR) receptors 23, 24. The ligand-activated forms of these receptors all recognize and bind to the same DNA sequence element 23. Attempts to isolate AR-inhibitory compounds acting downstream of receptor-ligand interaction (such as our screen) could, in theory, lead to isolation of molecules capable of inhibiting all of these SR. Inhibition of ER or PR is not expected to be a serious problem for PCa treatment, since these receptors do not control vital functions in men. However, the GR and MR systems are functional in men and their inhibition by a cross-reactive AR inhibitor could cause adverse side effects, especially with chronic administration.
To determine whether any of our AR-inhibitory compounds also inhibit GR or MR, we first tested whether the CWR22RARE-Luc reporter system used for our screen is responsive to steroids other than androgens, since the screening was done in the presence of FBS which might contain ligands of GR and MR. We treated CWR22RARE-Luc cells in SFM with either the GR ligand dexamethasone (Dex) or the MR ligand aldosterone (Ald) and measured luciferase activity in the presence or absence of the selected compounds. Both Dex and Ald induced activity of the ARE-Luc reporter in CWR22R cells. Moreover, all selected AR-inhibitors suppressed this activity and the extent of suppression was proportional to their effect on DHT-induced ARE-Luc activity (data not shown).
CWR22R cells express not only ARΔLBD, but also full length AR containing LBD mutations that could affect its ligand specificity (19 and Fig. 1). To test whether the observed induction of luciferase by Dex and Ald is due to activation of AR by these ligands, we used an AR-specific siRNA to block AR (both ΔLBD and full length) expression in CWR22R cells. ARE-Luc activation by DHT, Dex and Ald was suppressed in these cells as compared to cells transfected with a control siRNA, which illustrates the promiscuous nature of the LBD-mutated AR present in CWR22R cells (Fig. 5A). This experiment demonstrated that the selected hits are able to inhibit AR even if it is stimulated by ligands other than DHT. At the same time, the data do not completely exclude the possibility that the molecules affect GR and/or MR activity in other cells.
Figure 5. Effect of AR inhibitors on the activity of steroid receptors.

A. ARE-Luc reporter activity in CWR22R cells can be induced by different steroids (DHT – dehydrotestosterone, Dex – dexamethasone, Ald – aldosterone), but depends on AR expression. siRNAs targeting AR or GAPDH (control) were transfected into CWR22R-ARE-Luc cells in SFM. The indicated concentrations of steroids were added to the transfected cells and luciferase activity was measured 24 h later. B. Effect of hit compounds on luciferase activity in MDA-MB-453-MMTV-Luc cells induced by DHT, Dex or Ald shown as %of DMSO control. X-axis – concentration of the hits in mM. Error bars show standard deviation between replicates within experiment.
MDA-MB-453 breast cancer cells express GR, MR and AR 25. These cells with an MMTV-Luc reporter construct containing a DNA binding element recognized by all three SR in its promoter region were stimulated with different ligands in the presence of candidate compounds. In this system, all compounds except for c66 demonstrated specific inhibition of DHT-stimulated reporter activity, with no effect on Dex- or Ald-stimulated MMTV-Luc (Fig. 5B). Therefore, we conclude that most of the selected hit molecules are specific AR inhibitors that do not have activity even against the highly homologous GR and MR (Table 1).
Effects of AR-inhibitory compounds in vivo
Five compounds, representing both the ARTIS and ARTIK categories (c6, c11, c17, c52 and c85), were selected for in vivo tests based on all of the previous assays. Compound c5 was rejected due to very low microsomal stability (<5% after 1 hour incubation with rat microsomes). The toxicity of the compounds was evaluated in a dose escalation assay in which male NIH Swiss mice (n=4) received three sequential intraperitoneal (i.p.) injections of each chemical delivered in 3 day intervals. The first dose was 20-fold higher than the in vitro IC50 (20×IC50) for the compound in CWR22R cells recalculated based on mouse weight (see Materials and Methods) and the second and third doses were 50×IC50 and 100×IC50, respectively. On the day after the last injection, half of the mice in each group were sacrificed for gross pathology examination and collection of blood for serum biochemistry assays, which include total protein, glucose, bilirubin, and creatinine levels, activity of liver enzymes, and ion concentrations. The remaining mice were sacrificed and evaluated similarly 7 days after the last compound injection. No abnormalities were observed in any of the compound-injected mice upon either visual inspection of live mice or gross pathology examination. A minor decrease in serum glucose concentration (70–80% of control) was observed on the day after the last compound injection in almost all mice, but glucose levels were normal a week later (data not shown). Low serum glucose level might reflect of a weak transient GR-inhibitory effect of the selected compounds. Overall, however, this experiment demonstrated that the tested doses of compounds are non-toxic in mice and therefore suitable for in vivo efficacy testing.
