

Abstract
The aspartate receptor of the Escherichia coli and Salmonella typhimurium chemotaxis pathway generates a transmembrane signal that regulates the activity of the cytoplasmic kinase CheA. Previous studies have identified a region of the cytoplasmic domain that is critical to receptor adaptation and kinase regulation. This region, termed the adaptation subdomain, contains a high density of acidic residues, including specific glutamate residues that serve as receptor adaptation sites. However, the mechanism of signal propagation through this region remains poorly understood. This study uses site-directed mutagenesis to neutralize each acidic residue within the subdomain to probe the hypothesis that electrostatics in this region play a significant role in the mechanism of kinase activation and modulation. Each point mutant was tested for its ability to regulate chemotaxis in vivo and kinase activity in vitro. Four point mutants (D273N, E281Q, D288N, and E477Q) were found to superactivate the kinase relative to the wild-type receptor, and all four of these kinase-activating substitutions are located along the same intersubunit interface as the adaptation sites. These activating substitutions retained the wild-type ability of the attractant-occupied receptor to inhibit kinase activity. When combined in a quadruple mutant (D273N/E281Q/D288N/E477Q), the four charge-neutralizing substitutions locked the receptor in a kinase-superactivating state that could not be fully inactivated by the attractant. Similar lock-on character was observed for a charge reversal substitution, D273R. Together, these results implicate the electrostatic interactions at the intersubunit interface as a major player in signal transduction and kinase regulation. The negative charge in this region destabilizes the local structure in a way that enhances conformational dynamics, as detected by disulfide trapping, and this effect is reversed by charge neutralization of the adaptation sites. Finally, two substitutions (E308Q and E463Q) preserved normal kinase activation in vitro but blocked cellular chemotaxis in vivo, suggesting that these sites lie within the docking site of an adaptation enzyme, CheR or CheB. Overall, this study highlights the importance of electrostatics in signal transduction and regulation of kinase activity by the cytoplasmic domain of the aspartate receptor.
The aspartate receptor of bacterial chemotaxis is one of the best-characterized members of a large family of cell surface receptors that modulate two-component signaling pathways (reviewed in refs 1–6). Through regulation of the associated histidine kinase CheA, the aspartate receptor controls cellular chemotaxis in response to changing concentrations of the chemoattractant aspartate, allowing the cell to move up an attractant concentration gradient. The receptor is an oligomer composed of three homodimers that assemble to form a trimer of dimers, as illustrated in Figure 1A. The chemotaxis signaling pathway is triggered by aspartate binding to the receptor periplasmic domain, which sends a conformational transmembrane signal to the cytoplasmic domain. The highly elongated, α-helical cytoplasmic domain serves as a framework for the assembly of the cytoplasmic soluble pathway components and transmits the signal to the bound CheA kinase. The fundamental unit of kinase regulation is a ternary complex comprised of the receptor, the CheA kinase, and the coupling protein CheW, shown in Figure 1B. In the absence of attractant aspartate, the receptor is “on” and stimulates autophosphorylation of the CheA kinase. Subsequently, a response regulator protein, CheY, binds to CheA and catalyzes transfer of the same phospho group from CheA to its own active site. Phospho-CheY then dissociates from the complex and diffuses to the flagellar motor where it regulates cellular swimming behavior. By contrast, aspartate binding to the periplasmic domain switches the receptor into its “off” state in which it sends a transmembrane attractant signal to CheA that inhibits autophosphorylation.
Figure 1.

Model of aspartate receptor structure and function. (A) Full-length model based on crystallographic and chemical data (18, 29, 37–39). Recent data support the model, except for the indicated helical conformation of the linker region (40, 41). The two identical subunits are colored blue and gold, respectively, and sites of methylation are colored black. Also shown is the crystal structure of the cytoplasmic domain fragment illustrating the trimer of dimers (5). Structures were generated with MacPymol (DeLano Scientific). (B) Schematic view of the core ternary signaling complex consisting of the receptor trimer of dimers, the coupling protein (CheW), and the histidine kinase (CheA). Accurate stoichiometries of the latter two components are not yet known. Two types of signals regulate kinase activity: the transmembrane attractant signal which inhibits the kinase (red) and the cytoplasmic adaptation signal which activates the kinase (green). These two signals originate at the periplasmic aspartate binding site (small black circle) and at the cytoplasmic adaptation sites (larger black circles), respectively.
In addition to the transmembrane attractant signal, the receptor also generates a cytoplasmic adaptation signal that regulates CheA kinase activity (1–6). This adaptation signal is controlled by four specific glutamate residues on the cytoplasmic domain of the receptor. Each of the adaptation site glutamates can be methyl esterified or demethylated by two adaptation enzymes in response to the presence or absence of attractant, respectively. The first enzyme, the methyltransferase CheR, is constitutively active and is tethered to the receptor by its interaction with a pentapeptide sequence at the receptor C-terminus (7). The tethered CheR enzyme docks to the adaptation subdomain and methyl esterifies the adaptation site glutamates, thereby neutralizing their negative charges. The second enzyme, the methyl-esterase CheB, is phospho-activated by CheA and also docks to the receptor C-terminus, where it can more efficiently access the receptor adaptation sites to hydrolytically remove amides and methyl esters, thereby restoring the glutamate negative charge (8). The overall methylation level of the adaptation sites has a direct effect on the activity of the CheA kinase: a high level of methylation activates the kinase, while a low level of methylation inhibits it. Overall, this covalent adaptation provides an adaptation signal that travels through the receptor cytoplasmic domain from the adaptation sites to the CheA kinase. The resulting adaptation signal serves as a counterbalance to the attractant signal, enabling the receptor population to adapt to a background concentration of attractant. In addition, the adaptation signal serves as the chemical memory used by the chemotaxis pathway to record the attractant level of the recent past, enabling the receptor to compare the current and past attractant levels to determine the sign of its output signal (1–6).
The cytoplasmic domain of each receptor dimer has a coiled-coil structure, consisting of a four-helix bundle formed by two α-helical hairpins, one provided by each subunit (3, 5, 9). The two cytoplasmic helices of one subunit are termed CD11 and CD2, and those of the other identical subunit are termed CD1′ and CD2′, respectively. The cytoplasmic domain can be divided into two subdomains, an adaptation subdomain containing the adaptation sites and docking surfaces for CheB and CheR, and a signaling subdomain where CheA and CheW bind to form the ternary complex. The signaling subdomain also provides the contacts that stabilize formation of the trimer of dimers (5). The other soluble components of the chemotaxis pathway, CheY and CheZ, bind to receptor-associated CheA; thus, all of the pathway components are organized in a single supermolecular signaling complex. In living cells, the trimeric signaling complexes are localized to the cell poles, where they form limited higher-order associations (5, 10–14).
The signaling mechanism of the receptor is only partially defined. Biochemical and biophysical studies have identified the transmembrane signal as a small pistonlike displacement of the membrane-spanning α4/TM2 signaling helix (3, 15–17). Considerably less is known about signal propagation through the cytoplasmic domain to the associated CheA kinase. Previous studies have shown that the conformational signal in the cytoplasmic domain is small in magnitude and may modulate subunit packing within the adaptation and signaling subdomains (18, 19); however, further studies are necessary to define the exact mechanism. Intriguingly, the adaptation subdomain possesses a high density of conserved acidic residues on both CD1 and CD2. A reasonable hypothesis is that this negative charge is instrumental in kinase regulation; however, a rigorous examination of this electrostatic hypothesis has not yet been undertaken.
