

Abstract
Progression through the G2/M transition following DNA damage is linked to cytokinesis failure and mitotic death. In four different transformed cell lines and two human embryonic stem cell lines, we find that DNA damage triggers mitotic chromatin decondensation and global phosphorylation of histone H2AX, which has been associated with apoptosis. However, extended time-lapse studies in HCT116 colorectal cancer cells indicate that death does not take place during mitosis, but 72% of cells die within 3 days of mitotic exit. By contrast, only 11% of cells in the same cultures that remained in interphase died, suggesting that progression through mitosis enhances cell death following DNA damage. These time-lapse studies also confirmed that DNA damage leads to high rates of cytokinesis failure, but showed that cells that completed cytokinesis following damage died at higher rates than cells that failed to complete division. Therefore, post-mitotic cell death is not a response to cytokinesis failure or polyploidy. We also show that post-mitotic cell death is largely independent of p53 and is only partially suppressed by the apical caspase inhibitor Z-VAD-FMK. These findings suggest that progression through mitosis following DNA damage initiates a p53- and caspase-independent cell death response that prevents propagation of genetic lesions.
Keywords: anti-proliferative response, G2 checkpoint abrogation, DNA damage, post-mitotic death, mitotic catastrophe
Introduction
To maintain genome integrity, cells respond to DNA damage by inducing repair or cell death.1 DNA damage and replication checkpoints help maintain genome integrity by delaying cell cycle progression, which allows time for completion of repair or DNA synthesis.2 By contrast, apoptosis eliminates cells carrying irreparable damage.3 Both “salvage” and “disposal” strategies thus prevent propagation of damage-induced genetic lesions, and mutations that disrupt these responses are linked to developmental defects and cancer.4-8 Checkpoint activation and apoptosis function during the G1, S and G2 phases of the cell cycle and prevent progression into mitosis with damaged DNA.9,10 When these systems fail and mitosis is initiated prior to completion of DNA replication or repair, cells die during mitosis, or delay in mitosis before exiting with high rates of chromosome segregation and cytokinesis failures.11-13 This process, often referred to as “mitotic catastrophe”, is observed in a wide range of systems, and has been proposed to be a significant cause of chemotherapy-induced cell death in some human tumors.14
DNA damage-induced delays in mitosis require the spindle assembly checkpoint,15-18 but the cytological changes that accompany these delays have not been studied in detail, and the long-term fate of cells that exit mitosis following DNA damage has not been determined. We show that DNA damage triggers cell-type independent changes in chromatin organization, histone modification and Aurora B localization. Long-term time-lapse imaging in HCT116 cells shows that progress through mitosis following DNA damage leads to high rates of cell death and cell cycle arrest, and that post-mitotic cell death is largely independent of p53 and is only partially suppressed by an apical caspase inhibitor. By contrast, post-mitotic cell cycle arrest requires p53. Progression through mitosis with DNA damage thus appears to trigger cell death and cell cycle arrest. These anti-proliferative responses thus maintain genome integrity when checkpoint control fails and DNA damage persists into mitosis.
Results
DNA damage-induced changes in mitotic chromatin and mitosis progression
To characterize the mitotic response to DNA damage in human cells, we initially surveyed mitotic phase distribution and chromatin organization in human H9 and H1 embryonic stem cells (hESCs), U2OS osteosarcoma cells H460 lung cancer cells, HeLa cervical carcinoma cells and HCT116 colorectal cancer cells. Because of the availability of knock out mutations in several DNA damage response genes,19 we optimized conditions for DNA damage induction using HCT116 cells. Parental HCT116 cells were treated with a series of bleomycin concentrations, harvested at various times after treatment, and analyzed for cell cycle progression by flow cytometry. Bleomycin induces DNA breaks,20 and 10 μg/ml bleomycin consistently induced a block in G1 and G2, consistent with checkpoint activation at these two control points (Suppl. Fig. 1). However, this level of bleomycin did not trigger high levels of cell death, as indicated by the absence of a sub-G1 peak in flow cytometry analysis and by direct inspection of the cultured cells (not shown). We therefore used 10 μg/ml bleomycin to induce DNA damage.
Following DNA damage, a fraction of cells eventually adapt to the G2 DNA damage checkpoint and progress into mitosis.21 To determine the time needed for checkpoint adaptation, parental HCT116 cells were treated with 10 μg/ml bleomycin, fixed at various times after drug addition, labeled for DNA and microtubules, and analyzed by laser scanning confocal microscopy. Very few mitotic cells were observed before 12 hours, but mitotic cells were consistently observed at the 24 hour time point. In these cells, chromatin appeared less condensed and failed to form normal metaphase figures. The spindles appeared somewhat elongated, and very few anaphase or telophase figures were observed (Suppl. Fig. 2). However, the mitotic index was low (0.25%, compared to 2.79% in nontreated cultures), making systematic analysis difficult.
To increase the number of mitotic cells, we cotreated cells with bleomycin and the G2 checkpoint inhibitor UCN-01.22 Isolated cells entered mitosis during the first few hours following drug addition, but more significant numbers were observed by 8 h (cytological examination, not shown). In the case of hESCs, high numbers of mitotic cells were observed after 2 h. Using these conditions, the majority of hESC, U2OS, H460, HeLa and HCT116 cells that progressed into mitosis showed defects in chromatin condensation (Figs. 1A and 2A) and a reduction in the fraction of mitotic cells in anaphase and telophase, consistent with delays in prometaphase or metaphase (Fig. 1B). The mitotic defects observed in H1 and H9 hESC lines were identical, and thus only data from H9 are shown (Figs. 1 and 2).
Figure 1.
