

Abstract
Energy homeostasis is critical for the survival of species. Therefore, multiple and complex mechanisms have evolved to regulate energy intake and expenditure to maintain body weight. For weight maintenance, not only does energy intake have to match energy expenditure, but also macronutrient intake must balance macronutrient oxidation. However, this equilibrium seems to be particularly difficult to achieve in individuals with low fat oxidation, low energy expenditure, low sympathetic activity or low levels of spontaneous physical activity, as in addition to excess energy intake, all of these factors explain the tendency of some people to gain weight. Additionally, large variability in weight change is observed when energy surplus is imposed experimentally or spontaneously. Clearly, the data suggest a strong genetic influence on body weight regulation implying a normal physiology in an ‘obesogenic’ environment. In this study, we also review evidence that carbohydrate balance may represent the potential signal that regulates energy homeostasis by impacting energy intake and body weight. Because of the small storage capacity for carbohydrate and its importance for metabolism in many tissues and organs, carbohydrate balance must be maintained at a given level. This drive for balance may in turn cause increased energy intake when consuming a diet high in fat and low in carbohydrate. If sustained over time, such an increase in energy intake cannot be detected by available methods, but may cause meaningful increases in body weight. The concept of metabolic flexibility and its impact on body weight regulation is also presented.
Keywords: substrate oxidation, carbohydrate balance, fat balance, energy balance, food intake
Introduction
Throughout evolution, humans and animals have evolved redundant mechanisms promoting the accumulation of fat during periods of feast to survive during periods of famine. However, what was an asset during evolution has become a liability in the current ‘pathoenvironment’ or ‘obesogenic’ environment.1 This hypothesis of the ‘thrifty genotype’ has, however, recently been challenged by Speakman2 who offers an alternative explanation called the ‘predation release’ hypothesis. Speakman2 argues that around 2 million years ago, predation was removed as a significant factor by the development of social behaviors, weapons, and fire. The absence of predation led to a change in the population distribution of body fatness due to random mutations and genetic drift. According to Speakman,2 such random drift, rather than directed selection, explains why some individuals are able to remain thin while living in an obesogenic environment.
Regardless of the origin of the genetic predisposition to obesity, the recent abrupt change in environmental conditions in which high-fat food is readily available and in which there is little need for physical activity has allowed obesity to reach epidemic proportions in both industrialized countries and in urbanized populations around the world.3 This epidemic is the result of a normal physiology (genetic variability) in a pathoenvironment (Figure 1). In the United States, in the early 2000′s, two out of three adult Americans were overweight or obese.6 More alarming, the prevalence of obesity is drastically increasing among children.7 The World Health Organization has identified obesity as one of the major emerging chronic diseases of the 21st century.3 Obesity increases the risk for a number of noncommunicable diseases, such as type 2 diabetes mellitus, hypertension, dyslipidemias and cardiovascular disease, and reduces life expectancy.8 In the United States alone, the yearly cost of obesity to the public health system is estimated to be more than $100 billion, representing between 5 and 10% of the US health care budget.9
Figure 1.

This figure depicts the potential effect of genes and environment on adiposity assessed by body mass index (BMI). Some of the concepts described in this figure were recently proposed by Bouchard.4 Our environment has evolved over the past century from a ‘traditional’ environment to a new ‘westernized’ environment. On the left part of the figure is presented the ‘traditional’ environment in which food was rather scarce and energy expenditure was high, mostly related to occupational physical activity. Such an environment leads to ‘leptogenic’ behaviors in which the variability of BMI will be dependent on the genetic propensity to weight gain of individuals. On the right part of the figure, the more recent modern ‘social’ and ‘built’ environment leads to obesogenic behaviors characterized by plenty of cheap high-calorie density food and little need for physical activity. Similarly, the variability in BMI will also depend on the genetic propensity to weight gain of individuals. Compared with the ‘obesogenic’ environment, the distribution of BMI will have a higher mean and higher standard deviation than that in the ‘leptogenic’ environment. Such a paradigm can be applied to populations with similar genetic background living in drastically different environment like the Pima Indians in Arizona and in Mexico.5
Obesity results from a chronic imbalance between energy intake and energy expenditure. Hyperphagia, a low metabolic rate, low rates of fat oxidation and an impaired sympathetic nervous activity characterize animal models of obesity. Similar metabolic factors have been found to characterize humans who are susceptible to weight gain. In this manuscript, we review the current knowledge of the role of daily energy expenditure and nutrient oxidation in the regulation of energy and substrate balances, and therefore, in the etiology of obesity. We will first introduce the concept of substrate balance by opposition to energy balance and its implication on body weight homeostasis. Then, we review studies in which energy balance has been perturbed by overfeeding, with particular focus on the variability in weight change. Finally, we discuss evidence about the potential role of carbohydrate balance on energy intake and body weight regulation in humans.