This study directly examines the functional role of the acidic residues in the adaptation subdomain. In total, the adaptation subdomain contains approximately 100 residues per subunit, of which 21 residues per subunit are acidic (including the four adaptation site glutamates). Figure 2 summarizes the positions of these acidic residues within a structural model of the adaptation subdomain consistent with the chemically determined structure of the native cytoplasmic domain and the crystallographically determined structure of the signaling subdomain (5, 9, 18). Table 1 lists the acidic residues and indicates their high degree of conservation (20). Previous studies have shown that neutralization of each of the four adaptation sites by methyl esterification, or by site-directed mutagenesis from Glu to Gln, significantly increases receptor-regulated kinase activity (12, 13, 21, 22). Our study uses site-directed mutagenesis to neutralize each of the 17 remaining acidic residues, termed background acidic residues, by conservative substitution from Glu to Gln or Asp to Asn. Four of the new substitutions are found to superactivate the kinase, and like the adaptation sites, all four are located along the intersubunit interface between helices CD1 and CD2 ′. To examine these new activating positions further, site-directed mutagenesis was used to engineer receptors containing multiple charge-neutralizing mutations as well as point charge reversals to arginine. Some of these engineered receptors exhibit partial lock-on behavior since aspartate binding is unable to fully inhibit kinase activity. Finally, rates of formation of disulfide bonds between symmetric pairs of engineered Cys residues on different subunits were significantly higher in the EEEE than in the QQQQ adaptation state, suggesting that negative charges at the adaptation sites enhance structural dynamics in the adaptation subdomain. These results, together with previous evidence, strongly suggest that the intersubunit interface of the adaptation subdomain plays a central role in kinase regulation, and that electrostatic interactions modulate this critical signaling interface.
Figure 2.

Atomic model of the adaptation subdomain, highlighting the acidic side chains. The indicated structure is excerpted from the structural model for the full receptor (5). The helices of subunit I are colored yellow and pink, and the helices of subunit II are colored blue and white. Adaptation sites are colored red. The 17 acidic Asp and Glu side chains targeted in this study for neutralization by conservative mutation to Asn and Gln are colored black. (B) Alternate view obtained by rotating the subdomain 90° about its long axis. Structures were generated with MacPymol (DeLano Scientific).
Table 1.
Conservation of Acidic Residues in Class I Bacterial Receptorsa
| position | no. of acidic residues | no. of acidic and Gln residues | % acidic | % acidic and Gln |
|---|---|---|---|---|
| 270 | 5 | 7 | 38.5 | 53.8 |
| 273 | 9 | 10 | 69.2 | 76.9 |
| 281 | 12 | 13 | 92.3 | 100.0 |
| 288 | 11 | 12 | 84.6 | 92.3 |
| 294 | 11 | 11 | 84.6 | 84.6 |
| 295 | 2 | 10 | 15.4 | 76.9 |
| 301 | 12 | 12 | 92.3 | 92.3 |
| 302 | 9 | 13 | 69.2 | 100.0 |
| 308 | 12 | 12 | 92.3 | 92.3 |
| 309 | 1 | 11 | 7.7 | 84.6 |
| 319 | 9 | 9 | 69.2 | 69.2 |
| 452 | 8 | 8 | 61.5 | 61.5 |
| 456 | 11 | 11 | 84.6 | 84.6 |
| 462 | 9 | 9 | 69.2 | 69.2 |
| 463 | 11 | 12 | 84.6 | 92.3 |
| 469 | 7 | 7 | 53.8 | 53.8 |
| 477 | 7 | 10 | 53.8 | 76.9 |
| 479 | 10 | 10 | 76.9 | 76.9 |
| 491 | 11 | 12 | 84.6 | 92.3 |
| 500 | 7 | 7 | 53.8 | 53.8 |
| 501 | 7 | 7 | 53.8 | 53.8 |
Adaptation sites are underlined. Conservation of charge is reported for 13 class I bacterial receptors (20).
MATERIALS AND METHODS
Mutagenesis
Mutations of the plasmid pSCF6 were created using the previously described Kunkel method of site-directed mutagenesis (23). In all cases, the substitutions were aspartate to asparagine or arginine and glutamate to glutamine or arginine. Restriction digestion and DNA sequencing were used to confirm the presence of the desired mutations.
In Vivo Analysis of Receptor Function
Aspartate-specific chemotaxis of the mutant receptors was assessed using the previously described chemotaxis swarm assay (24). Briefly, Escherichia coli strain RP8611, a strain lacking the aspartate receptor but containing all other soluble pathway components, was transformed with the mutant plasmids. Liquid cultures were grown for 6 h in Luria broth at 37 °C with shaking; 5 μL of culture was spotted onto agar minimal medium plates containing Vogel Bonner Citrate medium supplemented with 0.1% glycerol, 20 mM lactate, 40 μg/mL DL-histidine, 20 μg/mL L-leucine, 1 μg/mL thiamine, and 100 μg/mL ampicillin, with or without 0.1 mM aspartate. After overnight incubation at 30 °C, measurements of the swarm diameters were taken at 3 h intervals and normalized to that of the wild type to determine the relative rate of cellular chemotaxis.
Protein Expression and Isolation
Confirmed mutant plasmids were transformed into E. coli strain RP3808 and expressed. Strain RP3808 lacks all major chemoreceptors and adaptation enzymes, allowing for the isolation of a homogeneous population of the mutant receptor (25). The overexpressed mutant receptors were isolated in native cell membranes by a previously described method (26) with the following modifications. Pellets from the overnight 500 mL cultures were resuspended in 5 mL of low-salt buffer [100 mM sodium phosphate (pH 7.0), 10% (w/v) glycerol, and 10 mM EDTA]. Resuspended cells were lysed by sonication (4 × 10 s bursts with a 60 s cooling between bursts). Immediately, DTT (5 mM), PMSF (1 mM), and 1,10-phenanthroline (2.5 mM) were added to the first wash buffer at the indicated final concentrations. Following membrane isolation, receptor expression levels were visualized on a Coomassie-stained 10% Laemmli SDS–PAGE gel (acrylamide:bisacrylamide ratio of 40:0.2). To equalize or normalize receptor levels, receptor bands were quantitated with a digital camera.
In Vitro Analysis of Receptor-Coupled Kinase Regulation
The ability of the mutant receptors to activate the CheA kinase was assessed using a previously described method (15). Briefly, the reconstituted receptor kinase signaling complex consisting of 2 μM receptor, 2 μM CheW, 0.5 μM CheA, and 10 μM CheY with and without aspartate was incubated for 45 min at room temperature. Reactions were carried out at room temperature in buffer containing 50 mM Tris-HCl (pH 7.5), 50 mM KCl, and 5 mM MgCl2. The reaction was initiated by addition of [γ-32P]ATP diluted with cold ATP to a total ATP concentration of 100 μM. The reaction was quenched after 10 s by the addition of 2× Laemmli sample buffer containing 25 mM EDTA. Kinase activity was measured by the rate of formation of phosphorylated CheY under conditions where receptor-regulated CheA autophosphorylation is the rate-limiting step, thereby ensuring that reactions measure the activity of the receptor–CheW–CheA ternary complex. Phospho-CheY was resolved on a 15% Laemmli SDS–PAGE gel (acrylamide:bisacrylamide ratio of 40:1.25) and quantitated by phosphorimaging.