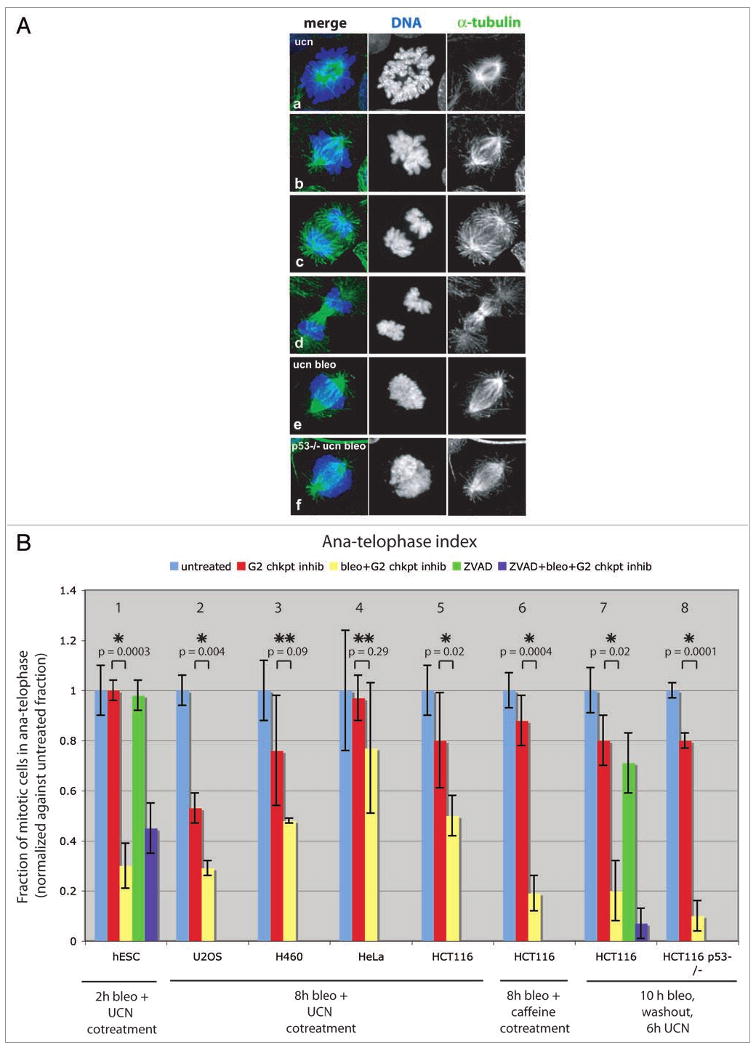
DNA damage blocks mitosis progression and disrupts mitotic chromatin. Prior to fixation, cells were grown 10 h in media with or without 10 μg/ml bleomycin, then washed, and released into 300 nM UCN-01 for 6 h (A and B, panels 7,8). Alternatively, cells were cotreated with 10 μg/ml bleomycin and G2 checkpoint inhibitor (300 nM UCN-01 or 5 mM caffeine) for 8 h (B, panels 2–6) or 2 h (B, panel 1). To address caspase dependency, 100 μM Z-VAD-FMK was added 2 h prior to bleomycin (B, panel 1) or simultaneously with bleomycin (B, panel 7) and kept in media throughout the experiment. (A) Confocal projections of α-tubulin (green) and DNA (blue) detected by immunolabeling during mitosis in HCT116 cells. In control cells, all mitotic phases were observed (a–d) and prometa-metaphase cells showed spindles organized around well-condensed, distinct chromosome arms (b). Mitotic cells in bleomycin treated cultures accumulated in a prometaphase-like stage with dispersed chromatin (e). Accumulation of prometaphase-like cells with abnormal chromatin structure also occurred in bleomycin treated HCT116 p53-/- cells (f). (B) Ana-telophase index determined by cytological examination. A normal progression through mitosis in control treated cultures was reflected by an ana-telophase index of 1 (blue = untreated, red = UCN-01 or caffeine treated). In response to bleomycin (yellow bars), cells accumulated as prometaphase-like stages (with morphology as shown in e). In all cell lines examined, except HeLa, a reduced ana-telophase index was observed in response to bleomycin. This block in mitotic progression was also observed in HCT116 p53-/- cells, and in cells treated with the poly-caspase inhibitor Z-VAD-FMK (hESC and HCT116: purple bars in panels 1 and 7). Bars show average of two or three independent experiments (n = 100–200 cells per experiment). p-values (UCN-01 versus bleomycin and UCN-01 treated) were calculated using a two-tailed Student's t-test. Statistically significant differences are marked by *(p-value <0.05), while p-values >0.05 are marked by **.
Figure 2.
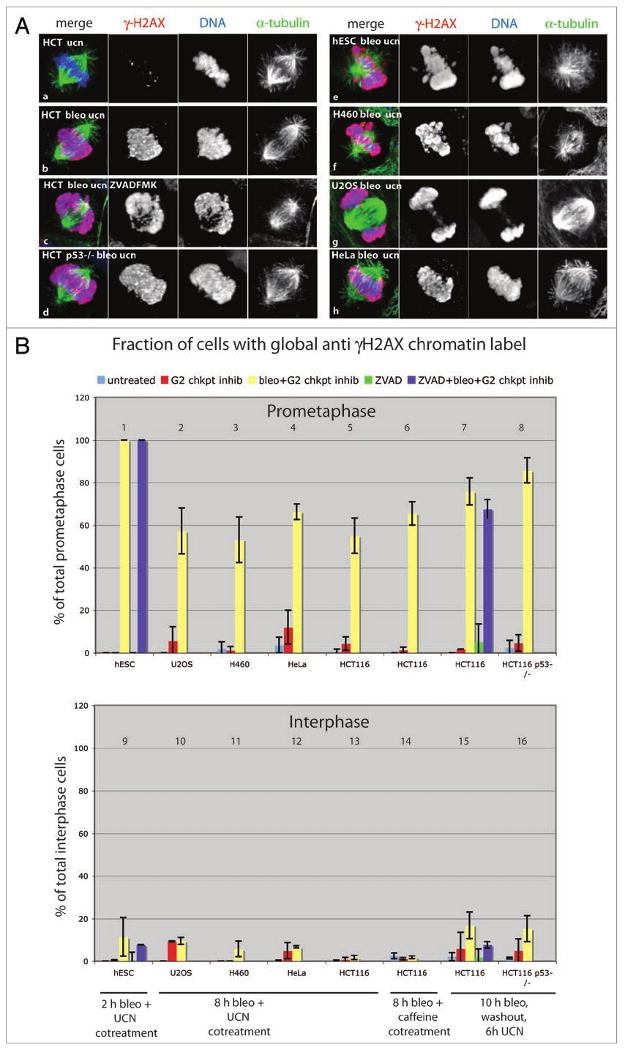
A mitosis specific amplification of γH2AX signal in response to DNA damage. Cells were grown 10 h in media with or without 10 μg/ml bleomycin and released into 300 nM UCN-01 for 6 h (A, B panels 7, 8, 15 and 16). Alternatively, cells were cotreated with 10 μg/ml bleomycin and G2 checkpoint inhibitor (300 nM UCN-01 or 5 mM caffeine) for 8 h (B, panels 2–6, 10–14) or 2 h (B, panels 1 and 9). To address caspase dependency, 100 μM Z-VAD-FMK was added 2 h prior to bleomycin (B, panels 1 and 9) or simultaneously with bleomycin (B, panels 7 and 15). (A) Confocal projections of mitotic cells immunolabeled for α-tubulin (green), γH2AX (red) and DNA (blue). In control cells, none or a few γH2AX foci were observed in interphase nuclei (not shown) and on mitotic chromatin (a). DNA double strand breaks introduced by bleomycin produced a dense distribution of γH2AX foci throughout interphase nuclei (not shown). In contrast, during mitosis the γH2AX signal localized throughout the dispersed chromatin during mitosis in all cell lines examined (b–h). Such global γH2AX signal was also observed in cells treated with poly-caspase inhibitor (c) and in p53-/- cells (d). The small fraction of bleomycin treated mitotic cells that did not show this global γH2AX labeling pattern had distinct foci distributed throughout the chromatin (not shown). (B) Quantification of global anti γH2AX label during mitosis and interphase. Cells with anti γH2AX label throughout chromatin (like in images b–h) were scored. In all cell lines, mitotic chromatin showed global anti γH2AX label when entering mitosis in the presence of DNA damage. This global γH2AX signal was mitosis specific, as very few cells with global γH2AX label were observed during interphase. Bars show average of two or three independent experiments (n = 100–200 cells per experiment).
The mitotic defects observed following bleomycin and UCN-01 treatment were essentially identical to those observed following bleomycin treatment and checkpoint adaptation without inhibitor (Suppl. Fig. 2). In addition, HCT116 cells treated with bleomycin and the pharmacologically distinct G2 checkpoint inhibitor caffeine show identical mitotic defects (Fig. 1B). Cells treated with the DNA damaging drugs adriamycin and etoposide also showed similar defects (Suppl. Fig. 2). DNA damage associated defects in mitotic progression and chromatin organization are therefore independent of cell type, mechanism of G2 checkpoint inhibition, and DNA damaging agent.
To further increase the number of cells entering mitosis with damage, HCT116 cells were treated with bleomycin for 10 h, which led to arrest of the majority of the cells in G2 (Suppl. Fig. 3A). Bleomycin was then removed and fresh medium containing the G2 checkpoint inhibitor UCN-01 was added. Under these conditions, the mitotic index increased to 14.5% 6 hours after UCN-01 addition (Suppl. Fig. 3B). Under these conditions, we observed the same defects in chromosome condensation and an absence of anaphase and telophase figures observed on checkpoint adaptation and caffeine treatment. This synchronization protocol was therefore used for the majority of the following studies.
