

Abstract
Human mesenchymal stem cells (hMSCs) are multipotent cells that can differentiate into many cell types. Chondrogenesis is induced in hMSCs cultured as a micromass pellet to mimic cellular condensation during cartilage development, and exposed to transforming growth factor β (TGFβ). Interestingly, TGFβ also can induce hMSC differentiation to smooth-muscle-like cell types, but it remains unclear what directs commitment between these two lineages. Our previous work revealed that cell shape regulates hMSC commitment between osteoblasts and adipocytes through RhoA signaling. Here we show that cell shape also confers a switch between chondrogenic and smooth muscle cell (SMC) fates. Adherent and well-spread hMSCs stimulated with TGFβ3 upregulated SMC genes, while cells allowed to attach onto micropatterned substrates, but prevented from spreading and flattening, upregulated chondrogenic genes. Interestingly, cells undergoing SMC differentiation exhibited little change in RhoA but significantly higher Rac1 activity than chondrogenic cells. Rac1 activation inhibited chondrogenesis, and was necessary and sufficient for inducing SMC differentiation. Furthermore, TGFβ3 and Rac1 signaling upregulated N-cadherin, which was required for SMC differentiation. These results demonstrate a chondrogenic-SMC fate decision mediated by cell shape, Rac1, and N-cadherin, and highlight the tight coupling between lineage commitment and the many changes in cell shape, cell-matrix adhesion, and cell-cell adhesion that occur during morphogenesis.
Keywords: Mesenchymal stem cells, Cell shape, Chondrogenesis, Smooth muscle cells, Rac1, N-cadherin
Introduction
Human mesenchymal stem cells (hMSCs) can differentiate into a variety of cell types, including osteoblasts, adipocytes, chondrocytes, and myocytes [1]. Like many stem cells, hMSCs differentiate into distinct lineages depending on which cues are present in the local tissue microenvironment. While much effort has focused on identifying soluble differentiation factors to mimic these cues, investigators only recently have begun to appreciate the potential importance of insoluble signals such as cell adhesion, cell shape, mechanical forces, and substrate rigidity in stem cell differentiation. [2–4].
During morphogenesis, cells undergo dramatic changes in cell shape as they differentiate into different lineages. Osteogenic cells initially take on a flattened morphology as they begin to calcify matrix, while chondrogenic and adipogenic cells round up in their early differentiation, and myogenic cells extend into elongated spindles to execute their muscular functions. It has largely been thought that these structural changes are a consequence of differentiation programs. However, several studies have noted that changes in cell shape itself can regulate the differentiation of mesenchymal progenitor cells. For example, fibronectin-induced cell flattening and spreading inhibits the adipogenic differentiation of 3T3-F442A pre-adipocytes, which can be reversed by keeping cells in a round configuration or upon disruption of the actin cytoskeleton [5]. Similarly, chondrocytes taken from condensed articular cartilage and allowed to attach on a planar substrate downregulate chondrocyte-specific markers [6]. In contrast, controlled cell spreading enhances differentiation in preosteoblastic progenitors [7]. Our previous work extended the osteogenic-adipogenic axis to hMSCs, identifying cell shape as an important regulator in hMSC commitment between an osteogenic or adipogenic fate [3].
Although accumulating data suggest a role for cell shape in many important biological processes such as cell division [8], apoptosis [9], nuclear organization [10], morphogenesis [11], as well as differentiation [12,13], the molecular basis of these cell shape-mediated effects has not been fully understood. It is well-known that Rho family small GTPases can regulate cell shape through modulating actin cytoskeleton [14]. Interestingly, it has been shown that cell shape may also affect the activity of Rho GTPases [15]. Our previous work has demonstrated that cell shape can regulate RhoA activity, and that this activity mediates the shape-dependent control of hMSC lineage commitment to osteoblasts or adipocytes [3]. These studies indicate a role for Rho family GTPase signaling in early cellular developmental processes.
In vitro chondrogenesis of MSCs is induced when cells are cultured as a high density micromass pellet, which mimics cellular condensation during cartilage development, and exposed to transforming growth factor β (TGFβ) [1,16]. Interestingly, it has been shown that TGFβ also induces differentiation of hMSCs to smooth muscle-like cell types in vitro [17,18], but it remains unclear what additional factors determine the TGFβ-induced commitment to a chondrogenic versus smooth muscle cell (SMC) lineage. While it has been assumed that the initial lack of cell-matrix adhesion and presence of cell-cell adhesion likely contribute to the chondrogenic effects of pellet culture, we hypothesized that cell shape plays an important role in regulating TGFβ-induced hMSC differentiation to chondrogenic versus SMC fate. Using a micropatterning technique to control cell shape and degree of cell spreading, we found that hMSCs allowed to attach onto micropatterned substrates, but prevented from spreading and flattening, upregulated chondrogenic genes in response to TGFβ3 stimulation, while cells allowed to spread on substrates underwent SMC differentiation, supporting the idea that cell shape confers a switch between chondrogenic and myogenic potential. We further show that cells undergoing SMC differentiation versus chondrogenesis exhibited little change in RhoA activity but significantly higher Rac1 activity during differentiation. This cell shape-dependent Rac1 activation was required for SMC differentiation and inhibited chondrogenesis, and further regulated changes in N-cadherin expression that appear to be also required for robust SMC differentiation. Thus, the many structural changes in cells that occur during morphogenesis may in fact play direct regulatory roles in modulating the differentiation of stem cells to appropriate tissue lineages.
Materials and Methods
Cell culture and reagents
Human mesenchymal stem cells were obtained from Lonza and maintained in growth medium (DMEM containing 10% fetal bovine serum, 0.3 mg/ml glutamine, 100 μg/ml streptomycin, 100 units/ml penicillin). Only early passage hMSCs (passage 4–6) were used for experimental studies. No difference was observed among these passages. For chondrogenic and smooth muscle cell differentiation, hMSCs were cultured in serum-free incomplete differentiation media (Lonza, hMSC Differentiation BulletKit® –Chondrogenic, cat#PT-3003) supplemented with 10 ng/ml TGFβ3 (Sigma) in pelleted form or on tissue culture plates (at 3,000 cells/cm2 unless specified otherwise), respectively. Pellet cultures were prepared according to protocol (Lonza). Briefly, 2.5×105 cells/pellet were transferred into 15ml polypropylene tubes and centrifuged at 150g for 5 min at room temperature. Then the pellets were rinsed with incomplete differentiation medium (without TGFβ3), centrifuged and resuspended in complete differentiation medium (with TGFβ3). The cells were centrifuged again and the pellets were incubated at 37°C with 5% CO2. Media were changed every three days.
