

Abstract
SLE is a chronic autoimmune inflammatory disorder that predominantly affects young women. Based on this observation, it has been speculated that sex steroids, particularly estrogens, contribute to SLE disease progression. Young women with SLE are at an increased risk for the development of hypertension yet the reasons for this are unclear. One potential mechanism for the increased risk of hypertension during SLE is the chronic inflammation caused by immune complex mediated tissue injury. Estrogens are known to have an immunomodulatory role that can lead to the production of characteristic autoantibodies important for immune complex formation. Therefore, it is conceivable that during SLE estrogens contribute to tissue injury, increased inflammation and hypertension. This brief review discusses the increased risk for hypertension during SLE, the role of estrogens in immune system function, evidence for estrogens in SLE, and a possible link between estrogens and SLE hypertension.
Introduction
Systemic lupus erythematosus (SLE) is a chronic, multifaceted inflammatory autoimmune disease of unknown etiology affecting an estimated 1.5 million people in the United States. The interaction between environmental factors and susceptibility genes, coupled with a loss of tolerance, trigger abnormal immune system activation. The hallmark of the disease is the production of autoantibodies leading to immune complex formation that can be deposited in any organ system and promote a generalized inflammatory response. The deposition of these immune complexes and subsequent inflammation promotes diverse clinical manifestations. These include non-specific symptoms such as fever, malaise, anorexia, weight loss, and fatigue, as well as disease-specific findings including the common malar rash, discoid rash, non-erosive polyarthritis, central nervous system involvement and hematologic disturbances such as anemia and thrombocytopenia. The kidneys are also prominently affected in the form of immune complex glomerulonephritis which occurs in approximately 50% of cases and the leading cause of mortality in patients with SLE is cardiovascular disease [1;2]. Hypertension is a major risk factor for cardiovascular disease and is highly prevalent in patients with SLE; however, the reasons for this have not been widely examined. Our laboratory has been focused on trying to understand mechanisms that promote hypertension during SLE with an emphasis on the kidney because of its prominent role in the long term regulation of blood pressure.
Although the exact underlying cause of SLE has not been fully elucidated, it is clear that SLE predominantly affects young women. The most commonly cited statistic is that the ratio of women to men with SLE is 9:1 during the time period ranging from menarche until the onset of menopause. Prior to the onset of puberty and after menopause, the ratio of women to men is markedly blunted [3]. The strong preference for women during this time suggests that sex steroids, specifically estrogens, play an important role in the pathology of SLE. This is of particular interest to us because the risk for cardiovascular disease, renal disease, and hypertension are dramatically increased in women with SLE at an age where they are otherwise thought to be protected. Therefore, this raises the question as to whether sex steroids such as estrogen play a causative role in the development of hypertension and renal disease during SLE. The purpose of this brief review is to discuss the increased risk for hypertension and cardiovascular disease during SLE, the role of estrogens in immune system function and its possible contribution to the progression of SLE, and to raise questions related to the potential role that estrogens could have in the development of hypertension during SLE.
Women with SLE are at increased CV risk
Women with SLE have a markedly reduced life expectancy and 10% of patients will die within five years after the initial diagnosis [4]. A bimodal pattern of mortality has been described in SLE patients with short-term mortality primarily due to lupus and infection and long-term mortality related to cardiovascular and vascular complications [1;2]. Women with SLE have an increased risk for virtually all cardiovascular diseases including atherosclerosis, stroke, and myocardial infarction [5–7]. This is supported by a study that compared the risk for cardiovascular events (myocardial infarction and angina pectoris) in a group of nearly 500 women with SLE from the University of Pittsburgh Medical Center to the cardiovascular risk in age matched women from the Framingham Offspring Study and found a more than 50-fold higher risk in women with SLE [8]. Importantly, hypertension is a major risk factor for cardiovascular disease and is highly prevalent in women with SLE.
The prevalence of hypertension is increased during SLE
In normal healthy young women (ages 15–44 years), the prevalence of hypertension is low (3–14%) [9]. In contrast, the prevalence of hypertension in women with SLE can be considerably higher ranging from 35–74% depending on the cohort studied [10–13]. The increased risk for hypertension during SLE can be erroneously dismissed as resulting from nephron loss due to glomerulonephritis. However, there are reports suggesting that lupus nephritis and hypertension can occur independently of each other. For example, in one study approximately one third of the patients had glomerulonephritis but no hypertension [14]. In another, 38% were hypertensive with no signs of glomerulonephritis [15]. These data suggest that nephron loss is likely not the only factor contributing to the increased prevalence of hypertension during SLE. To date, few studies have examined mechanisms of hypertension during SLE.
A role for inflammation in hypertension
One potential contributing factor to the high prevalence of hypertension during SLE is the increased level of inflammatory cytokines. It is well known that a number of circulating cytokines such as tumor necrosis factor alpha (TNF-α), interferon gamma (IFN-γ) and interleukin 6 (IL-6) are implicated in the pathogenesis of SLE [16]. Similarly, studies report a direct correlation between circulating inflammatory cytokines (TNF-α, IL-6) and blood pressure in patients with essential hypertension or in healthy men [17;18]. Experimental animal models have also been utilized to confirm the importance of chronic inflammation in several forms of hypertension including angiotensin II dependent and salt sensitive hypertension [19;20]. Therefore, it seems reasonable to predict that the increased inflammatory mediators during SLE may be an important mechanism leading to hypertension. Our laboratory is currently investigating the role for renal inflammation in the progression of SLE hypertension. We recently reported that treating a mouse model of SLE with a thiazolidinedione (rosiglitazone) reduces the number of inflammatory cells in the kidneys in part through reducing the expression of chemokines (Figure 1) [21]. Importantly, the reduced renal inflammation correlated directly with a reduction in blood pressure. Although estrogens are generally thought of as protective against cardiovascular disease, the immunomodulatory actions of estrogen could theoretically contribute to the increased risk for hypertension and cardiovascular disease found in young women with SLE.
Figure 1.
