

Abstract
The progression of prostate cancer from an organ-confined, androgen-sensitive disease to a metastatic one is associated with dysregulation of androgen receptor (AR)-regulated target genes and with a decrease in insulin-like growth factor-I receptor (IGF-IR) expression. To investigate the differential effects of wild type (wt) and mutant AR on IGF-IR levels we employed a series of isogenic prostate-derived cell lines and human xenografts. We show that basal and phosphorylated IGF-IR levels progressively decreased as prostate cancer cells became more tumorigenic and metastatic. In addition, we show that wt, but not mutant, AR along with dihydrotestosterone treatment increased IGF-IR promoter activity and endogenous IGF-IR levels. ChIP analysis show enhanced AR binding to the IGF-IR promoter in AR-overexpressing cells. Finally, wt AR-overexpressing cells display an enhanced proliferation rate. In summary, we provide evidence that activated wt AR enhances IGF-IR transcription in prostate cancer cells via a mechanism that involves AR binding to the IGF-IR promoter. AR mutations alter the ability of the mutated protein to regulate IGF-IR expression. Our results suggest that prostate cancer progression is associated with a decrease in IGF-IR expression that could be the result of impaired ability of AR to stimulate IGF-IR gene expression.
Keywords: insulin-like growth factor-I (IGF-I), IGF-I receptor, androgen receptor, prostate cancer
1. Introduction
Prostate cancer is a major health issue in the Western world (Deutsch et al., 2004). A key component of the androgen transduction cascade is the androgen receptor (AR). Alterations in AR structure and expression are responsible for the progression of the androgen-dependent (AD) tumor to a more aggressive, hormone-refractory, androgen-independent (AI) stage. Progression of the tumor from an organ-confined, androgen-sensitive disease to a metastatic one is associated with dysregulation of AR-regulated targets and up-regulation of AR expression (Culig et al., 1993; Schroder, 1993).
The insulin-like growth factors have important roles in normal growth as well as in tumor development (Khandwala et al., 2000; Samani et al., 2007; Werner and Maor, 2006). In the specific context of prostate cancer data has accumulated suggesting that IGFs play a role in prostate epithelium transformation (Cohen et al., 1991; DiGiovanni et al., 2000; Kaplan et al., 1999; Roddam et al., 2008; Ruan et al., 1999). The contribution of IGF action to prostate cancer is supported by epidemiological studies showing an increase in serum IGF-I levels in patients who later developed prostate cancer (Chan et al., 1998). Acquisition of the malignant phenotype is initially IGF-IR-dependent, however, the progression of prostate cancer from an AD to an AI disease is associated with a decrease in IGF-IR levels (Tennant et al., 1996; Wu et al., 2006b). Likewise, IGF-IR expression is extinguished in a majority of human cancer bone marrow metastases (Chott et al., 1999). In addition, Sutherland et al. (2008) showed that prostate epithelial-specific deletion of IGF-IR accelerated the emergence of aggressive prostate cancer. The molecular mechanisms responsible for regulation of the IGF-IR gene in prostate cancer, however, remain largely unidentified.
AR is a ligand-dependent transcription factor that belongs to the steroid receptors superfamily (Lee and Chang, 2003). Our understanding of the joint regulation of the androgen and IGF systems in prostate cancer is limited. Early reports indicated that IGF-I transactivates the AR in prostate cancer cells (Culig et al., 1994). Other studies have shown that IGF-I enhanced androgen-mediated AR transcriptional activity but was unable to transactivate the AR in the absence of androgens (Orio et al., 2002). Recently, one of us has shown that the effect of IGF-I on AR transcriptional activity is even more complex and depends on cell context (Plymate et al., 2004). Lin et al. (2001) have shown that IGF-I phosphorylates AR at Ser210 and Ser790. AR phosphorylation may inhibit AR-mediated apoptosis, possibly by inhibiting the interaction between AR and coregulators. In addition, IGF-I may sensitize the AR transcriptional complex to subphysiologic levels of androgens in LNCaP cells (Bakin et al., 2003). Little data, however, is available regarding the regulation of IGF-IR expression by androgens. Recently, Pandini et al. (2005) have shown that androgens induced IGF-IR up-regulation via a nongenomic AR pathway.
The mechanisms responsible for the differential regulation of IGF-IR gene expression in benign in comparison to malignant prostate tumors are unknown. Furthermore, little information is available regarding the differential control of IGF-IR gene expression by wild type (wt) and mutated AR. In view of the fact that progression to advanced stage disease is associated with acquisition of AI, understanding the involvement of AR in regulation of IGF-IR expression and action is of cardinal importance. The purpose of this study was to investigate the effect of wt and mutant AR on IGF-IR expression and signaling in a cellular model of tumor progression. Our results demonstrate that AR re-expression in the M12 human prostate cancer cell line led to a significant increase in IGF-IR expression and activation, proliferation rate, and IGF-IR promoter activity. On the other hand, mutant ARs were impaired in their ability to regulate IGF-IR expression. Furthermore, following castration, when AR expression increases significantly and drives prostate cancer progression in the absence of ligand, IGF-IR is suppressed (Chen et al., 2004). In addition, chromatin immunoprecipitation assays showed that regulation of IGF-IR expression by AR is mediated via direct binding of AR to the IGF-IR promoter region.
2. Materials and methods
2.1. Cell cultures
The human prostate cancer experimental system employed was previously described (Bae et al., 1994, 1998). The derivation of the cell lines is summarized in Figure 1A. Cells were maintained in RPMI-1640 medium (Biological Industries, Kibbutz Beit Haemek, Israel). The media were supplemented 5% fetal bovine serum (FBS), 2 mM glutamine, 50 μg/ml gentamicin sulfate, 10 ng/ml EGF, 0.1 nM dexamethasone, 5 μg/ml insulin, 5 μg/ml transferrin, and 5 ng/ml selenium (Damon et al., 2001). All reagents were purchased from Biological Industries.
Fig. 1.
