

Abstract
The kidney regulates body fluid, ion, and acid/base homeostasis via the interaction of a host of channels, transporters, and pumps within specific tubule segments, specific cell types, and specific plasma membrane domains. Furthermore, renal epithelial cells have adapted to function in an often unique and challenging environment that includes high medullary osmolality, acidic pHs, variable blood flow and constantly changing apical and basolateral “bathing” solutions. In this review, we focus on selected protein trafficking events by which kidney epithelial cells regulate body fluid, ion and acid-base homeostasis in response to changes in physiological conditions. We discuss aquaporin 2 and G-protein coupled receptors (GPCRs) in fluid and ion balance, the V-ATPase and intercalated cells (IC) in acid/base regulation, and acidification events in the proximal tubule degradation pathway. Finally, in view of its direct role in vesicle trafficking that we outline here, we propose that the V-ATPase itself should, under some circumstances, be considered as a fourth category of vesicle “coat” protein, alongside clathrin, caveolin, and COPs.
Keywords: Aquaporin 2, V-ATPase, phosphorylation, acidification, principal cell, intercalated cell, proximal tubule, endocytosis, exocytosis, vasopressin receptor
A) Trafficking of aquaporin 2 - the vasopressin-responsive water channel
Aquaporin 2 is the vasopressin (VP)-sensitive water channel that is involved in urinary concentration. After VP is released from the posterior pituitary upon an increase in plasma osmolality or decrease in volume, it interacts with the vasopressin type 2 receptor (V2R) on collecting duct principal cells, increases cAMP, induces PKA phosphorylation of AQP2 at serine 256 in the C-terminus, and shifts AQP2 localization from intracellular vesicles to the plasma membrane (Fig. 1)(1–3). This greatly increases epithelial water permeability, and allows urinary concentration to occur by osmotic equilibration of the luminal fluid with the hypertonic interstitium. Defects in the V2R/AQP2 signaling pathway lead to nephrogenic diabetes insipidus – a disease in which over 15 L of dilute urine can be produced each day (4, 5).
Fig. 1.
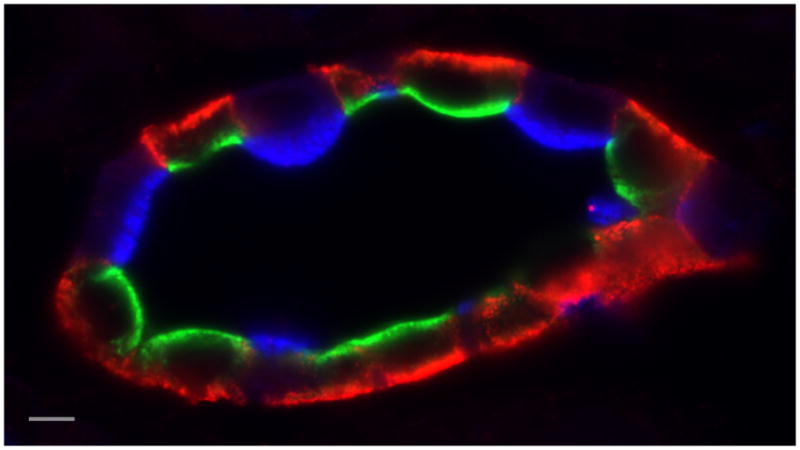
Section of kidney collecting duct triple-immunostained to show AQP2 (green – goat anti AQP2 antibody) and AQP4 (red – rabbit anti AQP4 antibody) in vasopressin-sensitive principal cells, and the proton-pumping V-ATPase (blue – chicken anti V-ATPase E-subunit antibody) in intercalated cells. AQP2 is located mainly in the apical domain of principal cells, where it shuttles between the plasma membrane and intracellular vesicles. Apical accumulation is induced by the action of the antidiuretic hormone, vasopressin. AQP4 is a basolateral water channel that works together with apical AQP2 to allow water to cross the epithelium, driven by interstitial hypertonicity. This is an essential component of the urinary concentrating mechanism. Finally, intercalated cells express high levels of the V-ATPase. In the region of the kidney shown here, the inner stripe of the outer medulla, A-IC express V-ATPase apically. In response to systemic acidosis, V-ATPase pumps accumulate in the apical plasma membrane and proton secretion is activated to help excrete the acid load (Bar = 5 μm).
A key question is how does phosphorylation lead to the relocation of AQP2 from cytoplasmic vesicles to the plasma membrane? AQP2 recycles constitutively between intracellular vesicles and the plasma membrane (Fig. 2 illustrates some aspects of the AQP2 recycling pathway), even in the absence of VP, similarly to the GLUT4 glucose transporter (3, 6, 7). Both the exocytotic and endocytotic steps of this recycling process may be regulated by AQP2 phosphorylation, with the S256 residue playing a critical role in this process (8), although other phosphorylation sites may also be involved (see below). The regulation of a phosphorylation-dephosphorylation cycle by the action of local kinases, kinase anchoring proteins and phosphatases is ultimately responsible for determining the cellular itinerary and localization of AQP2 (9).
Fig. 2.

Diagram showing some key features of AQP2 recycling. AQP2 recycles via a constitutive pathway through a trans-Golgi and/or the recycling endosome. Inhibiting clathrin-mediated endocytosis induces apical accumulation of AQP2 in a VP-independent manner. Upon VP interaction with the basolateral (V2R), cAMP elevation activates protein kinase A (PKA) and AQP2 phosphorylation increases on residues S256, S264 and S269. Increased activity of the soluble guanylyl cyclase (GC), for example via nitric oxide, also results in AQP2 phosphorylation and membrane accumulation. The MAP kinase pathway is essential for hypertonicity-induced AQP2 surface accumulation. AQP2 interacts with the SNARE regulatory protein Munc 18-2, and this interaction may be regulated by phosphorylation. Phosphorylated AQP2 at the plasma membrane is located in endocytosis-resistant domains; its interaction with hsc70, which is involved in clathrin-mediated endocytosis, is inhibited. Non-phosphorylated AQP2 interacts strongly with hsc70 and this may lead to AQP2 accumulation in clathrin coated pits, followed by endocytosis. MAL, the myeloid and lymphocyte associated protein, is also involved in AQP2 endocytosis by an as yet unknown mechanism. In addition, the actin cytoskeleton is centrally involved in AQP2 trafficking – actin depolymerization alone results in cell surface accumulation of AQP2 - but whether the endocytotic or exocytotic pathway is involved remains uncertain (Adapted from ref. 7).
Regulation of AQP2 exocytosis
Exocytosis of AQP2 involves, not surprisingly, SNARE proteins including VAMP2, SNAP23, and syntaxin 3 or 4. The syntaxin-binding protein Munc 18b negatively regulates AQP2 exocytosis. One report suggests that this occurs by the traditional Munc18/Q-SNARE regulatory mechanism at the plasma membrane (10), whereas another indicates that Munc18b regulates the action of an R-SNARE, VAMP2, at the level of AQP2 containing vesicles (11). The latter study shows that Munc18b detaches from a vesicular complex involving AQP2 and VAMP2 upon VP treatment of cultured cells, possibly as a result of AQP2 phosphorylation (11). Overexpression of Munc18b decreases AQP2 exocytosis (11), and Munc18b knockdown increases AQP2 exocytosis (10). Thus, regulation of SNARE protein interactions may contribute to VP-induced AQP2 exocytosis.