Caspase independent chromatin modification in damaged mitotic cells
To determine if the damage-associated defects in mitotic chromatin organization were linked to chromatin fragmentation, we immunolabeled for a phosphorylated form of histone H2AX (γH2AX) which accumulates in foci at sites of double strand breaks.23 In the absence of bleomycin, all six cell lines showed low levels of γH2AX labeling during mitosis (exemplified by HCT116 in Fig. 2A) and interphase (not shown). Following bleomycin treatment, interphase cells showed distinct γH2AX foci (not shown). However, 100% of mitotic human embryonic stem cells and 53% to 66% of the mitotic U2OS, H460 and HCT116 cells showed striking γH2AX labeling covering all of the chromatin (Fig. 2). By contrast, global γH2AX modification was rarely observed during interphase in any of the cell lines (Fig. 2B).
Global H2AX phosphorylation has been associated with caspase induced apoptosis, and we speculated that progression into mitosis following damage might trigger a form of apoptosis.24,25 However, in both hESC and HCT116 cells, bleomycin induced changes in chromatin condensation, suppression of anaphase and telophase, and global γH2AX labeling were not blocked by the poly-caspase inhibitor Z-VAD-FMK (Figs. 1 and 2) which efficiently suppressed deoxycholic acid (DCA) induced apoptosis in HCT116 cells (Suppl. Fig. 4). In addition, these defects were not suppressed by a p53 null mutation in HCT116 cells (Figs. 1 and 2). γH2AX accumulation during apoptosis has been associated with nucleosome level DNA fragmentation, which is readily detected using TUNEL assays that label DNA ends.24,25 However, HCT116 cells that initiated mitosis following bleomycin treatment were consistently TUNEL negative, while cells induced to undergo apoptosis with DCA26 were TUNEL positive (Suppl. Fig. 5). DNA damage associated global H2AX modification and delays in prometaphase thus appear to be caspase and p53 independent, and the global H2AX phosphorylation may reflect spreading of this modification from a limited number of DNA break sites.
To determine if DNA damage-induced mitotic delays were associated with defects in kinetochore alignment, we labeled for the inner kinetochore protein CENP-C.27 In control cells, kinetochores align at the spindle midzone during prometaphase and segregate during anaphase (Fig. 3). Following bleomycin treatment, the vast majority of mitotic cells showed CENP-C foci distributed throughout the spindle region, suggesting that kinetochore alignment is delayed (Fig. 3). Supporting this speculation and consistent with previous observations, the spindle checkpoint protein BubR1, which localizes to kinetochores until a proper bipolar attachment is achieved, was consistently present at a subset of the kinetochores following DNA damage (Fig. 3).28 Moreover, the bleomycin treated cells consistently showed high cyclin B levels (Fig. 3), consistent with delays in prometaphase.
Figure 3.
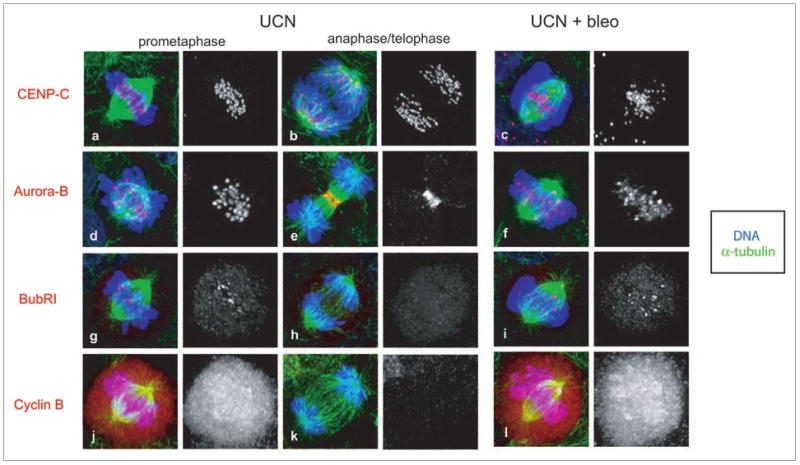
DNA damage-induced defect in mitotic HCT116 cells. Prior to fixation, HCT116 cells were grown for 10 h in media with or without 10 μg/ml bleomycin and released into 300 nM UCN-01 for 6 h. Confocal projections of α-tubulin (green), DNA (blue), and CENP-C (red, a–c), Aurora B (red, d–f), BubR1 (red, g–i) or Cyclin B (red, j–l) in immunolabeled mitotic HCT116 cells. While CENP-C labeled kinetochores in control cells aligned at the metaphase plate (a) and separated during anaphase (b), kinetochores in bleomycin treated cells were scattered throughout the spindle region (c). In control cells, the passenger protein Aurora B concentrated at kinetochores during prometaphase (d). Following transition to anaphase, Aurora B relocated to the central spindle and eventually to the midbody during telophase (e). In bleomycin treated mitotic cells, Aurora B labeled a subset of the kinetochores and showed a diffuse labeling of the bulk chromatin (f). Contrary to the relatively uniform Aurora B levels observed at kinetochores within a control cell (d), Aurora B levels at kinetochores in bleomycin treated cells were irregular (f). In control treated cells, the spindle checkpoint protein BubR1 accumulated at all kinetochores during prophase (not shown), and was progressively lost from these structures as bipolar attachment was achieved during prometaphase (g). By anaphase onset the BubR1 signal was undetectable (h). In response to bleomycin treatment, mitotic cells delayed in a prometaphase-like stage with dispersed chromatin and a subset of the kinetochores persistently labeled with BubR1 (i). In mitotic control cells, cyclin B levels were high prior to anaphase (j) and degraded upon anaphase onset (k). Bleomycin treated parental cells accumulated with high cyclin B levels (l).
Aurora B is a passenger protein required for chromosome alignment, segregation, and cytokinesis.29 Aurora B normally accumulates at kinetochores in prophase, disappears from kinetochores prior to anaphase onset, and relocates to the central spindle during telophase29 (Fig. 3). In bleomycin treated mitotic cells, by contrast, Aurora B localized to dispersed foci of irregular size and to bulk chromatin (Fig. 3). Chk1 has recently been implicated in Aurora B localization,30 raising the possibility that these defects were due to checkpoint inhibition by UCN-01. However, cells treated with UCN-01 alone do not show defects in Aurora B localization, and identical defects in Aurora B localization were observed in cells that adapt to the G2 checkpoint (not shown). DNA damage thus appears to disrupt Aurora B localization through a Chk1-independent mechanism.
DNA damage causes mitotic delays and division failure
To directly assay mitotic progression following DNA damage, control and bleomycin treated HCT116 cells were monitored by time-lapse video microscopy. In the presence of bleomycin, parental HCT116 cells delay at the G2/M transition, and relatively few cells progress into mitosis. However, the few cells that adapted to the G2 checkpoint subsequently delayed in mitosis and exited without completing cytokinesis (not shown). To increase the number of cells entering mitosis following DNA damage, cultures were treated with bleomycin for 10 h and released into UCN-01, as described above (Suppl. Fig. 3B). Cells that entered mitosis were scored for the duration of mitosis and completion of cytokinesis. To determine the long-term consequences of progression through mitosis following damage, the UCN-01 was washed away after 12 hours and the cells were filmed for an additional 60 hours, corresponding to 3–4 control cell cycles (Fig. 4A).
Figure 4.