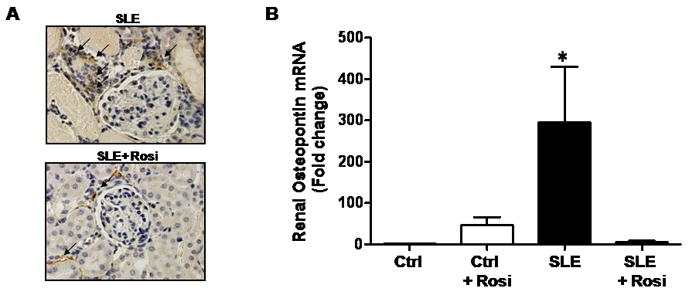
Treatment of a mouse model of SLE with rosiglitazone reduces renal inflammation. (A) Previous work from our laboratory show that macrophage and monocyte infiltration (noted by black arrows) is reduced in the kidneys of the NZBWF1 mouse model of SLE after treatment with rosiglitazone (image taken at 40× magnification) (B) The expression of the chemokine, osteopontin, is reduced in the renal cortex of NZBWF1 mice treated with rosiglitazone. (figures from reference 21)
A role for estrogen in immune system function
Estrogens are well known modulators of immune system function [22;23]; however, their role is complex and cannot be reviewed comprehensively here. This section will provide a brief summary of the effect of estrogen in three aspects of immune system function (inflammatory cytokines, T cell function, and B cell function) that have implications for the development of SLE.
With regard to cytokine production, estrogens are thought to suppress T helper 1 (Th1) and promote T helper 2 (Th2) cytokine profiles. The Th1 response typically leads to the production of inflammatory cytokines such as TNF-α and IFN-γ whereas the Th2 lymphocytes typically produce cytokines such as IL-4, IL-6, IL-10 that are important for promoting B-cell activity to increase antibody production (humoral immunity). The concept of an estrogen mediated shift to a Th2 response is exemplified during pregnancy since estrogen levels continually rise throughout gestation [24]. The exact mechanism by which estrogen promotes the Th2 response and suppresses Th1 cytokines remains unclear with some evidence suggesting an indirect effect of estrogen on T lymphocytes through its actions in dendritic cells [25;26]. In addition, this Th1/Th2 paradigm is oversimplified as it applies to SLE given that both Th1 and Th2 cytokines are implicated in disease progression [27]. For example, estrogens can increase the production of Th1 inflammatory cytokines indirectly through the stimulation of prolactin release [28]. Overall, the data suggest that estrogen tends to promote humoral immunity through Th2 cytokines, but can also increase the production of Th1 cytokines.
In addition to promoting cytokine release from T cells, estrogens can have direct effects on T cell maturation. Estrogen administration induces thymic atrophy by causing apoptosis in immature T cells [29]. The loss of immature T cells enriches the thymocyte population for mature CD5+ T lymphocytes [30] capable of promoting B cell mediated immunoglobulin class switching and therefore the production of antibodies. Maturation of CD4+ T helper cells (important for B cell function) is also enhanced in females [23;31] and recent evidence suggests that this may occur through estrogen mediated activation of the calcineurin signaling pathway. Interestingly, T cells from patients with SLE reportedly exhibit an enhanced calcineurin response to estrogen stimulation [32;33]. Taken together, the net effect of estrogens on T cell regulation is to promote humoral immunity that is prominent in SLE.
Finally, estrogens have direct effects on B cell production and function. While estrogens can attenuate the production of new B cells, they also promote B cell differentiation and an increased number of mature B cells capable of producing antibodies (reviewed in [34]), a response that can be blocked by tamoxifen. Several lines of evidence support these effects of estrogens. For example, 17β-estradiol permits B cells to escape negative selection by reducing apoptosis and also promotes autoantibody production from B cells [35]. The increased production of autoantibodies stems from an estradiol induced polyclonal expansion of B cells that are capable of producing IgG to double stranded DNA that are pathogenic for SLE. In summary, the immunomodultory role of estrogen can promote the production of autoantibodies critical for immune complex formation and subsequent tissue injury and inflammation.
Evidence for a role of estrogens in SLE
Although estrogens are known to promote antibody production, and SLE predominantly affects women, definitive proof of estrogen’s involvement in SLE progression remains elusive. In fact, whether serum levels of estrogens are actually increased in patients with SLE is not entirely clear. Some evidence suggests an altered aromatase-mediated production of estrogens in women with SLE [36]; however, evidence for increased estrogens during SLE has been conflicting. This is highlighted in a review/meta-analysis of 20 studies [37] that found populations of women with SLE had either increased, decreased, or normal physiological levels of circulating 17-β estradiol. In men with SLE there are similar equivocal findings related to serum estradiol levels [37]. On balance, the serum levels of estrogens in women with SLE appear to be in the normal physiological range; however, this does not exclude a possible role for estrogens in SLE. One potentially important factor to consider is the ratio estrogen to testosterone as this has been implicated in the pathogenesis of autoimmune diseases [38;39]. An altered ratio of estradiol to testosterone has also been considered as a potentially important factor in the development of postmenopausal hypertension [40]. Another interesting consideration is that, although serum levels of estradiol may not be different; there may be an increased conversion of 17-β estradiol to more feminizing estrogens like 16α-hydroyestrone in women with SLE [41] which has potent mitogenic activities that could increase inflammatory cell numbers [42].
Several genetic, physiologic and pharmacologic lines of evidence have been used to speculate about the role of estrogens in the progression of SLE; however, none of these definitively prove a link. One example is related to men with the rare genetic disorder Klinefelter’s Syndrome. Men with Klinefelter’s syndrome carry an extra X chromosome (47, XXY) and have increased serum levels of estradiol [43] as well as a 14-fold higher risk of developing SLE than genetically normal men (46,XY) [44]. In fact, men with Klinefelter’s have a similar risk for developing SLE as in women. Alternatively, it is possible that the presence of the extra X chromosome may simply increase SLE susceptibility by a gene dosage effect. This is partially supported by evidence in women with Turner’s syndrome who lack one X of their chromosomes (45,XO) where the prevalence of SLE in these women is low [45]. These patients also have low levels of estrogens and high levels of testosterone making it difficult to definitively know the cause for the reduced risk of SLE.