Interaction with the actin cytoskeleton is also critical for AQP2 recycling, and AQP2 directly interacts with G-actin (12). Actin depolymerization by RhoA inactivation or cytochalasin D treatment of cells results in VP-independent membrane accumulation of AQP2 (2). Phosphorylation of AQP2 at S256 decreases its affinity for G-actin, but stimulates interaction with tropomyosin 5B (TM5b) (12). This sequestration of TM5b locally depolymerizes F-actin in cells expressing AQP2 and results in membrane accumulation of AQP2 by an as yet unknown mechanism. Using LLC-PK1 cells expressing ssYFP, a secreted marker that partially colocalizes in AQP2 containing vesicles, we showed that VP treatment indeed induces an increase in ssYFP exocytosis, but only in cells that express AQP2, and not in native cells (13). Myosin Vb is also important for the AQP2 shuttle to occur, via its interaction with Rab11 and its binding partner Rab11-FIP2 (14). A dominant negative myosin Vb tail expressed in cultured CD8 cells causes accumulation of AQP2 in a Rab11 positive compartment and inhibits VP action (14). However, actin dynamics play a complex role in both exo- and endocytosis (15), so the precise point in the recycling pathway of AQP2 that is affected by regulating actin polymerization remains to be clarified.
Regulation of AQP2 endocytosis
Internalization of AQP2 is clathrin mediated, and AQP2 accumulation at the cell surface can be induced simply by inhibiting the endocytotic arm of the recycling pathway using dominant negative dynamin or methyl-β-cyclodextrin (7). In addition, VP treatment causes AQP2 to accumulate in “endocytosis-resistant” membrane domains (16) (Figs. 2, 3). Phosphorylation of AQP2 at S256 prevents its interaction with the heat shock protein hsc70 (17), which is critically involved in clathrin mediated endocytosis (18), and expression of ATPase-deficient hsc70 in cells also results in membrane accumulation of AQP2. In addition, phosphorylated AQP2 interacts strongly with MAL (myelin and lymphocyte associated protein) a tetraspanning membrane-associated protein that may also contribute to increased surface expression by restricting AQP2 internalization (19).
Fig. 3.

A–D) Spinning disk confocal microscopy showing internalization of the vasopressin type 2 receptor (V2R) after vasopressin treatment of LLC-PK1 cells expressing V2R-GFP. Under baseline conditions, the V2R is located mainly on the basolateral plasma membrane of these cells (A), and is progressively internalized into perinuclear vesicles (arrows) after VP stimulation (B–D). These vesicles have been identified as later endosomes and lysosomes (38).
E–F) Localization of V2R-GFP and AQP2 (red) by immunocytochemistry in cells treated with VP. After VP stimulation, endocytosis of the V2R ensues (E), but AQP2 accumulates at the cell surface (F) and resides in distinct membrane microdomains that are distinct from those containing the V2R. At higher magnification, the V2R appears to be forming endocytotic vesicles that bud from the plasma membrane (arrows) and in some cases appear free in the cytoplasm. AQP2 remains at the cell surface and does not internalize under these conditions – it is in “endocytosis-resistant” microdomains (16).
AQP2 has multiple phosphorylation sites
At least 13 actual and putative phosphorylation sites for multiple kinases (PKA, PKG, casein kinase II, calmodulin-dependent kinases and MAP kinases) are present in the AQP2 sequence (8). The phosphorylation status of four sites in the AQP2 C-terminus – S256, S261, S264 and S269 - is modified by VP action (20, 21). S256 phosphorylation is critical for AQP2 cell surface accumulation (8), and may be necessary for subsequent downstream phosphorylation of S264 and S269. S269 may be phosphorylated (but not by PKA) when AQP2 is already located at the cell surface: thus, it may play a role in cell surface retention rather than delivery (22). S261 phosphorylation is actually decreased upon VP action (23), but the phosphorylation status of this residue does not detectably affect AQP2 trafficking (24). Previous work also proposed a role of PKA-independent S256 phosphorylation in trafficking of AQP2 through the Golgi (25), although other work has shown no apparent effect of an S256A mutation on the ability of AQP2 to transit the Golgi and reach the cell surface during its constitutive recycling itinerary (12, 26). Further complicating the picture is that some sites are potential targets for multiple kinases – indeed S256 phosphorylation by PKA, PKG and casein kinase II have all been reported (8).
Finally, principal cells in the renal medulla can be exposed to hypertonic environments that are an essential component of the renal concentrating mechanism. Exposure of principal cells both in culture and in situ to hypertonicity causes an accumulation of AQP2 at the plasma membrane in a MAP kinase-dependent manner (Fig. 2), mainly by decreasing endocytosis (27). In contrast, hypotonicity decreases AQP2 cell surface expression in cultured cells and decreases the level of cAMP and S256 phosphorylation on AQP2 (28). In some kidney regions, AQP2 is also present on the basolateral plasma membrane together with other aquaporins (AQP3 and/or AQP4) (Fig. 1). One factor stimulating basolateral AQP2 targeting may be hypertonicity (29), but since this also occurs in the isotonic cortex of the kidney, other regulatory elements must be involved. Aldosterone treatment of rats increases basolateral AQP2 targeting (30), but the physiological importance of this process and its cell biological regulation remain unclear.
Together, the data outlined above indicate that the presence of AQP2 on intracellular vesicles allows them to interact with the cellular transport machinery in a phosphorylation-dependent manner. This occurs by the formation and dissolution of protein complexes that regulate phosphorylation and dephosphorylation of AQP2 and control its association with actin and actin related proteins, with proteins of the clathrin endocytotic apparatus, and with exocytotic SNARE-regulating proteins.
B) G-protein coupled receptor trafficking in the kidney
Many functions of renal epithelia are directed by the interactions of specific cell types with circulating hormones, including VP, parathyroid hormone (PTH), and angiotensin II (ANGII) which are ligands for G-protein coupled receptors. (31, 32). Emerging data have emphasized that while some features of GPCR trafficking, internalization and degradation are shared by multiple receptor types, the precise mechanisms that regulate individual receptors must be examined using appropriate conditions and cell systems (33, 34). Not only are a variety of clathrin adaptor molecules involved in the endocytotic process (35), but the cellular environments in which receptor/ligand coupling occurs can also lead to differential receptor signaling and processing (33). Nowhere is this more critical than in the kidney, where receptor-ligand interactions occur in environments that differ greatly from that in most other tissues and organs. For example, VP must bind to the V2R in a medullary environment that can be hypertonic (1200 mOsm/kg), contain up to 600 mOsm/kg urea, and have a pH as low as 5.5. Yet, VP still binds to the V2R and signals appropriately under these harsh conditions. An acidic pH dissociates many receptor/ligand pairs in the endosomal compartment, terminating signaling and allowing receptors to be recycled back to the cell surface, while ligands are delivered to lysosomes for degradation. So how can the V2R/VP pair function in the renal medulla?