Assessment of Disulfide Formation
Disulfide formation reactions were carried out as described in ref 27 with the following modifications. Receptors were diluted in a buffer containing 20 mM sodium phosphate (pH 7.0), 10% glycerol, 5 mM EDTA, and 150 mM KCl. Following addition of 2 mM Cu(II)(1,10-phenanthroline)3 to trigger disulfide formation, the reaction was allowed to proceed for the indicated length of time before being quenched with 2× Laemmli sample buffer supplemented with 10 mM N-ethylmaleimide. Finally, the fraction of the receptor population converted to covalent dimer was quantitated by SDS–PAGE. The presence of excess EDTA during the reaction ensured mild oxidation conditions.
Electrostatics Analysis
The electrostatic surface potential of the receptor adaptation subdomain was analyzed using GRASP (28). The potentials were calculated for an ionic strength of 110 mM, a protein core dielectric constant of 2, and a solvent dielectric constant of 80. Isopotential contours are shown for electrostatic energies of ±3 and ±1 kT/e.
RESULTS
Strategy for Probing the Role of Negative Charge in the Adaptation Subdomain
It is reasonable to hypothesize that the high density of negative charges within the adaptation subdomain of the aspartate receptor plays a role in the regulation of the CheA kinase. To test this hypothesis, each of the 17 background acidic aspartate and glutamate residues in this subdomain was neutralized by conservative mutation to asparagine and glutamine, respectively. The electrostatic hypothesis predicts that these charge-neutralizing mutations will affect the ability of the receptor to regulate kinase activity. Figure 2 depicts the modeled adaptation subdomain (5), illustrating the sites of the 17 point mutations. The effect of each mutation on receptor function was determined both in living cells and in the reconstituted receptor kinase signaling complex using an in vivo chemotaxis assay and an in vitro receptor-coupled kinase assay, respectively.
Design and Characterization of Mutant Receptors
Neutralizing substitutions at the 17 background acidic positions within the adaptation subdomain were generated by site-directed mutagenesis of the aspartate receptor gene in expression plasmid pSCF6 (29). The mutant plasmids were transformed into E. coli strain RP3808 (25), a chemoreceptor-deficient strain that lacks the aspartate receptor, the other major chemotaxis receptors, and the soluble pathway components, including adaptation enzymes CheB and CheR. The lack of adaptation enzymes ensures the isolation of a homogeneous population of aspartate receptors with identical adaptation states that have not been subjected to post-translational modification. Mutant receptors were overexpressed, and native membranes containing the overexpressed receptors were isolated (26). All mutant receptors exhibited expression levels similar to that of the wild type, indicating that the mutant receptors are stable, membrane-imbedded proteins.
Effect of Substitutions on Receptor Function in Vivo
The in vivo chemotactic ability of the mutant receptors was assessed by the previously described swarm plate assay (24), wherein mutant receptors were expressed in E. coli strain RP8611, an aspartate receptor-deficient strain. The swarm plate assay measures the mutant receptor’s ability to restore chemotaxis in response to a self-induced attractant gradient generated by metabolic depletion of aspartate in the medium. Because of the compensatory nature of the adaptation branch of the chemotaxis pathway, which corrects many minor receptor defects, this in vivo swarm plate assay of cellular chemotaxis only reveals major receptor perturbations that significantly inhibit either signaling or adaptation.
Figure 3 shows that of the 17 mutant receptors that have been tested, 14 demonstrate swarming behavior similar to that of the wild type. All 14 exhibit the ability to restore cellular chemotaxis in the swarm assay to within 50% of that of the wild type. The three remaining substitutions (E308Q, E463Q, and D479N) completely block swarming. In principle, the observed variability in swarm rates observed for the different mutants could be due to perturbation of kinase activation, or to perturbation of the adaptation system, or to variability in receptor expression levels, making it difficult to resolve the mechanism of individual perturbations. To more rigorously determine the effect of all 17 substitutions on receptor-regulated kinase activity, an in vitro approach was utilized.
Figure 3.

Effects of charge neutralization on cellular chemotaxis in vivo. Receptors were expressed in a strain lacking the aspartate receptor, and their ability to restore in vivo chemotaxis in a self-induced gradient of aspartate was assessed (see Materials and Methods). Liquid culture was spotted onto an agar minimal medium plate containing or lacking 0.1 mM aspartate. The diameter of the chemotactic swarm was measured at four to six time points to determine the swarm rate at 30 °C. Each bar represents the aspartate-specific swarm rate for a given receptor normalized relative to the wild-type aspartate-specific rate.
Effect of Substitutions on Receptor-Regulated Kinase Activity in Vitro
The in vitro receptor-coupled kinase assay is well-suited for detecting even subtle effects of mutations on receptor modulation of kinase. Using the reconstituted receptor–CheW–CheA ternary complex, the accumulation of phospho-CheY is assessed in the presence and absence of a saturating aspartate concentration, determining the abilities of the mutant receptors to couple to and activate the CheA kinase (15). Since this in vitro system lacks the adaptation enzymes CheR and CheB, the compensatory effects of the adaptation system will not mask subtle perturbations in receptor function. Moreover, the assay is carried out using a fixed receptor concentration, eliminating variations due to different receptor expression levels.
Figure 4 shows that four of the 17 engineered mutants produce kinase superactivation in the in vitro receptor-coupled kinase assay. Each of these mutants, D273N, E281Q, D288N, and E477Q, yields rates of phospho-CheY formation exceeding 150% of the wild-type rate in the absence of attractant aspartate. Such results suggest that charge neutralization at these positions shifts the signaling equilibrium of the receptor toward its kinase-activating on-state. However, aspartate binding is able to effectively inhibit the kinase activity stimulated by these mutants, as for the wild-type receptor, indicating that attractant binding still drives the equilibrium fully toward the off-state. Figure 5 illustrates the locations of these four activating sites, as well as the four adaptation sites, in the structural model of the adaptation subdomain. All four of the activating sites lie along the same face as the adaptation sites. In fact, E281Q, D288N, and E477Q are each separated from at least one adaptation site by an integral number of heptad repeats in the N-terminal direction. In the coiled-coil structure of the cytoplasmic domain, separation by heptad repeat units ensures a same-face location. Also of note, the face possessing the four activating and four adaptation sites lies along a specific interface between helix CD1 on one subunit and helix CD2′ on the other subunit. These observations are consistent with those previously obtained for the corresponding cysteine substitutions, E281C, D288C, and E477C, which yield rates of phospho-CheY formation that are 200, 150, and 160% of that of the wild type, respectively (30; also S. E. Winston and J. J. Falke, unpublished results). The similar effects of side chain amidation and conversion to cysteine suggest that it is the change in charge, not size, that accounts for kinase superactivation.
Figure 4.

Effects of charge neutralization on receptor-coupled kinase activity in vitro. Receptors were expressed in a strain lacking all soluble pathway components. Native membranes were isolated, and the ability of each modified receptor to activate the kinase in the reconstituted ternary complex was assessed. Each bar represents the observed receptor-coupled kinase activity in the presence (white bars) and absence (black bars) of aspartate, normalized to wild-type kinase activity in the absence of aspartate. Reaction mixtures at 22 °C contained 2 μM receptor monomer, 2 μM CheW monomer, 0.5 μM CheA monomer, and 10 μM CheY in a buffer containing 50 mM Tris-HCl (pH 7.5), 50 mM KCl, 5 mM MgCl2, and 100 μM ATP.
Figure 5.