One example of a normal physiological situation that has been used to bolster the argument for estrogens in SLE is pregnancy. Estrogen levels continually rise during the normal 40 week gestation and as a result some women with SLE have been discouraged from becoming pregnant. However, more recent studies report that the number and severity of flares are not increased during pregnancy in women with SLE [46;47]. One factor that may influence SLE flares is the timing of the pregnancy in relation to disease activity. For example, low complement levels, active renal flares, and not being in remission at the time of conception are predictive of worse pregnancy outcomes in women with SLE [48]. This may account for the increased risk of pregnancy related hypertension, preeclampsia (hypertension with albuminuria), premature birth and fetal growth restriction in women with SLE.
The use of oral contraceptives in women with SLE has also been largely avoided because of the fear of possible complications related to estrogens. However, it is now recognized that oral contraceptives can be effectively and safely used in women with SLE, specifically in women who have stable or improving SLE [49]. Patients with recently diagnosed SLE, active lupus nephritis, or high titers of antiphospholipid antibodies (present in approximately one third of patients with SLE) are often excluded from these studies, and therefore the risk of taking estrogen based oral contraceptives during SLE cannot be generalized to all populations. Similar to oral contraceptives, hormone replacement therapy (HRT) has been considered a risk factor for women with SLE; however, the scientific evidence does not seem to support this. For example, one study reported no difference in the rate of severe lupus flares between women who received ethynilestradiol/norethindine and those who received placebo [50]. Similar conclusions have been reported by other investigators examining the role of HRT in SLE [51;52]. In a recent study of post-menopausal women, it was reported that HRT did not increase the cardiovascular risk in women with SLE compared to women with SLE who had never taken hormone replacement [53].
The biological effects of estrogen and estrogen metabolites are mediated largely via interactions with two different estrogen receptors (ERα and ERβ). Few studies have examined the role of selective estrogen receptor modulators (SERM) in women with SLE. One small case study involving only 11 patients reported that treatment with the SERM tamoxifen did not improve disease activity or laboratory indices of SLE activity [54]. In another study, treatment with the SERM raloxifene in post-menopausal women with SLE was found to have beneficial effects to prevent bone loss but had no apparent effect to reduce SLE disease activity [55].
Finally, menstrual disturbances such as irregular cycling are associated with SLE disease activity. However, these are not attributable to altered serum estradiol levels when compared to normal cycling women with SLE [56]. There are also functional polymorphisms (Asp327Asn) in sex hormone-binding globulin that associate with an imbalance between estrogens and androgens during SLE [57] as well as polymorphisms in mitochondrial DNA (mtDNA) that associate with SLE. The mtDNA polymporphisms exhibit a sexual dimorphism, with the single nucleotide polymorphisms nt16189C and nt13708A associated with increased SLE disease activity in women and men, respectively [58].
Collectively, the data in humans does not appear to definitively prove or refute a role for estrogens in the development of SLE. However, much of the ambiguity can likely be attributed to the variable nature of populations studied and the specific inclusion criteria used in the studies that may focus mainly on women in remission or with stable SLE activity. Because of the variability in data from humans, the use of experimental animal models to study the pathogenesis of SLE and SLE hypertension becomes increasingly important.
The utility of animal models in investigating SLE hypertension
Our understanding of the potential role for estrogen in the progression of SLE has been greatly enhanced by the use of experimental animal models. There are several good genetic models of SLE in mice. The most commonly utilized are the NZBWF1 and the MRL/lpr, although there are several other models available. NZBWF1 represent the F1 offspring from a cross between New Zealand Black (NZB) and New Zealand White (NZW) mice. These mice produce the characteristic anti-double stranded DNA antibodies and develop immune complex glomerulonephritis similar to what is observed in humans with SLE. In addition, we recently demonstrated in conscious freely moving mice using direct methods (radio telemetry and indwelling catheters) that female NZBWF1 mice are hypertensive (Figure 2) [59;60]. Interestingly, there is no single genetic mutation that promotes SLE in these mice, thus mimicking the complex genetic contributions to SLE that are prevalent in humans. Moreover, female NZBWF1 mice are more prominently affected with most dying before one year of age (245 days) while the males live to an average of 406 days (The Jackson Laboratory, jaxmice.jax.org). This makes the NZBWF1 model useful for investigating the importance of estrogens in SLE hypertension. A second established and widely used model, the MRL/lpr, has a mutation in the lymphoproliferative gene (Fas Ligand) that accelerates features of SLE including autoantibody production, immune complex glomerulonephritis and arthritis. The average life span of male MRL/lpr is only moderately longer than females (120 vs 154 days) (The Jackson Laboratory, jaxmice.jax.org). In addition, it is not clear whether and to what extent these mice develop hypertension. Both of these models have been extensively studied to understand SLE and several studies have examined the possible role of estrogens.
Figure 2.
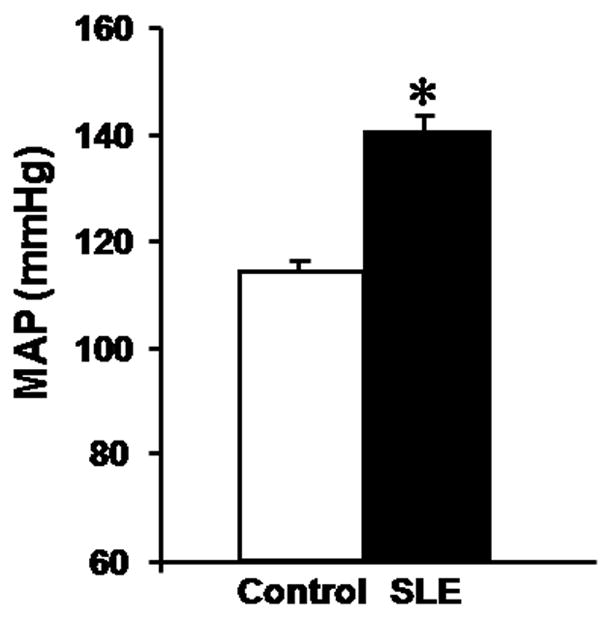
The NZBWF1 mouse model of SLE develops hypertension. The control used is the NZW/LacJ mouse. Pressure was measured in conscious freely moving mice at 36 weeks of age. (figure from reference 59)
A recent study elegantly tested the effect of a genetic deletion of the ERα receptor in NZBWF1 mice [61]. ERα NZBWF1 knockout mice exhibited less glomerulonephritis, reduced urinary albumin, and a delayed development of anti-double stranded DNA antibodies. ERα deficient NZBWF1 male mouse also presented increased survival and reduced anti-double stranded DNA antibodies. Importantly, survival was increased the ERα knockout NZBWF1. These data appear to unambiguously support a role for estrogens in the development of SLE that is mediated by the ERα receptor. In agreement with these findings, another study showed that ERα deficient NZM2410 mice (a less commonly utilized mouse model of SLE that originated from NZBWF1 mice in genetic crosses) exhibit reduced proteinuria and glomerulosclerosis and increased survival compared with wild-type mice [62]. The decreased renal damage occurred even as serum levels of anti-double stranded DNA antibodies, estrogen and testosterone were increased, thus further emphasizing the importance of the ERα receptor in murine lupus.