Repopulation of the plasma membrane by the V2R after downregulation occurs very slowly compared to many other GPCRs, implying that the V2R either does not recycle, or recycles very slowly (36). While phosphorylation of the V2R has been implicated in this “slow” recycling process (36), much of the internalized, liganded V2R is degraded in lysosomes (Fig. 3) (37, 38). The V2R that re-appears on the cell surface to restore vasopressin sensitivity to principal cells after downregulation is mainly newly-synthesized receptor (38). This ability of the V2R to bind to its ligand at acidic pH allows functional coupling to occur in the renal medullary environment. In contrast, the PTH receptor (PTHR), which regulates phosphate and calcium reabsorption in parts of the kidney where the interstitial environment is milder, fails to signal and does not internalize after ligand binding under conditions of hypertonicity and low pH (39). In addition, the response of cells to a given concentration of VP is also modulated by the presence of other hormones. For example, the plasma membrane targeting of AQP2 by VP in inner medullary collecting duct cells is “sensitized” to lower doses of VP by simultaneous exposure of cells to ANGII (40).
Another feature of GPCRs in the kidney is that they often are expressed on both the apical and basolateral plasma membranes of epithelial cells, and elicit responses from both membrane domains. An intriguing feature of the PTHR in proximal tubules is that receptors located in different membrane domains stimulate different signaling pathways. The apical receptor stimulates a PLC/PKC response, whereas the basolateral receptor drives an increase in intracellular cAMP, and a PKA-mediated effect (41). Both responses are, however, involved in provoking the endocytosis of the apical sodium/phosphate co-transporter, NaPiIIa and, thereby, increase phosphate excretion. The apical pathway involves NHERF1 mediated coupling of the PTHR to PLC (41, 42). There is also some evidence that the V2R is bipolar in collecting duct principal cells as well as in cultured cells in vitro, and can signal, perhaps differently, from these two membrane domains upon VP binding (42, 43). GPCRs can also signal from intracellular locations, including the nucleus, the Golgi, and the endoplasmic reticulum (44, 45). The ability of GPCRs including the V2R to signal from an endosomal location is the subject of much current interest (34, 46–48).
GPCR biology is of enormous physiological and pathophysiological importance, and GPCRs are major drug development targets. However, in vitro data obtained using cells cultured in normal, isotonic, neutral medium may not be strictly comparable to in vivo GPCR/ligand physiology. This is especially true in the kidney, in which GPCR responses are modified by extreme extracellular environments, the presence of other hormone/ligand interactions, and the interaction of ligands with both apical and basolaterally-expressed receptors.
C) Vacuolar H+-ATPase (V-ATPase) trafficking in intercalated cells
The V-ATPase acidifies many intracellular organelles in all eukaryotic cells, and is also expressed at high density in plasma membranes of some specialized cells that are involved in proton transport and extracellular pH regulation in different organs (Fig. 4)(49–52). In the kidney, these “intercalated cells” (IC) in the late distal tubule, the connecting segment and the collecting duct (Fig. 1) are key players in body acid/base regulation. Systemic acidosis causes accumulation of V-ATPase in the apical membrane of A-IC, which are responsible for proton secretion into the urine. Conversely, type B-IC (found only in the cortical collecting duct) can express the V-ATPase in their basolateral membrane, and they secrete bicarbonate (51). Alkalosis induces the endocytotic retrieval of V-ATPase from the apical membrane of all IC and accumulation of V-ATPase in the basolateral membrane of B-IC. The mechanisms by which IC respond to variations in acid/base status by changing the polarized expression of the V-ATPase remain poorly understood, but the Al-Awqati laboratory has shown that this process is mediated by a protein called hensin. This protein is secreted into the extracellular environment where it polymerizes and binds to cell surface integrins that in turn induce terminal differentiation signals resulting in transformation of B-IC into A-IC (53).
Fig. 4.

Composite plate showing the V-ATPase coat on vesicles in proton secreting cells, a schematic representation of its structure, and its interaction with Arf6, ARNO, and actin. By conventional electron microscopy, V-ATPase forms a dense coat that surrounds trafficking vesicles in kidney intercalated cells (A – arrows; Bar = 50 nm). This V-ATPase-coated vesicle is from a kidney intercalated cell that has endocytosed the fluid phase marker horseradish peroxidase from the tubule lumen, identifying this vesicle as being part of the endocytotic pathway. Panel B (Bar = 50 nm) shows a V-ATPase coated vesicle in a proton secreting cell from the amphibian urinary bladder. The densely-packed V-ATPase molecules form a coat that is visualized here by rapid-freeze, deep etch electron microscopy, and which represents the V1 sector of the V-ATPase (83). The arrows indicate an adjacent actin filament. The diagram shows the complex subunit structure of the V-ATPase and illustrates the binding sites for actin (subunits B and C), ARNO (subunit a) and Arf6 (subunit c). Further details on the structural organization of the V-ATPase can be found in recent comprehensive reviews (49–51).
The V-ATPase is divided into two distinct domains (Fig. 4), the transmembrane V0 domain which contains subunits a, c, d and e, and the peripheral V1 domain, which contains 8 subunits (A–H) (50, 51). Several subunits have more than one isoform, and the arrangement of one set of subunit isoforms in the holoenzyme is cell-type specific and partially determines the sub-cellular localization of the V-ATPase (50–52, 54). For example, in kidney proximal tubule cells, V-ATPase complexes containing the a2 isoform are located in endosomes while the a4 isoform is in the plasma membrane (55). IC express mainly the B1 isoform of the B subunit (but see below), whereas proximal tubule cells (and osteoclasts) express the B2 subunit. However, no difference in subunit composition of the apical versus basolateral V-ATPase in A- and B-IC has yet been revealed.
Isoform replacement and pathophysiology of V-ATPase dysfunction
Mutations of subunits B1 and a4 cause distal renal tubular acidosis (rDTA) due to an impairment of proton secretion by IC. Distal RTA is accompanied by deafness in patients with B1 mutations, but those harboring a4 subunit mutations usually have intact hearing despite the expression of both B1 and a4 in the inner ear (56). Similarly, a4 is localized in the brush-border membrane of proximal tubules (55) but patients with a4 mutations do not have proximal tubular acidosis. Other a subunit isoforms may, therefore, compensate for the lack of functional a4. Similarly, B1-subunit deficient mice unexpectedly maintain normal acid/base balance under non-stressed conditions (57), but their A-ICs had an atypically pronounced apical localization of the B2 V-ATPase isoform (58). However, when challenged with an acid load, these mice developed severe metabolic acidosis (57), suggesting that B2 cannot fully compensate for the absence of B1 under extreme conditions.
Recycling of the V-ATPase
Proton secretion in IC (and PT cells) is regulated by recycling of V-ATPase-containing vesicles to and from the plasma membrane, and involves calcium, SNARE proteins and the regulatory protein Munc 18b (see above for a role of Munc 18b in AQP2 exocytosis), microtubules and actin (52, 59). Subunits B1, B2 and C interact directly with actin, and increased association of the V-ATPase with F-actin correlates with its internationalization in osteoclasts (60). In addition, B1 interacts with the PDZ binding protein NHERF1 (61), which binds to the actin cytoskeleton via MERM (merlin, ezrin, radixin, moesin) proteins; this interaction might play a role in the stabilization and/or regulation of the V-ATPase in B-ICs.
sAC is a bicarbonate sensor that regulates apical insertion of the V-ATPase
Work using the male reproductive tract as a model has revealed that luminal bicarbonate is a key factor in the regulation of proton secreting cells (62). Bicarbonate induces apical accumulation of the V-ATPase in epididymal clear cells (which resemble kidney IC) via activation of the soluble adenylyl cyclase (sAC), which generates cAMP in response to increased bicarbonate, followed by PKA activation (62). sAC and the V-ATPase partially co-localize in both IC subtypes (63), and conditions that induce apical membrane insertion of V-ATPase in A-IC also cause the apical redistribution of sAC. Importantly, sAC co-immunoprecipitates with the V-ATPase B1 and A subunits (63), indicating that they form a signaling complex that contributes to the response of A-IC to systemic acidosis. Increased delivery of bicarbonate to the lumen of the collecting duct occurs in some acid base disorders and could activate sAC and lead to apical mobilization of the V-ATPase in IC. Proton secretion by IC can also be modulated by hormones: ANGII stimulates V-ATPase-dependent proton secretion by A-IC by increasing V-ATPase trafficking to the plasma membrane (64).