We evaluated the potential anti-tumor effect of the compounds using C4-2 and CWR22R xenograft models of androgen-independent PCa in nude mice. Groups of C4-2 tumor-bearing mice (4–5 mice per group, each with two tumors) were given 6 daily i.p. injections of compound (100×IC50 dose) or DMSO vehicle. Under this treatment regimen, compounds c6 and c11 demonstrated clear suppression of C4-2 tumor growth, while c17, c52 and c85 did not (Fig. 6A).
Figure 6. Anti-tumor activity of the selected hits in mouse models of androgen-independent PCa.

A. Compounds or DMSO vehicle were injected i.p. into nude mice carrying subcutaneous (s.c.) C4-2 tumors. Six daily injections were given starting when tumors were around 100mm3 in size. There were 4–5 mice per group with two tumors per mouse. Asterisks (*) and number signs (#) are placed under the means of measurement in the c6 and c11 groups respectively which are statistically different from the control group by ANOVA test (p<0.05) B. C52 (9mg/kg or 18mg/kg) or DMSO vehicle was injected into the tail vein of nude mice carrying s.c. CWR22R tumors. Five daily injections were given starting when tumors were around 40mm3 in size. There were 10 mice per group with one tumor per mouse. Asterisks (*) are placed under the means of measurement which are statistically different from the control group by ANOVA test (p<0.05). Error bars in both A and B – standard deviation between the size of tumors within a given treatment group.
ARTIK compounds, c52 and c85, were also tested in the CWR22R model. Intravenous administration of compounds was done to avoid first liver pass of metabolically not very stable compounds, c52 – 17% and c85 – 35% of compound left after 1 hour incubation with rat liver microsomes. Five daily injections of c52 at a dose of 18mg/kg/day (equivalent to 200×IC50) suppressed growth of subcutaneous CWR22R tumors (Fig. 6B) without any noticeable adverse side effects. Compound c85 did not demonstrate reliable suppression of tumor growth in this model (data not shown). Among 10 mice treated with 18mg/kg/day of c52, tumors completely disappeared in 2 mice, regressed in 1 mouse and did not grow in size in 5 mice. In contrast, 9 out of 10 control animals displayed progressively growing tumors and 1 mouse had a slowly growing tumor.
These in vivo results indicate that representatives of both ARTIK and ARTIS (compounds c6, c11 and c52) are generally non-toxic, yet block growth of androgen-independent PCa tumors and, therefore, have potential as candidate therapeutic agents against PCa. It is likely that pharmacological optimization could significantly improve the anti-tumor efficacy of these compounds as well as reveal in vivo efficacy of other hit compounds that did not suppress tumor growth in these experiments.
Discussion
The significance of AR as a target for PCa treatment has been appreciated for more than 60 years 6. However, the majority of drug discovery efforts has been focused on modulation of one particular aspect of AR function – androgen binding. Anti-androgen treatment of PCa, while often initially successful, almost always fails in the long-term due to selection of cells that have acquired AR mutations relaxing ligand specificity 2, 12, 14–17, 26. This, together with data from experiments using neutralizing antibodies, antisense RNA and RNAi to test the importance of AR in PCa cells of all disease stages 1–5, confirms the critical role played by AR in both androgen-dependent and – independent PCa and supports targeting of the AR pathway as a potentially effective PCa treatment.
The approach that we took to target AR was based on a different rationale and utilized different strategies than other reports describe. Attar et al. reported rational design of new non-steroidal AR inhibitors based on resolution of the crystal structure of bicalutamide bound to the LBD of AR 27. These new inhibitors have improved AR-binding properties as compared to bicalutamide and, while bicalutamide is active against some mutant forms of AR, the new compounds are more toxic to androgen-independent PCa cells 27. Nevertheless, this approach is a variant of therapy targeted against the AR LBD and is therefore likely to be limited by the mutability of the LBD.