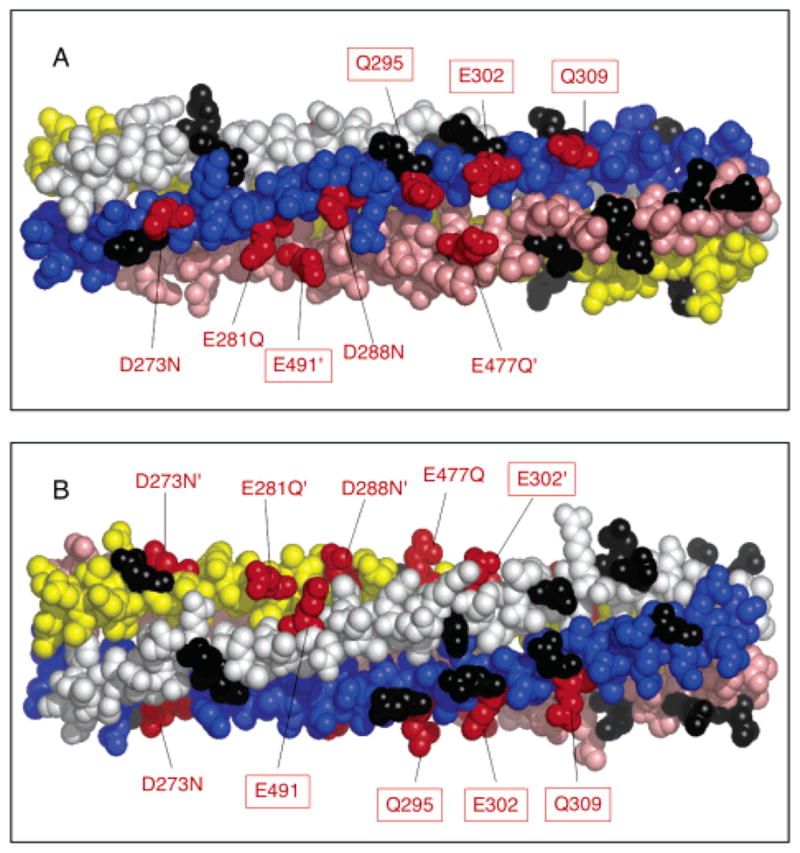
Locations of the four newly identified activating sites, and of the four adaptation sites. The helices of subunit I are colored yellow and pink, and the helices of subunit II are colored blue and white. Adaptation sites are denoted in red and boxed. Positions where charge-neutralizing substitutions activate or have no effect on kinase activity are colored red or black, respectively. (B) Alternate view obtained by rotating the subdomain 90° about its long axis. Structures were generated with MacPymol (DeLano Scientific).
Interestingly, of the three mutants that abolished chemotactic ability in the swarm plate assay, E308Q and E463Q show wild-type activation of the CheA kinase in the in vitro assay. The simplest explanation is that these positions play an important role in receptor adaptation rather than in kinase regulation. For example, they could be located within the CheB or CheR docking site. The result for E308Q is consistent with the previous finding that position E308 interacts with the methyltransferase, CheR (31), and is part of the CheR recognition sequence for adaptation site 3 on helix CD1 (32, 33). E463 is not part of the currently proposed adaptation site 4 recognition sequence on helix CD2; however, it is located close to adaptation site 3. Substitution D479N, on the other hand, completely blocks kinase activation. D479 is the most buried of the 17 positions; therefore, this result suggests that substitution at this position perturbs the subunit interface and blocks receptor-mediated kinase activation both in vivo and in vitro.
Additional Electrostatic Modifications at the Four Activating Positions
To further examine the role of electrostatics, the acidic residues at each of the four activating positions (D273, E281, D288, and E477) were targeted for two additional types of modification. Site-directed mutagenesis was used to engineer a charge reversal mutation at each position by substitution with arginine, as well as to create combinations of multiple charge neutralizations at the four positions. Altogether, nine new mutant receptors were created: D273R, E281R, D288R, E477R, E281Q/D288N, E281Q/E477Q, D288N/E477Q, E281Q/D288N/E477Q, and D273N/E281Q/D288N/E477Q.
Swarm assays were carried out to assess the abilities of these nine mutant receptors to restore in vivo chemotaxis. Figure 6 shows that seven of the nine mutant receptors display chemotactic functions similar to that of the wild type. One exception, the E281Q/D288N double mutant, yields a swarm rate 2-fold higher than that observed for the overexpressed wild-type receptor. Examination of isolated membranes reveals that this double mutant is expressed at significantly lower levels, yielding a receptor density closer to that of the native, non-overexpressed receptor. Such reduced levels of receptor overexpression have previously been observed to increase the swarm rate relative to that of the overexpressed receptor (J. A. Bornhorst and J. J. Falke, unpublished results), thereby providing a plausible explanation for the enhanced swarm rate of E281Q/D288N. The other perturbed receptor possesses all four charge-neutralizing substitutions, D273N, E281Q, D288N, and E477Q, and is overexpressed at normal levels but exhibits no swarm activity. Thus, together, these four substitutions completely abolish the ability of the receptor to restore chemotaxis.
Figure 6.

Effects of charge reversal, multisite neutralization, and cysteine substitution on cellular chemotaxis in vivo. Chemotactic swarm assays were carried out in vivo as described in the legend of Figure 3 at 30 °C. Each bar represents the aspartate-specific swarm rate for the indicated receptor normalized relative to the wild-type aspartate-specific swarm rate.
The in vitro receptor-coupled kinase assay was used to determine the effects of the nine mutations on receptor-mediated kinase regulation. Figure 7 shows that the three charge reversal mutants (D273R, E281R, and D288R), like their charge-neutralizing counterparts, yield rates of phospho-CheY formation 1.5–2.0-fold faster than the wild-type rate in the absence of aspartate. E477R, however, exhibits a rate of phospho-CheY formation that is 2-fold slower than that of the wild type. Interestingly, in the aspartate-occupied state, D273R retains some kinase activity, indicating that attractant binding no longer drives the receptor fully to the off-state. Figure 7 also summarizes the effects of multiple charge neutralizations on kinase regulation. In the absence of aspartate, mutants E281Q/D288N and E281Q/D288N/E477Q exhibit rates similar to those of their single mutant counterparts, 1.5–2.0-fold faster than that of the wild type. Mutants E281Q/E477Q and D288N/E477Q exhibit rates similar to the wild-type rate. Strikingly, however, the quadruple mutant D273N/E281Q/D288N/E477Q exhibits a rate fully 3-fold faster than that of the wild type. Moreover, in the aspartate-occupied state, this mutant exhibits considerable lock-on character, demonstrated by its nearly complete lack of attractant downregulation. It follows that the quadruple mutation drives the receptor equilibrium toward the on-state, and largely blocks the conformational change to the off-state triggered by attractant binding. The lock-on character is sufficient to explain the lack of activity of this receptor in the swarm assay, since it fails to generate adequate attractant regulation of kinase activity. Overall, the additional electrostatic modifications at the four activating positions place further constraints on the mechanism of electrostatic modulation of the adaptation subdomain.
Figure 7.

Effects of charge reversal and multisite neutralization on receptor-coupled kinase activity in vitro. Assays were carried out as described in the legend of Figure 4 at 22 °C. The bars represent the observed kinase activity in the presence (white bars) and absence (black bars) of aspartate, normalized to the wild-type kinase activity in the absence of aspartate.