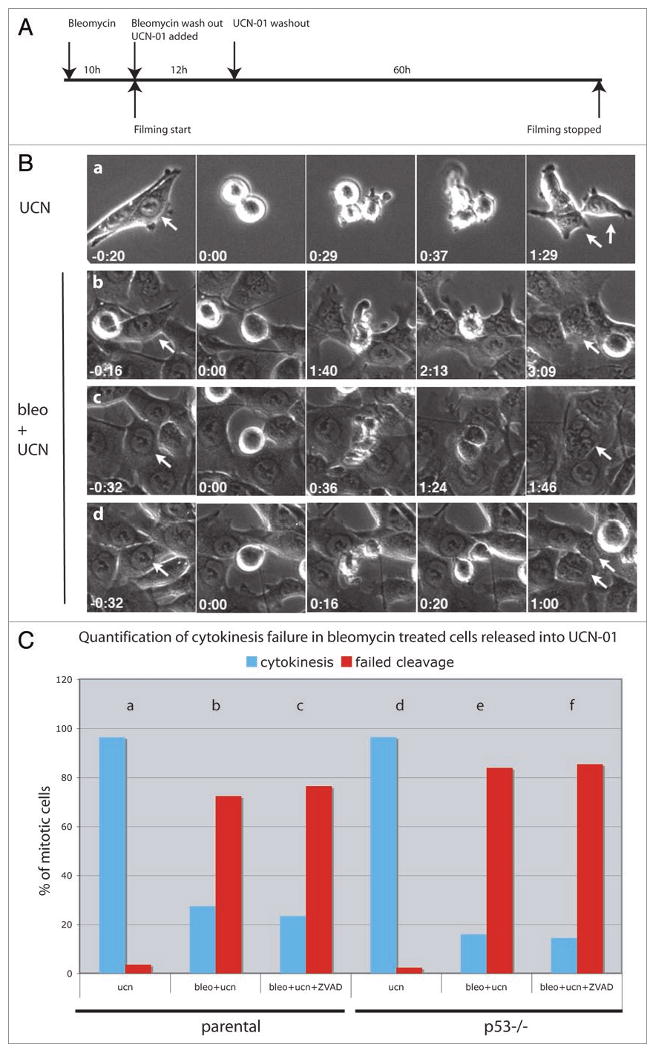
DNA damage induces mitotic delay and cytokinesis failure. (A) HCT116 cells were treated with 10 μg/ml bleomycin for 10 h and following washout of the drug released into 300 nM UCN-01. Filming was initiated shortly after addition of UCN-01 and continued for a total of 72 h. UCN-01 was washed out after 12–14 h. (B) Control (UCN-01 treated) cells rounded up as they entered mitosis and produced two daughter cells by successful completion of cytokinesis (a). Arrows point to pre-mitotic cells (first column) and their resulting daughter cells (last column). Bleomycin treated parental cells driven into mitosis by UCN-01 were frequently delayed and failed cytokinesis (b and c). These cells delayed in prometaphase, after which a short phase of amorphous membrane “blebbing” occurred, followed by mitotic exit without formation of a cytokinesis furrow (b). Alternatively, constriction was initiated but the cytokinesis furrow regressed prior to mitotic exit (c). A fraction of the bleomycin treated cells did cleave successfully, producing two separate daughter cells (d). (C) Quantification of cytokinesis failure in parental and p53-/- HCT116 cells. Parental or p53 mutant cells were treated with UCN-01 (ucn), bleomycin and UCN-01 (bleo + ucn) or with UCN-10, bleomycin and the polycaspase inhibitor Z-VAD-FMK (bleo + ucn + ZVAD). Blue bars represent cells that completed cytokinesis and red bars indicate cells that failed cytokinesis. Number of cell scored for each condition: 84 (a), 64 (b), 51 (c), 85 (d), 50 (e), 55 (f).
Time-lapse studies of cells treated with UCN alone showed that this inhibitor does not block mitotic progression. The cells rounded up as they initiated mitosis, and then split to produce two new daughter cells that eventually flattened on the substrate (Fig. 4B). The length of mitosis was variable, but division was generally completed in less than 45 min (Suppl. Fig. 6a). By contrast, 63% of the cells treated with bleomycin and then released into UCN-01 remained longer than 45 min in mitosis, and most cells (72%) failed cytokinesis (Suppl. Fig. 6b, Fig. 4C). Cells that completely failed cytokinesis showed a phase of amorphous “membrane blebbing” without cleavage furrow formation prior to exiting mitosis (Fig. 4). Other cells initiated constriction, but the furrow regressed, causing the two initially separated daughters to fuse (Fig. 4). 28% of UCN-01 and bleomycin treated cells did complete cytokinesis and produced two separate daughter cells (Fig. 4). The poly-caspase inhibitor Z-VAD-FMK did not suppress the bleomycin induced mitotic delays or division failures (Fig. 4C, Suppl. Fig. 6d). The majority of p53-/- cells (84%) treated with bleomycin and UCN-01 also delayed in mitosis and failed cytokinesis (Fig. 4C, Suppl. Fig. 6f). DNA damage thus causes p53 and caspase independent mitotic delays and cytokinesis failures.
DNA damage leads to post-mitotic cell death and cell division arrest
We next assayed the long-term viability and proliferation capacity of post-mitotic cells using extended (72 h) time-lapse imaging. In these experiments, 72% of HCT116 cells that exited mitosis following DNA damage died, as marked by cytoplasmic shrinkage associated with membrane blebbing (Fig. 5A and C), often followed by detachment from the substrate. Cell death occurred after interphase delays of 5–70 h. In addition, the cells that did not die failed to divide again within the 72 h window (Fig. 5D). By contrast, only 11% of cells in the same cultures that remained in interphase died (Fig. 5B). Progression through mitosis following bleomycin treatment is therefore linked to a significant increase in cell death.
Figure 5.
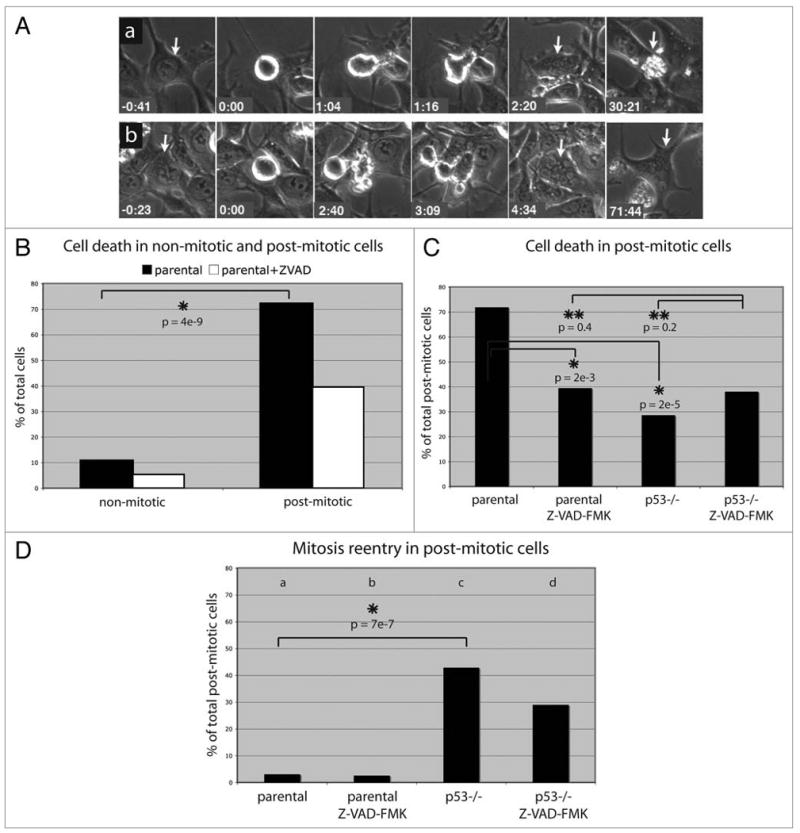
Long-term fate of post-mitotic, bleomycin treated cells. HCT116 cells were treated as described in legend of Figure 4. (A–D) show data of cells treated with bleomycin and UCN-01. (A) Post-mitotic, bleomycin treated cells either died after a prolonged interphase arrest (a), or alternatively, arrested in interphase (b). The time 0:00 corresponds to rounding up at mitosis onset. (B) 11% (n = 19) of bleomycin treated cells that did not go through mitosis died, but 72% (n = 37) of cells that progressed through mitosis died during the subsequent interphase. (C) Death in post-mitotic bleomycin treated cells (72%, n = 32) was only partly suppressed by poly-caspase inhibitor (40%, n = 38) or p53 knockout (29%, n = 42). The suppression observed under each of these conditions was similar to that observed in Z-VAD-FMK treated p53-/- cells (38%, n = 42), suggesting that p53 and caspase independent mechanisms contribute to cell death in the post-mitotic cells. (D) Fraction of parental and p53 mutant cells that initiated a second division following bleomycin treatment in the presence or absence of Z-VAD-FMK. Number of cells scored for each condition: 32 (a), 38 (b), 42 (c),42 (d). All p-values were based on a Z-score calculation.