Charles Saywers’ group found several years ago that upon selection of androgen-sensitive LNCaP cells for the ability to grow in the absence of androgens, the most prevalent genetic change was amplification of the AR gene13. The authors postulated that the observed amplification was selected since it compensated for the low activity of AR in the absence of ligand by augmenting the number of AR molecules per cell. The same group recently reported that they used these cells to select a small molecule with activity against androgen-independent PCa 10. This molecule, MDV3100, has demonstrated efficacy in decreasing PSA levels in a Phase I clinical trial in patients with castration resistant PCa 10. However, the mechanism of activity of MDV3100 against AR is different from what we expect for AR inhibitors isolated by our approach. MDV3100, as well as its close homologue RD162, are both non-competitive antagonists of AR that bind with high affinity to the AR LBD 10. The binding affinity of MDV3100 (and RD162) is the closest to that of androgen that has been achieved so far for an antagonist. Thus, these compounds prevent activation of AR by androgens with superior potency as compared to the AR antagonists available in the clinic, such as bicalutamide and flutamide 10. In addition, the latter molecules are hampered by a partial agonist activity which is not displayed by MDV3100 or RD162 10. Potential problem with these new compounds may come from the fact they also bind LBD and may induce selection of mutations in LBD as casodex does.
Jones et al. selected compounds in a cell-based assay by their ability to block conformational changes in AR needed for AR activation 28. They isolated two AR inhibitors with unknown mechanisms of activity. One blocks AR-DNA interactions and another inhibits AR-dependent gene expression. Another approach is the design of fusion molecules between two small molecules: selective androgen receptor modulator (SARM) and FK506. The FK506 chemical moiety brings to the candidate drug the ability to attract FK506 binding protein (FKBP), a known scaffolding protein. Thus, the SARM-FK506 fusion molecule brings FKBP into close proximity to the AR LBD where it is able to completely abrogate pro-activation conformational changes in AR including C-N terminus interaction and binding of coactivators. Several other approaches reported in the literature were also based on inhibition of the transcriptional activity of full length AR with wild type or mutant LBD 29, 30. Different types of AR inhibitors working through different mechanisms or targeting different parts of the AR pathway (including our new ones) could be used in combinations for improved anti-cancer effects.
One of the unexpected findings of our screen was that loss of AR transcriptional activity does not necessarily lead to inhibition of growth or survival of androgen-independent PCa cells. Almost all of the molecules that we identified as inhibitors of AR-dependent transcription also inhibited growth of LNCaP cells, thereby confirming that AR transcriptional activity is important for growth of ligand-dependent PCa cells. However, only some compounds had an effect on growth of androgen-independent PCa cells (C4-2 and CWR22R). Importantly, the few compounds that did suppress growth of all PCa cells expressing AR (both androgen-dependent and – independent) were not necessarily more potent as inhibitors of AR-dependent transcription than molecules showing no effect on growth of androgen-independent PCa cells. This suggests that another, so far uncharacterized, function of AR besides transcriptional regulation is important for survival of PCa cells. These results also suggest that the transcriptional regulation function of AR can be inhibited specifically using small molecules without elimination of this other postulated pro-survival function.
Further characterization of the AR-inhibitors displaying toxicity against androgen-independent PCa showed that they could be clearly separated into two categories, those that kill PCa cells and those that only suppress their proliferation. In addition, the effect of the compounds on AR protein levels correlated with the type of toxicity that they induced: compounds causing elimination of AR protein killed androgen-independent PCa cells while compounds having no effect on AR level only suppressed their growth. This data again may be explained by the presence of a transcription-independent function of AR that controls survival of PCa cells and is only completely abolished when AR protein is eliminated from the cells. The mechanism underlying the anti-proliferative effect of compounds that do not reduce AR protein levels remains to be elucidated. One potential explanation could be that they somehow affect an anti-proliferative function of AR, which is normally growth-suppressive for many types of cells including PCa cells 4. Alternatively, they could inhibit a pro-survival function of AR, but not strongly enough (as compared to complete elimination of AR protein) to kill PCa cells. It is possible that the postulated pro-survival function of AR requires AR interaction with a number of molecular partners and that compounds which disrupt interaction with just one of these partners cannot fully eliminate this function, but can attenuate it (producing growth suppression but not cell killing).
The idea that elimination of AR protein, not just inhibition of its transactivation function, is required to induce prostate cancer cell death is supported by the different effects of castration on normal prostate epithelial cells expressing wild type AR as compared to prostate tumor cells expressing AR with mutations in the LBD. Wild type AR is unstable in the absence of androgen, particularly if its binding partner HSP90 is limited 31. Thus, castration results in loss of AR protein and apoptosis in normal prostate epithelial cells 32. In contrast, since AR LBD mutations not only affect the specificity of ligand binding but also promote stability of AR in the absence of androgens 21, castration does not kill all PCa cells, but suppresses proliferation of cells with mutated LBD.
The existence of a transcription-independent role of AR that controls survival of PCa cells is further supported by our finding that shRNA-mediated elimination of AR reduced colony formation by PCa cells while expression of the transactivation-deficient dominant negative HD1-KRAB-AR122 AR mutant did not.