Fabrication of micropatterned substrates
Micropatterned substrates were prepared per Tan et al. (2002) [19]. In brief, PDMS (polydimethylsiloxane) stamps were cast, baked, and removed from master templates, which were previously created using photolithographic methods. Stamps were coated with fibronectin (25μg/ml; BD) for 2hr, washed with water, and dried with compressed nitrogen. Flat PDMS substrates were UV oxidized for 7 min (UVO-cleaner 342, Jelight Co.), stamped with fibronectin, blocked with Pluronic F127 for 1hr, and rinsed three times with PBS before cell seeding. Substrates without features (flat substrate), with 100×100μm (10,000μm2) islands, or with 32×32μm (1,024μm2) islands were prepared.
Cell differentiation assay
hMSCs were cultured in growth medium (GM) or differentiation medium (DM) for 7 days under different culture conditions (plate: tissue culture plastic; pellet: pellet culture; spread: unpatterned fibronectin-coated flat regions on PDMS; 10,000μm2/1,024μm2: patterned large/small fibronectin-coated islands on PDMS), then either fixed and stained for smooth muscle actin/calponin (SMC markers), or alcian blue/collagen II (chondrogenic markers) for immunohistology or immunofluorescence assays, or lysed in 1X Laemmli sample buffer (Bio-Rad Laboratories) to examine protein expression by western blot. For real-time RT-PCR analysis of SMC and chondrogenic markers, hMSCs were cultured in GM or DM under different culture conditions and harvested for RNA extraction at different time points (1 day for plate or patterned cultures, 7 days or 14 days for pellet cultures).
Immunohistology and Immunofluorescence
Pelleted cells were fixed with 3.7% formaldehyde, embedded in paraffin, processed, and sectioned according to standard histopathologic protocols (Johns Hopkins Surgical Pathology). Sections were deparaffinized using xylene followed by a graded alcohol series, and stained with Alcian Blue. Cells grown on plates or patterns were either fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100, or fixed and permeabilized with −20°C methanol: acetone (1:1), blocked with 33% goat serum, incubated with primary antibodies and followed by Alex 594-conjugated goat anti-mouse secondary antibody (Molecular Probes). The primary antibodies were obtained as follows: anti-smooth muscle actin α (Sigma), anti-calponin (Dako), anti-collagen II (MP Biomedicals). Cell nuclei were stained with DAPI. Cells were visualized using a Nikon Eclipse TE200.
Differential interference contrast (DIC) microscopy
DIC images of hMSC on patterns were acquired using Zeiss AxioVert200M. Images were shade-corrected per [20].
Real-time RT-PCR
Total RNA was isolated using an RNeasy Micro kit (QIAGEN) according to the manufacturer’s instructions. cDNA was transcribed with Oligo-dT primers using MMLV reverse transcriptase (Invitrogen) with 0.5 μg of total RNA per reaction. Quantitative PCR was performed in an ABI 7300 system (Applied Biosystems) using TaqMan gene expression assays according to the manufacturer’s instructions. Results were analyzed using Relative Quantitation method and all mRNA expression data were normalized to GAPDH expression in the corresponding sample and then to the control sample. All data are represented as mean±SEM; n≥3. TaqMan gene expression assays used were as follows: ACTA2 (Hs00426835_g1); CNN1 (Hs00154543_m1); SMTN (Hs00199489_m1); MYOCD (Hs00538076_m1); SOX9 (Hs00165814_m1); COL2A1 (Hs00264051_m1); OPGL (Hs00243522_m1); SOX5 (Hs00374709_m1); SOX6 (Hs00264525_m1); CALD (Hs00263998_m1); SM22 (Hs00162558_m1); MYH11 (Hs00224610_m1); MYOD (Hs00159528_m1).
Western blot
Cells were lysed in 1X Laemmli sample buffer (Bio-Rad Laboratories) and boiled. Proteins were separated by SDS-PAGE and electroblotted onto PVDF, blocked with 5% dry milk in TBST, immunoblotted with specific primary antibodies and HRP-conjugated secondary antibodies (Jackson Laboratories), and detected by ECL (Pierce Chemical Co.). Protein levels were quantified using a Versadoc imaging system (Bio-Rad Laboratories). The antibodies were obtained as follows: anti-α-tubulin (Sigma); anti-N-cadherin (BD Transduction Laboratories); anti-calponin (DakoCytomation); anti-smooth muscle actin α (Sigma); anti-Rac1 (Upstate). All data are represented as mean±SEM; n≥3.
Rho GTPase Assay
RhoA-GTP loading was measured by either pull-down assay as previously described [3] or G-LISA™ RhoA Activation Assay (Cytoskeleton) per manufacturer’s instructions.
Rac GTPase Assay
Rac1-GTP loading was measured by either pull-down assay using a commercially available kit (Upstate) or G-LISA™ Rac Activation Assay (Cytoskeleton) according to the manufacturers’ instructions. Pak1-PBD beads were used as supplied and also made using GST-tagged recombinant Pak1-PBD produced in BL21 cells containing the pGEX-PBD vector (a gift from L. Romer, Johns Hopkins University, Baltimore, MD). Protein levels were detected by Western blot and quantified using a Versadoc imaging system (Bio-Rad Laboratories).
Lentivirus production
The cDNAs of EGFP, EGFP-RacV12 (constitutively active mutant of Rac1 fused to EGFP), and EGFP-RacN17 (dominant negative mutant of Rac1 fused to EGFP) (gifts from M. Philips, New York University Medical Center, New York, NY) were subcloned into the pRRL-cPPT-CMV-X2-PRE-SIN vector and then co-transfected with pMDLg/pRRE, pCMV-VSVG and pRSV-REV packaging vectors (gifts from W. Osborne, University of Washington, Seattle, WA) into Ad293 cells using Lipofectamine 2000 (Invitrogen). Viral supernatants were collected after 48 hr, centrifuged at 1500rpm for 5 min, filtered through a 0.45μm filter (Nalgene), aliquoted and stored at −80°C. Viral titer was determined by serial dilution and infection of hMSCs. Cells were infected in the presence of 10μg/ml polybrene. The titer giving >90% transduction efficiency (by examining percentage of GFP-positive cells) and minimum cell death was used.