As an alternative to engineering knockout mice, ovariectomy, has been used in both NZBWF1 and MRL/lpr female mice as a means to test the importance of estrogen in SLE progression. Ovariectomy at 6–8 weeks of age delays the onset of renal disease, production of autoantibodies and mortality in NZBWF1 mice [63]. In another study, estradiol infusion into ovariectomized NZBWF1 increased the number of activated CD4+ T cells when compared to ovariectomized controls [64]. The limited studies that investigate the role of estrogens in MRL/lpr mice show that ovariectomy actually exacerbates autoimmune arthritis and that supplementation with estrogen protects against this damage [65]. Similar results were achieved when MRL/mp mice (control strain for lpr) where ovariectomized and an increase in parotid, lacrimal and submandibular gland lesions were observed [66]. The effects of ovariectomy on renal pathology in MRL/lpr has not been addressed to our knowledge and in neither model has the effects of ovariectomy on blood pressure been evaluated. It is also interesting to note that ovariectomy appears only to protect NZBWF1 given that SLE has a more pronounced effect in female NZBWF1 compared with males whereas life expectancy is similar in male and female MRL/lpr. Therefore, careful consideration should be given when selecting models to examine the possible role of estrogens in SLE.
Experiments in animal models of SLE have also provided additional insight as to the possible role for estrogen receptors through the use of SERMs. Tamoxifen decreases proteinuria and increases survival in female NZBWF1 mice with no changes in serum levels of anti-single stranded DNA and anti-double stranded DNA antibodies [67]. Female NZBWF1 mice treated with tamoxifen also have decreased circulating levels of B cells and CD5+ B cells as well as reduced serum levels of soluble tumor necrosis factor (TNF) receptor I and II molecules. When MRL/lpr mice were treated with an analog of the SERM raloxifene (LY139478), survival increased and glomerular injury was reduced. In the same study MRL/lpr mice treated with 17a-ethinylestradiol (EE2, an orally active estrogen) exhibited reduced glomerular injury with no change in mortality. Neither treatment reduced the production of autoantibodies [68].
One common underlying characteristic of the studies described here is that the inhibition of estrogen receptors, removal of ovaries, or genetic deletion of the estrogen receptors were all initiated at birth (knockout mice) or at the onset of sexual maturity in mice (6–8 weeks of age). This may also be an important consideration when designing experiments to test the effects of estrogens on SLE and SLE hypertension given evidence that hormone manipulation after sexual maturity has been reached may have minimal impact on SLE [69]. Therefore, one explanation for the conflicting data related to the role for estrogens in human SLE may be that the affected women are more susceptible as a result of sex hormone-induced imprinting of the immune system early in life [70]. Indeed, it will be important to examine the effect of estrogen blockade on the cardiovascular system in adult experimental mouse models of SLE to compare with studies using lifetime inhibition of this system.
Other hormones implicated in SLE pathogenesis
While the primary focus of this review has been on a possible role for estrogens in SLE and SLE hypertension, there are other hormones that may play a role in the pathogenesis of this disease. Dehydroepiandrostinedione (DHEA) is a weak androgen and the most abundant circulating androgen. Reduced levels of DHEA have been implicated in the progression of SLE [71] and there have been reports that DHEA supplementation can cause modest improvements in the quality of life for patients with SLE [72]. Another hormone that may be important to consider is prolactin. Prolactin is a small peptide hormone produced by the adenohypophysis that is commonly known for its role to promote lactation. Prolactin, which is stimulated by estrogens, is permissive for autoreactive B cells [73] and its activity is associated with worsening of SLE in the post-partum period [28;74]. The potential role for prolactin in murine lupus was evaluated in NZBWF1 mice. Mice were ovariectomized and supplemented with estradiol and/or prolactin in the presence or absence of bromocriptine, an inhibitor of prolactin. The mice with high levels of estradiol but low levels of prolactin had improved survival [75]. These data support an important contribution of prolactin to SLE progression in these mice. Interestingly, prolactin has also been implicated in various forms of hypertension. For example, men with essential hypertension have higher plasma prolactin concentration compared to normotensive men, and oral administration of bromocriptine, a dopaminergic agonist, lowers blood pressure in these patients [76]. Additionally, preeclampsia is characterized by increased urinary prolactin and urinary levels of this hormone correlate with the severity of the disease [77]. Finally, New Zealand rats treated with bromocriptine have significantly lower blood pressure compared to vehicle-treated animals, suggesting that prolactin may play a role in the pathogenesis of hypertension in this animal model [78].
Perspectives and questions
A major risk factor for developing SLE is the female sex with nine out of every ten cases being in young women. For this reason sex steroids have been implicated as a causative or permissive factor in disease progression. Although this association between SLE and the female sex is well established, data in humans has not yet definitively proven a mechanistic link between estrogen and SLE. This is likely due to the highly variable nature of SLE progression, as well as the diverse populations studied. Animal models of SLE, particularly murine, have been extremely valuable in delineating a role for estrogens in SLE progression. However, it is clear that even among different murine models there are variable roles for estrogens in the progression of SLE and that experimental design can substantially alter the outcome of the study and therefore interpretations of estrogen’s importance. Despite, some of the ambiguity, it seems very likely that estrogens play an important role in SLE progression, particularly given their known immunomodulatory effects to increase autoantibody production.