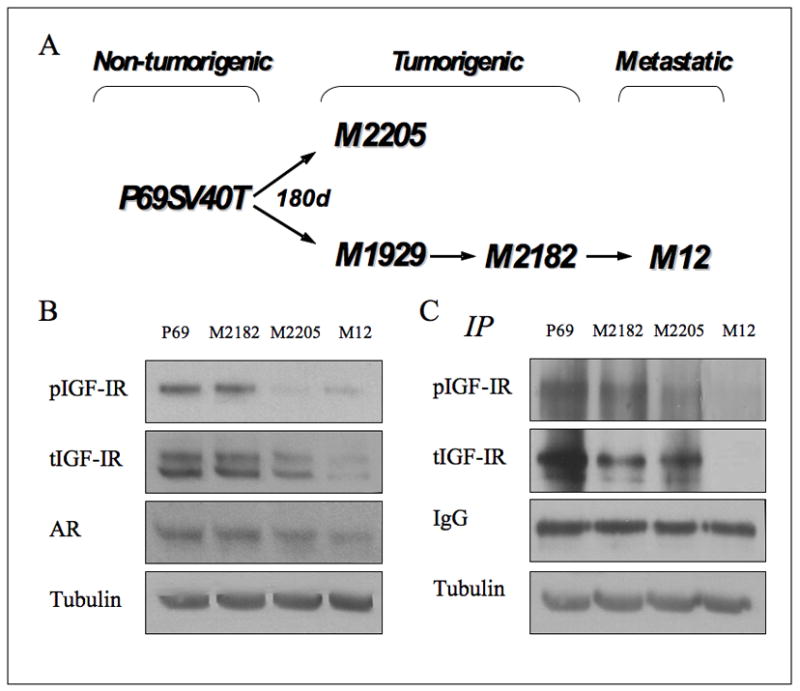
Expression of IGF-IR and AR in prostate cancer cell lines. (A) Derivation of SV40 Tag immortalized cell lines. Tumorigenic sublines of the SV40 Tag immortalized prostate epithelial cells (P69SV40T) were subjected to sequential cycles of in vivo passage in athymic nude mice. The parental line is rarely tumorigenic. The M2182 and M2205 cell lines are tumorigenic after s.c. injection, whereas the M12 line is tumorigenic and metastatic after intraprostatic injection. (B) Western blot analysis. Lysates (80 μg protein) were separated by 10% SDS-PAGE, transferred onto nitrocellulose filters, and blotted with anti-IGF-IR, anti-phospho (p)-IGF-IR, anti-AR, and anti-tubulin. Experiments were repeated at least three times, with similar results. (C) Immunoprecipitation analysis. Lysates were precipitated overnight with anti-IGF-IR and precipitates were incubated with protein A/G beads for 3 hr. The precipitates were washed, electrophoresed, transferred onto membranes, and probed with antibodies against phosphotyrosine and total-IGF-IR.
2.2. Prostate xenografts
The human prostate xenografts LuCaP 23.1 and LuCaP 35 have been described (Corey et al., 2002, 2003; Wu et al., 2005, 2006a). LuCaP 96 was derived and maintained in a similar manner to LuCaP 23.1 and LuCaP 35. LuCaP 96 has a wt AR and is maintained by passage in SCID mice. Briefly, tumor bits from the xenografts were implanted sc into the flanks of 20 male non-castrate SCID mice as previously described (Corey et al., 2003). Animals were followed and when tumor volume reached 100 mm3 half of the animals were castrated. Animals were followed until tumors reached a volume of 400 mm3. In the case of the castrated animals tumors initially regressed and then recurred. Mice were euthanized and tumors harvested when they reached a 1000 mm3 volume. All procedures were approved by the University of Washingto Animal Care and Use Committee.
2.3. Western immunoblots
Cells and xenograft tissues were processed as described (Damon et al., 2001). Samples were subjected to 10% SDS-PAGE. Blots were incubated with a polyclonal human IGF-IR β-subunit antibody (Santa Cruz Biotechnology), washed, and incubated with an horseradish peroxidase-conjugated secondary antibody. In addition, blots were incubated with antibodies against AR, tubulin, phospho-IGF-IR, and actin.
2.4. Immunoprecipitation (IP) assays
Cells were treated, harvested, and lysed as described above. Lysates were precipitated overnight with anti-human IGF-IR β-subunit. The precipitates were incubated with protein A/G beads for 3 hr. Precipitates were washed, subjected to 10% SDS-PAGE, and blotted with anti-phosphotyrosine.
2.5. Plasmids and DNA transfections
Expression vectors encoding wt mouse AR, and E231G (human E251G), and T857A (human T877A) mutants have been described (Han et al., 2005). For cotransfections, an IGF-IR promoter luciferase reporter construct, [p(−476/+640)LUC], was employed (Werner et al., 1994). P69 and M12 cells were transfected with 1 μg of the p(−476/+640)LUC reporter construct, along with 1 μg of the wtAR/T857A/E231G expression vectors (or pcDNA3) and 0.3 μg of a β-gal expression plasmid (pCMV-β, Clontech) using Jet-PEI™ (Polyplus, Illkirch, France). Twenty-four hours after transfection cells were treated with 10−8 M DHT and, after an additional 24 hr, cells were harvested and luciferase and β-gal activities were measured. Transfections were also performed in the LNCaP, PC3, DU-145, and C4-2 prostate cell lines. For stable transfections, M12 cells were plated in 6-well plates and transfected with wt AR, or T857A or E231G mutant expression vectors (or pcDNA3). After 24 hr, selection by 500 mg/ml of G418 was started and after 2 weeks independent colonies were picked up and AR expression was assessed by RT-PCR.
2.6. RT-PCR for IGF-IR and AR mRNA expression
RNA was prepared from M12-derived AR transfectants and prostate cancer xenografts. One microgram of RNA was reverse transcribed and amplified by PCR. The primers used for IGF-IR mRNA were: sense, GAA-GTG-GAA-CCC-TCC-CTC-TC; antisense, CTT-CTC-GGC-TTC-AGT-TTT-GG. The size of the band was 275 bp. The primers used to confirm wt AR expression were P1F (ATG-TGC-CAG-CAG-AAA-CG) and P1R (CGG-TAC-ACA-TTG-AAA-ACC-A). The presence of point mutations E231G and T857A was confirmed by sequencing. GAPDH mRNA levels were measured as controls.
2.7. Chromatin immunoprecipitation (ChIP) studies
AR-transfected (or pcDNA3) M12 cells were incubated with formaldehyde (1%) for 10 min, after which cells were washed and harvested. Pelleted cells were resuspended in 1% SDS-containing buffer, incubated on ice for 10 min, and sonicated. Lysates were immunoprecipitated with anti-AR for 18 hr at 4°C. For PCR analysis of AR-immunoprecipitated chromatin, a set of primers encompassing the IGF-IR proximal promoter region (nt −469 to +288) was employed: sense, CTT-TCC-AGC-CGC-GCT-GTT-GTT-G; anti-sense, GGT-AAA-CAA-GAG-CCC-CAG-CCT-C. PCR was performed using TermalAce™ DNA polymerase (Invitrogen).