Relationship between Electrostatics and Conformational Dynamics of the Adaptation Subdomain
To examine the effects of electrostatics changes on the conformational dynamics of the adaptation subdomain, formation of disulfide bonds between symmetric pairs of engineered cysteines was used to compare the conformational fluctuations of different adaptation states. Three cysteine point mutations (G278C, A304C, and T482C) were each engineered into the QQQQ or EEEE adaptation state background, each of which possesses all glutamine or glutamate side chains at the four native adaptation sites, respectively, and thus substantially alter the electrostatics of the adaptation subdomain. Moreover, the QQQQ adaptation state yields greater than wild-type (QEQE) stimulation of kinase activity, while the EEEE state yields near-zero kinase activity; thus, these two backgrounds drive the receptor equilibrium between its on-and off-states (13). Each cysteine mutation generates a unique, symmetric pair of cysteines in the receptor homodimer, which lacks intrinsic cysteines. Since the selected cysteine pairs lie at buried locations in the predicted structure of the signaling subdomain, collisions generated by conformational fluctuations that bring the two cysteines into direct contact can be trapped by disulfide bond formation to yield a covalent dimer (34). The structural model of the cytoplasmic domain (5) indicates that each of the three cysteine pairs requires translational motion ranging from 6 to 9 Å in length to achieve a collision and disulfide formation. Thus, perturbations that increase conformational dynamics are expected to increase the rate of disulfide bond formation.
The disulfide bond formation reaction was triggered by the addition of the redox catalyst Cu(II)(1,10-phenanthroline)3, and was allowed to proceed for a predetermined time until quenching, and then was quantitated by the detection of disulfide-linked dimers on SDS–PAGE. Analysis of the time dependence of each reaction (not shown) revealed that the G278C and A304C disulfide formation reactions were too fast to measure. Thus, instead of measuring initial rates, we allowed all three reactions to proceed to completion, and the final extent of disulfide formation is presented in Figure 8. The final extent of disulfide formation is defined by the relative rates of the disulfide formation reaction and a competing reaction that oxidatively damages free cysteines (34). For all three cysteine positions, the final extent of disulfide formation is 2-fold higher in the EEEE background than in the QQQQ background, suggesting that the rate of the disulfide formation reaction is significantly faster in the EEEE background. Alternatively, the competing reaction could be slower in the EEEE background for all three reactions, or the helices could be closer together in the average structure of the EEEE state than in the QQQQ state. These possibilities are disfavored by the observation that the competing reaction is typically less sensitive to environmental changes than the disulfide formation reaction (34), and by the fact that a simple electrostatic argument would predict that the charge repulsion would push the helices apart in the EEEE state. Overall, the simplest explanation for the results is that the EEEE adaptation state is more dynamic than the QQQQ adaptation state, leading to its higher final extent of disulfide bond formation. It follows that conformational dynamics of the adaptation subdomain are sensitive to the electrostatic character of the adaptation sites.
Figure 8.

Effect of receptor adaptation state on intersubunit disulfide bond formation. Each receptor possessed the indicated cysteine mutation engineered into the QQQQ or EEEE adaptation state background. The receptor was overexpressed in an E. coli strain lacking the adaptation enzymes and was isolated in native cell membranes. The disufide formation reaction was triggered by addition of a redox catalyst, and the reaction was allowed to proceed for 10 s (G278C and A304C) or 90 s (T482C) at 25 °C before being quenched and prior to analysis of the fraction of receptor converted to disulfide-linked dimer. These reaction times ensured each reaction reached completion. Reaction mixtures contained 2 μM receptor in 150 mM KCl, 20 mM sodium phosphate (pH 7.0), 10% glycerol, 5 mM EDTA, and 2 mM Cu(II)(1,10-phenanthroline)3 as a redox catalyst.
Examination of the Adaptation Subdomain Electrostatic Surface Potential
The molecular imaging program GRASP (28) was used to examine the electrostatic surface potential of the aspartate receptor adaptation subdomain. Analysis of surface potential and isopotential patterns can be a useful tool in the interpretation of biochemical data (35). To that end, panels A and B of Figure 9 show a visual representation of the isopotential contours generated for the adaptation subdomain for energies of ±1 and ±3 kT/e, respectively, calculated for physiological ionic strength. Notably, there is a distinct red surface of negative potential that contains the adaptation sites and superactivating mutations. These observations are consistent with the hypothesis that electrostatics play an important role in the signaling mechanism in this region of the receptor.
Figure 9.

Isopotential contours for the adaptation subdomain. GRASP (28) was used to calculate isopotential contours for the EEEE adaptation state at an ionic strength of 110 mM, based on the modeled structure of the adaptation subdomain (5). Red is negative, and blue is positive. Sites of adaptation are represented by black circles: (A) contours for ±1 kT/e, where the negative contour extends at most ~10 Å from the domain surface, and (B) contours for ±3 kT/e.
The isopotential contours for ±1 kT/e represent the maximum distance at which a unit charge approaching from infinity would experience either an attraction to or a repulsion from charges located on the surface of the receptor, where the energy of the interaction is equal to the thermal energy. Although an adjacent receptor would possess multiple charges, adaptation in vivo proceeds by enzymatic modification of one charge at a time; thus, the unit charge picture is most useful here. The GRASP calculation effectively yields an upper limit on the electrostatic interaction distance for this unit charge, because (1) it assumes full ionization of charged side chains while in reality the high negative charge density in the adaptation subdomain will likely cause some acidic residues to be partially protonated and (2) it does not take into account the screening of surface negative charges by Mg2+ ions while in reality these metal ions will likely bind to the high density of carboxylates on the subdomain surface and neutralize some of the surface charge. Beyond this upper limit, at distances at or beyond the ±1 kT/e isopotential contours, the electrostatic interaction energy of a unit charge on an adjacent receptor will be smaller than the thermal energy, and thus can be neglected.
Examination of the ±1 kT/e isopotential contour indicates that the relevant electrostatic field extends at most 10 Å from the surface of the protein. The fact that this distance is an upper limit is consistent with the results presented above for charge neutralization and reversal mutations at the four activating positions. In the in vitro receptor-coupled kinase assay, conversion of the native anionic side chain (Asp/Glu) to a neutral side chain (Asn/Gln) has the virtually same effect on kinase activity as conversion to a cationic side chain (Arg) in six of the eight cases tested (four positions, each with and without attractant). The simplest explanation for the failure to observe larger effects for arginine substitution, at least in most cases, is that the positive charge on the long arginine side chain lies outside the region of strong electrostatic effects. Since a fully extended arginine side chain is 7 Å in length, the positive charge could project outside the relevant electrostatic field, for example, by forming a salt bridge distal to the subunit interface. Together, these observations have important implications for the issue of whether electrostatic modulation of kinase activity arises from the interactions between charges within the same receptor dimer, or on different dimers.
DISCUSSION
These findings further demonstrate the importance of the adaptation subdomain in the regulation and activation of the CheA kinase and directly establish the importance of electrostatic interactions involving the intersubunit interface between adjacent helices CD1 and CD2′ of the same dimer. Altogether, eight acidic residues, including the four adaptation sites, lie along this interface and generate significant kinase activation when neutralized. The degree of kinase activation observed for neutralization of the four background residues (D273N, E281Q, D288N, and E477Q) falls within the range of activation observed for neutralization of the four adaptation sites (E295Q, E302Q, E309Q, and E491Q), as illustrated in Figure 10. Notably, residues E281, E288, E295, E302, and E309 on CD1 and residues E477 and E491 on CD2 are separated by integral numbers of heptad repeats, indicating that they fall on the same face of the coiled-coil CD1 and CD2 helices, respectively. Because of the 2-fold symmetry of the receptor, a symmetric surface containing the same eight activating positions is found on the opposite side of the homodimer (through the page in Figure 5A), representing a 180° rotation about the long axis of the subdomain. When the subdomain is instead rotated 90° about its long axis to reveal the face lying between the two symmetric activation faces (Figure 5B), a high density of negative charges is also observed, but neutralization of these charges does not activate the kinase. Thus, the positions at which neutralization of a negative charge generates kinase activation are highly localized along the CD1–CD2′ interface, and the symmetric CD1′–CD2 interface on the opposite side of the subdomain.