Energy balance vs substrate balance
Energy balance
The balance between energy intake and energy expenditure determines energy stores (Figure 2, upper panel). As living organisms must obey the first law of thermodynamics, the energy balance equation has been used to predict changes in body weight when energy intake or expenditure is changed. However, the classic equation of energy balance, which states that the body energy store is equal to energy intake minus energy expenditure, has provided both insight and confusion in the understanding of energy balance in humans. Much of the confusion comes from inappropriate use of the static equation of energy balance. Clearly, energy intake equates energy expenditure, when body weight and body composition are maintained. However, during energy imbalance, most scientists are using a static energy balance equation, which states that ‘change in energy stores=energy intake–energy expenditure’. However, Alpert10 has elegantly shown that this equation is inadequate, because it does not take into account the increasing energy expenditure with increasing weight or the reverse during weight loss.11–13 Thus, a small initial increase in energy intake sustained over a number of years ‘cannot lead to large weight gains’, as is often claimed.14,15 The valid equation to use is dynamic, and states that the ‘rate of change of energy stores=rate of energy intake–rate of energy expenditure’. The use of ‘rates’ in this equation introduces time dependency, thereby allowing the effect of changing energy stores (especially fat-free mass and weight) on energy expenditure to enter into the calculation.10
Figure 2.

The major components of body weight regulation in an obesogenic environment are described in this figure. Body weight in adulthood is most likely to be the result of two key components; (a) changes in the environment of subsequent generations that influence genetic and epigenetic propensity for weight gain, and (b) the current habitual lifestyle that promotes sedentary behaviors and provides an oversupply of energy dense foods. The daily energy and nutrient balance of a 70-kg man (20% body fat) in relationship to macronutrient energy stores, intake and oxidation. Each macronutrient intake and oxidation on a 2500 kcal day−1 standard American diet (composition 40% fat, 40% carbohydrate and 20% protein) is shown on the left as absolute intake in kilocalories and on the right as a percentage of its respective nutrient store. Because carbohydrate, protein and alcohol intakes and oxidation rates are tightly regulated on a daily basis, any inherent differences between energy intake and energy expenditure therefore predominantly impact body fat stores. During overfeeding (shown in red), the oxidation of carbohydrate and protein is increased to compensate for the increased intake at the expense of fat intake, yet the increase in oxidation is not equally coupled with intake. Thus, if sustained fat kilocalories are stored, fat stores will expand and the body weight is gained.
Substrate balance
If the origins of a positive energy balance lie in the chronic imbalance of energy intake and expenditure, an appropriate question is ‘what conditions allow a long-lasting imbalance between intake and expenditure?’ An examination of each nutrient balance to determine whether a chronic imbalance between nutrient intake and oxidation exists is valid only if each nutrient has a separate balance equation, implying separate regulation (Figure 2, lower panel). In practical terms, is each nutrient either oxidized or stored in its own compartment (separate regulation)?, or does it get converted into another compartment for storage? This applies particularly to the issue of whether dietary carbohydrate is stored as fat by de novo lipogenesis. In contrast to animals,16 de novo lipogenesis is very limited in humans and occurs only when large excesses of carbohydrate are ingested.17–21 As a consequence, one should consider each nutrient balance equation as a separate entity.
Protein balance
Protein intake is usually about 15% of dietary energy and the protein stores in the body represent about one-third of the total stored energy in a 70 kg man. The daily protein intake amounts to a little over 1% of the total protein stores22,23 (Figure 2, lower panel). The protein stores increase in size only in response to growth stimuli, such as growth hormone, androgens, physical training and weight gain, but not simply from increased dietary protein. Protein stores are, therefore, tightly controlled and, on a day-to-day basis, protein balance is achieved.24 As a consequence, protein imbalance cannot be implicated as a direct cause of obesity, but protein intake may affect fat balance.24
Carbohydrate balance
Carbohydrate is usually the main source of dietary energy, yet the body stores of glycogen are very limited: 500–1000 g.21 Daily intake of carbohydrate corresponds to about 50–100% of the carbohydrate stores compared with about 1% for protein and fat25 (Figure 2, lower panel), so that over a period of hours and days, the carbohydrate stores fluctuate markedly compared with those of protein and fat. However, similar to the protein stores, they are tightly controlled,24 even though the mechanisms of this control (humoral and/or nervous) remain to be established. This implies that excess carbohydrate intake cannot be the basis of weight gain because the storage capacity is limited and controlled, and conversion to fat only occurs under extreme conditions in humans.21
Fat balance
In marked contrast to the other nutrients, body fat stores are large, and fat intake has no or very little influence on fat oxidation.25,26 As with protein, the daily fat intake represents less than 1% of the total fat stores; however, the fat stores contain about six times the energy of the protein stores23 (Figure 2, lower panel). The fat stores represent an energy buffer for the body, and the slope of the relationship between energy balance and fat balance is equal to one in conditions of day-to-day small positive or negative energy imbalances.24 A deficit of 200 kcal over 24 h means 200 kcal comes from fat stores, and the same holds true for an excess of 200 kcal, which ends up in fat stores. In conditions of spontaneous overfeeding, the entire excess fat intake is stored as body fat.27
What promotes fat oxidation if it is not dietary fat intake? The amount of total body fat exerts a small, but significant, effect on fat oxidation, and this promotion of fat oxidation at higher body fat levels may represent a mechanism for attenuating the rate of weight gain in response to chronic overfeeding.28
Alcohol balance
There is an inconsistent relationship between reported alcohol intake and body mass index.29,30 However, it has been shown that in healthy individuals, the fate of ingested alcohol is oxidation and not storage, and therefore perfect alcohol balance is achieved.31 Similar to dietary carbohydrate and protein, alcohol diverts dietary fat away from oxidation and toward storage and inhibits lipolysis. Therefore, a chronic imbalance between alcohol intake and oxidation cannot directly cause obesity, although it may indirectly influence fat balance.