At present there is no consensus mechanism for the dramatically increased risk of hypertension in patients with SLE. Several lines of evidence now support an important role for chronic inflammation and inflammatory cytokines in numerous forms of hypertension. We have been investigating the importance of chronic inflammation during SLE is an important mechanism that leads to renal injury, impaired renal function and ultimately hypertension. We hypothesize, therefore, that one mechanism by which women are more susceptible to hypertension through SLE is via estrogen mediated increases in autoantibody production. Increased autoantibody production promotes immune complex formation and deposition that promotes a local inflammatory response in the kidney to promote hypertension. This is a hypothesis that can be experimentally tested using murine models of SLE and could provide important insight into the progression of SLE hypertension.
Acknowledgments
M.V-P. is the recipient of an American Heart Association Greater Southeast Affiliate Postdoctoral Fellowship (2260874). M.J.R. is supported by the National Institutes of Health, National Heart Lung and Blood Institute (HL085907, HL085907S3, HL092284).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Bernatsky S, Boivin JF, Joseph L, et al. Mortality in systemic lupus erythematosus. Arthritis Rheum. 2006 Aug;54(8):2550–7. doi: 10.1002/art.21955. [DOI] [PubMed] [Google Scholar]
- 2.Urowitz MB, Bookman AA, Koehler BE, Gordon DA, Smythe HA, Ogryzlo MA. The bimodal mortality pattern of systemic lupus erythematosus. Am J Med. 1976 Feb;60(2):221–5. doi: 10.1016/0002-9343(76)90431-9. [DOI] [PubMed] [Google Scholar]
- 3.Tucker LB, Menon S, Schaller JG, Isenberg DA. Adult- and childhood-onset systemic lupus erythematosus: a comparison of onset, clinical features, serology, and outcome. Br J Rheumatol. 1995 Sep;34(9):866–72. doi: 10.1093/rheumatology/34.9.866. [DOI] [PubMed] [Google Scholar]
- 4.Smolen JS. Therapy of systemic lupus erythematosus: a look into the future. Arthritis Res. 2002;4 (Suppl 3):S25–S30. doi: 10.1186/ar579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kitagawa Y, Gotoh F, Koto A, Okayasu H. Stroke in systemic lupus erythematosus. Stroke. 1990 Nov;21(11):1533–9. doi: 10.1161/01.str.21.11.1533. [DOI] [PubMed] [Google Scholar]
- 6.Riboldi P, Gerosa M, Luzzana C, Catelli L. Cardiac involvement in systemic autoimmune diseases. Clin Rev Allergy Immunol. 2002 Dec;23(3):247–61. doi: 10.1385/CRIAI:23:3:247. [DOI] [PubMed] [Google Scholar]
- 7.Thomas GN, Tam LS, Tomlinson B, Li EK. Accelerated atherosclerosis in patients with systemic lupus erythematosus: a review of the causes and possible prevention. Hong Kong Med J. 2002 Feb;8(1):26–32. [PubMed] [Google Scholar]
- 8.Manzi S, Meilahn EN, Rairie JE, et al. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: comparison with the Framingham Study. Am J Epidemiol. 1997 Mar 1;145(5):408–15. doi: 10.1093/oxfordjournals.aje.a009122. [DOI] [PubMed] [Google Scholar]
- 9.Health, United States. 2007 With Chartbook on Trends in Health of Americans. Hyattsville, MD: National Center for Health Statistics; 2007. [PubMed] [Google Scholar]
- 10.Budman DR, Steinberg AD. Hypertension and renal disease in systemic lupus erythematosus. Arch Intern Med. 1976 Sep;136(9):1003–7. [PubMed] [Google Scholar]
- 11.Doria A, Shoenfeld Y, Wu R, et al. Risk factors for subclinical atherosclerosis in a prospective cohort of patients with systemic lupus erythematosus. Ann Rheum Dis. 2003 Nov;62(11):1071–7. doi: 10.1136/ard.62.11.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petri M. Detection of coronary artery disease and the role of traditional risk factors in the Hopkins Lupus Cohort. Lupus. 2000;9(3):170–5. doi: 10.1191/096120300678828226. [DOI] [PubMed] [Google Scholar]
- 13.Selzer F, Sutton-Tyrrell K, Fitzgerald S, Tracy R, Kuller L, Manzi S. Vascular stiffness in women with systemic lupus erythematosus. Hypertension. 2001 Apr;37(4):1075–82. doi: 10.1161/01.hyp.37.4.1075. [DOI] [PubMed] [Google Scholar]
- 14.Petrin J, Rozman B, Dolenc P, et al. The dissociation of arterial hypertension and lupus glomerulonephritis in systemic lupus erythematosus. Blood Press. 1993 Jun;2(2):108–12. doi: 10.3109/08037059309077537. [DOI] [PubMed] [Google Scholar]
- 15.Ward MM, Studenski S. Clinical prognostic factors in lupus nephritis. The importance of hypertension and smoking. Arch Intern Med. 1992 Oct;152(10):2082–8. [PubMed] [Google Scholar]
- 16.Kelley VR, Wuthrich RP. Cytokines in the pathogenesis of systemic lupus erythematosus. Semin Nephrol. 1999 Jan;19(1):57–66. [PubMed] [Google Scholar]
- 17.Bautista LE, Vera LM, Arenas IA, Gamarra G. Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-alpha) and essential hypertension. J Hum Hypertens. 2005 Feb;19(2):149–54. doi: 10.1038/sj.jhh.1001785. [DOI] [PubMed] [Google Scholar]