2.8. Cell proliferation assays
The growth rate of M12-derived AR-overexpressing cells was determined by cell counting as a function of time. Cells (1×105 cells/well) were seeded, allowed to attach for 24 h, and cultured for 72 hr with daily medium changes. Cells were trypsinized after 24, 48, and 72 hr and counted using an hemocytometer.
2.9. Statistical analysis
The statistical significance of the differences observed between groups was assessed using the T-test (two samples, equal variance). P < 0.05 was considered statistically significant.
3. Results
3.1. Basal IGF-IR levels in lineage-derived prostate cancer cell lines
To investigate the potential regulation of IGF-IR gene expression by AR during prostate cancer progression, the human prostate epithelial cell lines P69, M2182, M2205, and M12 were used. These cell lines provide a defined genetic lineage in which to study changes that occur during cancer progression (Akalin et al., 2001). Results of Western blots (Figure 1B) and IP (Figure 1C) showed that basal IGF-IR levels decrease as prostate cancer cells become more tumorigenic and metastatic. Specifically, IGF-IR levels were higher in the non-tumorigenic prostate epithelial cell line P69 compared to its metastatic derivative, the M12 line. Intermediate IGF-IR levels were seen in the tumorigenic, but non-metastatic, M2182 and M2205 lines. These results replicate, in part, previously published data (Bae et al., 1994; Plymate et al., 1996). In addition, corresponding decreases in basal pIGF-IR levels were noticed, consistent with a reduction in IGF-IR activation during progression. Finally, basal AR levels were very low in all four cells lines (Figure 1B).
3.2. Differential effects of wt and mutant AR on IGF-IR protein levels
To investigate the effects of wt and mutant AR on IGF-IR levels, M12 cells were stably transfected with vectors encoding wt AR or one of two different mutants: AR-T857A, that includes a mutation in the ligand-binding domain and, hence, shows a promiscuous ligand response, and AR-E231G, that includes a mutation in the conserved N-terminal domain, involved in interactions with coregulators. AR mRNA expression in stable-transfected M12-derived clones was monitored by RT-PCR. As shown in Figure 2A, wt AR, AR-T857A, and AR-E231G mRNAs were expressed at similar levels in M12-derived transfectants. Furthermore, Western blots revealed that IGF-IR levels were largely enhanced in wt AR-expressing, but not mutant AR-expressing, M12 cells (Figures 2B and 2C). These results were corroborated by IP, which confirmed that wt, but not mutant, AR increased IGF-IR levels (Figure 2D).
Fig. 2.
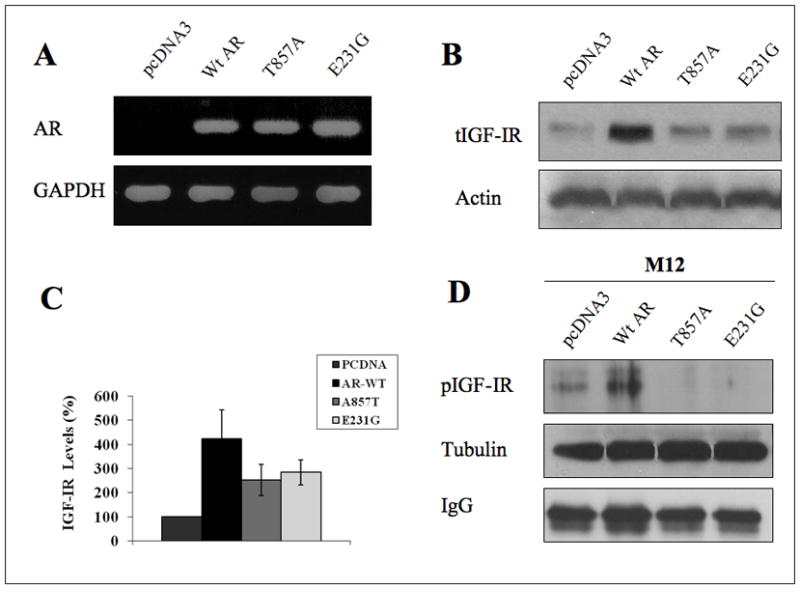
Regulation of IGF-IR levels by wt and mutant AR. (A) M12 cells were stably transfected with vectors expressing the full length wt AR or one of two different mutant versions (AR-T857A and AR-E231G). AR mRNA levels were evaluated by RT-PCR. (B) IGF-IR levels were assessed by Western blots using anti-IGF-IR β-subunit. Results shown are representative of a typical experiment repeated three times with similar results. Blots were stripped and blotted with anti-actin. (C) The bar graph denotes the densitometric scanning of the total IGF-IR bands normalized to actin. The bars represent the mean ± S.E.M of three experiments. (D) Wt and mutant AR-overexpressing M12 cells were lysed, precipitated with anti-IGF-IR, and incubated with protein A/G beads for 3 hr. The precipitates were washed, electrophoresed, transferred, and probed with antibodies against phosphotyrosine. Results shown are representative of an experiment repeated three times with similar results.
3.3. Effect of wt and mutant AR on IGF-IR promoter activity
To examine the hypothesis that AR regulation of IGF-IR expression is mediated at the transcription level, P69 and M12 cells were transiently transfected with a proximal IGF-IR promoter-luciferase reporter [p(−476/+640)LUC], in the absence or presence of a wt AR vector, along with a β-gal vector. After 24 hr cells were treated with 10−8 M DHT (or left untreated) for 24 hr, after which cells were harvested and luciferase and β-gal activities were measured (Rubinstein et al., 2004; Werner et al., 2007). Coexpression of AR (in the absence of androgen treatment) had a paradoxical inhibitory effect on IGF-IR promoter activity (~20% and ~50% reductions in P69 and M12 cells, respectively) compared to the untreated ceontrol cells (Figures 3A, 3B). In contrast, DHT treatment enhanced promoter activity by ~160% in wt AR-transfected P69 cells (Figure 3A) and by ~210% in M12 cells (Figure 3B). DHT had no effect in pcDNA3-transfected cells. Similar results (i.e., repression of promoter activity by AR without androgen treatment, and enhancement after DHT treatment) were seen in LNCaP, DU-145, C4-2, and PC3 prostate cancer cells (data not shown).
Fig. 3.