We speculated that polyploidy resulting from cleavage failure could lead to the observed post-mitotic death. However, 47% of the cells that failed cleavage died, while 94% of the daughters arising from a complete cleavage died (Suppl. Fig. 7B). DNA damage induces mitotic delays, and we speculated that postmitotic death could be secondary to prolonged time in mitosis. However, our single cell tracking studies showed that DNA damage induced post-mitotic death was observed in cells that did not delay in mitosis and in cells that showed significant delays (data not shown). Therefore, the high rates of post-mitotic cell death in HCT116 cells are not due to cleavage failure, polyploidy, or mitotic delays, but appear to be a consequence of progression through mitosis with DNA lesions.
Post-mitotic cell death is accompanied by extensive membrane blebbing, which is also observed during caspase-dependent apoptosis.31 However, the poly-caspase inhibitor Z-VAD-FMK only partially suppressed post-mitotic death (from 72% to 40%) (Fig. 5C). Similarly, 29% of p53 null mutant HCT116 cells showed damage induced postmitotic cell death (Fig. 5C). In addition, 38% of p53 mutant cells treated with the pan-caspase inhibitor Z-VAD-FMK showed post-mitotic cell death (Fig. 5C, 38%). These findings suggest that progression through mitosis following damage leads to conventional apoptosis that depends on p53 and caspase activation, as well as cell death through a process that is independent of p53 and caspase activation.
The vast majority of parental HCT116 cells that did not die following progression through mitosis failed to divide again within 72 h (Fig. 5D). Cells that escape post-mitotic cell death may therefore undergo terminal cell cycle arrest. However, 43% of post-mitotic p53-/- cells divided again (Fig. 5D). p53 may therefore play a critical role in blocking proliferation of cells that progress through mitosis following DNA damage.
These data strongly suggested that progression through mitosis enhances damage induced cell death. To test this hypothesis, we assayed the effect of the Cdk1 inhibitor RO3306 on cell death using the live assay described above (Fig. 6). Only 2% of HCT116 cells treated with UNC-01 alone died, and only 4% of cells treated with bleomycin alone died (these cells arrested in G1, and G2). 11% of cells treated with RO3306 alone died, perhaps due to non-specific toxicity. RO3306 blocked mitotic entry under all conditions assayed (Fig. 6). 54% of all cells treated with both bleomycin and UCN-01 entered mitosis and 49% died. By contrast, cells treated with RO3306, bleomycin and UCN-01 failed to progress through mitosis and only 24% died (Fig. 6). This reduction in cell death is statistically significant (p = 0.0002), and supports the hypothesis that progression through mitosis enhances cell death.
Figure 6.
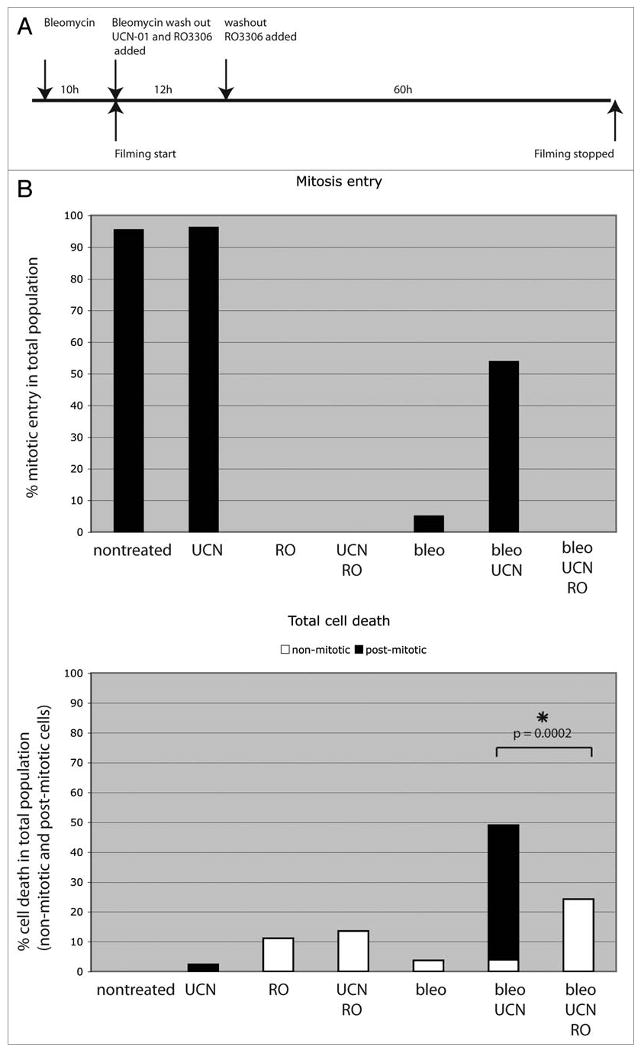
Death following DNA damage depends on mitosis. (A) Parental HCT116 cells were treated as described in the legend of Figure 4, except that 9 μM of the cdk1 inhibitor RO3306 (RO) was added simultaneously with UCN-01 and left in the media throughout the experiment. (B) Two parameters were scored: (top) the percentage of cells that enter mitosis and (bottom) the percentage of the total population, including both non-mitotic (white) and post-mitotic (black) cells, that die during the acquisition. 97% of UCN-01 treated control cells entered mitosis and only 2% died during the course of the experiment. No cells treated with RO3306 entered mitosis, but cell death increased to 11% in RO3306 treated control cells, likely due to toxic effects of the Cdk1 inhibitor. Only 5% of cells treated with bleomycin alone intiated mitosis, and 4% died. By contrast, 54% of cells treated with bleomycin and UCN-01 intiated mitosis, and cell death increased to 49%, which mainly occurred in post-mitotic cells. The cdk1 inhibitor RO3306 blocked mitotic entry in damaged cells and significantly reduced the death rate (24%) (p-value = 0.0002, based on a Z-score calculation). At least 50 cells per drug condition were included in analysis.
To determine if the post-mitotic cell death is a non-specific effect of the Chk1 kinase inhibitor UCN-01, we followed the long-term fate of p53-/- HCT116 cells, which have a compromised G2 checkpoint and progress into mitosis following DNA damage In the absence of inhibitor.19 For these studies, p53-/- cells were treated with bleomycin for 10 h, the drug was removed, and the long-term cell fate was assayed by time-lapse imaging. 83% of the p53-/- cells entered mitosis, typically between 30–90 hours after bleomycin removal. 90% delayed in mitosis, 72% failed cytokinesis and 81% of the post-mitotic cells died following a prolonged interphase arrest (Suppl. Fig. 8). Therefore, post-mitotic death is not a consequence of transient UCN-01 treatment.