Phosphorylation of the V-ATPase
Whether direct phosphorylation of V-ATPase subunits is involved in V-ATPase recycling remains uncertain. An early study showed that the brain isoform of the V-ATPase B subunit (presumably the B2 isoform) is phosphorylated in vitro by AP50, a subunit of the clathrin assembly protein AP-2 (65). However, phosphorylation of V-ATPase subunits in mammals has not been demonstrated in vivo, although some V-ATPase subunits, including A, B1, B2 and C, contain putative phosphorylation sites (54), and V-ATPase subunit phosphorylation has been described in insects (66). Alternatively, regulation of V-ATPase recycling by PKA may occur indirectly by modulation of the actin cytoskeleton. Kidney IC and epididymal clear cells contain high levels of the actin remodeling protein, gelsolin, which plays a key role in the regulation of V-ATPase recycling in clear cells (67). In addition, the actin cytoskeleton is markedly regulated by PKA, via phosphorylation of RhoA, leading to actin disassembly and reduced endocytosis of the V-ATPase.
D) Regulation of the endosomal-lysosomal protein degradative pathway in proximal tubules
In this section, we will focus on recent insights into the function and regulation of the endosomal/lysosomal protein degradation pathway using kidney proximal tubule (PT) epithelial cells as an example. Kidney PT cells have an extensive apical endocytotic apparatus that is critical for the reabsorption and degradation of filtered proteins via the endosomal/lysosomal pathway, as well as for the extensive recycling of many membrane transport proteins (68, 69). From early endosomes, the internalized material (ligands) and receptors can be either recycled, exocytosed back into the PT lumen, or delivered to organelles including late endosomes and lysosomes for degradation. Many low-molecular weight plasma proteins (such as albumin, hormones, vitamin-binding proteins and cytokines among others)(70–72) as well as some drugs (including gentamicin and amikacin)(73) are reabsorbed in the PT by receptor-mediated endocytosis and are delivered to lysosomes for degradation. This process involves three apically-located multiligand-binding receptors, megalin, cubilin and amnionless (68, 74, 75). Megalin is a 600 kDa transmembrane glycoprotein that belongs to the low-density lipoprotein (LDL) receptor gene family and functions as a low-selectivity, high-capacity scavenger receptor. Cubilin is a 460 kDa peripherally attached glycoprotein which interacts and functions with megalin in a dual-receptor complex (68).
Megalin: an endocytic scavenger receptor involved in intracellular signaling
Unconventional functions of the megalin receptor in intracellular signaling have recently emerged in kidney PT. First, the cytoplasmic C-terminal domain of megalin is subjected to regulated intramembrane proteolysis, followed by its translocation to the nucleus where it regulates the expression of specific proteins in PT epithelial cells. These data implicate megalin as a central element in a Notch-like pathway that could interconnect the function of the endosomal/lysosomal protein degradation pathway with regulated gene expression and signaling (76, 77). Second, megalin was suggested to be the albumin-sensor that mediates an equilibrium between albumin-induced apoptosis and cell survival. Megalin, through a novel mechanism, binds the serine/threonine kinase PKB which controls phosphorylation of Bad and, thus, determines the anti- or pro-apoptotic outcome as a result of low and high albumin reabsorption via the endosomal/lysosomal pathway in these cells (78). While the function of megalin and cubilin receptors as well as their ligand specificity have been intensively studied during last decade, the role of amnionless in protein reabsorption and degradation is just emerging. Furthermore, novel and unexpected signaling pathways involving these proteins are providing exciting new insights into PT function and regulation in the kidney.
Role of endosomal acidification in the protein degradative pathway
In all eukaryotic cells, including kidney PT cells, endocytic trafficking is associated with increasing acidification of the lumen of the organelles as internalized content passes from early endosomes to late endosomes and finally to lysosomes (69). Intra-vesicular acidification along the endosomal-lysosomal pathway is mainly accomplished by the proton pumping V-ATPase that has been described earlier in this review (50–52). This electrogenic pump translocates protons from the cytoplasm to the organelle lumen which, in conjunction with a parallel chloride conductance (via CLC-5 and CLC-4 electrogenic Cl−/H+-exchangers) generates the acidic milieu (pH between 5.0–6.0) of the endosomal/lysosomal compartments (79). However, an important role of Na+/H+-exchangers in organelle acidification is also currently emerging (80). Importantly, perturbation of endosomal acidification in PT cells leads to pathophysiological dysfunction typified by diminished reabsorption and urinary wasting of albumin and other low-molecular proteins (69). The physiological importance of endosomal acidification is also underlined by our recent finding that V-ATPase inhibitors (bafilomycin and concanamycin) as well as acidification uncouplers (NH4CL and FCCP) strongly abolish albumin uptake by mouse proximal tubule cells (MTC) (55). Interestingly, a functionally disruptive mutation in CLC-5 results in Dent disease, whose manifestations include Fanconi syndrome in humans, which is accompanied by diminished function of the endosomal/lysosomal pathway, deficient protein reabsorption, proteinuria and kidney stone formation. CLC-5 knockout mice have similar defects in the PT endosomal/lysosomal protein degradative pathway (79). Thus, impaired function of the PT endocytic pathway and diminished albumin reabsorption is explained by a reduced chloride-driven electrical shunt that inhibits endosomal V-ATPase-dependent acidification. On the other hand, kidney stone formation in Dent disease may be caused by impaired megalin-dependent reabsorption of filtered parathyroid hormone (PTH) in early PT cells and the resultant excessive stimulation of apical PTH-receptors in the late PT, which triggers both the endocytic removal NaPiIIa phosphate transporters and an augmentation of the transcription of 1a-25(OH)-VitD3-hydrolase (79). Recently, using both CLC-5 and megalin-knockout mouse models, megalin-mediated endocytosis was implicated in the reabsorption of filtered cathepsin B and its delivery to lysosomes, thus providing a novel alternative pathway that could contribute to the biogenesis of lysosomes in kidney PT cells (81).