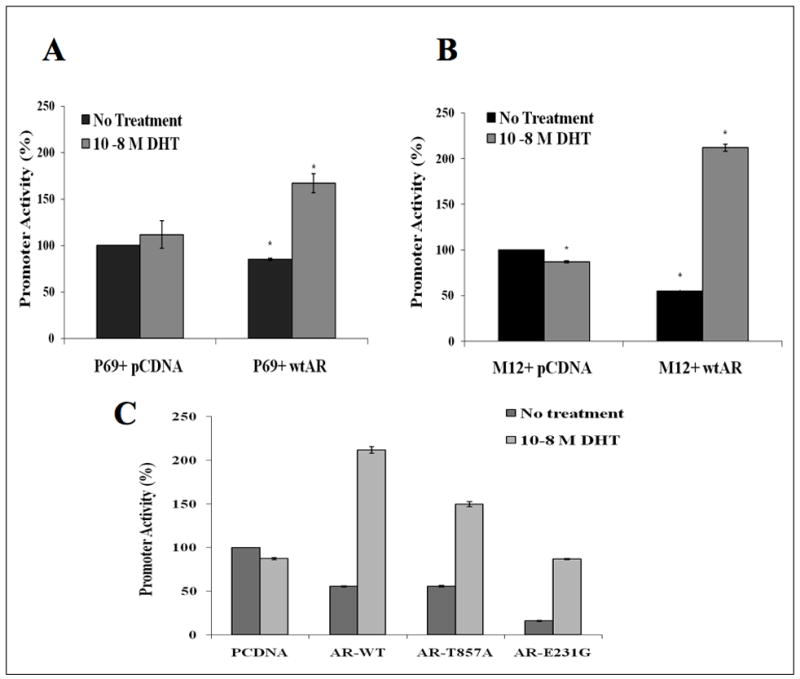
Regulation of IGF-IR promoter activity by unligated and ligand-activated AR. P69 (A) and M12 (B) cells were cotransfected with the p(−476/+640)LUC IGF-IR promoter-luciferase reporter, along with an AR vector (or pcDNA3) and a β-gal vector. Twenty-four hours after transfection cells were treated with 10−8M DHT and, after 24 hr, cells were harvested and luciferase and β-gal activities were measured. C) Effect of AR mutations on IGF-IR promoter activity. M12 cells were cotransfected with 1 μg of the p(−476/+640)LUC IGF-IR promoter, along with 1 μg of AR expression vector (wt AR, AR-T857A, or AR-E231G, or pcDNA3) and 0.3 μg of pCMVβ. After 24 hr cells were treated with 10−8M DHT and, after 24 hr, cells were harvested and luciferase and β-gal activities were measured. Promoter activities are expressed as luciferase normalized for β-gal levels. Results are mean ± S.E.M. of three independent experiments, performed in duplicate dishes.
Next, we investigated whether AR mutation can alter its ability to enhance IGF-IR promoter activity. For this purpose, M12 cells were transiently cotransfected with the IGF-IR promoter reporter, along with AR-T857A or AR-E231G vectors (or wt AR or pcDNA3) and a β-gal plasmid. After 24 hr cells were treated with DHT and, after an additional 24 hr, cells were harvested and luciferase and β-gal activities were measured. As with wt AR, coexpression of mutant AR in the absence of androgens had an inhibitory effect on IGF-IR promoter activity (Figure 3C). In contrast, addition of DHT increased promoter activity in mutant AR-transfected cells compared to the untreated control cells, however the extent of transactivation was significantly reduced in comparison to wt AR-transfected cells (~1.5-fold for T857A and ~0.9-fold for E231G, in comparison to ~2.2-fold for wt AR).
3.4. ChIP analysis of physical interactions between AR and the IGF-IR promoter
Next, the potential physical interaction between wt AR and the IGF-IR promoter was assessed using ChIP. Briefly, AR-overexpressing and control pcDNA3-transfected M12 cells were treated with 1% formaldehyde for 10 min, after which cells were lysed and immunoprecipitated with anti-AR or normal mouse serum. The AR-precipitated chromatin was amplified by PCR with a set of primers encompassing the proximal IGF-IR promoter region extending from nt −469 in the 5′ flanking region to nt +288 in the 5′ untranslated region. Results obtained showed that AR binding to the IGF-IR promoter was largely enhanced in AR-overexpressing, in comparison to control, cells (Figure 4A).
Fig. 4.
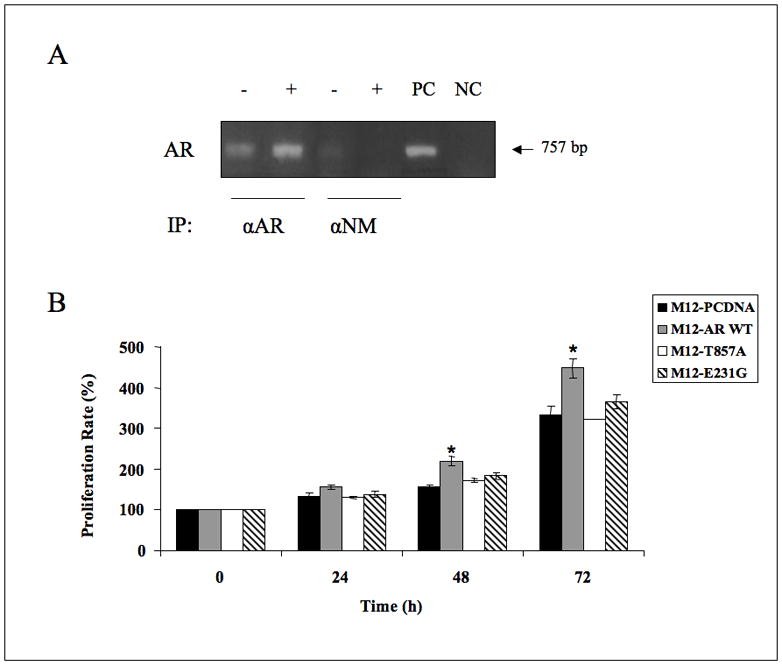
(A) ChIP analysis of AR interaction at the IGF-IR promoter. M12 cells were transfected with a wt AR vector (lanes 2, 4, denoted with a + symbol) or with pcDNA3 (lanes 1, 3, denoted with a − symbol). After 48 hr, cells were lysed and immunoprecipitated with anti-AR (lanes 1, 2) or with normal mouse serum (αNM) (lanes 3, 4), followed by PCR amplification of precipitated chromatin using primers encompassing the IGF-IR promoter. The position of the 757 bp fragment is indicated. Lane 5 represents the PCR product of M12 DNA (positive control, PC). Lane 6 represents the PCR product without template (negative control, NC). (B) Proliferation rate of M12-AR stable transfectants. Wt and mutant AR-expressing M12 cells were plated in 6-well plates (1×105 cells/well). Cells were trypsinized every 24 hr and counted with a hemocytometer. The number of cells at time 0 was assigned a value of 100%. The y axis denotes cell numbers (percentage of cells compared to time 0. Bars are mean ± S.D. (n=3 experiments). Proliferation rates of wt AR-expressing M12 cells at 72 hr were significantly higher than pcDNA3-transfected cells (p < 0.05).