In summary, when one considers energy balance in humans under physiological conditions, fat is the only nutrient capable of causing a chronic imbalance between intake and oxidation, and thus directly contributing to the increase in adipose tissue. The other nutrients will indirectly influence adiposity by their contribution to overall energy balance, and thus fat balance, as emphasized by Frayn.32 The use of the fat balance equation instead of the energy balance equation offers a new framework for understanding the pathogenesis of obesity.
Energy metabolism as a predictor of weight gain
Cross-sectional studies that compare lean and obese individuals have added little to our understanding of the physiological mechanisms predisposing to weight gain.11 An understanding of the etiology of human obesity requires longitudinal studies to identify predictors or risk factors for weight gain. Several studies have prospectively examined these predictors in the Pima Indian population in south western Arizona, a population where obesity is extremely prevalent33 and weight gain is common among young adults. In these individuals, at least four metabolic parameters have been found to be predictive of weight gain—low metabolic rate, low spontaneous physical activity, low sympathetic nervous system (SNS) activity and low fat oxidation.
Low metabolic rate
Obesity is associated with a high absolute metabolic rate, both in resting conditions and over 24 h,12,13 and therefore, truly cannot be caused by a low absolute metabolic rate as often proposed. However, it is important to note that there is wide variability in the relationship between metabolic rate and body size suggesting that at any given body size, individuals can have high, normal or low relative metabolic rates. Studies in adult nondiabetic Pima Indians identified that a low relative metabolic rate (adjusted for differences in fat-free mass, fat mass, age and sex) was a risk factor for body weight gain.34 Over a 4-year follow-up, we found that the risk of gaining 10 kg in body weight was approximately eight times greater in those individuals within the lowest tertile of resting metabolic rate (RMR) compared with those within the highest tertile of RMR. These findings were later confirmed in an independent group of nondiabetic Pima Indians where weight change ( −9 to 26 kg) over 4 years of follow-up was negatively associated with adjusted RMR.35 Moreover, a meta-analysis of 12 studies showed that high rates of weight regain were related to a 3–5% lower RMR in formerly obese compared with control individuals.36 In contrast, relative low energy expenditure was not found to be a predictor of weight gain in other adult populations.37,38 In studies of the Pima Indians, it is important to note that average weight gain could not entirely be accounted for by lower rates of energy expenditure and that only 30–40% of the increase in body energy stores was attributable to a deficit in energy expenditure. These results indicate that energy intake and physical activity also contribute to the observed variability in weight gain. Interestingly, in response to weight gain, the new adjusted metabolic rate increases to the normal levels and is comparable to the metabolic rates of individuals who remained weight-stable. Therefore, weight gain seems to be the price to pay to normalize energy metabolism.
Low spontaneous activity
Another component of 24-h energy expenditure is the energy cost of spontaneous physical activity, which accounts for 8–15% of total daily expenditure.12 Consistent with the cross-sectional observation of a decreased spontaneous physical activity in obese individuals, longitudinal studies in Pima Indians showed that spontaneous physical activity is a familial trait inversely related to weight and fat mass gain at least in men.39 Although it could be argued that spontaneous physical activity during a respiratory chamber study is limited, it was found to be highly correlated with habitual physical activity in free-living conditions.40 However, in a prospective study in which free-living physical activity was measured by doubly labeled water in 92 nondiabetic Pima Indians, activity energy expenditure or the level of physical activity was not associated with changes in body weight.35 Levine et al.41 fed 16 sedentary individuals an extra 1000 kcal day−1 for 8 weeks and measured free-living activity and changes in body weight. Despite no changes in voluntary physical activity or energy wastage through fecal losses, fat gain varied more than 10-fold among individuals, ranging from 0.36 to 4.23 kg, and was inversely related to the increase in total energy expenditure. Because changes in RMR and thermic effect of food were small, the resistance to weight and fat gain with overfeeding was attributed to changes in spontaneous physical activity ranging from −98 to 692 kcal day−1. We recently reviewed the literature on spontaneous physical activity and the control of body weight.42
Low SNS activity
Studies in Caucasians indicate that the activity of the SNS is related to each of the major components of energy expenditure: RMR,43 the thermic effect of food,44 spontaneous physical activity45 and 24-h respiratory quotient.46 Importantly, cross-sectional studies indicate that individuals (Pima Indians) prone to obesity have lower rates of muscle sympathetic activity compared with weight-matched Caucasians.43 Consequently, we prospectively studied the role of impaired SNS activity and/or adrenal medullary function in the etiology of human obesity in Pima Indian men. At follow-up, body-weight change was negatively correlated with baseline urinary norepinephrine excretion rate, whereas the changes in waist-to-thigh circumference ratio negatively correlated with baseline epinephrine excretion rate.47 Together, these results show that an impaired SNS activity and a low activity of the adrenal medulla axis are associated with body weight gain and central obesity. Consistently, a low SNS activity was associated with poor weight loss outcomes in obese individuals treated with a dietary restriction intervention.48
Low fat oxidation
As reviewed above, the composition of nutrient intake is an important factor in the development of obesity, and consequently, one expects that the composition of nutrient oxidation also plays a role in its etiology. The nonprotein respiratory quotient (RQ) is an index of the ratio of carbohydrate to fat oxidation, and fasting values of 0.80 after an overnight fast indicate a major reliance on fat oxidation,49 whereas values approaching 1.00 following ingestion of a carbohydrate meal indicate reliance on carbohydrate as the major energy substrate.26,50 Apart from the obvious impact of diet composition, the RQ is also influenced by recent energy balance, sex, adiposity, and importantly family membership, suggesting genetic determinants.50,51
A longitudinal study of Pima Indians showed that a high 24-h RQ predicted weight gain, with low fat oxidizers (90th percentile for respiratory quotient) having a 2.5 times greater risk of gaining 5 kg or more body weight than high fat oxidizers (10th percentile for respiratory quotient).50 This effect was independent of a relatively low or high 24-h metabolic rate. Similar results have been reported in Caucasians.52 Others have shown that postobese volunteers have low rates of fat oxidation,53,54 and those successful at maintaining weight loss have higher fat oxidation rates than those experiencing weight relapse.55
As shown in Figure 3, together these four predictors of weight gain explained approximately 30% of the variability in weight gain in Pima Indians.