Figure 10.

Comparing the effects of charge neutralization at activating sites and adaptation sites on receptor-coupled kinase activity. The four left-most bars indicate the increased kinase activity, relative to that of the wild type, observed for the four activating site mutations (data from Figure 4). The four right-most bars indicate the increase in kinase activity, relative to that of the indicated background receptor, observed upon neutralization of each of the four adaptation site glutamates (data from ref 13). The background receptor chosen for each of the right-most bars is the background closest to that of the wild type (QEQE) in which the indicated adaptation site can be neutralized, yielding the following set of receptor pairs: EEQE and QEQE for site 1, QEQE and QQQE for site 2, QEEE and QEQE for site 3, and QEQE and QEQQ for site 4. All values are for reaction mixtures lacking aspartate.
There are several possible explanations for the importance of this face to kinase activation. The negative charges on the face could modulate (i) the stability of the individual CD1 or CD2′ helix, (ii) the stability of the subunit–subunit interaction within the subdomain of a single dimer, (iii) interactions between dimers within the same trimer of dimers, or (iv) interactions between adjacent trimers of dimers. The latter two possibilities are disfavored because of the relatively large distances between the adaptation subdomains of different dimers, illustrated in Figure 11. Specifically, nearest-neighbor adaptation subdomains on different dimers are predicted to be approximately 14 or 27 Å in length in a simple geometric model that uses the current picture of the full-length dimer (5) to generate a schematic trimer of dimers or a pair of adjacent trimers of dimers, respectively (Figure 11). (Note that the 14 Å distance is effectively a lower limit since the adaptation site residues and the activating residues described herein are facing out from the center of the trimer of dimers where the adaptation sites can be accessed by the adaptation enzymes; therefore, these residues are not facing the adjacent dimer as implied by the 14 Å distance.) Such distances are significantly larger than the upper limit of 10 Å estimated by GRASP to be the maximum separation for significant electrostatic interactions, defined as interactions as large or larger than the thermal energy, when a unit charge change occurs on a nearby receptor dimer under the relevant ionic strength conditions (see Figure 9A). It follows that receptor dimers in the same or different trimers of dimers are likely too far apart to allow significant electrostatic interactions due to point charge changes at adaptation sites or at the additional activating positions described herein.
Figure 11.
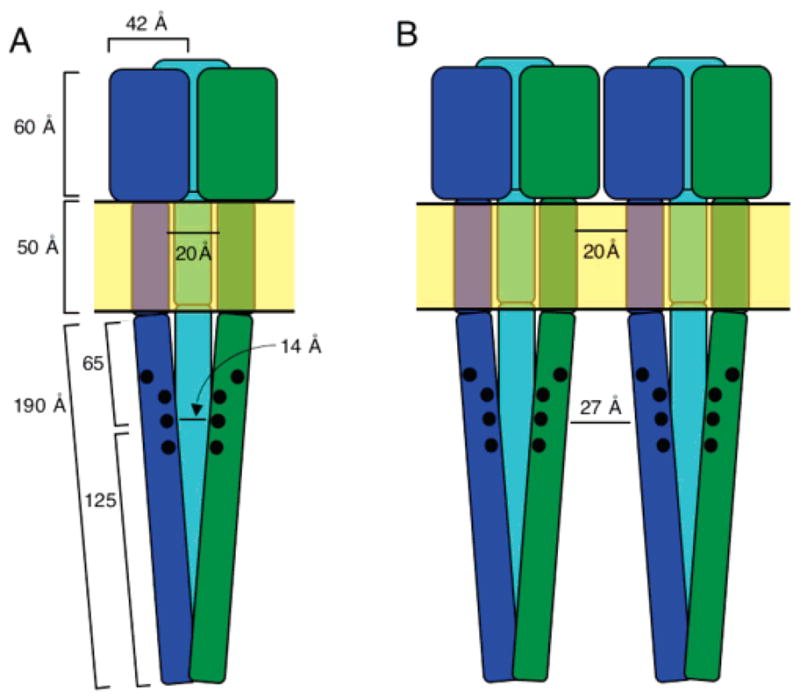
Schematic model of interdimer distances. (A) Estimated minimum distance between the adaptation subdomains of two dimers within the same trimer of dimers. (B) Estimated minimum distance between the adaptation subdomains of two adjacent dimers in different trimers of dimers. Dimensions are from refs 5 and 41.
The most likely mechanism of the electrostatic switch in the adaptation subdomain, then, involves electrostatic interactions between charges within the same receptor dimer. This mechanism could modulate the stabilities and packing interactions of adjacent helices or subunits. Figure 9B indicates that the electrostatic interactions between negative charges in the adaptation site region are significantly stronger than the thermal energy at physiological ionic strength, consistent with an electrostatic switch which triggers changes in helix stability or packing. We propose that this electrostatic switching generates a conformational change within the dimer arising from small displacements of the helices in the four-helix bundle of the adaptation subdomain. Thus, when an adaptation or activating site negative charge is neutralized, the decrease in the extent of electrostatic repulsion between the negative charges at the CD1–CD2′ helix interface will increase the stability of this interface between subunits. The resulting enhancement of the subunit–subunit interaction would decrease the average separation between the helices and could also alter the helical supercoiling in the four-helix bundle. Such small changes in helix packing and supercoiling could easily be transmitted the entire length of the cytoplasmic domain to the docking site for CheA kinase, and thus could regulate kinase activity.
The electrostatic switch is also proposed to modulate the conformational dynamics of the cytoplasmic domain. This study reports a decrease in helix–helix disulfide formation rates within the cytoplasmic domain when the EEEE adaptation state is changed to the QQQQ state, indicating that the QQQQ state is less dynamic. This decrease in dynamics is consistent with a model in which charge neutralization stabilizes the subunit interface and thereby reduces the level of conformational freedom of the helices within the four-helix bundle. Similarly, the QQQQ state yields a lower B factor in crystals (43), and charge neutralization by decreasing pH is observed to reduce conformational dynamics of the isolated cytoplasmic domain in an NMR hydrogen exchange study (44). In principle, either the change in average conformation or the change in conformational dynamics triggered by the electrostatic switch could dominate signal transmission. Previous models have pointed out the possible functional role of dynamical changes in signaling by the receptor (43, 44).
The greatest difference between the activating sites and the true adaptation sites is the lock-on behavior exhibited by certain modifications at the activating sites. Simultaneous neutralization of all four adaptation sites in the QQQQ receptor stimulates kinase activity to a similar extent as the quadruple activation site mutant D273N/E281Q/D288N/E477Q. However, while binding of the attractant to the QQQQ receptor (or to any other modified receptor possessing a combination of glutamines and glutamates at the adaptation sites) yields full kinase inhibition (12, 13), the D273N/E281Q/D288N/E477Q receptor is locked in this on-state such that attractant binding fails to generate the normal inhibition of kinase activity. Partial lock-on character is also observed for the D273R substitution at the first activating site. Such lock-on behavior could arise from electrostatic stabilization of the on-state of the aspartate-occupied receptor, or from destabilization of the off-state, since either type of perturbation would shift the equilibrium toward the on-state with its higher kinase activity.