3.5. Effect of wt and mutant AR expression on cell proliferation
To assess the impact of AR on cell proliferation, wt or mutant AR-overexpressing M12 cells were plated in 6-well plates (1×105 cells/well) and counted after 24, 48, and 72 hr. Results obtained indicate that wt AR-overexpressing M12 cells consistently displayed an enhanced proliferation rate in comparison to pcDNA3-transfected cells (approximately 1.3-fold increase at 72 hr, p < 0.05, in three independent experiments). On the other hand, no significant enhancement in proliferation rates were seen in AR-T857A or AR-E231G overexpressing, compared to pcDNA3-transfected, cells (Figure 4B).
3.6. Effect of castration on xenografts
Following castration and regrowth of the tumor there was an increase in both AR mRNA and protein expression (Figure 5). The levels of receptor and magnitude of change varied between xenografts, however, within each xenograft the level of AR expression and change with castration were consistent. Also, while AR levels increase after castration, IGF-IR levels decrease significantly, p <0.01.
Fig. 5.
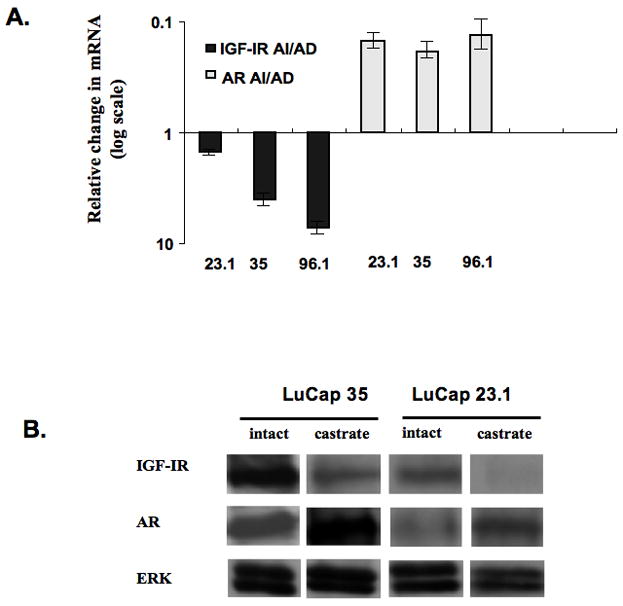
Effects of castration on IGF-IR and AR expression in prostate xenografts. (A) Ratio of IGF-IR and AR mRNA determined by qRT-PCR in human prostate xenografts grown in SCID mice and removed when the tumor volume reached ca. 400 mm3 either without castration (AD) or when the tumors recurred following castration and reached a volume of 400 mm3 (AI). There was a significant decrease in IGF-IR mRNA in each of the tumors following castration, p < 0.01, and an increase in AR expression, p < 0.001. Measurements were done in tumors from 5 animals per group for each xenograft. The numbers correspond to LuCaP lines, each from a different patient. Error bars = +/− SEM. (B) Representative Western blots from LuCaP 35 and 23.1 xenografts confirming changes in protein that follow the direction of mRNA. Note that the changes occur regardless of protein levels. Loading was controlled for total ERK expression.
4. Discussion
The involvement of IGF-IR in the initiation and progression of prostate cancer has been the subject of extensive investigation. Contradictory reports, however, have been presented regarding the pattern of IGF-IR expression throughout the various stages of the disease. Thus, while progression to AI in xenografts was associated with an increase in IGF-IR mRNA (Nickerson et al., 2001), expression levels were higher in the benign prostate epithelial cell line P69, compared to its metastatic derivative, the M12 line (Damon et al., 2001). In addition, while IGF-IR mRNA levels were shown to be reduced in bone marrow metastases (Chott et al., 1999), other reports showed a persistent IGF-IR expression in prostate metastases (Hellawell et al., 2002). These seemingly paradoxical results may reflect the ability of IGF-IR to mediate both differentiative and proliferative effects.
Two important features in the progression of prostate cancer from an organ-confined to a metastatic disease are the dysregulation of AR-regulated targets and a change in IGF-IR levels (Kaplan et al., 1999; Tennant et al., 1996). Although these changes could be considered independent epigenetic phenomena, evidence indicates that there is a relationship between IGF-IR signaling and AR action (Lin et al., 2001). Prostate cancer-associated alterations of AR function, including AR gene amplifications, mutations, altered interaction with coactivators, and ligand-independent AR activation by growth factors, may contribute to cancer progression (Gelmann, 2002; Grossmann et al., 2001; Shi et al., 2002; Veldscholte et al., 1990). However, while AR mutations were correlated with cancer progression in humans (Gelmann, 2002; Grossmann et al., 2001; Shi et al., 2002) and transgenic mice (Han et al., 2005), the functional impact of these changes is not clear.
In initial experiments we showed that basal and pIGF-IR levels progressively decrease as prostate cancer cells become more tumorigenic and metastatic. Next, we showed that wt AR transfection followed by DHT treatment increased IGF-IR promoter activity in P69 and M12 cells, whereas mutant ARs are impaired in this respect. ChIP analysis showed enhanced AR binding to the IGF-IR promoter in AR-overexpressing M12 cells. To examine the differential regulation of the endogenous IGF-IR gene by wt or mutant AR, M12 cells were stably transfected with vectors encoding wt AR or one of two different mutated versions (AR-T857A and AR-E231G). Western blots showed that wt AR enhanced IGF-IR levels in tumorigenic M12 cells while mutant AR had no effect. In addition, we showed by IP that wt AR-overexpressing cells had increased pIGF-IR levels, while AR mutants are unable to activate the IGF-IR. Finally, proliferation assays indicate that wt AR-overexpressing cells consistently displayed an enhanced proliferation rate. No significant enhancements in proliferation were seen in AR-T857A- or AR-E231G-overexpressing M12 cells.
Our prostate cancer xenografts data shows that following castration and return of the lethal castrate resistant tumor (lethal phenotype), there has been a significant increase in AR expression. These data are consistent with those of Chen et al. (2004) and Scher and Sawyers (2005) indicating that increased AR expression following castration is associated with, if not responsible for, cancer progression. Furthermore, data are also consistent with the decrease in IGF-IR expression that occurs following castration. When viewed in the light of the data with mutant receptors as well as the effects of transfection of wt AR without added DHT, it would appear that the AR alone suppresses IGF-IR expression in the absence of ligand, manifest clinically as the postcastration recurrence. However, when enough ligand is available to occupy the AR, IGF-IR expression is stimulated.