Figure 3.
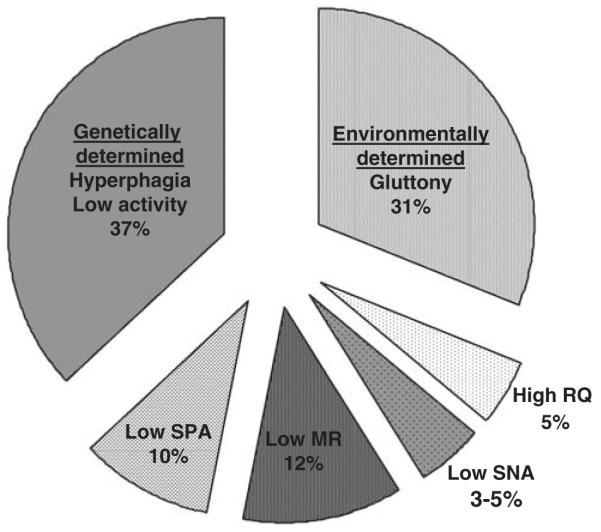
Studies of monozygotic twins reared apart indicate that approximately one-third of the variability in body mass index (BMI) is attributable to nongenetic factors and two-thirds to genetic factors. In this figure, we broke down the genetic part of the variability in BMI into the effects of metabolic rate (MR), respiratory quotient (RQ), spontaneous physical activity (SPA; fidgeting) and sympathetic nervous activity (SNA). The remaining genetic variability is assumed to be related to the variability in food intake and physical activity. Values for MR, RQ, SPA and SNA were calculated from prospective studies conducted among the Pima Indians of Arizona.
Weight changes in response to overfeeding
The weight gain observed after overfeeding is quite variable across individuals and varies up to four times.56 Dietary compliance is an important factor to explain such variability, but well-controlled studies have shown similar variability in weight gain. Such differences in weight gain can first be explained by our inability to assess weight-maintenance energy requirements, and therefore the actual energy excess. Secondly, differences in digestion and absorption may modify the amount of ‘bioavailable’ energy, affecting the actual positive energy balance. Third, the composition of weight gain (fat mass and lean mass) makes a difference in weight gain, as the energy cost of protein deposition is higher than that of adipose tissue. Fourth, dietary protein content was suggested by Stock56 to be a critical determinant of weight gain during overfeeding, as low and high protein diets seem to increase the energy cost to deposit 1 kg of body weight. Finally, differences in mitochondrial energy efficiency may also represent an underlying cause of the variability in weight gain.
Bouchard et al.57 undertook a very well-controlled study to determine whether there are true differences in the responses among individuals to long-term overfeeding and to assess the possibility that genotypes are involved in such differences. In response to 84 days of 1000 kcal day−1 of overfeeding, 12 pairs of monozygotic twins gained on average 8.1 kg, but the range was from 4.3 to 13.3 kg. However, the similarity within each twin pair in response to overfeeding was significant with respect to body weight gain, percentage of fat and total fat mass with about three times more variance among pairs than within pairs. The similarity in the adaptation to long-term overfeeding within the pairs of twins clearly indicated that genetic factors are involved in the partitioning between fat and lean mass deposition and in determining the energy expenditure response.