To determine if high rates of post-mitotic death are specific to HCT116 cells, we used identical conditions to assay U2OS cells. U2OS cells treated with bleomycin and released into UCN-01 delayed in mitosis, 67% failed cytokinesis, and 45% died after mitotic exit (Suppl. Fig. 9A–C). As observed with HCT116 cells, post-mitotic cell death occurred in cells that failed cytokinesis and in cells that completed cytokinesis (Suppl. Fig. 9E). Of the cells that did not die after mitosis exit, 29% re-entered mitosis (Suppl. Fig. 9D). The post-mitotic division block is therefore less robust than in HCT116 cells. However, in both U2OS and HCT116 cells, progression through mitosis following DNA damage leads to high rates of cell death and cell cycle arrest.
Following treatment with UCN-01 alone, approximately half of U2OS cells progressed through mitosis and only 7% of these cells died during the extended time-lapse studies (Suppl. Fig. 9C). Surprisingly, 60% of the cells that failed to initiate mitosis died, and interphase cell death was not increased by bleomycin and UCN-01 treatment (40% death, Suppl. Fig. 9C). In U2OS cells, UCN-01 has been reported to induce DNA damage during S-phase.32 The high rates of interphase death may be related to damage during DNA replication. It is unclear why some cells escape death in response to UCN-01 and progress through mitosis, but the post-mitotic DNA damage response appears to be intact in these escapers.
Discussion
DNA damage disrupts mitotic chromatin condensation and Aurora B localization
In human cells, the spindle assembly checkpoint, inactivation of Polo-like kinase 1, and Chk1 signaling have all been implicated in DNA damage induced delays in mitotic exit.15,16,28,33,34 However, the structural changes associated with damage induced division failure remain poorly understood, and the long-term consequences of progression through mitosis following DNA damage have not been characterized. Our analysis of four transformed human cell lines and two human embryonic stem cell lines shows that damage disrupts kinetochore alignment, Aurora B localization and chromatin morphology, and that these defects are linked to chromosome-wide accumulation of γH2AX, a phosphorylated form of histone H2AX associated with DNA double strand breaks23 (Fig. 7). During apoptosis, similar γH2AX accumulation has been linked to nucleosome-level DNA fragmentation, which can be detected by TUNEL assays.24,25 However, cells that enter mitosis following DNA damage are consistently TUNEL negative, suggesting that the DNA is not highly fragmented. H2AX phosphorylation often extends well beyond the double strand break site,23,35 and our data suggest that the mitotic DNA damage response may lead to significant further spreading of this modification. Under the conditions used here, essentially all of the interphase cells showed only distinct γH2AX foci, and our time-lapse studies indicated that the mitotic cells, which showed global γH2AX labeling, exit mitosis. These findings suggest that global H2AX phosphorylation may be reversed on mitotic exit. The functional significance of this chromatin modification is not clear,36 but it could contribute to the apparent chromosome condensation and segregation defects.
Figure 7.
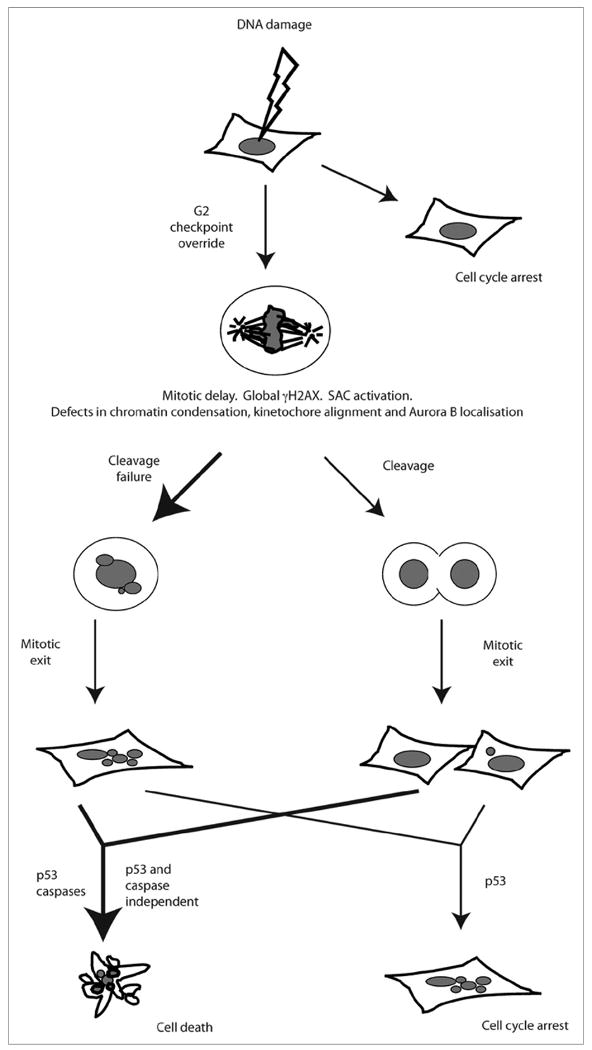
G2/M transition in the presence of DNA damage initiates post-mitotic anti-proliferative responses. Following DNA damage, cells with functional checkpoints arrest. In response to G2 checkpoint defects or adaptation, cells enter mitosis, undergo mitotic delay, and ultimately exit mitosis. Most damaged mitotic cells show cytokinesis failure, but some do cleave successfully. Post-mitotic cells, regardless of whether they arose from a failed or successful cleavage, die after a prolonged interphase arrest. This post-mitotic death occurs via mechanisms that are either dependent or independent of p53 and caspases. The cells that do not die show p53 dependent cell cycle arrest. Thus, mitosis entry with DNA damage results in cell death or cell cycle arrest, preventing propagation of DNA lesions.
We find that DNA damage also disrupts localization of Aurora B, which is a passenger protein required for chromosome alignment and cytokinesis.29 DNA damage induced disruption of Aurora B localization could lead to the cell cycle delays, chromosome congression defects, and cytokinesis failures observed in response to DNA damage. However, the defects in Aurora B localization could be secondary to defects in chromosome condensation. The available data cannot distinguish between these alternatives.
G2/M transition with DNA damage initiates robust anti-proliferative responses
In response to DNA damage, cells activate checkpoints that arrest cell cycle progression until the lesions have been cleared by repair, cell death or senescence. When checkpoint control is compromised, either due to mutations in checkpoint proteins or pharmacological inhibition, cells can enter mitosis with DNA lesions. This initiates a poorly understood mitotic response that includes cytokinesis failure, cell death, and cell cycle arrest.11,14,37 While this response might play a minor role in cells with normal cell cycle control, it has been hypothesized to be the major anti-proliferative response in cancer cells, which often have defective cell cycle control.37 We have used long-term time-lapse imaging to test this hypothesis, and find that the majority of HCT116 and U2OS cells that progress through mitosis following DNA damage either die or fail to divide again (Fig. 7). By contrast, cells that remain in interphase under the same conditions show only low rates of cell death. Progression through mitosis thus appears to significantly enhance damage induced cell death, at least in some cell types.