The V-ATPase as an endosomal pH sensor involved in coat protein recruitment
While many studies have linked an inhibition of endosomal acidification to perturbations of intracellular trafficking pathways, the biochemical mechanism(s) linking deficient endosomal pH regulation to impaired vesicle trafficking were poorly understood. Recent data from our laboratory have now provided new insights into this process by showing the direct involvement of the endosomal V-ATPase in acidification-dependent transmembrane signaling. This involves a direct, acidification-dependent interaction of some V-ATPase subunits with cytosolic small GTPases and their regulatory proteins that modulate vesicular trafficking along the protein degradative pathway (55). In particular, the transmembrane a2 subunit isoform of the V-ATPase is targeted to early endosomes in the proximal tubule and directly interacts with the guanine-nucleotide exchange factor ARNO, while the Arf6 small GTPase specifically interacts with the c-subunit of the V-ATPase Vo-sector. Thus, the V-ATPase has a novel function as an endosomal pH-sensor: the acidification-dependent interaction between V-ATPase subunits and small GTPases is crucial for protein trafficking between early and late endosomes (55, 82). Although this work showed that the V-ATPase could, in an acidification-dependent manner, modulate vesicular trafficking by scaffolding and recruiting small GTPases, the downstream biochemical events and functional significance of this rendezvous remain unclear. Our recent data suggest that V-ATPase may not simply recruit and scaffold small GTPases to their target membranes but could also, via its interaction with regulatory elements in ARNO, have a novel role as an activator of Arf-GEF activity during its function as an endosomal pH-sensor and regulator of the endosomal/lysosomal pathway.
E) The V-ATPase as a fourth class of membrane/vesicle “coat” protein
The uncovering of a critical role for the V-ATPase itself as a regulator of vesicle trafficking, in addition to its well-known role in vesicle acidification, has significant implications for all cell types, not only proximal tubule cells. V-ATPase-transporting endo- and exocytotic vesicles in ICs have an extensive cytoplasmic coat formed by the V-ATPase V1 sector subunits (Fig. 4)(83), but the coat does not contain clathrin or caveolin1 (52) or any COP proteins (Brown and Breton, unpublished data). The V-ATPase is clearly involved in regulating vesicle trafficking by the downstream recruitment of functionally important cytosolic proteins, and by interacting directly with the actin cytoskeleton. This suggests that the V-ATPase forms a fourth category of vesicle “coat” and regulates its own trafficking and recycling. Several other lines of evidence point in this direction: 1) Subunit B of the V-ATPase interacts with the SNARE protein syntaxin 1a (59); 2) Subunit H is homologous to the adaptor protein β-adaptin, which is involved in clathrin-mediated endocytosis (84); 3) The V-ATPase plays a direct role in the acidification-dependent recruitment of cytoplasmic accessory proteins to endosomes (55)(and see above). In addition, the transmembrane V0 domain of the V-ATPase may itself be directly involved in the process of membrane fusion (51), presumably following complete or partial dissociation of the bulky cytoplasmic V1 sectors of the V-ATPase.
Based on these considerations, we propose that the V-ATPase itself should be considered as a fourth type of vesicle and membrane “coat” protein, on a level with clathrin, caveolins, and COP proteins, particularly when expressed at high levels within a given membrane domain. It may, therefore, play a role in mediating “clathrin-independent endocytosis” (85).
Acknowledgments
We are grateful to the National Institutes of Health for supporting the work from our own laboratory that is included in this review. Much of this work was carried out under the auspices of an NIH Program Project Grant, DK38452, as well as individual RO1 grants DK42956 (DB) and HD40793 (SB). We are grateful to our many excellent colleagues who have made such important contributions to our work on trafficking processes in the kidney over the years. We apologize that we could not cite many important original articles due to space constraints.
References
- 1.Brown D. The ins and outs of aquaporin-2 trafficking. Am J Physiol Renal Physiol. 2003;284:F893–901. doi: 10.1152/ajprenal.00387.2002. [DOI] [PubMed] [Google Scholar]
- 2.Valenti G, Procino G, Tamma G, Carmosino M, Svelto M. Minireview: aquaporin 2 trafficking. Endocrinology. 2005;146:5063–5070. doi: 10.1210/en.2005-0868. [DOI] [PubMed] [Google Scholar]
- 3.Sasaki S, Noda Y. Aquaporin-2 protein dynamics within the cell. Curr Opin Nephrol Hypertens. 2007;16:348–352. doi: 10.1097/MNH.0b013e32818b27bf. [DOI] [PubMed] [Google Scholar]
- 4.Bichet DG. Vasopressin receptor mutations in nephrogenic diabetes insipidus. Semin Nephrol. 2008;28:245–251. doi: 10.1016/j.semnephrol.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 5.Loonen AJ, Knoers NV, van Os CH, Deen PM. Aquaporin 2 mutations in nephrogenic diabetes insipidus. Semin Nephrol. 2008;28:252–265. doi: 10.1016/j.semnephrol.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 6.Huang S, Czech MP. The GLUT4 glucose transporter. Cell Metab. 2007;5:237–252. doi: 10.1016/j.cmet.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 7.Bouley R, Hasler U, Lu HA, Nunes P, Brown D. Bypassing vasopressin receptor signaling pathways in nephrogenic diabetes insipidus. Semin Nephrol. 2008;28:266–278. doi: 10.1016/j.semnephrol.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown D, Hasler U, Nunes P, Bouley R, Lu HA. Phosphorylation events and the modulation of aquaporin 2 cell surface expression. Curr Opin Nephrol Hypertens. 2008;17:491–498. doi: 10.1097/MNH.0b013e3283094eb1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szaszak M, Christian F, Rosenthal W, Klussmann E. Compartmentalized cAMP signalling in regulated exocytic processes in non-neuronal cells. Cell Signal. 2008;20:590–601. doi: 10.1016/j.cellsig.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 10.Procino G, Barbieri C, Tamma G, De Benedictis L, Pessin JE, Svelto M, Valenti G. AQP2 exocytosis in the renal collecting duct -- involvement of SNARE isoforms and the regulatory role of Munc18b. J Cell Sci. 2008;121:2097–2106. doi: 10.1242/jcs.022210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ross J, Li G, Freiburg B, Singh S, Matzuzaki T, Lu HAJ, Brown D, Alexander EA, Schwartz J. Munc 18–2 regulates exo- and endocytotic pathways involved in aquaporin-2 trafficking. Am J Physiol Renal Physiol. 2008:300A. in press. [Google Scholar]