The fact that the increase in body weight was not proportional to the excess energy intake led Neumann58 to propose that some of the excess ingested energy is dissipated as heat or ‘luxuskonsumption’. This concept was revived later by Miller et al.,59 and was supported by the findings of the Vermont studies in prisoners in whom almost 50% more energy intake was necessary to maintain their new body weights.60 However, at present, the role of thermogenesis in body weight regulation is still a matter of debate, and the mechanisms underlying the variability in thermogenesis have not been identified. Recently, Befroy et al.61 provided insightful data about differences in muscle energy efficiency between trained and untrained people. They found that the in vivo muscle fuel oxidation (Krebs cycle flux) was higher in trained than in untrained individuals. Using the ATP synthesis flux to Krebs cycle flux ratio as a marker of mitochondrial coupling,62,63 lower energy efficiency was observed in trained vs untrained individuals. Such results are in line with the observation that after intense endurance exercise, in vitro muscle energy efficiency was decreased in humans.64 Similarly, Rosenbaum et al.65 showed that maintenance of reduced or elevated body weight results in respective decrease or increase in energy expended in physical activity. Furthermore, at reduced body weight, muscle work efficiency was increased by approximately 25% measured by both cycle ergometry and magnetic resonance spectroscopy (ATP flux change). In contrast, weight gain resulted in decreased muscle work efficiency by ergometry. Changes in muscle efficiency at altered body weight accounted for 35% of the change in daily energy expended in physical activity.
Understanding the cellular and molecular mechanisms behind these metabolic processes influencing energy efficiency of weight maintenance or weight gain may be fruitful to uncover novel therapeutic targets.
Carbohydrate balance and energy homeostasis
As described above, body mass is stable when macronutrient ingestion is equivalent to macronutrient oxidation. When food supply is restricted, a number of circulating substrates, peptides and hormones act on the hypothalamus, the brain stem and the autonomic nervous system to increase appetite in an effort to reestablish macronutrient balance.66 It is suggested that carbohydrate imbalance may be responsible for generating this compensatory response67,68 due to the body’s limited ability to store excess carbohydrate intake. Reduced carbohydrate intake or increased carbohydrate oxidation can rapidly decrease the body glycogen content. Interestingly, a reduction in glycogen stores in liver or muscle stimulates AMP-activated protein kinase, a cellular energy sensor,69 which in turn causes an increase in lipid oxidation, and thus decreasing carbohydrate oxidation and preserving carbohydrate stores. Furthermore, depleted glycogen stores are associated with increased ad libitum food intake in mice.68 In summary, there is evidence suggesting that low carbohydrate stores may trigger adaptive changes in fuel oxidation and eventually in energy intake.
Carbohydrate balance and energy intake in humans
Energy deficit is a potent stimulus promoting energy intake. Following a 1-day energy deficit by removing fat or carbohydrate, Goldberg et al.70 assessed 24-h ad libitum energy intake. They found a compensatory increase in food intake after the 1-day energy restriction period, but without any difference between fat or carbohydrate restriction. As lower carbohydrate intake was quickly compensated by reduced carbohydrate oxidation, carbohydrate balance was maintained after the carbohydrate-restricted diet. Several other studies have been conducted to investigate the role of carbohydrate or glycogen depletion on food intake assessed for 1 or 2 days after the carbohydrate balance manipulation.71–73 For instance, using low- and high-carbohydrate diets combined with or without intense exercise, changes by at least 40% in carbohydrate balance or muscle glycogen content were observed.71–73 However, such manipulation in carbohydrate stores was insufficient to induce changes in ad libitum energy intake. Common to all these studies is that changes in macronutrient intake were mostly compensated for by rapid adjustments in carbohydrate oxidation. In contrast, when ad libitum food intake was assessed for a longer time period (3 days), and no manipulation of carbohydrate store was intended besides spontaneous differences under weight-maintenance conditions, Pannacciulli et al.74 observed that ad libitum energy intake was inversely related with 24-h carbohydrate balance (Table 1).
Table 1.
Studies assessing the role of carbohydrate stores on energy intake or body weight
| Study | Study design (n) | Difference in carbohydrate balance or glycogen store |
Difference in EI or BW |
|---|---|---|---|
| Goldberg et al.70 | Crossover; 1-day energy restriction by removing fat or carbohydrate (9) |
120±38 kcal day−1 (P=0.09) | 86±72 kcal day−1 (NS) |
| Snitker et al.71 | Crossover; 2-day isoenergetic diet, low or high in carbohydrate (8) |
130mmol glucose per kg dry muscle (46%; P<0.001) |
244 kcal day−1 (NS) |
| Shetty et al.72 | Crossover; 2-day isoenergetic diet high, medium or low in carbohydrate (6) |
1233 kcal 48 h−1 (P<0.05)a | 36 kcal 36 h−1 (P=NS) |
| Stubbs et al.73 | Crossover; 1-day isoenergetic diet, high or low in carbohydrate (9) |
576±53 kcal day−1 (P<0.05) | 2 kcal day−1 (P=NS) |
| Pannacciulli et al.74 | Cross-sectional; 3-day isoenergetic diet followed for a 3-day ad libitum food intake period (112) |
−476−533 kcal day−1 (range) | Carbohydrate balance inversely correlated with ad libitum energy intake (r=−0.34; P<0.001). |
| Eckel et al.76 | Longitudinal; Measurement of carbohydrate balance after 15-day isoenergetic, high-carbohydrate diet followed by weight gain determination after 4 years (36) |
642±85 kcal day−1 (end of high-carbohydrate diet) |
For each 100 kcal day−1 in carbohydrate balance, weight gain was 81±37 g per year lower (P=0.04) |
Abbreviations: BW, body weight; EI, energy intake; NS, not significant.