Our findings suggest that post-mitotic cell death is mediated by both p53 dependent and p53 independent processes. The post-mitotic block to cell division, by contrast, appears to require p53, which may induce senescence or an alternative exit from the cell cycle.38 Previous studies have reported activation of pro-apoptotic proteins during DNA damage induced mitotic delays, and have proposed that a balance of pro- and anti-apoptotic signals activated during mitosis regulate post-mitotic survival.17,39 For example, Cdk1 dependent phosphorylation of the initiator protease caspase-9 appears to suppress apoptosis during mitosis.39 Under the conditions used here, DNA damage does not induce cell death during mitosis, suggesting that this control pathway may be engaged. However, post-mitotic cell death occurs hours to days after exit from mitosis, and reflects both caspase dependent and independent processes. This time course strongly suggests that cell death following mitotic progression is unlikely to be a direct response to reversal of a Cdk1 dependent block to caspase activity, which should occur immediately on mitotic exit. Instead, mitotic exit may trigger a more complex process, which could include a transcriptional cascade that requires hours to days to execute.
Chemotherapy that combines DNA damaging drugs with G2 checkpoint inhibition by the non-specific Chk1 inhibitor UCN-01 are currently in clinical trials.40 The anti-proliferative responses documented here could contribute to tumor cell death and division arrest during this and other chemotherapeutic treatments. Gaining a full understanding of post-mitotic cell death could therefore lead to more effective cancer therapies.
Materials and Methods
Cell culture
HCT116 cells (B. Vogelstein, Johns Hopkins, Baltimore, USA): McCoy's 5A medium (GIBCO) supplemented with 10% FBS (Hyclone). The human embryonic stem cell lines H9 and H1 (WA09, WiCell Institute, University of Wisconsin): 80% DMEM-F12 (Invitrogen) and 20% KO Serum Replacer (Invitrogen) supplemented with non-essential amino acids, L-glutamine/β-mercaptoethanol (SIGMA) and basic FGF (R&D Systems) in the presence of mouse embryonic fibroblast feeder cells. U2OS and HeLa cells: DMEM (high glucose, GIBCO) supplemented with 10% FBS (Hyclone). H460 cells: RPMI 1640 media (GIBCO), supplemented with 1 mM sodium pyruvate (Sigma), 1.5 g/L sodium bicarbonate (Sigma) and 10% FBS (HyClone).
Drug treatments
The cytological assays used for studying the mitotic DNA damage response in fixed samples are described in the relevant figure legends. Drugs used: bleomycin, adriamycin and etoposide (Sigma). G2 checkpoint inhibitors UCN-01 (National Cancer Institute, Maryland, USA)22,41 and caffeine (Sigma). For apoptosis induction, deoxycholic acid (DCA) (Sigma). To address caspase dependence, Z-VAD-FMK (R&D Systems, Inc.,).
Immunocytochemistry
Cells were fixed in -20°C methanol (anti α-tubulin, anti γH2AX, anti BubR1), or 4% paraformaldehyde in 0.25% TritonX-100 (anti CENP-C), or 4% paraformaldehyde in PBS (anti cyclin B). For Aurora B labeling, cells were extracted with 0.25% TritonX-100 in PBS (5 min) prior to fixation in 4% paraformaldehyde in 0.25% TritonX-100 in PBS (10 min). Primary antibodies used, diluted in PBS-T: mouse anti α-tubulin (clone DM1A, Sigma, 1:100), mouse anti γH2AX (S139) (Upstate, 1:400), rabbit anti CENP-C (Bill Earnshaw, Johns Hopkins, Baltimore, USA, 1:1,000), rabbit anti cyclin B (Santa Cruz, 1:200), mouse anti BubR1 (BD Biosciences, 1:300), rabbit anti Aurora B (Abcam, 1:200). Secondary antibodies goat anti mouse Alexa-568, goat anti rabbit Alexa-568, goat-anti-rabbit Alexa 488 (Molecular Probes), all diluted 1:1,000 in PBS-T. DNA was counterstained in PBST containing 0.5 μM TOTO-3 iodide (Molecular Probes) for 15 min. Cells were imaged by confocal microscopy.
TUNEL
The TUNEL DNA fragmentation assay and DNase treatment was performed using a TUNEL Apoptosis Detection Kit (Upstate) according to manufacturer's protocol.
Flow cytometry
For cell cycle analysis: cells were trypsinized, washed with PBS, fixed in ice cold ethanol, stored in ethanol at 4°C, labeled by propidium iodide within three days and analyzed immediately by flow cytometry. For scoring of mitotic index: cells were fixed in 70% ice cold ethanol, rehydrated in PBS, blocked in PBST containing 1% BSA, incubated 1 h with mouse anti MPM-2 (Upstate, 1:800), washed three times in PBST, and incubated 1 h with goat-anti-mouse Alexa 488 (Molecular Probes, 1:1,000). Cells were then labeled by propidium iodide and immediately analyzed by flow cytometry.
Time-lapse microscopy
For phase contrast video microscopy, nonsynchronized cells were grown in glass bottom microwell dishes (MatTek) and filmed on a Zeiss Axiovert S100 equipped with an open microincubator (Harvard Apparatus, Inc.) with CO2 and a cooled CCD camera (Photometrics). Cells were treated as described in the legend of Figure 4. An image was captured every 4 minutes. To address caspase dependency, 100 μM Z-VAD-FMK was added after release from bleomycin and kept in the media throughout the experiment. Mitosis entry was blocked by adding 9 μM RO3306 (Calbiochem) simultaneously with UCN-01 and kept in the media throughout the experiment (Fig. 6A). Mitotic timing was measured from the first frame of the cell rounding up to the first frame of membrane constriction during telophase, or, in the case of cells not undergoing constriction to the first frame of membrane distortion, which takes place prior to mitotic exit.
Supplementary Material
Acknowledgments
The authors thank Bert Vogelstein (Johns Hopkins, Baltimore, USA) for the HCT116 cell lines, Bill Earnshaw (Johns Hopkins, Baltimore, USA) for the CENP-C antibody, and the UMMS Stem Cell Facility for stemcells. This work was supported by grants to William Theurkauf and Greenfield Sluder from the National Institute of General Medical Sciences, National Institutes of Health (RO1 GM50898 and RO1 GM30758, respectively).