Based on the literature, there are several proteins that interact with AR and have the potential to mediate an effect of AR on survival of PCa cells. For example, p53 is a strong pro-apoptotic factor in PCa cells and its binding to AR could suppress p53 pro-apoptotic function 33. Elimination of AR may “unleash” p53 in PCa leading to apoptosis.
Another hypothesis may be that AR control PCa survival by repression of some genes. Toxic compounds block both the transactivation and transrepression functions of AR, while non-toxic compounds affect only the transactivation function. Additional studies are needed to test this hypothesis.
Thus, our data support the possibility that AR has a previously unrecognized transactivation-independent function that impacts prostate cell survival. Recent study demonstrated that protection of PCa cells from apoptosis by AKT involves phosphorylation and inhibition of FKHR and related FOXO forkhead transcription factors 34. AR represses FKHR function in PCa cells by blocking its DNA binding activity and impairing its ability to induce Fas ligand expression and PCa cell apoptosis and cell cycle arrest 34. By this mechanism AR can impact PCa cell survival independently of transactivation. It will be important to assess the effect of our newly identified AR inhibitors on the AKT/FKHR pathway to determine if this is the mechanism underlying their anti-PCa activity.
A number of compounds that were toxic to androgen-independent PCa cells in vitro did not have any noticeable effect on in vivo tumor growth in the mouse models that we examined. Two potential explanations for this are: (i) these compounds have not been pharmacologically optimized and we do not have data on their bioavailability; and/or (ii) the mechanism of action of these compounds is not as important in vivo as in vitro. Compound C52 did not show in vivo efficacy in our experiment with the C4-2 model, but was effective in our experiment with CWR22R tumors. This difference may be due to our use of a higher compound dose and different route of administion (i.v. versus i.p.) in the latter experiment. Future studies will aim to clarify this by optimizing the pharmacological properties of the compounds and schedules and routes for their administration
In summary, we have identified several new compounds, which represent a new class of AR inhibitors since they do not target AR-ligand interaction. Therefore, it is likely that they will be active against AR regardless of LBD mutations that develop in response to standard anti-androgen therapy and may present new hope for patients with advanced PCa. With the ultimate goal of moving this class of AR inhibitors into clinical use, our next studies will focus on pharmacological optimization of the selected molecules and further investigation of the biological effects and mechanism(s) of action of growth suppressive and pro-apoptotic compounds.
Experimental Procedures
Cell lines
LNCaP, DU145, MDA-MB-453-MMTV-Luc, HeLa, ACHN, MRC5 and HT1080 cells were obtained from ATCC. RCC45 and normal kidney epithelial cell (NKE) were described in 35. Immortalized NKE-hTERT cells were obtained by transduction of human telomerase subunit in pBabe-puro vector. CWR22R and C4-2 cells were provided by Dr. Warren Heston (Department of Cancer Biology, Cleveland Clinic). The pARE-Luc reporter construct and CWR22R-ARE-Luc reporter cells were described already 4. Although the consensus DNA element bound by AR is also recognized by several other steroid receptors, the pARE-Luc reporter utilizes a region of the probasin promoter that is known as one of the most AR-specific promoter regions 4
All prostate cell lines and MDA-MB-453-MMTV-Luc were cultured in RPMI 1640 media supplemented with 10% FBS, 1mM sodium pyruvate, 10mM HEPES buffer, 55nM β-mercaptoethanol and antibiotics. HeLa and HT1080 cells were maintained in DMEM media with 10% FBS and antibiotics. Phenol red-free media and charcoal-stripped serum (CSS) were used to generate steroid-free media (SFM). CSS was purchased from Biosource (Rockville, MD).
Plasmids
The pTZV-wtAR and pTZV-ARΔLBD lentiviral expression constructs were obtained by subcloning of wild type full length (1–919 aa) AR or the AR fragment corresponding to the first 639 aa of human AR into the pTZV-CMV vector 4. The pLPCPw-shAR lentiviral vector directing expression of shRNA against human AR was generated as described 4. The shRNA sequence corresponding to the non-translated region of human AR was TGATCCTCATATGGCCCAG. pHD1-KRAB-AR122 was obtained from Dr. G. Jenster (Department of Urology, Josephine Nefkens Institute, Erasmus MC, Rotterdam, the Netherlands) and was already described 22. The pLV-CMV-Luc vector was provided by Dr. Peter Chumakov (Department of Molecular Genetics, Cleveland Clinic).