Recombinant adenovirus construction
The cDNA fragment encoding human N-cadherin lacking 105 bps at the C-terminus (the β-catenin-binding domain) was amplified by PCR from full length N-cadherin (a gift from B. McKay, University of Arizona) using forward primer GAGGCGGCCGCACCATGTGCCGGATAGCGGGA and reverse primer GAGCTCGAGTCAGCTCAAGGACCCAGCAGT, and was cloned into pShuttle-IRES-hrGFP-1 using NotI/XhoI sites. Recombinant adenoviruses were prepared using the AdEasy XL system (Stratagene) as previously described [21].
Measurement of the spreading area of hMSCs
MSCs were transduced with low titer lentivirus expressing GFP so that only about 10% of cells were infected. Fluorescent images of infected cells were taken and areas were measured with AxioVision software. About 30 cells were measured for each condition and data was averaged across 3 independent experiments.
Statistical analysis
Statistical analysis was performed by Student’s t test, 1-way ANOVA, or 2-way ANOVA. Tukey’s honestly significant difference (HSD) test was used for pairwise comparisons. Sample sizes and data analysis specific to individual experiments are indicated in the associated text or figure legends.
Results
Cell shape regulates hMSC differentiation into smooth muscle cells vs. chondrocytes
When grown as micromass pellets in a chondrogenic differentiation medium containing TGFβ3 for 7 days, hMSCs underwent chondrogenesis in culture as indicated by Alcian Blue staining for glycosaminoglycans and real-time RT-PCR analysis of chondrogenic markers (Figure 1A and 1B, Figure S1), as has been also reported by others [1]. Interestingly, when these same cells were plated on tissue culture dishes at subconfluent density (3,000 cell/cm2) in the same differentiation medium, smooth muscle cell (SMC) but not chondrogenic markers were rapidly upregulated (Figure 1A and 1C). The upregulation of SMC gene mRNAs was most robust at one day after induction and then the mRNA expression gradually decreased as the protein expression increased and peaked between 4–7 days after induction (Figure S2). TGFβ is the major growth factor present in the differentiation medium critical to MSC chondrogenesis [16,22], and we found that TGFβ3 alone is also sufficient for upregulating SMC genes (Figure S3), consistent with previous reports suggesting a role for TGFβ in SMC differentiation [17,18]. However, we did not observe the upregulation of late markers of SMC differentiation such as myosin heavy chain (Figure S2) even after 14 days of induction, which presumably requires additional factors. In addition, the expression of MyoD1, a skeletal muscle marker, was not detected after 14 days of induction, suggesting hMSCs are not undergoing striated muscle differentiation (Figure S2). Together, these findings suggest an important role for the differences in culture configuration (pellet versus plate culture) in driving a stem cell fate switch between chondrogenesis and SMC differentiation in the presence of the same soluble cytokines.
Figure 1.

Cell shape regulates hMSC differentiation into smooth muscle cells (SMCs) vs. chondrocytes. (A) hMSCs were grown on tissue culture plates (3,000 cells/cm2) or as micromass pellets in growth medium (GM) or differentiation medium (DM) for 7 days. Cells on plates were fixed and stained for α-smooth muscle actin (red) and DAPI (blue). Cell pellets were fixed, embedded in paraffin, sectioned and stained with Alcian Blue. Upper panels: bar =300μm; lower panels: bar =50μm. (B) Real-time PCR analysis of chondrogenic gene expression of hMSCs grown under different culture conditions for 7 days (data are represented as mean±SEM; n=3). SOX9: SRY-box 9; COL2A1: collagen II; OPGL: osteoprotegerin ligand. Although osteoprotegerin ligand (OPGL) is not commonly used as a chondrogenic marker, we found this gene was specifically and robustly upregulated only during hMSC chondrogenesis (Figure S1). (C) Real-time PCR analysis of SMC gene expression of hMSCs grown under different culture conditions for 1 day (data are represented as mean±SEM; n=3). ACTA2: α-smooth muscle actin; CNN1: calponin 1; MYOCD: myocardin; SMTN: smoothelin. (D) hMSCs were grown on either unpatterned fibronectin-coated PDMS substrate, or PDMS substrates patterned with large (10,000μm2) or small (1024μm2) fibronectin islands in GM or DM for 7 days. Cells were fixed, stained for calponin or collagen II (green) and DAPI (blue), and counted (data are represented as mean±SEM; n=3). Bar=50μm. (E) Real-time PCR analysis of SMC and chondrogenic gene expression of hMSCs grown on either flat substrate or 1024μm2 islands after 1 day (data are represented as mean±SEM; n=6). * (P<0.05) and ns (not significant), as calculated by 2-way ANOVA and Tukey’s HSD test.
It has largely been assumed that absence of cell-matrix adhesion in the pellet culture acts as a permissive cue for chondrogenesis, but other differences also exist between the chondrogenic and myogenic cultures. We postulated that differences in cell shape, induced in pellet versus plated cultures, may play a role in modulating whether TGFβ3 would drive hMSCs toward a chondrogenic versus SMC lineage. To examine directly a role for cell shape in specifying chondrogenic versus SMC fate, we plated hMSCs on polydimethylsiloxane (PDMS) substrates patterned with either small fibronectin islands (1,024μm2), or large fibronectin islands (10,000μm2), or unpatterned fibronectin-coated flat regions (spread). Cells on large islands and flat regions attached and spread, while those on small islands were restricted from spreading and maintained a round morphology (Figure 1D). Following TGFβ3 treatment for 7 days, nearly 40% of cells on large islands and flat regions expressed calponin, a SMC marker, while more than 40% of cells on small islands expressed collagen II, a chondrogenic marker (Figure 1D). Real-time RT-PCR analysis showed that SMC and chondrogenic genes were rapidly upregulated (after 1 day of induction) in spread cells and round cells, respectively (Figure 1E). These results suggested that cell shape indeed regulates hMSC lineage commitment between SMC and chondrogenic fates.