Difference between high- and low-carbohydrate diet. Values are mean±s.e.
Unfortunately, even together these studies do not definitively answer whether carbohydrate balance plays a significant role in the regulation of food intake in humans. A common limitation to all these studies is the relatively artificial conditions under which ad libitum food intake is assessed. Most studies report much higher energy intake relative to energy requirement when food intake is measured in laboratory conditions.27 Furthermore, suppression of carbohydrate oxidation seems to be the main mechanism by which further carbohydrate depletion is prevented in response to reduced carbohydrate intake. Importantly, the regulation of energy intake relies on multiple and redundant homeostatic mechanisms involved in the maintenance of body weight and energy balance. Finally, more complexity has to be considered for the regulation of energy balance in humans, as factors such as cognition, emotion and reward all affect food intake behaviors (Figure 2, upper panel). Therefore, it can be anticipated that any role of carbohydrate balance on food intake will be difficult to observe when energy intake is assessed over a short period of time.
So far, no signals in the liver and/or muscle have been identified to explain the potential effect of glycogen content on food intake. Most of the known circulating signals involved in food intake regulation are coming from tissues with no glycogen stores, such as the adipose tissue, the gut or the pancreas. If glycogen content plays a role in the regulation of food intake, this must be accompanied by the release of neuronal and/or humoral signals from the liver and/or muscle. The presence of vagal afferences from the liver to the central nervous system has been suggested to play this role.75
Carbohydrate balance and weight gain in humans
Another way to assess the potential impact of carbohydrate balance on energy homeostasis is to evaluate long-term changes in body weight. Experimentally, undetectable changes in daily food intake could, however, be meaningful after months or years. We, therefore, hypothesized that individuals with reduced carbohydrate stores should gain more weight than those with larger carbohydrate stores. Accordingly, we evaluated the impact of 24-h RQ under controlled energy balance and diet composition on spontaneous weight gain over a 25±11-month period.50 Even after controlling for energy balance and body fat content, a significant association between 24-h RQ and weight gain was observed. Even though carbohydrate balance was not measured in this study, higher carbohydrate oxidation as indicated by the increased 24-h RQ can reduce carbohydrate stores and increase energy intake.
In support of Zurlo’s data,50 Eckel et al.76 reported that individuals with increased carbohydrate balance at the end of a 15-day high-carbohydrate diet gained less body weight and fat mass over the next 4 years than those with lower carbohydrate balances. For each additional daily 25 g (100 kcal day−1) in carbohydrate balance, participants gained 80 g less weight per year, whereas a difference by one standard deviation for carbohydrate balance (510 kcal day−1) could explain a difference of 400 g per year in body weight gain. Such estimates show that the energy excess required to account for a change in body weight is very small and probably impossible to detect when energy intake is evaluated for a few days.
Metabolic flexibility and body weight regulation
Metabolic flexibility is defined by the capacity of the body or cells to match fuel oxidation to fuel availability and the endocrine environment. It is typically assessed by (1) the increase in RQ from fasting to glucose- and insulin-stimulated conditions; (2) the reduction in RQ during overnight fasting or (3) the macronutrient oxidation adaptation in response to isoenergetic changes in diet composition.77,78 In the context of food intake regulation, an impaired metabolic flexibility may induce differential macronutrient imbalance. Under body weight stability, the food quotient (RQ of the diet) matches 24-h RQ, and therefore macronutrient oxidation rates. When the macronutrient composition of the diet is modified, fuel oxidation must be adjusted to achieve a new equilibrium. The rate at which this adjustment is achieved is faster in the transition from a low-carbohydrate to high-carbohydrate diet (~2 days); however, in response to an increase in dietary fat content, fat oxidation can take more than 1 week to match fat intake.79 Additionally, there is a large interindividual variability in the time required to achieve this new macronutrient equilibrium.80–82 Thus, increased dietary fat may cause more depletion of the carbohydrate stores in those individuals with impaired capacity to upregulate lipid oxidation, and may therefore be a signal promoting food intake. Even though the proper studies to prove this hypothesis have not been conducted, one can anticipate that differences in metabolic (in)flexibility may influence energy intake, and therefore be relevant to the regulation of body weight gain over the course of years.
Conclusions
The recent large increase in the prevalence of obesity has been mostly caused by our modern urban societies in which demand for physical activity is extremely reduced, and highly palatable and relatively cheap food is ubiquitously available. However, in this ‘obesogenic’ environment, there are still many people who are protected against obesity. As reviewed above, high levels of fat oxidation, metabolic rate, spontaneous physical activity and sympathetic activity are all protective factors against weight gain. It is, however, not clear how these factors are altering the balance between intake and expenditure of energy. For instance, subjects with low fat oxidation have increased carbohydrate oxidation to sustain the ATP demand, and therefore have decreased carbohydrate stores. A low carbohydrate reserve has been associated with increased weight gain, probably mediated through small but significant changes in energy intake. The mechanisms underlying the genetic variability in substrate oxidation are not known, but may explain why there is such a wide interindividual variability in weight gain in our modern ‘westernized’ lifestyle. Of course, factors such as cognition, emotion and reward all affect food intake behaviors and may explain why some individuals are resistant to large weight gains.