Since prostate cancer rarely develops in the absence of androgens, it is assumed that androgens are at least permissive in the transformation process. However, AR expression is necessary for the development of the normal luminal prostate epithelium. Tennant et al. (1996) suggested that maintaining a certain level of IGF-IR may be necessary for normal differentiation, whereas increased levels may be required for epithelial transformation, and decreased expression may be required for malignant progression. This concept has been corroborated by the observation that re-expression of IGF-IR in xenografts is associated with a decrease in tumor metastases and an increase in apoptosis (Plymate et al., 1997).
In the presence or possibly absence of androgens, AR translocates from the cytosol to the nucleus and functions as a transcription factor, which may be necessary or even crucial for cancer progression (Scher and Sawyers, 2005). Classically, in the absence of androgens, AR remains in the cytosol and is not active (Wu et al., 2006c). Our results are consistent with a report (Pandini et al., 2005) showing that wt AR enhanced IGF-IR transcription in prostate cells in the presence of ligand, though the mechanism described in this report was a non-genomic one. In our work we showed that impaired AR function as a result of mutation alters the ability of AR to regulate IGF-IR expression. In the absence of a functional AR, IGF-IR levels are decreased and the tumor becomes more aggressive. Our hypothesis is supported by a study (Moehren et al., 2008) which suggested that wt AR inhibits expression of the telomerase reverse transcriptase (hTERT) via inhibition of hTERT promoter activity in the presence of AR agonists, which is indicative of a protective mechanism. On the other hand, the T877A mutant not only broadens the ligand spectrum of the receptor but also abrogates this inhibitory mechanism. In addition, somatic AR mutations have been found in only 2% of patients with localized prostate cancer (Marcelli et al., 2000), but the frequency of mutations seems to increase with stage, being the highest in metastatic disease. In addition, a study by Sun et al. (2006) supports the causal link of the AR-T877A mutation to prostate cancer progression by showing that this mutation conferred increased cell growth.
The data presented here also help address the issue of decreased IGF-IR expression in prostate cancer progression in the presence of increased AR. For example, in non-castrate AD prostate cancer, AR functions in a ligand-bound state and the AR is not mutated. Therefore, as we show, AR enhances IGF-IR expression. However, as prostate cancer progresses following castration, mutations occur in the AR, splice variants in the ligand binding domain increase, and unligated AR are increased. Each of these three states decreases IGF-IR expression. Since the IGF-IR is necessary for prostate epithelial cell differentiation (Sutherland et al., 2008), it is plausible that the AR-induced down regulation of IGF-IR leads to further tumor dedifferentiation and cancer progression. However, these data do not contradict the role of IGF-IR in counteracting the effects of chemotherapeutic agents and castration induced apoptosis and its potential role as a therapeutic target (Plymate et al., 2007; Wu et al., 2006b). In summary, in this work we provide evidence that an active wt AR enhanced IGF-IR transcription in prostate cancer cells via a mechanism that involves binding to the IGF-IR promoter and requires ligand binding, while AR mutations and splice variants alter the ability of the mutated protein to regulate IGF-IR expression. Our results suggest that progression from early to advanced stage disease is associated with a decrease in IGF-IR expression which could be the result of impairment in the ability of AR to induce IGF-IR levels.
Acknowledgments
This work was performed in partial fulfillment of the requirements for a Ph.D. degree by Hagit Schayek in the Sackler Faculty of Medicine, Tel Aviv University. The authors wish to thank Dr. Joy L. Ware for providing the P69-derived prostate cell lines, and Ms. Tal Ohayon for help with manuscript preparation. This research was supported by Grant 2003341 of the United States-Israel Binational Science Foundation (to H.W. and S.R.P.) and NIH-NCI P01 CA 97186, NIH-NCI P01 CA 085859, and Veterans Affairs Research Service (to S.R.P.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akalin A, Elmore LW, Forsythe HL, Amaker BA, McCollum ED, Nelson PS, Ware JL, Holt SE. A novel mechanism for chaperone-mediated telomerase regulation during prostate cancer progression. Cancer Res. 2001;61:4791–4796. [PubMed] [Google Scholar]
- Bae VL, Jackson-Cook CK, Brothman AR, Maygarden SJ, Ware JL. Tumorigenicity of SV40 T antigen immortalized human prostate epithelial cells: association with decreased epidermal growth factor receptor (EGFR) expression. Int J Cancer. 1994;58:721–729. doi: 10.1002/ijc.2910580517. [DOI] [PubMed] [Google Scholar]
- Bae VL, Jackson-Cook CK, Maygarden SJ, Plymate SR, Chen J, Ware JL. Metastatic sublines of an SV40 large T antigen immortalized human prostate epithelial cell line. Prostate. 1998;34:275–282. doi: 10.1002/(sici)1097-0045(19980301)34:4<275::aid-pros5>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Bakin RE, Gioeli D, Sikes RA, Bissonette EA, Weber MJ. Constitutive activation of the Ras/mitogen-activated protein kinase signaling pathway promotes androgen hypersensitivity in LNCaP prostate cancer cells. Cancer Res. 2003;63:1981–1989. [PubMed] [Google Scholar]
- Chan JM, Stampfer MJ, Giovannucci E, Gann PH, Ma J, Wilkinson P, Hennekens CH, Pollak M. Plasma insulin-like growth factor-I and prostate cancer risk: a prospective study. Science. 1998;279:563–566. doi: 10.1126/science.279.5350.563. [DOI] [PubMed] [Google Scholar]