Rac1 activation is necessary and sufficient for inducing SMC differentiation
Prior work described the involvement of RhoA signaling in stem cell commitment [3,23]. We therefore examined the level of endogenous RhoA activity in hMSCs cultured either at subconfluent density on plates or as micromass pellets, in growth medium as well as differentiation medium. Levels of RhoA activity did not vary substantially among the different culture conditions (Figure 2A). Unexpectedly, in screening the activity level of other small GTPases, we discovered that Rac1 activity was significantly higher in well-spread cells during early differentiation than in cells grown as pellets (Figure 2B, Figure S4). When cells were grown on patterned substrates in growth medium, spread and unspread cells showed similar levels of Rac1 activity, but exposure to differentiation medium increased Rac1 activity only in the spread cells (Figure 2C). These data implicated the involvement of Rac1 and not RhoA in the commitment of hMSCs to smooth muscle cells.
Figure 2.

Rac1 activation is necessary and sufficient for inducing SMC differentiation. (A) Western blot and quantification of total and active RhoA in hMSCs under different culture conditions for 2 days. (B) Western blot and quantification of total and active Rac1 in hMSCs under different culture conditions for 2 days. (C) Western blot and quantification of total and active Rac1 in hMSCs grown on patterned substrates in growth or differentiation medium for 8 hours. (D) Real-time PCR analysis of SMC gene expression in hMSCs transduced with GFP, RacV12, or RacN17 after 1 day in growth or differentiation medium on plate. (E) Western blot and quantification of SMAα and calponin expression in hMSCs transduced with GFP, RacV12, or RacN17 after 7 days in growth or differentiation medium on plate. (F) Real-time PCR analysis of chondrogenic gene expression in hMSCs transduced with GFP, RacV12, or RacN17 and grown as pellets for 14 days in growth or differentiation medium. All data are represented as mean±SEM; n≥3. * (P<0.05) and ns (not significant), as calculated by 2-way ANOVA and Tukey’s HSD test (A, B and C) or student t test (D, E and F).
To further examine the role of Rac1, we constructed lentivirus encoding constitutively active (V12) or dominant negative (N17) Rac1 fused to GFP and infected hMSCs prior to plating. The infection of control GFP lentivirus did not promote hMSC SMC differentiation or chondrogenesis in the absence of induction media (growth medium), nor did it inhibit differentiation in the presence of induction media (differentiation medium), compared with non-infected cells (Figure S5). Interestingly, constitutively active Rac1 induced SMC differentiation when cells were grown in non-differentiating (growth) medium, while dominant negative Rac1 inhibited SMC differentiation when cells were grown in differentiation medium, as seen by real-time PCR analysis of SMC markers at mRNA levels and western blot at the protein levels (Figure 2D and 2E), indicating that Rac1 activity is both necessary and sufficient for inducing hMSC differentiation into the SMC lineage. Conversely, constitutively active Rac1 inhibited chondrogenesis when transduced cells were cultured as pellets in the differentiation medium (Figure 2F), though dominant negative Rac1 did not affect chondrogenesis, suggesting that inhibiting Rac1 activity is required, but not sufficient for inducing chondrogenesis.
Because cell shape was previously shown to regulate an adipogenic-osteogenic fate switch in these same hMSCs [3], we also measured Rac1 activity and the effects of Rac1 mutants during hMSC differentiation into osteoblasts or adipocytes. Moderate (twofold) changes in Rac1 activity were observed during osteogenesis and adipogenesis, respectively (Figure S6). Under pro-osteogenic conditions, inhibiting Rac1 activity by either dominant negative Rac1 or a small molecule Rac1 inhibitor, NSC23766 (10μm), suppressed osteogenesis (Figure S7A), and under pro-adipogenic conditions expressing constitutively active Rac1 inhibited adipogenesis (Figure S7B). While modulating Rac1 activity could suppress adipogenic or osteogenic fates, these manipulations did not lead to increased adipogenesis or osteogenesis, and were not sufficient to induce such lineage fates in non-differentiating growth media (Figure S7 C, D). Therefore, unlike RhoA, which is the major regulator in hMSC osteo- and adipo-differentiation, Rac1 seems to play a key role in directing hMSC differentiation into smooth muscle or chondrogenic lineages.
N-cadherin expression is upregulated during hMSC differentiation into SMCs
Interestingly, in addition to the effects of increased cell spreading on SMC differentiation, we also observed greater SMC differentiation in hMSCs seeded at increasingly higher densities (Figure 3A). When cells were seeded at very low density (1,000 cells/cm2) without cell-cell contacts, we observed that ~30% of cells underwent SMC differentiation after 7 days induction (by calponin staining). Meanwhile, 64% of cells underwent SMC differentiation when they were seeded at high density and formed a confluent monolayer (10,000 cells/cm2). Since our previous reports have shown cell spreading to decrease with increasing density [21], this result appears to contradict a model where increased cell spreading induces SMC differentiation. However, measurements of cell spreading in hMSCs seeded at different densities in the presence of TGFβ3 revealed that cells were still well-spread at the highest density examined (10,000 cells/cm2), compared with cells in growth media (Figure S8).
Figure 3.

N-cadherin expression is upregulated during SMC differentiation. (A) hMSCs were plated at different densities and cultured in growth or differentiation medium on plate for 7 days. Cells were fixed, stained for calponin, and counted. (B) Real-time PCR and western blot analysis of N-cadherin mRNA (1 day) and protein expression (7 days) in hMSCs under different culture conditions. (C) Real-time PCR and western blot analysis of N-cadherin mRNA (1 day) and protein expression (7 days) in hMSCs grown on either flat substrate or 1024μm2 islands. All data are represented as mean±SEM; n≥3. * (P<0.05) and ns (not significant), as calculated by 2-way ANOVA and Tukey’s HSD test.
Given that decreased cell spreading was not observed with increasing density in this setting, we explored whether the increased cell-cell contact might be involved in further enhancing SMC differentiation. Cell adhesion molecules such as N-cadherin have been reported to play a role in skeletal muscle and osteoblast differentiation [24,25]. In order to examine whether N-cadherin is also involved in hMSC differentiation into SMCs and chondrocytes, we first looked at the N-cadherin expression under different culture conditions. Cells in myogenic conditions (adherent on dishes) showed increased N-cadherin expression, while cells cultured as pellets expressed barely detectable levels of N-cadherin (Figure 3B). When cells were grown on patterned substrates in growth medium, spread and unspread cells expressed similar levels of N-cadherin, but exposure to differentiation medium increased expression of N-cadherin only in the spread cells (Figure 3C), a pattern very similar to Rac1 activation. These data suggested a strong correlation between N-cadherin expression and smooth muscle myogenesis.