Multiple lines of evidence from previous studies provide further support for the importance of the dimer subunit interface in regulating kinase activity. To date, six lock-on disulfides that constitutively activate the receptor-bound kinase have been identified in the cytoplasmic domain (18, 19, 30). Of these, five are located at the intersubunit interface within the adaptation subdomain. Thus, disulfide bonds that covalently stabilize the subunit interface within the adaptation subdomain lock the receptor in the kinase-activating state. In addition, position G278 within the adaptation subdomain at the intersubunit interface has been identified as a hot spot for kinase activation, where many side chain substitutions superactivate the kinase (19). Also of note, mutations at positions E281 and D288 within the adaptation subdomain were initially identified as second-site revertants that restored chemotactic ability to the defective receptor bearing the A19K substitution (36). These second-site revertants presumably restore function by increasing the kinase activity, as observed for the E281Q and D288N mutants in our study. Finally, as previously mentioned (see the Results), cysteine substitutions at three of the positions identified here as activating positions (E281C, D288C, and E477C) yield kinase activation to levels similar to those observed herein for the more conservative charge-neutralizing substitutions (30; also S. E. Winston and J. J. Falke, unpublished results). These findings provide further support for the hypothesis that it is the charge of the activating side chains, rather than their size or shape, that modulates kinase activity. At first glance, the observation that arginine substitutions and neutralizing substitutions at the activating positions often yield the same effects on in vitro kinase activation appears to contradict a simple electrostatic model. However, this finding can be explained by the long arginine side chain, which could place the positive charge outside the critical region of the electrostatic field, perhaps by forming a salt bridge with an anionic side chain distant from the subunit interface.
Analysis of the sequence alignment for the 13 class I bacterial receptors reveals a high degree of conservation for the 17 background acidic residues and the four adaptation site residues (Table 1) (20). At 16 of the 17 positions probed in this study, the degree of conservation of an acidic residue is ≥50%, and at 14 of the 17 positions, the degree of conservation exceeds 75%. This finding is further evidence that the negative charges within the adaptation subdomain play an important role in receptor function. Examination of class I receptors indicates that the homologous position corresponding to activating position D288 in the aspartate receptor is sometimes occupied by a glutamate residue that putatively serves as a true adaptation site in other receptors.
More generally, other, more distantly related classes of prokaryotic receptors also possess adaptation subdomains that exhibit high densities of acidic residues and utilize glutamate residues as adaptation sites (20). Presumably, the adaptation signals of these receptors will share the electrostatic mechanism proposed for class I receptors, although there could be exceptions. For example, the haloarchaeal taxis receptors exhibit a spatial distribution of adaptation sites within their cytoplasmic domains that differs significantly from the typical class I pattern (20), and these receptors function in a cytoplasmic ionic enviroment that contains high levels of salt (4 M KCl and NaCl) where the resulting high ionic strength would weaken long-range electrostatic interactions (42). These haloarchaeal receptors may use a nonelectrostatic mechanism to trigger adaptation signals, or they may be specialized to enable electrostatic signals to be generated in a high-ionic strength environment.
Overall, the previous and current observations are consistent with the hypothesis that electrostatics play an important role in the signaling mechanism in the adaptation region of the aspartate receptor. This subdomain processes input signals from ligand binding and adaptation, sending them to the signaling subdomain and eventually to the associated kinase enzyme. While this study identifies the electrostatics at the intersubunit interface as critical in this process, further structural studies are necessary to better define the conformational or dynamical changes within the cytoplasmic domain that are responsible for driving kinase regulation.
Footnotes
Abbreviations: CD1 and CD2, α-helices 1 and 2 of the cytoplasmic domain, respectively; QQQQ, QEQE, and EEEE, receptors with the indicated amino acids at the four adaptation site positions; DTT, dithiothreitol; EDTA, ethylenediaminetetraacetic acid; PMSF, phenyl-methanesulfonyl fluoride.
Support provided by NIH Grant GM R01-40731 (to J.J.F.).
References
- 1.Szurmant H, Ordal GW. Diversity in chemotaxis mechanisms among the bacteria and archaea. Microbiol Mol Biol Rev. 2004;68:301–319. doi: 10.1128/MMBR.68.2.301-319.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bourret RB, Stock AM. Molecular information processing: Lessons from bacterial chemotaxis. J Biol Chem. 2002;277:9625–9628. doi: 10.1074/jbc.R100066200. [DOI] [PubMed] [Google Scholar]
- 3.Falke JJ, Hazelbauer GL. Transmembrane signaling in bacterial chemoreceptors. Trends Biochem Sci. 2001;26:257–265. doi: 10.1016/s0968-0004(00)01770-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Armitage JP. Bacterial tactic responses. Adv Microb Physiol. 1999;41:229–289. doi: 10.1016/s0065-2911(08)60168-x. [DOI] [PubMed] [Google Scholar]
- 5.Kim KK, Yokota H, Kim SH. Four-helical-bundle structure of the cytoplasmic domain of a serine chemotaxis receptor. Nature. 1999;400:787–792. doi: 10.1038/23512. [DOI] [PubMed] [Google Scholar]
- 6.Falke JJ, Bass RB, Butler SL, Chervitz SA, Danielson MA. The two-component signaling pathway of bacterial chemotaxis: A molecular view of signal transduction by receptors, kinases, and adaptation enzymes. Annu Rev Cell Dev Biol. 1997;13:457–512. doi: 10.1146/annurev.cellbio.13.1.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu J, Li J, Li G, Long DG, Weis RM. The receptor binding site for the methyltransferase of bacterial chemotaxis is distinct from the sites of methylation. Biochemistry. 1996;35:4984–4993. doi: 10.1021/bi9530189. [DOI] [PubMed] [Google Scholar]
- 8.Barnakov AN, Barnakova LA, Hazelbauer GL. Allosteric enhancement of adaptational demethylation by a carboxyl-terminal sequence on chemoreceptors. J Biol Chem. 2002;277:42151–42156. doi: 10.1074/jbc.M206245200. [DOI] [PubMed] [Google Scholar]
- 9.Falke JJ, Kim SH. Structure of a conserved receptor domain that regulates kinase activity: the cytoplasmic domain of bacterial taxis receptors. Curr Opin Struct Biol. 2000;10:462–469. doi: 10.1016/s0959-440x(00)00115-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ames P, Studdert CA, Reiser RH, Parkinson JS. Collaborative signaling by mixed chemoreceptor teams in Escherichia coli. Proc Natl Acad Sci USA. 2002;99:7060–7065. doi: 10.1073/pnas.092071899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sourjik V, Berg HC. Functional interactions between receptors in bacterial chemotaxis. Nature. 2004;428:437–441. doi: 10.1038/nature02406. [DOI] [PubMed] [Google Scholar]