- 12.Noda Y, Horikawa S, Kanda E, Yamashita M, Meng H, Eto K, Li Y, Kuwahara M, Hirai K, Pack C, Kinjo M, Okabe S, Sasaki S. Reciprocal interaction with G-actin and tropomyosin is essential for aquaporin-2 trafficking. J Cell Biol. 2008;182:587–601. doi: 10.1083/jcb.200709177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nunes P, Hasler U, McKee M, Lu HAJ, Bouley R, Brown D. A novel fluorimetry-based secretion assay to monitor vasopressin-induced exocytosis in LLC-PK1 cells expressing aquaporin-2 (AQP2) Am J Physiol Renal Physiol. 2008 doi: 10.1152/ajpcell.00344.2008. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nedvetsky PI, Stefan E, Frische S, Santamaria K, Wiesner B, Valenti G, Hammer JA, 3rd, Nielsen S, Goldenring JR, Rosenthal W, Klussmann E. A Role of myosin Vb and Rab11-FIP2 in the aquaporin-2 shuttle. Traffic. 2007;8:110–123. doi: 10.1111/j.1600-0854.2006.00508.x. [DOI] [PubMed] [Google Scholar]
- 15.Lanzetti L. Actin in membrane trafficking. Curr Opin Cell Biol. 2007;19:453–458. doi: 10.1016/j.ceb.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 16.Bouley R, Hawthorn G, Russo LM, Lin HY, Ausiello DA, Brown D. Aquaporin 2 (AQP2) and vasopressin type 2 receptor (V2R) endocytosis in kidney epithelial cells: AQP2 is located in ‘endocytosis-resistant’ membrane domains after vasopressin treatment. Biol Cell. 2006;98:215–232. doi: 10.1042/BC20040054. [DOI] [PubMed] [Google Scholar]
- 17.Lu HA, Sun TX, Matsuzaki T, Yi XH, Eswara J, Bouley R, McKee M, Brown D. Heat shock protein 70 interacts with aquaporin-2 and regulates its trafficking. J Biol Chem. 2007;282:28721–28732. doi: 10.1074/jbc.M611101200. [DOI] [PubMed] [Google Scholar]
- 18.Eisenberg E, Greene LE. Multiple roles of auxilin and hsc70 in clathrin-mediated endocytosis. Traffic. 2007;8:640–646. doi: 10.1111/j.1600-0854.2007.00568.x. [DOI] [PubMed] [Google Scholar]
- 19.Kamsteeg EJ, Duffield AS, Konings IB, Spencer J, Pagel P, Deen PM, Caplan MJ. MAL decreases the internalization of the aquaporin-2 water channel. Proc Natl Acad Sci U S A. 2007;104:16696–16701. doi: 10.1073/pnas.0708023104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoffert JD, Pisitkun T, Wang G, Shen RF, Knepper MA. Quantitative phosphoproteomics of vasopressin-sensitive renal cells: Regulation of aquaporin-2 phosphorylation at two sites. Proc Natl Acad Sci U S A. 2006;103:7159–7164. doi: 10.1073/pnas.0600895103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fenton RA, Moeller HB, Hoffert JD, Yu MJ, Nielsen S, Knepper MA. Acute regulation of aquaporin-2 phosphorylation at Ser-264 by vasopressin. Proc Natl Acad Sci U S A. 2008;105:3134–3139. doi: 10.1073/pnas.0712338105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoffert JD, Fenton RA, Moeller HB, Simons B, Tchapyjnikov D, McDill BW, Yu MJ, Pistikun T, Chen F, Knepper MA. Vasopressin-stimulated increase in phosphorylation at ser-269 potentiates plasma membrane retention of aquaporin-2. J Biol Chem. 2008;283:24617–24627. doi: 10.1074/jbc.M803074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoffert JD, Nielsen J, Yu MJ, Pisitkun T, Schleicher SM, Nielsen S, Knepper MA. Dynamics of aquaporin-2 serine-261 phosphorylation in response to short-term vasopressin treatment in collecting duct. Am J Physiol Renal Physiol. 2007;292:F691–700. doi: 10.1152/ajprenal.00284.2006. [DOI] [PubMed] [Google Scholar]
- 24.Lu HJ, Matsuzaki T, Bouley R, Hasler U, Qin QH, Brown D. The phosphorylation state of serine 256 is dominant over that of serine 261 in the regulation of AQP2 trafficking in renal epithelial cells. Am J Physiol Renal Physiol. 2008;295:F290–294. doi: 10.1152/ajprenal.00072.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Procino G, Carmosino M, Marin O, Brunati AM, Contri A, Pinna LA, Mannucci R, Nielsen S, Kwon TH, Svelto M, Valenti G. Ser-256 phosphorylation dynamics of Aquaporin 2 during maturation from the ER to the vesicular compartment in renal cells. Faseb J. 2003;17:1886–1888. doi: 10.1096/fj.02-0870fje. [DOI] [PubMed] [Google Scholar]
- 26.Lu H, Sun TX, Bouley R, Blackburn K, McLaughlin M, Brown D. Inhibition of endocytosis causes phosphorylation (S256)-independent plasma membrane accumulation of AQP2. Am J Physiol Renal Physiol. 2004;286:F233–243. doi: 10.1152/ajprenal.00179.2003. [DOI] [PubMed] [Google Scholar]
- 27.Hasler U, Nunes P, Bouley R, Lu HA, Matsuzaki T, Brown D. Acute hypertonicity alters aquaporin-2 trafficking and induces a MAP kinase-dependent accumulation at the plasma membrane of renal epithelial cells. J Biol Chem. 2008 doi: 10.1074/jbc.M801071200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tamma G, Procino G, Strafino A, Bononi E, Meyer G, Paulmichl M, Formoso V, Svelto M, Valenti G. Hypotonicity induces aquaporin-2 internalization and cytosol-to-membrane translocation of ICln in renal cells. Endocrinology. 2007;148:1118–1130. doi: 10.1210/en.2006-1277. [DOI] [PubMed] [Google Scholar]
- 29.van Balkom BW, van Raak M, Breton S, Pastor-Soler N, Bouley R, van der Sluijs P, Brown D, Deen PM. Hypertonicity is involved in redirecting the aquaporin-2 water channel into the basolateral, instead of the apical, plasma membrane of renal epithelial cells. J Biol Chem. 2003;278:1101–1107. doi: 10.1074/jbc.M207339200. [DOI] [PubMed] [Google Scholar]
- 30.de Seigneux S, Nielsen J, Olesen ET, Dimke H, Kwon TH, Frokiaer J, Nielsen S. Long-term aldosterone treatment induces decreased apical but increased basolateral expression of AQP2 in CCD of rat kidney. Am J Physiol Renal Physiol. 2007;293:F87–99. doi: 10.1152/ajprenal.00431.2006. [DOI] [PubMed] [Google Scholar]
- 31.Dong C, Filipeanu CM, Duvernay MT, Wu G. Regulation of G protein-coupled receptor export trafficking. Biochim Biophys Acta. 2007;1768:853–870. doi: 10.1016/j.bbamem.2006.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marchese A, Paing MM, Temple BR, Trejo J. G protein-coupled receptor sorting to endosomes and lysosomes. Annu Rev Pharmacol Toxicol. 2008;48:601–629. doi: 10.1146/annurev.pharmtox.48.113006.094646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gurevich VV, Gurevich EV. Rich tapestry of G protein-coupled receptor signaling and regulatory mechanisms. Mol Pharmacol. 2008;74:312–316. doi: 10.1124/mol.108.049015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hanyaloglu AC, von Zastrow M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol. 2008;48:537–568. doi: 10.1146/annurev.pharmtox.48.113006.094830. [DOI] [PubMed] [Google Scholar]