Lentiviral transduction and siRNA transfection were performed as described 35
Chemicals
The 34,000 DiverSet Chemical Library (consisting of small polycyclic molecules of molecular weight around 500 Da) was purchased from Chembridge Chemical Corporation (San Diego, CA). DHT was obtained from Cleveland Clinic Pharmacy Department (Cleveland, OH). Aldosterone, dexamethasone, BSA, RNase A and propidium iodide were purchased from Sigma-Aldrich (St. Louis, MO). Casodex (bicalutamide) was purchased from TRC (Toronto, Canada).
Primary Chemical Screening
Chemicals from the DiverSet Library were applied to a monolayer of CW22R-ARE-Luc cells in 96-well plates at a final concentration of 20μM. The next day, luciferase activity was read using BrightGlo Luciferase Assay System (Promega, Madison, WI) and the percentile of the reporter activity in chemical-treated cells was calculated (as compared to cells treated with DMSO taken as 100%). Compounds for which this parameter was 50% or lower were considered primary hits. The list of hits was compared with those obtained from other screenings of the same library which included inhibitors of reporters driven by promoters containing p53, NF-κb, or E-box binding elements and compounds toxic to HeLa, melanoma and neuroblastoma cells. AR-inhibitory hits that were also present on any of these other lists were excluded from further consideration. Non-specific inhibition of transcription/translation or luciferase activity was tested in HT1080 cells with the CMV-luc reporter and hit compounds active in this system were eliminated from further consideration.
Dose-dependent inhibition of reporter activity
CWR22R cells were plated at 5×104 cells per well in 96-well plates. Compounds were added in a dose range from 1 to 20000nM in two-fold increments. Each dose was tested in duplicate. Controls were 0.1% DMSO and 15μg/ml actinomycin D. Luciferase activity was measured 24h after compound addition by Bright Glo assay (Promega, Madison, WI). The assay was run two times and the effective concentration of compound inhibiting reporter activity by 50% as compared to DMSO (EC50) was calculated by the sigmoid approximation method using CalcuSyn software (Biosoft, Chembridge, UK). Confidence intervals were calculated for each sigmoid.
Cell Colony Assay
Cells were plated in 24-well plates at a density allowing 6–7 days of exponential growth before confluency. Medium and compound was replaced every 48 hours to minimize the effect of potential compound instability. After 7 days of incubation in the presence of compound, cells were fixed and stained with methylene blue, followed by extraction of the stain with 1% SDS and spectrophotometry at 650nm. Each assay was run in triplicate and the difference between control and compound-treated wells was determined using Student’s t-test.
Dose-dependent inhibition of cell growth
Cells were plated at 1×103 cells per well in 96-well plates. Compounds were added in a dose range from 1 to 20000nM in two-fold increments. Each dose was tested in triplicate. Controls were 0.1% DMSO (no effect on cell viability) and 50μM 9-aminoacridine (complete cell death). Cells were incubated for 6 days and then fixed and stained with methylene blue, followed by extraction of the stain with 1% SDS and spectrophotometry at 650nm. The assay was run two times and, based on the results; the inhibitory concentration of compound suppressing cell growth by 50% as compared to DMSO (IC50) was calculated by the sigmoid approximation method using CalcuSyn software (Biosoft, Chembridge, UK). Confidence intervals were calculated for each sigmoid.
Selection of compound-resistant variants of CWR22R cells was done by incubation of 107 cells for 3 days in the presence of 10×IC50 of a compound. Then cells were kept in drug-free medium until they reached a pre-confluent level. The procedure was repeated several times until no dead cells were observed in the presence of a compound.
PSA protein levels in conditioned media were determined after incubation of cells with or without 10μM test compound for 72h using “Human PSA ELISA Kit” from Antigenix America (Huntington, NY).
Western blot analysis
Cells were lysed in Cell Culture Lysis Reagent (Promega, Madison, WI). Total protein concentration was determined using Dc Protein Assay (BioRad, Hercules, CA). Western blotting was performed using precast 4–12% SDS Novex gels (Invitrogen) and PVDF membrane (Pharmacia BioTech). The following antibodies were used: against AR (BD PharMingen, San Diego, CA; #554225, 1μg/ml); against GAPDH as a loading control (Santa Cruz Biotechnology Inc, Santa Cruz, CA).
Analysis of cell cycle distribution was based on staining of DNA content by propidium iodide. 105 cells were removed from culture dishes using trypsin, washed with PBS and resuspended in 300 μl 3% BSA in PBS, followed by addition of 5μl 70% ethanol. Cells were kept at −20°C for several hours and then stained with 10 μg/ml propidium iodide in the presence of 30 μg/ml RNase A for 2 hrs at 37°C. DNA content was assessed using a FACS Calibur instrument and CellQuest software (Becton Dickinson, Franklin Lakes, NJ).