- 18.Chae CU, Lee RT, Rifai N, Ridker PM. Blood pressure and inflammation in apparently healthy men. Hypertension. 2001 Sep;38(3):399–403. doi: 10.1161/01.hyp.38.3.399. [DOI] [PubMed] [Google Scholar]
- 19.Guzik TJ, Hoch NE, Brown KA, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007 Oct 1;204(10):2449–60. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mattson DL, James L, Berdan EA, Meister CJ. Immune suppression attenuates hypertension and renal disease in the Dahl salt-sensitive rat. Hypertension. 2006 Jul;48(1):149–56. doi: 10.1161/01.HYP.0000228320.23697.29. [DOI] [PubMed] [Google Scholar]
- 21.Venegas-Pont M, Sartori-Valinotti JC, Maric C, et al. Rosiglitazone decreases blood pressure and renal injury in a female mouse model of systemic lupus erythematosus. Am J Physiol Regul Integr Comp Physiol. 2009 Apr;296(4):R1282–R1289. doi: 10.1152/ajpregu.90992.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kovacs EJ, Messingham KA, Gregory MS. Estrogen regulation of immune responses after injury. Mol Cell Endocrinol. 2002 Jul 31;193(1–2):129–35. doi: 10.1016/s0303-7207(02)00106-5. [DOI] [PubMed] [Google Scholar]
- 23.Lang TJ. Estrogen as an immunomodulator. Clin Immunol. 2004 Dec;113(3):224–30. doi: 10.1016/j.clim.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 24.Doria A, Iaccarino L, Sarzi-Puttini P, et al. Estrogens in pregnancy and systemic lupus erythematosus. Ann N Y Acad Sci. 2006 Jun;:1069, 247–56. doi: 10.1196/annals.1351.022. [DOI] [PubMed] [Google Scholar]
- 25.Uemura Y, Liu TY, Narita Y, Suzuki M, Matsushita S. 17 Beta-estradiol (E2) plus tumor necrosis factor-alpha induces a distorted maturation of human monocyte-derived dendritic cells and promotes their capacity to initiate T-helper 2 responses. Hum Immunol. 2008 Mar;69(3):149–57. doi: 10.1016/j.humimm.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 26.Salem ML. Estrogen, a double-edged sword: modulation of TH1- and TH2-mediated inflammations by differential regulation of TH1/TH2 cytokine production. Curr Drug Targets Inflamm Allergy. 2004 Mar;3(1):97–104. doi: 10.2174/1568010043483944. [DOI] [PubMed] [Google Scholar]
- 27.Druet P, Sheela R, Pelletier L. Th1 and Th2 cells in autoimmunity. Clin Exp Immunol. 1995 Jul;101(Suppl 1):9–12. doi: 10.1111/j.1365-2249.1995.tb06153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McMurray RW. Estrogen, prolactin, and autoimmunity: actions and interactions. Int Immunopharmacol. 2001 Jun;1(6):995–1008. doi: 10.1016/s1567-5769(01)00045-5. [DOI] [PubMed] [Google Scholar]
- 29.Yao G, Hou Y. Thymic atrophy via estrogen-induced apoptosis is related to Fas/FasL pathway. Int Immunopharmacol. 2004 Feb;4(2):213–21. doi: 10.1016/j.intimp.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 30.Screpanti I, Meco D, Morrone S, Gulino A, Mathieson BJ, Frati L. In vivo modulation of the distribution of thymocyte subsets: effects of estrogen on the expression of different T cell receptor V beta gene families in CD4-, CD8- thymocytes. Cell Immunol. 1991 May;134(2):414–26. doi: 10.1016/0008-8749(91)90314-2. [DOI] [PubMed] [Google Scholar]
- 31.Lang TJ, Nguyen P, Papadimitriou JC, Via CS. Increased severity of murine lupus in female mice is due to enhanced expansion of pathogenic T cells. J Immunol. 2003 Dec 1;171(11):5795–801. doi: 10.4049/jimmunol.171.11.5795. [DOI] [PubMed] [Google Scholar]
- 32.Rider V, Foster RT, Evans M, Suenaga R, Abdou NI. Gender differences in autoimmune diseases: estrogen increases calcineurin expression in systemic lupus erythematosus. Clin Immunol Immunopathol. 1998 Nov;89(2):171–80. doi: 10.1006/clin.1998.4604. [DOI] [PubMed] [Google Scholar]
- 33.Rider V, Jones SR, Evans M, Abdou NI. Molecular mechanisms involved in the estrogen-dependent regulation of calcineurin in systemic lupus erythematosus T cells. Clin Immunol. 2000 May;95(2):124–34. doi: 10.1006/clim.2000.4844. [DOI] [PubMed] [Google Scholar]
- 34.Carlsten H. Immune responses and bone loss: the estrogen connection. Immunol Rev. 2005 Dec;208:194–206. doi: 10.1111/j.0105-2896.2005.00326.x. [DOI] [PubMed] [Google Scholar]
- 35.Grimaldi CM, Michael DJ, Diamond B. Cutting edge: expansion and activation of a population of autoreactive marginal zone B cells in a model of estrogen-induced lupus. J Immunol. 2001 Aug 15;167(4):1886–90. doi: 10.4049/jimmunol.167.4.1886. [DOI] [PubMed] [Google Scholar]
- 36.Folomeev M, Dougados M, Beaune J, et al. Plasma sex hormones and aromatase activity in tissues of patients with systemic lupus erythematosus. Lupus. 1992 May;1(3):191–5. doi: 10.1177/096120339200100312. [DOI] [PubMed] [Google Scholar]
- 37.McMurray RW, May W. Sex hormones and systemic lupus erythematosus: review and meta-analysis. Arthritis Rheum. 2003 Aug;48(8):2100–10. doi: 10.1002/art.11105. [DOI] [PubMed] [Google Scholar]
- 38.Lahita RG, Bradlow HL, Ginzler E, Pang S, New M. Low plasma androgens in women with systemic lupus erythematosus. Arthritis Rheum. 1987 Mar;30(3):241–8. doi: 10.1002/art.1780300301. [DOI] [PubMed] [Google Scholar]