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nature Med. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- Chott A, Sun Z, Morganstern D, Pan J, Li T, Susani M, Mosberger I, Upton MP, Bubley GJ, Balk SP. Tyrosine kinases expressed in vivo by human prostate cancer bone marrow metastases and loss of type 1 insulin-like growth factor receptor. Am J Pathol. 1999;155:1271–1279. doi: 10.1016/S0002-9440(10)65229-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Peehl DM, Lamson G, Rosenfeld RG. Insulin-like growth factors (IGFs), IGF receptors, and IGF-binding proteins in primary cultures of prostate epithelial cells. J Clin Endocrinol Metab. 1991;73:401–407. doi: 10.1210/jcem-73-2-401. [DOI] [PubMed] [Google Scholar]
- Corey E, Quinn JE, Bladou F, Brown LG, Roudier MP, Brown JM, Buhler KR, Vessella RL. Establishment and characterization of osseous prostate cancer models: intra-tibial injection of human prostate cancer cells. Prostate. 2002;52:20–33. doi: 10.1002/pros.10091. [DOI] [PubMed] [Google Scholar]
- Corey E, Quinn JE, Buhler KR, Nelson PS, Macoska JA, True LD, Vessella RL. LuCaP 35: a new model of prostate cancer progression to androgen independence. Prostate. 2003;55:239–246. doi: 10.1002/pros.10198. [DOI] [PubMed] [Google Scholar]
- Culig Z, Hobisch A, Cronauer M, Radmayr C, Hittmair A, Bartsch G, Klocker H. Andogen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994;54:5474–5478. [PubMed] [Google Scholar]
- Culig Z, Hobisch A, Cronauer MV, Cato AC, Hittmair A, Radmayr C, Eberle J, Bartsch G, Klocker H. Mutant androgen receptor detected in an advanced-stage prostatic carcinoma is activated by adrenal androgens and progesterone. Mol Endocrinol. 1993;7:1541–1550. doi: 10.1210/mend.7.12.8145761. [DOI] [PubMed] [Google Scholar]
- Damon SE, Plymate SR, Carroll JM, Sprenger CC, Dechsukhum C, Ware JL, Roberts CT., Jr Transcriptional regulation of insulin-like growth factor-I receptor gene expression in prostate cancer cells. Endocrinology. 2001;142:21–27. doi: 10.1210/endo.142.1.7890. [DOI] [PubMed] [Google Scholar]
- Deutsch E, Maggiorella L, Eschwege P, Bourhis J, Soria JC, Abdulkarim B. Environmental, genetic and molecular features of prostate cancer. Lancet Oncol. 2004;5:303–313. doi: 10.1016/S1470-2045(04)01468-8. [DOI] [PubMed] [Google Scholar]
- DiGiovanni J, Kiguchi K, Frijhoff A, Wilker E, Bol DK, Beltran L, Moats S, Ramirez A, Jorcano J, Conti C. Deregulated expression of insulin-like growth factor I in prostate epithelium leads to neoplasia in transgenic mice. Proc Natl Acad Sci USA. 2000;97:3455–3460. doi: 10.1073/pnas.97.7.3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol. 2002;20:3001–3015. doi: 10.1200/JCO.2002.10.018. [DOI] [PubMed] [Google Scholar]
- Grossmann ME, Huang H, Tindall DJ. Androgen receptor signaling in androgen-refractory prostate cancer. J Natl Cancer Inst. 2001;93:1687–1697. doi: 10.1093/jnci/93.22.1687. [DOI] [PubMed] [Google Scholar]
- Han G, Buchanan G, Ittmann M, Harris JM, Yu X, Demayo FJ, Tilley WD, Greenberg NM. Mutation of the androgen receptor causes oncogenic transformation of the prostate. Proc Natl Acad Sci USA. 2005;102:1151–1156. doi: 10.1073/pnas.0408925102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellawell GO, Turner GD, Davies DR, Poulsom R, Brewster SF, Macaulay VM. Expression of the type 1 insulin-like growth factor receptor is up-regulated in primary prostate cancer and commonly persists in metastatic disease. Cancer Res. 2002;62:2942–2950. [PubMed] [Google Scholar]
- Kaplan PJ, Mohan S, Cohen P, Foster BA, Greenberg NM. The insulin-like growth factor axis and prostate cancer: lessons from the transgenic adenocarcinoma of mouse prostate (TRAMP) model. Cancer Res. 1999;59:2203–2209. [PubMed] [Google Scholar]
- Khandwala HM, McCutcheon IE, Flyvbjerg A, Friend KE. The effects of insulin-like growth factors on tumorigenesis and neoplastic growth. Endocrine Rev. 2000;21:215–244. doi: 10.1210/edrv.21.3.0399. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Chang C. Recent advances in androgen receptor action. Cell Mol Life Sci. 2003;60:1613–1622. doi: 10.1007/s00018-003-2309-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HK, Yeh S, Kang HY, Chang C. Akt suppresses androgen-induced apoptosis by phosphorylating and inhibiting androgen receptor. Proc Natl Acad Sci USA. 2001;98:7200–7205. doi: 10.1073/pnas.121173298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelli M, Ittmann M, Mariani S, Sutherland R, Nigam R, Murthy L, Zhao Y, DiConcini D, Puxeddu E, Esen A, Eastham J, Weigel NL, Lamb DJ. Androgen receptor mutations in prostate cancer. Cancer Res. 2000;60:944–949. [PubMed] [Google Scholar]
- Moehren U, Papaioannou M, Reeb CA, Grasselli A, Nanni S, Asim M, Roell D, Prade I, Farsetti A, Baniahmad A. Wild-type but not mutant androgen receptor inhibits expression of the hTERT telomerase subunit: a novel role of AR mutation for prostate cancer development. FASEB J. 2008;22:1258–1267. doi: 10.1096/fj.07-9360com. [DOI] [PubMed] [Google Scholar]
- Nickerson T, Chang F, Lorimer D, Smeekens SP, Sawyers CL, Pollak M. In vivo progression of LAPC-9 and LNCaP prostate cancer models to androgen independence is associated with increased expression of insulin-like growth factor I (IGF-I) and IGF-I receptor (IGF-IR) Cancer Res. 2001;61:6276–6280. [PubMed] [Google Scholar]
- Orio F, Terouanne B, Georget V, Lumbroso S, Avances C, Siatka C, Sultan C. Potential action of IGF-I and EGF on androgen receptor nuclear transfer and transactivation in normal and cancer human prostate cell lines. Mol Cell Endocrinol. 2002;198:105–114. doi: 10.1016/s0303-7207(02)00374-x. [DOI] [PubMed] [Google Scholar]
- Pandini G, Mineo R, Frasca F, Roberts CT, Jr, Marcelli M, Vigneri R, Belfiore A. Androgens up-regulate the insulin-like growth factor-I receptor in prostate cancer cells. Cancer Res. 2005;65:1849–1857. doi: 10.1158/0008-5472.CAN-04-1837. [DOI] [PubMed] [Google Scholar]