Testing of compound safety in mice was done according to an IACUC-approved protocol. The experiment was performed using outbred 8 week old NIH Swiss male mice from Harlan (Indianapolis, IN), 4 mice per group. IC50 concentrations were used to calculate in vivo doses based on mouse weight (IC50 in vivo dose equivalent in mg/kg = IC50 in mg/ml ×1000, based on the assumption that 1g of mouse tissues have an approximate volume of 1ml). The compounds were injected intraperitoneally (i.p.) in 25% DMSO-75% PBS starting from 20×IC50 equivalent of in vitro cytotoxic dose of compounds. The mice were observed for three days before each of two subsequent injections of 50×IC50 (dose 2) and 100×IC50 (dose 3). The control group of mice received 3 injections of 25% DMSO-75% PBS in intervals of three days. Blood biochemistry analysis was performed by BioReliance (Rockville, MD).
Testing of the anti-tumor effect of compounds in mice was done according to an IACUC-approved protocol in 8 week old male athymic nude mice from Harlan (Indianapolis, IN). For the C4-2 xenograft model, 106 C4-2 cells were injected subcutaneously (s.c.) in 2 sites of each mouse in 50% matrigel (BD Biosciences, Bradford, MA) in PBS. Upon development of visible tumors, they were measured using digital caliper. Tumor volume was calculated according to the formula: Volume=Length × Width2/2. Mice were injected with compounds i.p. daily for 6 days starting on the day when at least one tumor per mouse reached 100 mm3 in size. Five mice per group were used. For the CWR22R xenograft model, CWR22R cells were inoculated (105 cells per inoculum) in 50% matrigel, one site per mouse. Treatment was started when the tumor grew to a size of at least 25 mm3. Compounds were diluted in captisol (CyDex, Lenexa, KS) and delivered intravenously (to reduce “first pass” liver effect) once a day for 5 days. Ten mice per group were used. Mice were monitored daily and tumors were measured every other day. Mice were euthanized according to institutional regulations when the size of tumor reached 1000 mm3. Comparison of the tumor growth in control and treated mice was done using ANOVA test.
Acknowledgments
We thank Dr. G. Jenster (Department of Urology, Josephine Nefkens Institute, Erasmus MC, Rotterdam, the Netherlands), Dr. Peter Chumakov (Department of Molecular Genetics, Cleveland Clinic), and Dr Oskar Rokhlin (Department of Pathology, Iowa University) for sharing plasmids; Dr Warren Heston for sharing cell lines and Dr. Patti Baker and Dr. James Mohler (RPCI) for the help with manuscript discussion and editing. This work was supported by Prostate Cancer Foundation Award to KG and NIH NCI Grant R42 CA 110400-01.
Abbrevations
- AR
androgen receptor
- LBD
ligand binding domain
- PCa
prostate cancer
- HRPC
hormone refractory prostate cancer
- ARTIS
androgen receptor transcription inhibitor, suppressive
- ARTIK
androgen receptor transcription inhibitor, killing
- ER
estrogen receptor
- PR
progesterone receptor
- MR
mineralcorticoid receptor
- GR
glucicirticoid receptor
- SR
steroid receptors
Contributor Information
Natalia V. Narizhneva, Email: nnarizhneva@cbiolabs.com.
Natalia D. Tararova, Email: natalia@dapcel.com.
Petro Ryabokon, Email: pryabokon@cbiolabs.com.
Inna Shyshynova, Email: ishyshynova@cbiolabs.com.
Anatoly Prokvolit, Email: aprokvolit@gmail.com.
Pavel G. Komarov, Email: pkomarov@cbiolabs.com.
Andrei A. Purmal, Email: apurmal@cbiolabs.com.
Andrei V. Gudkov, Email: andrei.gudkov@roswellpark.rog.
Katerina V. Gurova, Email: katerina.gurova@roswellpark.org.