- 35.Wolfe BL, Trejo J. Clathrin-dependent mechanisms of G protein-coupled receptor endocytosis. Traffic. 2007;8:462–470. doi: 10.1111/j.1600-0854.2007.00551.x. [DOI] [PubMed] [Google Scholar]
- 36.Madziva MT, Birnbaumer M. A role for ADP-ribosylation factor 6 in the processing of G-protein-coupled receptors. J Biol Chem. 2006;281:12178–12186. doi: 10.1074/jbc.M601357200. [DOI] [PubMed] [Google Scholar]
- 37.Robben JH, Knoers NV, Deen PM. Cell biological aspects of the vasopressin type-2 receptor and aquaporin 2 water channel in nephrogenic diabetes insipidus. Am J Physiol Renal Physiol. 2006;291:F257–270. doi: 10.1152/ajprenal.00491.2005. [DOI] [PubMed] [Google Scholar]
- 38.Yi X, Bouley R, Lin HY, Bechoua S, Sun TX, Del Re E, Shioda T, Raychowdhury MK, Lu HA, Abou-Samra AB, Brown D, Ausiello DA. Alix (AIP1) is a vasopressin receptor (V2R)-interacting protein that increases lysosomal degradation of the V2R. Am J Physiol Renal Physiol. 2007;292:F1303–1313. doi: 10.1152/ajprenal.00441.2005. [DOI] [PubMed] [Google Scholar]
- 39.Zalyapin EA, Bouley R, Hasler U, Vilardaga JP, Lin HY, Brown D, Ausiello DA. Effectg of renal medullary pH and ioinic environment on vasopressin binding and signaling. Kidney Int. 2008 doi: 10.1038/ki.2008.412. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee YJ, Song IK, Jang KJ, Nielsen J, Frokiaer J, Nielsen S, Kwon TH. Increased AQP2 targeting in primary cultured IMCD cells in response to angiotensin II through AT1 receptor. Am J Physiol Renal Physiol. 2007;292:F340–350. doi: 10.1152/ajprenal.00090.2006. [DOI] [PubMed] [Google Scholar]
- 41.Murer H, Hernando N, Forster I, Biber J. Regulation of Na/Pi transporter in the proximal tubule. Annu Rev Physiol. 2003;65:531–542. doi: 10.1146/annurev.physiol.65.042902.092424. [DOI] [PubMed] [Google Scholar]
- 42.Nonoguchi H, Owada A, Kobayashi N, Takayama M, Terada Y, Koike J, Ujiie K, Marumo F, Sakai T, Tomita K. Immunohistochemical localization of V2 vasopressin receptor along the nephron and functional role of luminal V2 receptor in terminal inner medullary collecting ducts. J Clin Invest. 1995;96:1768–1778. doi: 10.1172/JCI118222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bouley R, Sun TX, Chenard M, McLaughlin M, McKee M, Lin HY, Brown D, Ausiello DA. Functional role of the NPxxY motif in internalization of the type 2 vasopressin receptor in LLC-PK1 cells. Am J Physiol Cell Physiol. 2003;285:C750–762. doi: 10.1152/ajpcell.00477.2002. [DOI] [PubMed] [Google Scholar]
- 44.Watson PH, Pickard BW. Nuclear trafficking of the G-protein-coupled parathyroid hormone receptor. Crit Rev Eukaryot Gene Expr. 2008;18:151–161. doi: 10.1615/critreveukargeneexpr.v18.i2.40. [DOI] [PubMed] [Google Scholar]
- 45.Boivin B, Vaniotis G, Allen BG, Hebert TE. G protein-coupled receptors in and on the cell nucleus: a new signaling paradigm? J Recept Signal Transduct Res. 2008;28:15–28. doi: 10.1080/10799890801941889. [DOI] [PubMed] [Google Scholar]
- 46.Shenoy SK, Lefkowitz RJ. Receptor-specific ubiquitination of beta-arrestin directs assembly and targeting of seven-transmembrane receptor signalosomes. J Biol Chem. 2005;280:15315–15324. doi: 10.1074/jbc.M412418200. [DOI] [PubMed] [Google Scholar]
- 47.Sneddon WB, Friedman PA. Beta-arrestin-dependent parathyroid hormone-stimulated extracellular signal-regulated kinase activation and parathyroid hormone type 1 receptor internalization. Endocrinology. 2007;148:4073–4079. doi: 10.1210/en.2007-0343. [DOI] [PubMed] [Google Scholar]
- 48.Tzafriri AR, Edelman ER. Endosomal receptor kinetics determine the stability of intracellular growth factor signalling complexes. Biochem J. 2007;402:537–549. doi: 10.1042/BJ20060756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brown D, Breton S. Mitochondria-rich, proton-secreting epithelial cells. J Exp Biol. 1996;199:2345–2358. doi: 10.1242/jeb.199.11.2345. [DOI] [PubMed] [Google Scholar]
- 50.Beyenbach KW, Wieczorek H. The V-type H+ATPase: molecular structure and function, physiological roles and regulation. J Exp Biol. 2006;209:577–589. doi: 10.1242/jeb.02014. [DOI] [PubMed] [Google Scholar]
- 51.Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol. 2007;8:917–929. doi: 10.1038/nrm2272. [DOI] [PubMed] [Google Scholar]
- 52.Wagner CA, Finberg KE, Breton S, Marshansky V, Brown D, Geibel JP. Renal vacuolar H+ATPase. Physiol Rev. 2004;84:1263–1314. doi: 10.1152/physrev.00045.2003. [DOI] [PubMed] [Google Scholar]
- 53.Al-Awqati Q. 2007 Homer W. Smith award: control of terminal differentiation in epithelia. J Am Soc Nephrol. 2008;19:443–449. doi: 10.1681/ASN.2007111195. [DOI] [PubMed] [Google Scholar]
- 54.Sun-Wada GH, Wada Y, Futai M. Diverse and essential roles of mammalian vacuolar-type proton pump ATPase: toward the physiological understanding of inside acidic compartments. Biochim Biophys Acta. 2004;1658:106–114. doi: 10.1016/j.bbabio.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 55.Hurtado-Lorenzo A, Skinner M, El Annan J, Futai M, Sun-Wada GH, Bourgoin S, Casanova J, Wildeman A, Bechoua S, Ausiello DA, Brown D, Marshansky V. V-ATPase interacts with ARNO and Arf6 in early endosomes and regulates the protein degradative pathway. Nat Cell Biol. 2006;8:124–136. doi: 10.1038/ncb1348. [DOI] [PubMed] [Google Scholar]
- 56.Stover EH, Borthwick KJ, Bavalia C, Eady N, Fritz DM, Rungroj N, Giersch AB, Morton CC, Axon PR, Akil I, Al-Sabban EA, Baguley DM, Bianca S, Bakkaloglu A, Bircan Z, et al. Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J Med Genet. 2002;39:796–803. doi: 10.1136/jmg.39.11.796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Finberg KE, Wagner CA, Bailey MA, Paunescu TG, Breton S, Brown D, Giebisch G, Geibel JP, Lifton RP. The B1-subunit of the H+ATPase is required for maximal urinary acidification. Proc Natl Acad Sci U S A. 2005;102:13616–13621. doi: 10.1073/pnas.0506769102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Paunescu TG, Russo LM, Da Silva N, Kovacikova J, Mohebbi N, Van Hoek AN, McKee M, Wagner CA, Breton S, Brown D. Compensatory membrane expression of the V-ATPase B2 subunit isoform in renal medullary intercalated cells of B1-deficient mice. Am J Physiol Renal Physiol. 2007;293:F1915–1926. doi: 10.1152/ajprenal.00160.2007. [DOI] [PubMed] [Google Scholar]
- 59.Schwartz JH, Li G, Yang Q, Suri V, Ross JJ, Alexander EA. Role of SNAREs and H+ATPase in the targeting of proton pump-coated vesicles to collecting duct cell apical membrane. Kidney Int. 2007;72:1310–1315. doi: 10.1038/sj.ki.5002500. [DOI] [PubMed] [Google Scholar]