- 39.Cutolo M, Capellino S, Sulli A, et al. Estrogens and autoimmune diseases. Ann N Y Acad Sci. 2006 Nov;1089:538–47. doi: 10.1196/annals.1386.043. [DOI] [PubMed] [Google Scholar]
- 40.Reckelhoff JF, Fortepiani LA. Novel mechanisms responsible for postmenopausal hypertension. Hypertension. 2004 May;43(5):918–23. doi: 10.1161/01.HYP.0000124670.03674.15. [DOI] [PubMed] [Google Scholar]
- 41.Weidler C, Harle P, Schedel J, Schmidt M, Scholmerich J, Straub RH. Patients with rheumatoid arthritis and systemic lupus erythematosus have increased renal excretion of mitogenic estrogens in relation to endogenous antiestrogens. J Rheumatol. 2004 Mar;31(3):489–94. [PubMed] [Google Scholar]
- 42.Cutolo M, Sulli A, Capellino S, et al. Sex hormones influence on the immune system: basic and clinical aspects in autoimmunity. Lupus. 2004;13(9):635–8. doi: 10.1191/0961203304lu1094oa. [DOI] [PubMed] [Google Scholar]
- 43.Lanfranco F, Kamischke A, Zitzmann M, Nieschlag E. Klinefelter’s syndrome. Lancet. 2004 Jul 17;364(9430):273–83. doi: 10.1016/S0140-6736(04)16678-6. [DOI] [PubMed] [Google Scholar]
- 44.Scofield RH, Bruner GR, Namjou B, et al. Klinefelter’s syndrome (47,XXY) in male systemic lupus erythematosus patients: support for the notion of a gene-dose effect from the X chromosome. Arthritis Rheum. 2008 Aug;58(8):2511–7. doi: 10.1002/art.23701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pan HF, Li WX, Yuan H, et al. Susceptibility to systemic lupus erythematosus may be related to gene dosage effect of the X chromosome. Med Hypotheses. 2009 Jan;72(1):104–5. doi: 10.1016/j.mehy.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 46.Tandon A, Ibanez D, Gladman DD, Urowitz MB. The effect of pregnancy on lupus nephritis. Arthritis Rheum. 2004 Dec;50(12):3941–6. doi: 10.1002/art.20638. [DOI] [PubMed] [Google Scholar]
- 47.Moroni G, Quaglini S, Banfi G, et al. Pregnancy in lupus nephritis. Am J Kidney Dis. 2002 Oct;40(4):713–20. doi: 10.1053/ajkd.2002.35678. [DOI] [PubMed] [Google Scholar]
- 48.Imbasciati E, Tincani A, Gregorini G, et al. Pregnancy in women with pre-existing lupus nephritis: predictors of fetal and maternal outcome. Nephrol Dial Transplant. 2009 Feb;24(2):519–25. doi: 10.1093/ndt/gfn348. [DOI] [PubMed] [Google Scholar]
- 49.Petri M, Kim MY, Kalunian KC, et al. Combined oral contraceptives in women with systemic lupus erythematosus. N Engl J Med. 2005 Dec 15;353(24):2550–8. doi: 10.1056/NEJMoa051135. [DOI] [PubMed] [Google Scholar]
- 50.Buyon JP, Petri MA, Kim MY, et al. The effect of combined estrogen and progesterone hormone replacement therapy on disease activity in systemic lupus erythematosus: a randomized trial. Ann Intern Med. 2005 Jun 21;142(12 Pt 1):953–62. doi: 10.7326/0003-4819-142-12_part_1-200506210-00004. [DOI] [PubMed] [Google Scholar]
- 51.Mok CC, Lau CS, Ho CT, Lee KW, Mok MY, Wong RW. Safety of hormonal replacement therapy in postmenopausal patients with systemic lupus erythematosus. Scand J Rheumatol. 1998;27(5):342–6. doi: 10.1080/03009749850154357. [DOI] [PubMed] [Google Scholar]
- 52.Kreidstein S, Urowitz MB, Gladman DD, Gough J. Hormone replacement therapy in systemic lupus erythematosus. J Rheumatol. 1997 Nov;24(11):2149–52. [PubMed] [Google Scholar]
- 53.Hochman J, Urowitz MB, Ibanez D, Gladman DD. Hormone replacement therapy in women with systemic lupus erythematosus and risk of cardiovascular disease. Lupus. 2009 Apr;18(4):313–7. doi: 10.1177/0961203308097475. [DOI] [PubMed] [Google Scholar]
- 54.Sturgess AD, Evans DT, Mackay IR, Riglar A. Effects of the oestrogen antagonist tamoxifen on disease indices in systemic lupus erythematosus. J Clin Lab Immunol. 1984 Jan;13(1):11–4. [PubMed] [Google Scholar]
- 55.Mok CC, To CH, Mak A, Ma KM. Raloxifene for postmenopausal women with systemic lupus erythematosus: a pilot randomized controlled study. Arthritis Rheum. 2005 Dec;52(12):3997–4002. doi: 10.1002/art.21477. [DOI] [PubMed] [Google Scholar]
- 56.Pasoto SG, Mendonca BB, Bonfa E. Menstrual disturbances in patients with systemic lupus erythematosus without alkylating therapy: clinical, hormonal and therapeutic associations. Lupus. 2002;11(3):175–80. doi: 10.1191/0961203302lu163oa. [DOI] [PubMed] [Google Scholar]
- 57.Piotrowski P, Gasik R, Lianeri M, et al. Asp327Asn polymorphism of sex hormone-binding globulin gene is associated with systemic lupus erythematosus incidence. Mol Biol Rep. 2009 Aug 1; doi: 10.1007/s11033-009-9639-7. [DOI] [PubMed] [Google Scholar]
- 58.Jonsen A, Yu X, Truedsson L, et al. Mitochondrial DNA polymorphisms are associated with susceptibility and phenotype of systemic lupus erythematosus. Lupus. 2009 Apr;18(4):309–12. doi: 10.1177/0961203308097477. [DOI] [PubMed] [Google Scholar]