References
- 1.Balk SP. Androgen receptor as a target in androgen-independent prostate cancer. Urology. 2002;60:132–8. doi: 10.1016/s0090-4295(02)01593-5. discussion 8–9. [DOI] [PubMed] [Google Scholar]
- 2.Chen Y, Sawyers CL, Scher HI. Targeting the androgen receptor pathway in prostate cancer. Curr Opin Pharmacol. 2008;8:440–8. doi: 10.1016/j.coph.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haag P, Bektic J, Bartsch G, Klocker H, Eder IE. Androgen receptor down regulation by small interference RNA induces cell growth inhibition in androgen sensitive as well as in androgen independent prostate cancer cells. J Steroid Biochem Mol Biol. 2005;96:251–8. doi: 10.1016/j.jsbmb.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 4.Tararova ND, Narizhneva N, Krivokrisenko V, Gudkov AV, Gurova KV. Prostate cancer cells tolerate a narrow range of androgen receptor expression and activity. Prostate. 2007;67:1801–15. doi: 10.1002/pros.20662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zegarra-Moro OL, Schmidt LJ, Huang H, Tindall DJ. Disruption of androgen receptor function inhibits proliferation of androgen-refractory prostate cancer cells. Cancer Res. 2002;62:1008–13. [PubMed] [Google Scholar]
- 6.Huggins C. Effect of Orchiectomy and Irradiation on Cancer of the Prostate. Ann Surg. 1942;115:1192–200. doi: 10.1097/00000658-194206000-00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaliks RA, Del Giglio A. Management of advanced prostate cancer. Rev Assoc Med Bras. 2008;54:178–82. doi: 10.1590/s0104-42302008000200025. [DOI] [PubMed] [Google Scholar]
- 8.Namiki M, Kitagawa Y, Mizokami A, Koh E. Primary combined androgen blockade in localized disease and its mechanism. Best Pract Res Clin Endocrinol Metab. 2008;22:303–15. doi: 10.1016/j.beem.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 9.Lee JT, Lehmann BD, Terrian DM, Chappell WH, Stivala F, Libra M, Martelli AM, Steelman LS, McCubrey JA. Targeting prostate cancer based on signal transduction and cell cycle pathways. Cell Cycle. 2008;7:1745–62. doi: 10.4161/cc.7.12.6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, Scher HI, Jung ME, Sawyers CL. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–90. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amato RJ, Teh BS, Henary H, Khan M, Saxena S. A retrospective review of combination chemohormonal therapy as initial treatment for locally advanced or metastatic adenocarcinoma of the prostate. Urol Oncol. 2009;27:165–9. doi: 10.1016/j.urolonc.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 12.Bohl CE, Gao W, Miller DD, Bell CE, Dalton JT. Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc Natl Acad Sci U S A. 2005;102:6201–6. doi: 10.1073/pnas.0500381102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–9. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 14.Hara T, Miyazaki J, Araki H, Yamaoka M, Kanzaki N, Kusaka M, Miyamoto M. Novel mutations of androgen receptor: a possible mechanism of bicalutamide withdrawal syndrome. Cancer Res. 2003;63:149–53. [PubMed] [Google Scholar]
- 15.Koivisto P, Kolmer M, Visakorpi T, Kallioniemi OP. Androgen receptor gene and hormonal therapy failure of prostate cancer. Am J Pathol. 1998;152:1–9. [PMC free article] [PubMed] [Google Scholar]
- 16.Marques RB, Erkens-Schulze S, de Ridder CM, Hermans KG, Waltering K, Visakorpi T, Trapman J, Romijn JC, van Weerden WM, Jenster G. Androgen receptor modifications in prostate cancer cells upon long-termandrogen ablation and antiandrogen treatment. Int J Cancer. 2005;117:221–9. doi: 10.1002/ijc.21201. [DOI] [PubMed] [Google Scholar]
- 17.Tilley WD, Buchanan G, Hickey TE, Bentel JM. Mutations in the androgen receptor gene are associated with progression of human prostate cancer to androgen independence. Clin Cancer Res. 1996;2:277–85. [PubMed] [Google Scholar]
- 18.Vander Griend DJ, Karthaus WL, Dalrymple S, Meeker A, DeMarzo AM, Isaacs JT. The role of CD133 in normal human prostate stem cells and malignant cancer-initiating cells. Cancer Res. 2008;68:9703–11. doi: 10.1158/0008-5472.CAN-08-3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tepper CG, Boucher DL, Ryan PE, Ma AH, Xia L, Lee LF, Pretlow TG, Kung HJ. Characterization of a novel androgen receptor mutation in a relapsed CWR22 prostate cancer xenograft and cell line. Cancer Res. 2002;62:6606–14. [PubMed] [Google Scholar]