- 60.Chen SH, Bubb MR, Yarmola EG, Zuo J, Jiang J, Lee BS, Lu M, Gluck SL, Hurst IR, Holliday LS. Vacuolar H+ATPase binding to microfilaments: regulation in response to phosphatidylinositol 3-kinase activity and detailed characterization of the actin-binding site in subunit B. J Biol Chem. 2004;279:7988–7998. doi: 10.1074/jbc.M305351200. [DOI] [PubMed] [Google Scholar]
- 61.Breton S, Wiederhold T, Marshansky V, Nsumu NN, Ramesh V, Brown D. The B1 subunit of the H+ATPase is a PDZ domain-binding protein. Colocalization with NHE-RF in renal B-intercalated cells. J Biol Chem. 2000;275:18219–18224. doi: 10.1074/jbc.M909857199. [DOI] [PubMed] [Google Scholar]
- 62.Pastor-Soler N, Beaulieu V, Litvin TN, Da Silva N, Chen Y, Brown D, Buck J, Levin LR, Breton S. Bicarbonate-regulated adenylyl cyclase (sAC) is a sensor that regulates pH-dependent V-ATPase recycling. J Biol Chem. 2003;278:49523–49529. doi: 10.1074/jbc.M309543200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Paunescu TG, Da Silva N, Russo LM, McKee M, Lu HA, Breton S, Brown D. Association of soluble adenylyl cyclase with the V-ATPase in renal epithelial cells. Am J Physiol Renal Physiol. 2008;294:F130–138. doi: 10.1152/ajprenal.00406.2007. [DOI] [PubMed] [Google Scholar]
- 64.Rothenberger F, Velic A, Stehberger PA, Kovacikova J, Wagner CA. Angiotensin II stimulates vacuolar H+ATPase activity in renal acid-secretory intercalated cells from the outer medullary collecting duct. J Am Soc Nephrol. 2007;18:2085–2093. doi: 10.1681/ASN.2006070753. [DOI] [PubMed] [Google Scholar]
- 65.Myers M, Forgac M. The coated vesicle vacuolar H+ATPase associates with and is phosphorylated by the 50-kDa polypeptide of the clathrin assembly protein AP-2. J Biol Chem. 1993;268:9184–9186. [PubMed] [Google Scholar]
- 66.Voss M, Vitavska O, Walz B, Wieczorek H, Baumann O. Stimulus-induced phosphorylation of vacuolar H+ATPase by protein kinase A. J Biol Chem. 2007;282:33735–33742. doi: 10.1074/jbc.M703368200. [DOI] [PubMed] [Google Scholar]
- 67.Beaulieu V, Da Silva N, Pastor-Soler N, Brown CR, Smith PJ, Brown D, Breton S. Modulation of the actin cytoskeleton via gelsolin regulates vacuolar H+ATPase recycling. J Biol Chem. 2005;280:8452–8463. doi: 10.1074/jbc.M412750200. [DOI] [PubMed] [Google Scholar]
- 68.Christensen EI, Birn H. Megalin and cubilin: multifunctional endocytic receptors. Nat Rev Mol Cell Biol. 2002;3:256–266. doi: 10.1038/nrm778. [DOI] [PubMed] [Google Scholar]
- 69.Marshansky V, Ausiello DA, Brown D. Physiological importance of endosomal acidification: potential role in proximal tubulopathies. Curr Opin Nephrol Hypertens. 2002;11:527–537. doi: 10.1097/00041552-200209000-00009. [DOI] [PubMed] [Google Scholar]
- 70.Birn H, Willnow TE, Nielsen R, Norden AG, Bonsch C, Moestrup SK, Nexo E, Christensen EI. Megalin is essential for renal proximal tubule reabsorption and accumulation of transcobalamin-B(12) Am J Physiol Renal Physiol. 2002;282:F408–416. doi: 10.1152/ajprenal.00206.2000. [DOI] [PubMed] [Google Scholar]
- 71.Cui S, Verroust PJ, Moestrup SK, Christensen EI. Megalin/gp330 mediates uptake of albumin in renal proximal tubule. Am J Physiol. 1996;271:F900–907. doi: 10.1152/ajprenal.1996.271.4.F900. [DOI] [PubMed] [Google Scholar]
- 72.Orlando RA, Rader K, Authier F, Yamazaki H, Posner BI, Bergeron JJ, Farquhar MG. Megalin is an endocytic receptor for insulin. J Am Soc Nephrol. 1998;9:1759–1766. doi: 10.1681/ASN.V9101759. [DOI] [PubMed] [Google Scholar]
- 73.Schmitz C, Hilpert J, Jacobsen C, Boensch C, Christensen EI, Luft FC, Willnow TE. Megalin deficiency offers protection from renal aminoglycoside accumulation. J Biol Chem. 2002;277:618–622. doi: 10.1074/jbc.M109959200. [DOI] [PubMed] [Google Scholar]
- 74.Kozyraki R, Gofflot F. Multiligand endocytosis and congenital defects: roles of cubilin, megalin and amnionless. Curr Pharm Des. 2007;13:3038–3046. doi: 10.2174/138161207782110507. [DOI] [PubMed] [Google Scholar]
- 75.Ahuja R, Yammani R, Bauer JA, Kalra S, Seetharam S, Seetharam B. Interactions of cubilin with megalin and the product of the amnionless gene (AMN): effect on its stability. Biochem J. 2008;410:301–308. doi: 10.1042/BJ20070919. [DOI] [PubMed] [Google Scholar]
- 76.Biemesderfer D. Regulated intramembrane proteolysis of megalin: linking urinary protein and gene regulation in proximal tubule? Kidney Int. 2006;69:1717–1721. doi: 10.1038/sj.ki.5000298. [DOI] [PubMed] [Google Scholar]
- 77.Li Y, Cong R, Biemesderfer D. The COOH terminus of megalin regulates gene expression in opossum kidney proximal tubule cells. Am J Physiol Cell Physiol. 2008;295:C529–537. doi: 10.1152/ajpcell.00037.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Caruso-Neves C, Pinheiro AA, Cai H, Souza-Menezes J, Guggino WB. PKB and megalin determine the survival or death of renal proximal tubule cells. Proc Natl Acad Sci U S A. 2006;103:18810–18815. doi: 10.1073/pnas.0605029103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jentsch TJ. Chloride and the endosomal-lysosomal pathway: emerging roles of CLC chloride transporters. J Physiol. 2007;578:633–640. doi: 10.1113/jphysiol.2006.124719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Orlowski J, Grinstein S. Emerging roles of alkali cation/proton exchangers in organellar homeostasis. Curr Opin Cell Biol. 2007;19:483–492. doi: 10.1016/j.ceb.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nielsen R, Courtoy PJ, Jacobsen C, Dom G, Lima WR, Jadot M, Willnow TE, Devuyst O, Christensen EI. Endocytosis provides a major alternative pathway for lysosomal biogenesis in kidney proximal tubular cells. Proc Natl Acad Sci USA. 2007;104:5407–5412. doi: 10.1073/pnas.0700330104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Marshansky V. The V-ATPase a2-subunit as a putative endosomal pH-sensor. Biochem Soc Trans. 2007;35:1092–1099. doi: 10.1042/BST0351092. [DOI] [PubMed] [Google Scholar]
- 83.Brown D, Gluck S, Hartwig J. Structure of the novel membrane-coating material in proton-secreting epithelial cells and identification as an H+ATPase. J Cell Biol. 1987;105:1637–1648. doi: 10.1083/jcb.105.4.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Geyer M, Fackler OT, Peterlin BM. Subunit H of the V-ATPase involved in endocytosis shows homology to beta-adaptins. Mol Biol Cell. 2002;13:2045–2056. doi: 10.1091/mbc.02-02-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mayor S, Pagano RE. Pathways of clathrin-independent endocytosis. Nat Rev Mol Cell Biol. 2007;8:603–612. doi: 10.1038/nrm2216. [DOI] [PMC free article] [PubMed] [Google Scholar]