Footnotes
Note: Supplementary materials can be found at: www.landesbioscience.com/supplement/VarmarkCC8-18-Sup.pdf
References
- 1.Bartek J, Lukas J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol. 2007;19:238–45. doi: 10.1016/j.ceb.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 2.Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science. 1996;274:1664–72. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- 3.Norbury CJ, Zhivotovsky B. DNA damage-induced apoptosis. Oncogene. 2004;23:2797–808. doi: 10.1038/sj.onc.1207532. [DOI] [PubMed] [Google Scholar]
- 4.Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–53. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 5.Sibon OC, Laurencon A, Hawley R, Theurkauf WE. The Drosophila ATM homologue Mei-41 has an essential checkpoint function at the midblastula transition. Curr Biol. 1999;9:302–12. doi: 10.1016/s0960-9822(99)80138-9. [DOI] [PubMed] [Google Scholar]
- 6.de Klein A, Muijtjens M, van Os R, Verhoeven Y, Smit B, Carr AM, et al. Targeted disruption of the cell cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol. 2000;10:479–82. doi: 10.1016/s0960-9822(00)00447-4. [DOI] [PubMed] [Google Scholar]
- 7.Takai H, Tominaga K, Motoyama N, Minamishima YA, Nagahama H, Tsukiyama T, et al. Aberrant cell cycle checkpoint function and early embryonic death in Chk1(-/-) mice. Genes Dev. 2000;14:1439–47. [PMC free article] [PubMed] [Google Scholar]
- 8.Brodsky MH, Sekelsky JJ, Tsang G, Hawley RS, Rubin GM. mus304 encodes a novel DNA damage checkpoint protein required during Drosophila development. Genes Dev. 2000;14:666–78. [PMC free article] [PubMed] [Google Scholar]
- 9.Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–96. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- 10.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 11.Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23:2825–37. doi: 10.1038/sj.onc.1207528. [DOI] [PubMed] [Google Scholar]
- 12.Chan TA, Hermeking H, Lengauer C, Kinzler KW, Vogelstein B. 14-3-3Sigma is required to prevent mitotic catastrophe after DNA damage. Nature. 1999;401:616–20. doi: 10.1038/44188. [DOI] [PubMed] [Google Scholar]
- 13.Andreassen PR, Lacroix FB, Lohez OD, Margolis RL. Neither p21WAF1 nor 14-3-3sigma prevents G2 progression to mitotic catastrophe in human colon carcinoma cells after DNA damage, but p21WAF1 induces stable G1 arrest in resulting tetraploid cells. Cancer Res. 2001;61:7660–8. [PubMed] [Google Scholar]
- 14.Roninson IB, Broude EV, Chang BD. If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resist Updat. 2001;4:303–13. doi: 10.1054/drup.2001.0213. [DOI] [PubMed] [Google Scholar]
- 15.Nitta M, Kobayashi O, Honda S, Hirota T, Kuninaka S, Marumoto T, et al. Spindle checkpoint function is required for mitotic catastrophe induced by DNA-damaging agents. Oncogene. 2004;23:6548–58. doi: 10.1038/sj.onc.1207873. [DOI] [PubMed] [Google Scholar]
- 16.Mikhailov A, Cole RW, Rieder CL. DNA damage during mitosis in human cells delays the metaphase/anaphase transition via the spindle-assembly checkpoint. Curr Biol. 2002;12:1797–806. doi: 10.1016/s0960-9822(02)01226-5. [DOI] [PubMed] [Google Scholar]
- 17.Vogel C, Hager C, Bastians H. Mechanisms of mitotic cell death induced by chemotherapy-mediated G2 checkpoint abrogation. Cancer Res. 2007;67:339–45. doi: 10.1158/0008-5472.CAN-06-2548. [DOI] [PubMed] [Google Scholar]
- 18.Morrison C, Rieder CL. Chromosome damage and progression into and through mitosis in vertebrates. DNA Repair (Amst) 2004;3:1133–9. doi: 10.1016/j.dnarep.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 19.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 20.Byrnes RW, Templin J, Sem D, Lyman S, Petering DH. Intracellular DNA strand scission and growth inhibition of Ehrlich ascites tumor cells by bleomycins. Cancer Res. 1990;50:5275–86. [PubMed] [Google Scholar]
- 21.Syljuasen RG, Jensen S, Bartek J, Lukas J. Adaptation to the ionizing radiation-induced G2 checkpoint occurs in human cells and depends on checkpoint kinase 1 and Polo-like kinase 1 kinases. Cancer Res. 2006;66:10253–7. doi: 10.1158/0008-5472.CAN-06-2144. [DOI] [PubMed] [Google Scholar]
- 22.Graves PR, Yu L, Schwarz JK, Gales J, Sausville EA, O'Connor PM, Piwnica-Worms H. The Chk1 protein kinase and the Cdc25C regulatory pathways are targets of the anticancer agent UCN-01. J Biol Chem. 2000;275:5600–5. doi: 10.1074/jbc.275.8.5600. [DOI] [PubMed] [Google Scholar]
- 23.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 24.Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J Biol Chem. 2000;275:9390–5. doi: 10.1074/jbc.275.13.9390. [DOI] [PubMed] [Google Scholar]
- 25.Lu C, Zhu F, Cho YY, Tang E, Zykova T, Ma WY, et al. Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol Cell. 2006;23:121–32. doi: 10.1016/j.molcel.2006.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yui S, Saeki T, Kanamoto R, Iwami K. Characteristics of apoptosis in HCT116 colon cancer cells induced by deoxycholic acid. J Biochem (Tokyo) 2005;138:151–7. doi: 10.1093/jb/mvi106. [DOI] [PubMed] [Google Scholar]
- 27.Saitoh H, Tomkiel J, Cooke CA, Ratrie H, 3rd, Maurer M, Rothfield NF. Earnshaw WC CENP-C, an autoantigen in scleroderma, is a component of the human inner kinetochore plate. Cell. 1992;70:115–25. doi: 10.1016/0092-8674(92)90538-n. [DOI] [PubMed] [Google Scholar]
- 28.Vogel C, Kienitz A, Muller R, Bastians H. The mitotic spindle checkpoint is a critical determinant for topoisomerase-based chemotherapy. J Biol Chem. 2005;280:4025–8. doi: 10.1074/jbc.C400545200. [DOI] [PubMed] [Google Scholar]
- 29.Carmena M, Earnshaw WC. The cellular geography of aurora kinases. Nat Rev Mol Cell Biol. 2003;4:842–54. doi: 10.1038/nrm1245. [DOI] [PubMed] [Google Scholar]
- 30.Peddibhotla S, Lam MH, Gonzalez-Rimbau M, Rosen JM. The DNA-damage effector checkpoint kinase 1 is essential for chromosome segregation and cytokinesis. Proc Natl Acad Sci USA. 2009;106:5159–64. doi: 10.1073/pnas.0806671106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nicholson DW. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 1999;6:1028–42. doi: 10.1038/sj.cdd.4400598. [DOI] [PubMed] [Google Scholar]
- 32.Syljuasen RG, Sorensen CS, Hansen LT, Fugger K, Lundin C, Johansson F, et al. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol. 2005;25:3553–62. doi: 10.1128/MCB.25.9.3553-3562.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smits VA, Klompmaker R, Arnaud L, Rijksen G, Nigg EA, Medema RH. Polo-like kinase-1 is a target of the DNA damage checkpoint. Nat Cell Biol. 2000;2:672–6. doi: 10.1038/35023629. [DOI] [PubMed] [Google Scholar]
- 34.Huang X, Tran T, Zhang L, Hatcher R, Zhang P. DNA damage-induced mitotic catastrophe is mediated by the Chk1-dependent mitotic exit DNA damage checkpoint. Proc Natl Acad Sci USA. 2005;102:1065–70. doi: 10.1073/pnas.0409130102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–16. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Skoufias DA, Lacroix FB, Andreassen PR, Wilson L, Margolis RL. Inhibition of DNA decatenation, but not DNA damage, arrests cells at metaphase. Mol Cell. 2004;15:977–90. doi: 10.1016/j.molcel.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 37.Vakifahmetoglu H, Olsson M, Zhivotovsky B. Death through a tragedy: mitotic catastrophe. Cell Death Differ. 2008;15:1153–62. doi: 10.1038/cdd.2008.47. [DOI] [PubMed] [Google Scholar]
- 38.Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–40. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 39.Allan LA, Clarke PR. Phosphorylation of caspase-9 by CDK1/cyclin B1 protects mitotic cells against apoptosis. Mol Cell. 2007;26:301–10. doi: 10.1016/j.molcel.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 40.Tse AN, Carvajal R, Schwartz GK. Targeting checkpoint kinase 1 in cancer therapeutics. Clin Cancer Res. 2007;13:1955–60. doi: 10.1158/1078-0432.CCR-06-2793. [DOI] [PubMed] [Google Scholar]
- 41.Yu L, Orlandi L, Wang P, Orr MS, Senderowicz AM, Sausville EA, et al. UCN-01 abrogates G2 arrest through a Cdc2-dependent pathway that is associated with inactivation of the Wee1Hu kinase and activation of the Cdc25C phosphatase. J Biol Chem. 1998;273:33455–64. doi: 10.1074/jbc.273.50.33455. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.