- 20.Wu HC, Hsieh JT, Gleave ME, Brown NM, Pathak S, Chung LW. Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: role of bone stromal cells. Int J Cancer. 1994;57:406–12. doi: 10.1002/ijc.2910570319. [DOI] [PubMed] [Google Scholar]
- 21.Gregory CW, Johnson RT, Jr, Mohler JL, French FS, Wilson EM. Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res. 2001;61:2892–8. [PubMed] [Google Scholar]
- 22.Bramlett KS, Dits NF, Sui X, Jorge MC, Zhu X, Jenster G. Repression of androgen-regulated gene expression by dominant negative androgen receptors. Mol Cell Endocrinol. 2001;183:19–28. doi: 10.1016/s0303-7207(01)00636-0. [DOI] [PubMed] [Google Scholar]
- 23.Geserick C, Meyer HA, Haendler B. The role of DNA response elements as allosteric modulators of steroid receptor function. Mol Cell Endocrinol. 2005;236:1–7. doi: 10.1016/j.mce.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 24.Tenbaum S, Baniahmad A. Nuclear receptors: structure, function and involvement in disease. Int J Biochem Cell Biol. 1997;29:1325–41. doi: 10.1016/s1357-2725(97)00087-3. [DOI] [PubMed] [Google Scholar]
- 25.Zhou C, Wu G, Feng Y, Li Q, Su H, Mais DE, Zhu Y, Li N, Deng Y, Yang D, Wang MW. Discovery and biological characterization of a novel series of androgen receptor modulators. Br J Pharmacol. 2008;154:440–50. doi: 10.1038/bjp.2008.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bohl CE, Miller DD, Chen J, Bell CE, Dalton JT. Structural basis for accommodation of nonsteroidal ligands in the androgen receptor. J Biol Chem. 2005;280:37747–54. doi: 10.1074/jbc.M507464200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salvati ME, Balog A, Shan W, Wei DD, Pickering D, Attar RM, Geng J, Rizzo CA, Gottardis MM, Weinmann R, Krystek SR, Sack J, An Y, Kish K. Structure based approach to the design of bicyclic-1H-isoindole-1,3(2H)-dione based androgen receptor antagonists. Bioorg Med Chem Lett. 2005;15:271–6. doi: 10.1016/j.bmcl.2004.10.085. [DOI] [PubMed] [Google Scholar]
- 28.Jones JO, Bolton EC, Huang Y, Feau C, Guy RK, Yamamoto KR, Hann B, Diamond MI. Non-competitive androgen receptor inhibition in vitro and in vivo. Proc Natl Acad Sci U S A. 2009;106:7233–8. doi: 10.1073/pnas.0807282106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gunther JR, Parent AA, Katzenellenbogen JA. Alternative inhibition of androgen receptor signaling: peptidomimetic pyrimidines as direct androgen receptor/coactivator disruptors. ACS Chem Biol. 2009;4:435–40. doi: 10.1021/cb900043e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mohler ML, Bohl CE, Jones A, Coss CC, Narayanan R, He Y, Hwang DJ, Dalton JT, Miller DD. Nonsteroidal selective androgen receptor modulators (SARMs): dissociating the anabolic and androgenic activities of the androgen receptor for therapeutic benefit. J Med Chem. 2009;52:3597–617. doi: 10.1021/jm900280m. [DOI] [PubMed] [Google Scholar]
- 31.Segnitz B, Gehring U. The function of steroid hormone receptors is inhibited by the hsp90-specific compound geldanamycin. J Biol Chem. 1997;272:18694–701. doi: 10.1074/jbc.272.30.18694. [DOI] [PubMed] [Google Scholar]
- 32.English HF, Kyprianou N, Isaacs JT. Relationship between DNA fragmentation and apoptosis in the programmed cell death in the rat prostate following castration. Prostate. 1989;15:233–50. doi: 10.1002/pros.2990150304. [DOI] [PubMed] [Google Scholar]
- 33.Sengupta S, Wasylyk B. Physiological and pathological consequences of the interactions of the p53 tumor suppressor with the glucocorticoid, androgen, and estrogen receptors. Ann N Y Acad Sci. 2004;1024:54–71. doi: 10.1196/annals.1321.005. [DOI] [PubMed] [Google Scholar]
- 34.Li P, Lee H, Guo S, Unterman TG, Jenster G, Bai W. AKT-independent protection of prostate cancer cells from apoptosis mediated through complex formation between the androgen receptor and FKHR. Mol Cell Biol. 2003;23:104–18. doi: 10.1128/MCB.23.1.104-118.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gurova KV, Hill JE, Razorenova OV, Chumakov PM, Gudkov AV. p53 pathway in renal cell carcinoma is repressed by a dominant mechanism. Cancer Res. 2004;64:1951–8. doi: 10.1158/0008-5472.can-03-1541. [DOI] [PubMed] [Google Scholar]