Additionally, differences in energy efficiency of growth and/or of weight maintenance are most likely to play a role in the susceptibly to weight gain in our present environment. Unlike measurements of food intake, we have reasonably accurate and precise methods to measure energy metabolism in humans. Whether the precision is sufficient to detect the necessary small chronic imbalances leading to obesity is debatable, especially with a lack of means to measure food intake. Uncovering the mechanisms underlying intersubject variability in energy metabolism (expenditure and fuel partitioning) will probably enhance our ability to identify people at risk for weight gain. As an example, recent investigations have provided evidence about differences in skeletal muscle efficiency to generate ATP as a function of training status or in response to weight perturbation. More research is needed to confirm these preliminary findings and to assess its impact on whole-body energy efficiency and body weight regulation. Perhaps, the evidence showing that low whole-body metabolic rate, low sympathetic activity and low spontaneous physical activity are predictors of weight gain will converge on new discoveries regarding the energy efficiency of the maintenance of tissues and organs. But, we absolutely need accurate and precise novel creative methods to measure energy intake from fat, carbohydrate and protein in free-living conditions.
Conversely, one must acknowledge that remarkable progress has been made in the past two decades regarding our understanding of both the complex disease called obesity and the regulation of body weight. Now, we need to translate this new knowledge into the prevention of obesity by establishing innovative public health policies and redesigning our built environment. Even if we do not have the reliable technology to identify those subjects at risk for obesity, one can expect new health policies to curb the present pandemic of obesity and its future cost to health care systems. However, we will need major technological break-throughs to identify the susceptibility of individuals to obesity even though they may live in a less ‘obesogenic’ environment. We hope to attend the 40-year celebration of the Pennington Biomedical Research Center to witness the results of another 20 years of important research.
Acknowledgements
Jose Galgani is supported by a fellowship from the International Nutrition Foundation/Ellison Medical Foundation. Eric Ravussin is supported by grants funded by the National Institutes of Health, U01-AG020478 and RO1-DK60412-06A1.
Footnotes
Conflict of interest The authors have declared no financial interests.
References
- 1.Ravussin E. Obesity in britain. Rising trend may be due to ‘pathoenvironment’. BMJ. 1995;311:1569. doi: 10.1136/bmj.311.7019.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Speakman JR. A nonadaptive scenario explaining the genetic predisposition to obesity: the ‘predation release’ hypothesis. Cell Metab. 2007;6:5–12. doi: 10.1016/j.cmet.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 3.Obesity: preventing and managing the global epidemic. Report of a WHO consultation. World Health Organ Tech Rep Ser. 2000;894:i–xii. 1–253. [PubMed] [Google Scholar]
- 4.Bouchard C. The biological predisposition to obesity: beyond the thrifty genotype scenario. Int J Obes (Lond) 2007;31:1337–1339. doi: 10.1038/sj.ijo.0803610. [DOI] [PubMed] [Google Scholar]
- 5.Schulz LO, Bennett PH, Ravussin E, Kidd JR, Kidd KK, Esparza J, et al. Effects of traditional and western environments on prevalence of type 2 diabetes in Pima Indians in Mexico and the US. Diabetes Care. 2006;29:1866–1871. doi: 10.2337/dc06-0138. [DOI] [PubMed] [Google Scholar]
- 6.Flegal KM, Carroll MD, Ogden CL, Johnson CL. Prevalence and trends in obesity among US adults, 1999–2000. JAMA. 2002;288:1723–1727. doi: 10.1001/jama.288.14.1723. [DOI] [PubMed] [Google Scholar]
- 7.Troiano RP, Flegal KM. Overweight children and adolescents: description, epidemiology, and demographics. Pediatrics. 1998;101:497–504. [PubMed] [Google Scholar]
- 8.Must A, Spadano J, Coakley EH, Field AE, Colditz G, Dietz WH. The disease burden associated with overweight and obesity. JAMA. 1999;282:1523–1529. doi: 10.1001/jama.282.16.1523. [DOI] [PubMed] [Google Scholar]
- 9.Wolf AM, Colditz GA. Social and economic effects of body weight in the United States. Am J Clin Nutr. 1996;63:466S–469S. doi: 10.1093/ajcn/63.3.466. [DOI] [PubMed] [Google Scholar]
- 10.Alpert SS. Growth, thermogenesis, and hyperphagia. Am J Clin Nutr. 1990;52:784–792. doi: 10.1093/ajcn/52.5.784. [DOI] [PubMed] [Google Scholar]