N-Cadherin is required for TGFβ3- and Rac1-induced SMC differentiation
To further investigate the role of N-cadherin in hMSC myogenesis, we constructed a recombinant adenovirus expressing the C-terminal cytoplasmic deletion mutant of N-cadherin (NΔ), which acts as a dominant negative to native N-cadherin (Figure S9). Expression of NΔ in hMSCs abrogated TGFβ3-induced SMC differentiation (Figure 4A). Importantly, cadherin engagement has previously been shown to affect Rac1 activity [24,26,27], and Rac1 signaling has been shown to modulate cadherin-based junction formation [28,29]. Interestingly, in our system, N-cadherin upregulation appears to be downstream of Rac1 signaling. Constitutively active Rac1, which promoted SMC differentiation, induced an increase in N-cadherin expression in the absence of induction medium (Figure 4B, C), although it did not promote a further increase of N-cadherin in the induction medium, which might be a result of self-limiting mechanism to avoid overexpression. Conversely, NΔ had little effect on Rac1 activity in either growth or differentiation medium (Figure 4D). Furthermore, Rac1-induced SMC differentiation could be inhibited by NΔ (Figure 4E). These results suggest both TGFβ3- and Rac1-induced SMC differentiation are mediated at least partially through N-cadherin.
Figure 4.

Dominant negative N-Cadherin mutant (NΔ) blocks TGFβ3 and Rac1-induced SMC differentiation. hMSCs were cultured on plates at 3,000 cells/cm2 in growth or differentiation medium. (A) Real-time PCR analysis of SMC gene expression in hMSCs transduced with GFP or NΔ after 1 day. (B) Real-time PCR analysis of N-cadherin mRNA level in hMSCs transduced with GFP, RacV12, or RacN17 after 1 day. (C) Western blot and quantification of N-cadherin protein level in hMSCs transduced with GFP, RacV12, or RacN17 after 7 days. (D) MSCs were transduced with GFP or NΔ and cultured in growth or differentiation media for 2 days. Rac1 activity was examined by G-LISA™ Rac Activation Assay. (E) Real-time PCR analysis of SMC gene expression in hMSCs co-transduced with different viruses as indicated after 1 day. All data are represented as mean±SEM; n≥3. * (P<0.05) and ns (not significant), as calculated by student t test.
Discussion
It has been shown that TGFβ can induce differentiation of hMSCs to either smooth muscle cells [17,18] or chondrocytes [1,16], depending on their culture configuration. In vitro chondrogenesis is induced when hMSCs are cultured as a micromass pellet and exposed to TGFβ, while SMC differentiation is induced when hMSCs are cultured as freely spread cells on tissue culture plates and exposed to TGFβ. An interesting question thus is how the difference in culture configuration determines TGFβ-induced commitment to a chondrogenic versus SMC lineage. We now report that cell shape acts as a key regulator of TGFβ3-induced hMSC differentiation to a chondrogenic versus smooth muscle cell fate. By demonstrating a need for pellet culture, previous studies had suggested a role for absence of cell-matrix adhesion in chondrogenesis [16,30], but the use of specialized micropatterned substrates here demonstrate the unique importance of cell shape at least in early chondrogenic lineage commitment from the multipotent MSC. Constraining hMSCs to assume a spherical morphology, whether by suspending them in pellet culture or plating onto micropatterned substrates, triggered a chondrogenic fate, while cell adhesion and spreading on plates induced SMC differentiation. Since TGFβ has been shown to regulate early steps of smooth muscle cell differentiation during vasculogenesis, it is not surprising that we did not observe the upregulation of late markers of SMC differentiation such as myosin heavy chain even after 14 days of induction (Figure S2), which presumably requires additional factors. One possibility is that the substrates used in the experiments are not of optimal rigidity—both tissue culture plastics and PDMS (~2.5MPa) have a modulus significantly higher than that of native muscle tissues (~10KPa) and it has been shown that matrix rigidity regulates stem cell lineage specification [2]. Interestingly, the upregulation of chondrogenic genes was much faster in patterned unspread cells (robust expression was detected within 1 day following induction, Figure 1E) than in pelleted cells (similar level expression was achieved only after 14 days, Figure S1), suggesting cells in pellets may need to integrate many more different cues besides cell shape in order to determine their fate, or they simply experience lower TGFβ3 concentrations due to the transport limitation.
Cell shape could regulate the differential responses to TGFβ3 through multiple mechanisms. For example, it has been shown that TGFβ signaling can be modulated by changes in receptor expression, focal adhesion signaling, and nuclear signaling [31–33], each of which could be impacted by cell shape. Changes in cell shape, via rearrangements in the architecture and mechanics of the cytoskeleton, can physically distort the nucleus and alter cell differentiation [7,34]; mechanically induce focal adhesion assembly and signaling [35,36]; and even directly impact receptor-mediated signaling by altering the spatial distribution of signaling intermediaries relative to the plasma membrane [37]. It would not be surprising that multiple parallel mechanisms are therefore in place for cells to transduce changes in cell shape to drive stem cell fate, especially in settings where such changes in cell shape are themselves important, such as during tissue morphogenesis. Interestingly, cell shape regulation of MSCs is not unique to the chondro-myogenic differentiation axis, as our previous work demonstrated that cell shape in the presence of other (non-TGFβ) factors can also regulate stem cell commitment between adipogenic and osteogenic fates [3]. These findings suggest that certain soluble factors can restrict the differentiation potential of MSCs to a subset of lineages (e.g. TGFβ permitting myogenic and chondrogenic differentiation), while shape restricts an orthogonal set (e.g. round shape favoring adipogenesis or chondrogenesis). Together, the combination of soluble and mechanical cues then specifies cell fate.