- Plymate S, Bae V, Madison L, Quinn L, Ware JL. Re-expression of the type I IGF receptor inhibits the malignant phenotype of simian virus 40 T antigen inmortalized human prostate epithelial cells. Endocrinology. 1997;138:1728–1735. doi: 10.1210/endo.138.4.5071. [DOI] [PubMed] [Google Scholar]
- Plymate SR, Haugk K, Coleman I, Woodke L, Vessella R, Nelson P, Montgomery RB, Ludwig DL, Wu JD. An antibody targeting the type I insulin-like growth factor receptor enhances the castration-induced response in androgen-dependent prostate cancer. Clin Cancer Res. 2007;13:6429–6439. doi: 10.1158/1078-0432.CCR-07-0648. [DOI] [PubMed] [Google Scholar]
- Plymate SR, Tennant M, Birnbaum RS, Thrasher JB, Chatta G, Ware JL. The effect on the insulin-like growth factor system in human prostate epithelial cells of immortalization and transformation by simian virus-40 T antigen. J Clin Endocrinol Metab. 1996;81:3709–3716. doi: 10.1210/jcem.81.10.8855827. [DOI] [PubMed] [Google Scholar]
- Plymate SR, Tennant MK, Culp SH, Woodke L, Marcelli M, Coleman I, Nelson PS, Carroll JM, Roberts CT, Jr, Ware JL. Androgen receptor (AR) expression in AR-negative prostate cancer cells results in differential effects of DHT and IGF-I on proliferation and AR activity between localized and metastatic tumors. Prostate. 2004;61:276–290. doi: 10.1002/pros.20099. [DOI] [PubMed] [Google Scholar]
- Roddam AW, Allen NE, Appleby P, Key TJ, Ferrucci L, Carter HB, Metter EJ, Chen C, Weiss NS, Fitzpatrick A, et al. Insulin-like growth factors, their binding proteins, and prostate cancer risk: analysis of individual patient data from 12 prospective studies. Ann Intern Med. 2008;149:461–471. doi: 10.7326/0003-4819-149-7-200810070-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan W, Powell-Braxton L, Kopchick JJ, Kleinberg DL. Evidence that insulin-like growth factor I and growth hormone are required for prostate gland development. Endocrinology. 1999;140:1984–1989. doi: 10.1210/endo.140.5.6721. [DOI] [PubMed] [Google Scholar]
- Rubinstein M, Idelman G, Plymate SR, Narla G, Friedman SL, Werner H. Transcriptional activation of the IGF-I receptor gene by the Kruppel-like factor-6 (KLF6) tumor suppressor protein: potential interactions between KLF6 and p53. Endocrinology. 2004;145:3769–3777. doi: 10.1210/en.2004-0173. [DOI] [PubMed] [Google Scholar]
- Samani AA, Yakar S, LeRoith D, Brodt P. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocrine Rev. 2007;28:20–47. doi: 10.1210/er.2006-0001. [DOI] [PubMed] [Google Scholar]
- Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253–8261. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- Schroder FH. Endocrine therapy for prostate cancer: recent developments and current status. Br J Urol. 1993;71:633–640. doi: 10.1111/j.1464-410x.1993.tb16056.x. [DOI] [PubMed] [Google Scholar]
- Shi XB, Ma AH, Xia L, Kung H, de Vere White RW. Functional analysis of 44 mutant androgen receptors from human prostate cancer. Cancer Res. 2002;62:1496–1502. [PubMed] [Google Scholar]
- Sun C, Shi Y, Xu LL, Nageswararao C, Davis LD, Segawa T, Dobi A, McLeod DG, Srivastava S. Androgen receptor mutation (T877A) promotes prostate cancer cell growth and cell survival. Oncogene. 2006;25:3905–3913. doi: 10.1038/sj.onc.1209424. [DOI] [PubMed] [Google Scholar]
- Sutherland BW, Knoblaugh SE, Kaplan-Lefko PJ, Wang F, Holzenberger M, Greenberg NM. Conditional deletion of insulin-like growth factor-I receptor in prostate epithelium. Cancer Res. 2008;68:3495–3504. doi: 10.1158/0008-5472.CAN-07-6531. [DOI] [PubMed] [Google Scholar]
- Tennant MK, Thrasher JB, Twomey PA, Drivdahl RH, Birnbaum RS, Plymate SR. Protein and mRNA for the type 1 insulin-like growth factor (IGF) receptor is decreased and IGF-II mRNA is increased in human prostate carcinoma compared to benign prostate epithelium. J Clin Endocrinol Metab. 1996;81:3774–3782. doi: 10.1210/jcem.81.10.8855837. [DOI] [PubMed] [Google Scholar]
- Veldscholte J, Ris-Stalpers C, Kuiper GG, Jenster G, Berrevoets C, Claassen E, van Rooij HC, Trapman J, Brinkmann AO, Mulder E. A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem Biophys Res Commun. 1990;173:534–540. doi: 10.1016/s0006-291x(05)80067-1. [DOI] [PubMed] [Google Scholar]
- Werner H, Adamo M, Roberts CT, Jr, LeRoith D. Molecular and cellular aspects of insulin-like growth factor action. Vitamins and Hormones. 1994;48:1–58. doi: 10.1016/s0083-6729(08)60495-1. [DOI] [PubMed] [Google Scholar]
- Werner H, Idelman G, Rubinstein M, Pattee P, Nagalla SR, Roberts CT., Jr A novel EWS-WT1 gene fusion product in desmoplastic small round cell tumor is a potent transactivator of the insulin-like growth factor-I receptor (IGF-IR) gene. Cancer Lett. 2007;247:84–90. doi: 10.1016/j.canlet.2006.03.027. [DOI] [PubMed] [Google Scholar]
- Werner H, Maor S. The insulin-like growth factor-I receptor gene: a downstream target for oncogene and tumor suppressor action. Trends Endocrinol Metab. 2006;17:236–242. doi: 10.1016/j.tem.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Wu JD, Haugk K, Coleman I, Woodke L, Vessella R, Nelson P, Montgomery RB, Ludwig DL, Plymate SR. Combined in vivo effect of A12, a type 1 insulin-like growth factor receptor antibody, and docetaxel against prostate cancer tumors. Clin Cancer Res. 2006a;12:6153–6160. doi: 10.1158/1078-0432.CCR-06-0443. [DOI] [PubMed] [Google Scholar]
- Wu JD, Haugk K, Woodke L, Nelson P, Coleman I, Plymate SR. Interaction of IGF signaling and the androgen receptor in prostate cancer progression. J Cell Biochem. 2006b;99:392–401. doi: 10.1002/jcb.20929. [DOI] [PubMed] [Google Scholar]
- Wu JD, Odman A, Higgins LM, Haugk K, Vessella R, Ludwig DL, Plymate SR. In vivo effects of the human type I insulin-like growth factor receptor antibody A12 on androgen-dependent and androgen-independent xenograft human prostate tumors. Clin Cancer Res. 2005;11:3065–3074. doi: 10.1158/1078-0432.CCR-04-1586. [DOI] [PubMed] [Google Scholar]