- 12.Li G, Weis RM. Covalent modification regulates ligand binding to receptor complexes in the chemosensory system of Escherichia coli. Cell. 2000;100:357–365. doi: 10.1016/s0092-8674(00)80671-6. [DOI] [PubMed] [Google Scholar]
- 13.Bornhorst JA, Falke JJ. Attractant regulation of the aspartate receptor-kinase complex: Limited cooperative interactions between receptors and effects of the receptor modification state. Biochemistry. 2000;39:9486–9493. doi: 10.1021/bi0002737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lybarger SR, Maddock JR. Polarity in action: Asymmetric protein localization in bacteria. J Bacteriol. 2001;183:3261–3267. doi: 10.1128/JB.183.11.3261-3267.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller AS, Falke JJ. Side Chains at the Membrane-Water Interface Modulate the Signaling State of a Transmembrane Receptor. Biochemistry. 2004;43:1763–1770. doi: 10.1021/bi0360206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chervitz SA, Falke JJ. Molecular mechanism of transmembrane signaling by the aspartate receptor: A model. Proc Natl Acad Sci USA. 1996;93:2545–2550. doi: 10.1073/pnas.93.6.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hughson AG, Hazelbauer GL. Detecting the conformational change of transmembrane signaling in a bacterial chemoreceptor by measuring effects on disulfide cross-linking in vivo. Proc Natl Acad Sci USA. 1996;93:11546–11551. doi: 10.1073/pnas.93.21.11546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bass RB, Falke JJ. The aspartate receptor cytoplasmic domain: In situ chemical analysis of structure, mechanism and dynamics. Struct Folding Des. 1999;7:829–840. doi: 10.1016/s0969-2126(99)80106-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trammell MA, Falke JJ. Identification of a site critical for kinase regulation on the central processing unit (CPU) helix of the aspartate receptor. Biochemistry. 1999;38:329–336. doi: 10.1021/bi981964u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le Moual H, Koshland DE., Jr Molecular evolution of the C-terminal cytoplasmic domain of a superfamily of bacterial receptors involved in taxis. J Mol Biol. 1996;261:568–585. doi: 10.1006/jmbi.1996.0483. [DOI] [PubMed] [Google Scholar]
- 21.Borkovich KA, Alex LA, Simon MI. Attenuation of sensory receptor signaling by covalent modification. Proc Natl Acad Sci USA. 1992;89:6756–6760. doi: 10.1073/pnas.89.15.6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ninfa EG, Stock A, Mowbray S, Stock J. Reconstitution of the bacterial chemotaxis signal transduction system. J Biol Chem. 1991;266:9764–9770. [PubMed] [Google Scholar]
- 23.Kunkel TA, Bebenek K, McClary J. Efficient site-directed mutagenesis using uracil-containing DNA. Methods Enzymol. 1991;204:125–139. doi: 10.1016/0076-6879(91)04008-c. [DOI] [PubMed] [Google Scholar]
- 24.Adler J. Bacterial chemotaxis. Science. 1966;153:708–716. doi: 10.1126/science.153.3737.708. [DOI] [PubMed] [Google Scholar]
- 25.Liu JD, Parkinson JS. Role of CheW protein in coupling membrane receptors to the intracellular signaling system of bacterial chemotaxis. Proc Natl Acad Sci USA. 1989;86:8703–8707. doi: 10.1073/pnas.86.22.8703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bass RB, Coleman MD, Falke JJ. Signaling domain of the aspartate receptor is a helical hairpin with a localized kinase docking surface: Cysteine and disulfide scanning studies. Biochemistry. 1999;38:9317–9327. doi: 10.1021/bi9908179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bass RB, Falke JJ. Detection of a conserved α-helix in the kinase-docking region of the aspartate receptor by cysteine and disulfide scanning. J Biol Chem. 1998;273:25006–25014. doi: 10.1074/jbc.273.39.25006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nicholls A, Sharp KA, Honig B. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 29.Chervitz SA, Lin CM, Falke JJ. Transmembrane signaling by the aspartate receptor: Engineered disulfides reveal static regions of the subunit interface. Biochemistry. 1995;34:9722–9733. doi: 10.1021/bi00030a010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Danielson MA, Bass RB, Falke JJ. Cysteine and disulfide scanning reveals a regulatory α-helix in the cytoplasmic domain of the aspartate receptor. J Biol Chem. 1997;272:32878–32888. doi: 10.1074/jbc.272.52.32878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shiomi D, Zhulin IB, Homma M, Kawagishi I. Dual recognition of the bacterial chemoreceptor by chemotaxis-specific domains of the CheR methyltransferase. J Biol Chem. 2002;277:42325–42333. doi: 10.1074/jbc.M202001200. [DOI] [PubMed] [Google Scholar]
- 32.Terwilliger TC, Wang JY, Koshland DE., Jr Surface structure recognized for covalent modification of the aspartate receptor in chemotaxis. Proc Natl Acad Sci USA. 1986;83:6707–6710. doi: 10.1073/pnas.83.18.6707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Terwilliger TC, Wang JY, Koshland DE., Jr Kinetics of receptor modification. The multiply methylated aspartate receptors involved in bacterial chemotaxis. J Biol Chem. 1986;261:10814–10820. [PubMed] [Google Scholar]
- 34.Careaga CL, Falke JJ. Structure and dynamics of E. coli chemosensory receptors. Engineered sulfhydryl studies. Biophys J. 1992;62:209–216. doi: 10.1016/S0006-3495(92)81806-4. 217–219 (discussion) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 36.Oosawa K, Simon M. Analysis of mutations in the transmembrane region of the aspartate chemoreceptor in Escherichia coli. Proc Natl Acad Sci USA. 1986;83:6930–6934. doi: 10.1073/pnas.83.18.6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chervitz SA, Falke JJ. Lock on/off disulfides identify the transmembrane signaling helix of the aspartate receptor. J Biol Chem. 1995;270:24043–24053. doi: 10.1074/jbc.270.41.24043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Milburn MV, Prive GG, Milligan DL, Scott WG, Yeh J, Jancarik J, Koshland DE, Jr, Kim SH. Three-dimensional structures of the ligand-binding domain of the bacterial aspartate receptor with and without a ligand. Science. 1991;254:1342–1347. doi: 10.1126/science.1660187. [DOI] [PubMed] [Google Scholar]
- 39.Lee GF, Burrows GG, Lebert MR, Dutton DP, Hazelbauer GL. Deducing the organization of a transmembrane domain by disulfide cross-linking. The bacterial chemoreceptor Trg. J Biol Chem. 1994;269:29920–29927. [PubMed] [Google Scholar]
- 40.Butler SL, Falke JJ. Cysteine and disulfide scanning reveals two amphiphilic helices in the linker region of the aspartate chemoreceptor. Biochemistry. 1998;37:10746–10756. doi: 10.1021/bi980607g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weis RM, Hirai T, Chalah A, Kessel M, Peters PJ, Subramaniam S. Electron microscopic analysis of membrane assemblies formed by the bacterial chemotaxis receptor Tsr. J Bacteriol. 2003;185:3636–3643. doi: 10.1128/JB.185.12.3636-3643.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spudich JL, Luecke H. Sensory rhodopsin II: Functional insights from structure. Curr Opin Struct Biol. 2002;12:540–546. doi: 10.1016/s0959-440x(02)00359-7. [DOI] [PubMed] [Google Scholar]
- 43.Kim SH, Wang W, Kim KK. Dynamic and clustering model of bacterial chemotaxis receptors. Proc Natl Acad Sci USA. 2002;99:11611–11615. doi: 10.1073/pnas.132376499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murphy JO, Yi X, Weis RM, Thompson LK. Hydrogen exchange reveals a stable and expandable core within the aspartate receptor cytoplasmic domain. J Biol Chem. 2001;276:43262–43269. doi: 10.1074/jbc.M105585200. [DOI] [PubMed] [Google Scholar]