The Rho GTPases have previously been implicated in regulating stem cell differentiation. Activation of Rho signaling in the p190RhoGAP knockout mouse led to alterations in adipogenesis and myogenesis [23], and in culture, changes in RhoA signaling has been shown to trigger an adipogenic-osteogenic switch in MSC differentiation [3]. In the current study, we demonstrated the importance of another small GTPase, Rac1, in regulating hMSC differentiation into smooth muscle cells or chondrocytes. While Rac1 has been implicated in late stage skeletal myotube fusion [26], its role in multipotent stem cell commitment has not been widely explored. Both RhoA and Rac1 appear to act as part of a central signaling hub that can integrate adhesive and soluble cues into a specific cellular response – in the context of our studies, lineage specification. Given that hMSCs have potential for yet more lineages, it will be interesting to identify other master regulators. Since cell shape is determined by the cytoskeletal organization and Rho family small GTPases have well-established roles in cytoskeleton remodeling, it is not surprising that Rho GTPases are involved in mediating the signals from structural/cytoskeletal changes to cell fate determination.
The intricate link between cell-matrix adhesion, changes in cell shape, and TGFβ signaling is further complicated by the observed involvement of N-cadherin in MSC lineage commitment. During skeletal and cardiac muscle differentiation, the interactions between cadherins and Rho GTPases have been shown to play an important role. It has been reported that active RhoA inhibits myoblast fusion by downregulating M-cadherin expression and localization [38]. On the other hand, M- and N-cadherin-dependent adhesion can activate Rac1 which is required for myoblast fusion [26] [24]. Here, we found increased N-cadherin expression downstream of TGFβ3 and Rac1 that was required for a much earlier step—MSC commitment to the SMC fate. However, N-cadherin expression may be regulated by additional signaling other than Rac1, since dominant negative Rac1 (RacN17) was not sufficient to inhibit N-cadherin upregulation in the presence of TGFβ3 (differentiation medium, Figure 4B, C). The cytoplasmic domain of N-cadherin is indirectly linked to actin cytoskeleton via the catenin complex, which consists of at least α-catenin, β-catenin, γ-catenin, and p120-catenin [39,40]. The dominant negative mutant of N-cadherin (NΔ) used in our study lacks β-catenin binding site, but can still bind to p120-catenin. This mutant changed β-catenin localization from cell-cell junctions and cell membrane to peri-nuclear regions (Figure S9). Thus, the inhibitory effect of NΔ on SMC differentiation may be the result of mislocalization of β-catenin and/or sequestering endogenous p120-catenin. It will be interesting to examine whether β-catenin and/or p120-catenin are involved in TGFβ3-induced SMC differentiation. Alternatively, it has been shown that N-cadherin modulates FGF receptor signaling [41,42] and can also negatively regulate Wnt/β-catenin signaling [43]. It is very likely that crosstalk exists between different pathways.
Interestingly, cadherins are also implicated in other stages of cartilage development. In embryonic limb chondrogenesis, N-cadherin is required for the initial cellular condensation [44], a process that also involves Rac1 signaling [45]; during chondrocytic differentiation, however, N-cadherin expression then shuts down [46]. In our studies, we also observed the loss of N-cadherin during hMSC chondrogenesis, but aside from preventing SMC differentiation, the role of this downregulation in chondrogenic commitment remains unclear. It is interesting that N-cadherin and Rac1 signaling are involved in multiple stages in both myogenic and chondrogenic differentiation, perhaps acting as checkpoints to ensure continuous cell-cell contact in the case of myogenic differentiation to produce functional muscle tissue, and to be only transiently required in cartilage development to allow cellular condensation, becoming unnecessary as cells separate into single cells encased in matrix during cartilage tissue formation.
In summary, our results implicate cell shape as an important microenvironmental cue that drives hMSC differentiation into different lineages in response to the same soluble factor. When cells are spread, TGFβ3 activates Rac1 and increases N-cadherin expression, which ultimately leads to the upregulation of SMC genes. When cell spreading is restricted, TGFβ3 fails to activate Rac1 or increase N-cadherin expression and instead drives induction of chondrogenic genes (Figure 5). These findings demonstrate an intricate link between cell shape, cell-cell adhesion, and TGFβ3 signaling, in this case guided by Rac1 signaling. Taken together with our previous findings implicating cell shape and RhoA in driving osteogenic and adipogenic fates, it appears that cells have evolved an elaborate mechanism to integrate adhesive, structural, mechanical, and soluble cues in the microenvironment. Understanding the complex interplay between these cues in different microenvironments is a critical step toward a greater appreciation of how stem cells choose their fates.
Figure 5.
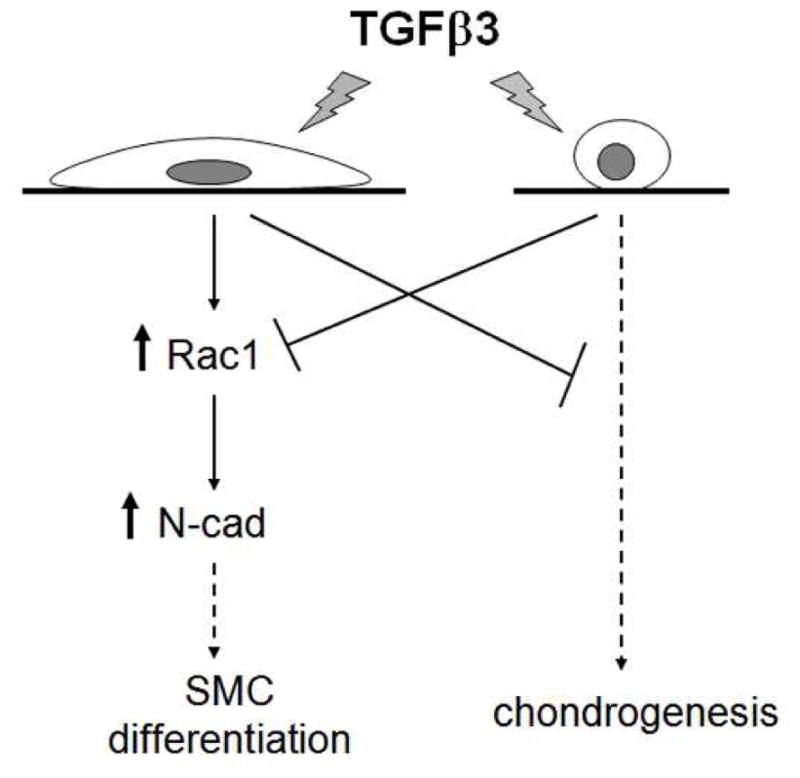
Model of cell shape-regulated hMSC differentiation to SMC or chondrogenic lineage. When cells are well-spread, TGFβ3 activates Rac1 and increases N-cadherin expression, which leads to the upregulation of SMC genes. When cells are in round shape, TGFβ3 fails to activate Rac1 signaling and instead upregulates chondrogenic genes.