- 59.Ryan MJ, McLemore GR, Jr, Hendrix ST. Insulin Resistance and Obesity in a Mouse Model of Systemic Lupus Erythematosus. Hypertension. 2006;48(5):988–993. doi: 10.1161/01.HYP.0000243612.02929.df. [DOI] [PubMed] [Google Scholar]
- 60.Ryan MJ, McLemore GR., Jr Hypertension and Impaired Vascular Function in a Female Mouse Model of Systemic Lupus Erythematosus. Am J Physiol Regul Integr Comp Physiol. 2007;292(2):R736–R742. doi: 10.1152/ajpregu.00168.2006. [DOI] [PubMed] [Google Scholar]
- 61.Bynote KK, Hackenberg JM, Korach KS, Lubahn DB, Lane PH, Gould KA. Estrogen receptor-alpha deficiency attenuates autoimmune disease in (NZB × NZW)F1 mice. Genes Immun. 2008 Mar;9(2):137–52. doi: 10.1038/sj.gene.6364458. [DOI] [PubMed] [Google Scholar]
- 62.Svenson JL, EuDaly J, Ruiz P, Korach KS, Gilkeson GS. Impact of estrogen receptor deficiency on disease expression in the NZM2410 lupus prone mouse. Clin Immunol. 2008 Aug;128(2):259–68. doi: 10.1016/j.clim.2008.03.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sobel ES, Gianini J, Butfiloski EJ, Croker BP, Schiffenbauer J, Roberts SM. Acceleration of autoimmunity by organochlorine pesticides in (NZB × NZW)F1 mice. Environ Health Perspect. 2005 Mar;113(3):323–8. doi: 10.1289/ehp.7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang F, Sobel ES, Butfiloski EJ, Roberts SM. Comparison of chlordecone and estradiol effects on splenic T-cells in (NZB×NZW)F(1) mice. Toxicol Lett. 2008 Dec 15;183(1–3):1–9. doi: 10.1016/j.toxlet.2008.08.020. [DOI] [PubMed] [Google Scholar]
- 65.Yoneda T, Ishimaru N, Arakaki R, et al. Estrogen deficiency accelerates murine autoimmune arthritis associated with receptor activator of nuclear factor-kappa B ligand-mediated osteoclastogenesis. Endocrinology. 2004 May;145(5):2384–91. doi: 10.1210/en.2003-1536. [DOI] [PubMed] [Google Scholar]
- 66.Ishimaru N, Haneji N, Hamano H, Kumiko Y, Takahashi M, Hayashi Y. Accelerated onset of age-related autoimmune lesions in MRL/+ mice by ovariectomy. Mech Ageing Dev. 1997 Feb;93(1–3):145–56. doi: 10.1016/s0047-6374(96)01823-4. [DOI] [PubMed] [Google Scholar]
- 67.Wu WM, Lin BF, Su YC, Suen JL, Chiang BL. Tamoxifen decreases renal inflammation and alleviates disease severity in autoimmune NZB/W F1 mice. Scand J Immunol. 2000 Oct;52(4):393–400. doi: 10.1046/j.1365-3083.2000.00789.x. [DOI] [PubMed] [Google Scholar]
- 68.Apelgren LD, Bailey DL, Fouts RL, et al. The effect of a selective estrogen receptor modulator on the progression of spontaneous autoimmune disease in MRL lpr/lpr mice. Cell Immunol. 1996 Oct 10;173(1):55–63. doi: 10.1006/cimm.1996.0251. [DOI] [PubMed] [Google Scholar]
- 69.Ansar AS, Penhale WJ, Talal N. Sex hormones, immune responses, and autoimmune diseases. Mechanisms of sex hormone action. Am J Pathol. 1985 Dec;121(3):531–51. [PMC free article] [PubMed] [Google Scholar]
- 70.Lockshin MD. Sex differences in autoimmune disease. Lupus. 2006;15(11):753–6. doi: 10.1177/0961203306069353. [DOI] [PubMed] [Google Scholar]
- 71.Sawalha AH, Kovats S. Dehydroepiandrosterone in systemic lupus erythematosus. Curr Rheumatol Rep. 2008 Aug;10(4):286–91. doi: 10.1007/s11926-008-0046-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Crosbie D, Black C, McIntyre L, Royle PL, Thomas S. Dehydroepiandrosterone for systemic lupus erythematosus. Cochrane Database Syst Rev. 2007;(4):CD005114. doi: 10.1002/14651858.CD005114.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Peeva E, Venkatesh J, Michael D, Diamond B. Prolactin as a modulator of B cell function: implications for SLE. Biomed Pharmacother. 2004 Jun;58(5):310–9. doi: 10.1016/j.biopha.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 74.Jara-Quezada L, Graef A, Lavalle C. Prolactin and gonadal hormones during pregnancy in systemic lupus erythematosus. J Rheumatol. 1991 Mar;18(3):349–53. [PubMed] [Google Scholar]
- 75.Elbourne KB, Keisler D, McMurray RW. Differential effects of estrogen and prolactin on autoimmune disease in the NZB/NZW F1 mouse model of systemic lupus erythematosus. Lupus. 1998;7(6):420–7. doi: 10.1191/096120398678920352. [DOI] [PubMed] [Google Scholar]
- 76.Stumpe KO, Kolloch R, Higuchi M, Kruck F, Vetter H. Hyperprolactinaemia and antihypertensive effect of bromocriptine in essential hypertension. Identification of abnormal central dopamine control. Lancet. 1977 Jul 30;2(8031):211–4. doi: 10.1016/s0140-6736(77)92832-x. [DOI] [PubMed] [Google Scholar]
- 77.Leanos-Miranda A, Marquez-Acosta J, Cardenas-Mondragon GM, et al. Urinary prolactin as a reliable marker for preeclampsia, its severity, and the occurrence of adverse pregnancy outcomes. J Clin Endocrinol Metab. 2008 Jul;93(7):2492–9. doi: 10.1210/jc.2008-0305. [DOI] [PubMed] [Google Scholar]
- 78.Tan BK, Hutchinson JS. Blood pressure, plasma and pituitary prolactin responses to bromocriptine in New Zealand genetically hypertensive and normotensive rats. Clin Exp Pharmacol Physiol. 1989 Jan;16(1):13–8. doi: 10.1111/j.1440-1681.1989.tb01903.x. [DOI] [PubMed] [Google Scholar]