Supplementary Material
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (EB00262, HL73305, GM74048), the University of Pennsylvania Institute for Regenerative Medicine and Center for Musculoskeletal Disorders. R.M. acknowledges support from the NIH Medical Scientist Training Program.
We thank D. Cohen, R. Desai, and Y-K. Wang for helpful discussions. We are grateful to B. McKay (University of Arizona) for the N-cadherin cDNA and W. Liu for making NΔ adenovirus.
Footnotes
Author contributions: L. G.: conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript
R. M.: conception and design, collection and/or assembly of data, data analysis and interpretation
C. S. C.: conception and design, financial support, data analysis and interpretation, manuscript writing, final approval of manuscript
References
- 1.Pittenger MF, Mackay AM, Beck SC, et al. Multilineage Potential of Adult Human Mesenchymal Stem Cells. Science. 1999;284(5411):143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 2.Engler AJ, Sen S, Sweeney HL, et al. Matrix Elasticity Directs Stem Cell Lineage Specification. Cell. 2006;126(4):677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 3.McBeath R, Pirone DM, Nelson CM, et al. Cell Shape, Cytoskeletal Tension, and RhoA Regulate Stem Cell Lineage Commitment. Developmental Cell. 2004;6(4):483–495. doi: 10.1016/s1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]
- 4.Ruiz SA, Chen CS. Emergence of Patterned Stem Cell Differentiation Within Multicellular Structures. Stem Cells. 2008;26(11):2921–2927. doi: 10.1634/stemcells.2008-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spiegelman BM, Ginty CA. Fibronectin modulation of cell shape and lipogenic gene expression in 3t3-adipocytes. Cell. 1983;35(3 Part 2):657–666. doi: 10.1016/0092-8674(83)90098-3. [DOI] [PubMed] [Google Scholar]
- 6.Benya PD, Padilla SR, Nimni ME. Independent regulation of collagen types by chondrocytes during the loss of differentiated function in culture. Cell. 1978;15(4):1313–1321. doi: 10.1016/0092-8674(78)90056-9. [DOI] [PubMed] [Google Scholar]
- 7.Thomas CH, Collier JH, Sfeir CS, et al. Engineering gene expression and protein synthesis by modulation of nuclear shape. PNAS. 2002;99(4):1972–1977. doi: 10.1073/pnas.032668799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Folkman J, Moscona A. Role of cell shape in growth control. Nature. 1978;273(5661):345–349. doi: 10.1038/273345a0. [DOI] [PubMed] [Google Scholar]
- 9.Chen CS, Mrksich M, Huang S, et al. Geometric control of cell life and death. Science. 1997;276(5317):1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- 10.Le Beyec J, Xu R, Lee SY, et al. Cell shape regulates global histone acetylation in human mammary epithelial cells. Exp Cell Res. 2007;313(14):3066–3075. doi: 10.1016/j.yexcr.2007.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nelson CM, Vanduijn MM, Inman JL, et al. Tissue geometry determines sites of mammary branching morphogenesis in organotypic cultures. Science. 2006;314(5797):298–300. doi: 10.1126/science.1131000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roskelley CD, Desprez PY, Bissell MJ. Extracellular matrix-dependent tissue-specific gene expression in mammary epithelial cells requires both physical and biochemical signal transduction. Proc Natl Acad Sci U S A. 1994;91(26):12378–12382. doi: 10.1073/pnas.91.26.12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watt FM, Jordan PW, O’Neill CH. Cell shape controls terminal differentiation of human epidermal keratinocytes. Proc Natl Acad Sci U S A. 1988;85(15):5576–5580. doi: 10.1073/pnas.85.15.5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420(6916):629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 15.Ren XD, Kiosses WB, Schwartz MA. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. Embo J. 1999;18(3):578–585. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mackay AM, Beck SC, Murphy JM, et al. Chondrogenic Differentiation of Cultured Human Mesenchymal Stem Cells from Marrow. Tissue Engineering. 1998;4(4):415–428. doi: 10.1089/ten.1998.4.415. [DOI] [PubMed] [Google Scholar]
- 17.Kinner B, Zaleskas JM, Spector M. Regulation of Smooth Muscle Actin Expression and Contraction in Adult Human Mesenchymal Stem Cells. Experimental Cell Research. 2002;278(1):72–83. doi: 10.1006/excr.2002.5561. [DOI] [PubMed] [Google Scholar]
- 18.Jeon ES, Moon HJ, Lee MJ, et al. Sphingosylphosphorylcholine induces differentiation of human mesenchymal stem cells into smooth-muscle-like cells through a TGF-{beta}-dependent mechanism. J Cell Sci. 2006;119(23):4994–5005. doi: 10.1242/jcs.03281. [DOI] [PubMed] [Google Scholar]
- 19.Tan JL, Tien J, Chen CS. Microcontact Printing of Proteins on Mixed Self-Assembled Monolayers. Langmuir. 2002;18(2):519–523. [Google Scholar]
- 20.Leong FJ, Brady M, McGee JO. Correction of uneven illumination (vignetting) in digital microscopy images. J Clin Pathol. 2003;56(8):619–621. doi: 10.1136/jcp.56.8.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nelson CM, Pirone DM, Tan JL, et al. Vascular endothelial-cadherin regulates cytoskeletal tension, cell spreading, and focal adhesions by stimulating RhoA. Mol Biol Cell. 2004;15 (6):2943–2953. doi: 10.1091/mbc.E03-10-0745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnstone B, Hering T, Caplan A, et al. In Vitro Chondrogenesis of Bone Marrow-Derived Mesenchymal Progenitor Cells. Experimental Cell Research. 1998;238(1):265–272. doi: 10.1006/excr.1997.3858. [DOI] [PubMed] [Google Scholar]
- 23.Sordella R, Jiang W, Chen G-C, et al. Modulation of Rho GTPase Signaling Regulates a Switch between Adipogenesis and Myogenesis. Cell. 2003;113(2):147–158. doi: 10.1016/s0092-8674(03)00271-x. [DOI] [PubMed] [Google Scholar]
- 24.Charrasse S, Meriane M, Comunale F, et al. N-cadherin-dependent cell-cell contact regulates Rho GTPases and beta-catenin localization in mouse C2C12 myoblasts. J Cell Biol. 2002;158 (5):953–965. doi: 10.1083/jcb.200202034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferrari SL, Traianedes K, Thorne M, et al. A Role for N-Cadherin in the Development of the Differentiated Osteoblastic Phenotype. Journal of Bone and Mineral Research. 2000;15 (2):198–208. doi: 10.1359/jbmr.2000.15.2.198. [DOI] [PubMed] [Google Scholar]
- 26.Charrasse S, Comunale F, Fortier M, et al. M-Cadherin Activates Rac1 GTPase through the Rho-GEF Trio during Myoblast Fusion. Mol Biol Cell. 2007;18 (5):1734–1743. doi: 10.1091/mbc.E06-08-0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakagawa M, Fukata M, Yamaga M, et al. Recruitment and activation of Rac1 by the formation of E-cadherin-mediated cell-cell adhesion sites. J Cell Sci. 2001;114(10):1829–1838. doi: 10.1242/jcs.114.10.1829. [DOI] [PubMed] [Google Scholar]
- 28.Braga VMM, Betson M, Li X, et al. Activation of the Small GTPase Rac Is Sufficient to Disrupt Cadherin-dependent Cell-Cell Adhesion in Normal Human Keratinocytes. Mol Biol Cell. 2000;11(11):3703–3721. doi: 10.1091/mbc.11.11.3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takaishi K, Sasaki T, Kotani H, et al. Regulation of Cell-Cell Adhesion by Rac and Rho Small G Proteins in MDCK Cells. J Cell Biol. 1997;139(4):1047–1059. doi: 10.1083/jcb.139.4.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoo JU, Barthel TS, Nishimura K, et al. The Chondrogenic Potential of Human Bone-Marrow-Derived Mesenchymal Progenitor Cells. J Bone Joint Surg Am. 1998;80(12):1745–1757. doi: 10.2106/00004623-199812000-00004. [DOI] [PubMed] [Google Scholar]
- 31.Petridou S, Maltseva O, Spanakis S, et al. TGF-beta Receptor Expression and Smad2 Localization Are Cell Density Dependent in Fibroblasts. Invest Ophthalmol Vis Sci. 2000;41 (1):89–95. [PubMed] [Google Scholar]
- 32.Thannickal VJ, Lee DY, White ES, et al. Myofibroblast Differentiation by Transforming Growth Factor-beta 1 Is Dependent on Cell Adhesion and Integrin Signaling via Focal Adhesion Kinase. J Biol Chem. 2003;278 (14):12384–12389. doi: 10.1074/jbc.M208544200. [DOI] [PubMed] [Google Scholar]
- 33.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF- family signalling. Nature. 2003;425(6958):577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 34.Maniotis AJ, Chen CS, Ingber DE. Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc Natl Acad Sci U S A. 1997;94(3):849–854. doi: 10.1073/pnas.94.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen CS, Alonso JL, Ostuni E, et al. Cell shape provides global control of focal adhesion assembly. Biochem Biophys Res Commun. 2003;307(2):355–361. doi: 10.1016/s0006-291x(03)01165-3. [DOI] [PubMed] [Google Scholar]
- 36.Pirone DM, Liu WF, Ruiz SA, et al. An inhibitory role for FAK in regulating proliferation: a link between limited adhesion and RhoA-ROCK signaling. J Cell Biol. 2006;174(2):277–288. doi: 10.1083/jcb.200510062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meyers J, Craig J, Odde DJ. Potential for control of signaling pathways via cell size and shape. Curr Biol. 2006;16(17):1685–1693. doi: 10.1016/j.cub.2006.07.056. [DOI] [PubMed] [Google Scholar]
- 38.Charrasse S, Comunale F, Grumbach Y, et al. RhoA GTPase Regulates M-Cadherin Activity and Myoblast Fusion. Mol Biol Cell. 2006;17(2):749–759. doi: 10.1091/mbc.E05-04-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gumbiner BM. Proteins associated with the cytoplasmic surface of adhesion molecules. Neuron. 1993;11(4):551–564. doi: 10.1016/0896-6273(93)90068-3. [DOI] [PubMed] [Google Scholar]
- 40.Kemler R. From cadherins to catenins: cytoplasmic protein interactions and regulation of cell adhesion. Trends Genet. 1993;9(9):317–321. doi: 10.1016/0168-9525(93)90250-l. [DOI] [PubMed] [Google Scholar]
- 41.Suyama K, Shapiro I, Guttman M, et al. A signaling pathway leading to metastasis is controlled by N-cadherin and the FGF receptor. Cancer Cell. 2002;2(4):301–314. doi: 10.1016/s1535-6108(02)00150-2. [DOI] [PubMed] [Google Scholar]
- 42.Williams E-J, Williams G, Howell FV, et al. Identification of an N-cadherin Motif That Can Interact with the Fibroblast Growth Factor Receptor and Is Required for Axonal Growth. J Biol Chem. 2001;276(47):43879–43886. doi: 10.1074/jbc.M105876200. [DOI] [PubMed] [Google Scholar]
- 43.Hay E, Laplantine E, Geoffroy V, et al. N-Cadherin Interacts with Axin and LRP5 To Negatively Regulate Wnt/{beta}-Catenin Signaling, Osteoblast Function, and Bone Formation. Mol Cell Biol. 2009;29(4):953–964. doi: 10.1128/MCB.00349-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oberlender SA, Tuan RS. Expression and functional involvement of N-cadherin in embryonic limb chondrogenesis. Development. 1994;120(1):177–187. doi: 10.1242/dev.120.1.177. [DOI] [PubMed] [Google Scholar]
- 45.Woods A, Wang G, Dupuis H, et al. Rac1 Signaling Stimulates N-cadherin Expression, Mesenchymal Condensation, and Chondrogenesis. J Biol Chem. 2007;282(32):23500–23508. doi: 10.1074/jbc.M700680200. [DOI] [PubMed] [Google Scholar]
- 46.Oberlender SA, Tuan RS. Spatiotemporal profile of N-cadherin expression in the developing limb mesenchyme. Cell Adhes Commun. 1994;2(6):521–537. doi: 10.3109/15419069409014216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.