- 11.Ravussin E, Swinburn BA. Metabolic predictors of obesity: crosssectional versus longitudinal data. Int J Obes Relat Metab Disord. 1993;17:S28–S31. [PubMed] [Google Scholar]
- 12.Ravussin E, Lillioja S, Anderson TE, Christin L, Bogardus C. Determinants of 24-h energy expenditure in man. Methods and results using a respiratory chamber. J Clin Invest. 1986;78:1568–1578. doi: 10.1172/JCI112749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weyer C, S Snitker, Rising R, Bogardus C, Ravussin E. Determinants of energy expenditure and fuel utilization in man: effects of body composition, age, sex, ethnicity and glucose tolerance in 916 subjects. Int J Obes Relat Metab Disord. 1999;23:715–722. doi: 10.1038/sj.ijo.0800910. [DOI] [PubMed] [Google Scholar]
- 14.Hill JO, Peters JC. Environmental contributions to the obesity epidemic. Science. 1998;280:1371–1374. doi: 10.1126/science.280.5368.1371. [DOI] [PubMed] [Google Scholar]
- 15.Hill JO, Wyatt HR, Reed GW, Peters JC. Obesity and the environment: where do we go from here? Science. 2003;299:853–855. doi: 10.1126/science.1079857. [DOI] [PubMed] [Google Scholar]
- 16.Zelewski M, Swierczynski J. Comparative studies on lipogenic enzyme activities in the liver of human and some animal species. Comp Biochem Physiol B. 1990;95:469–472. doi: 10.1016/0305-0491(90)90004-d. [DOI] [PubMed] [Google Scholar]
- 17.Chascione C, Elwyn DH, Davila M, Gil KM, Askanazi J, Kinney JM. Effect of carbohydrate intake on de novo lipogenesis in human adipose tissue. Am J Physiol. 1987;253:E664–E669. doi: 10.1152/ajpendo.1987.253.6.E664. [DOI] [PubMed] [Google Scholar]
- 18.Just B, Messing B, Darmaun D, Rongier M, Camillo E. Comparison of substrate utilization by indirect calorimetry during cyclic and continuous total parenteral nutrition. Am J Clin Nutr. 1990;51:107–111. doi: 10.1093/ajcn/51.1.107. [DOI] [PubMed] [Google Scholar]
- 19.Manji S, Shikora S, McMahon M, Blackburn GL, Bistrian BR. Peritoneal dialysis for acute renal failure: overfeeding resulting from dextrose absorbed during dialysis. Crit Care Med. 1990;18:29–31. doi: 10.1097/00003246-199001000-00008. [DOI] [PubMed] [Google Scholar]
- 20.Acheson KJ, Schutz Y, Bessard T, Flatt JP, Jequier E. Carbohydrate metabolism and de novo lipogenesis in human obesity. Am J Clin Nutr. 1987;45:78–85. doi: 10.1093/ajcn/45.1.78. [DOI] [PubMed] [Google Scholar]
- 21.Acheson KJ, Schutz Y, Bessard T, Anantharaman K, Flatt JP, Jequier E. Glycogen storage capacity and de novo lipogenesis during massive carbohydrate overfeeding in man. Am J Clin Nutr. 1988;48:240–247. doi: 10.1093/ajcn/48.2.240. [DOI] [PubMed] [Google Scholar]
- 22.Snyder WS, Cook NJ, Nasset ES, Karhausen LR, Howells GP, Tipton IH. Report of the task group on reference man. The International Commission on Radiological Protection. no. 23 edn Pergamon Press; New York: 1974. [Google Scholar]
- 23.Bray GA. Treatment for obesity: a nutrient balance/nutrient partition approach. Nutr Rev. 1991;49:33–45. doi: 10.1111/j.1753-4887.1991.tb02990.x. [DOI] [PubMed] [Google Scholar]
- 24.Abbott WG, Howard BV, Christin L, Freymond D, Lillioja S, Boyce VL, et al. Short-term energy balance: relationship with protein, carbohydrate, and fat balances. Am J Physiol. 1988;255:E332–E337. doi: 10.1152/ajpendo.1988.255.3.E332. [DOI] [PubMed] [Google Scholar]
- 25.Schutz Y, Flatt JP, Jequier E. Failure of dietary fat intake to promote fat oxidation: a factor favoring the development of obesity. Am J Clin Nutr. 1989;50:307–314. doi: 10.1093/ajcn/50.2.307. [DOI] [PubMed] [Google Scholar]
- 26.Flatt JP, Ravussin E, Acheson KJ, Jequier E. Effects of dietary fat on postprandial substrate oxidation and on carbohydrate and fat balances. J Clin Invest. 1985;76:1019–1024. doi: 10.1172/JCI112054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rising R, Alger S, Boyce V, Seagle H, Ferraro R, Fontvieille AM, et al. Food intake measured by an automated food-selection system: relationship to energy expenditure. Am J Clin Nutr. 1992;55:343–349. doi: 10.1093/ajcn/55.2.343. [DOI] [PubMed] [Google Scholar]
- 28.Schutz Y, Tremblay A, Weinsier RL, Nelson KM. Role of fat oxidation in the long-term stabilization of body weight in obese women. Am J Clin Nutr. 1992;55:670–674. doi: 10.1093/ajcn/55.3.670. [DOI] [PubMed] [Google Scholar]
- 29.Hellerstedt WL, Jeffery RW, Murray DM. The association between alcohol intake and adiposity in the general population. Am J Epidemiol. 1990;132:594–611. doi: 10.1093/oxfordjournals.aje.a115703. [DOI] [PubMed] [Google Scholar]
- 30.Colditz GA, Giovannucci E, Rimm EB, Stampfer MJ, Rosner B, Speizer FE, et al. Alcohol intake in relation to diet and obesity in women and men. Am J Clin Nutr. 1991;54:49–55. doi: 10.1093/ajcn/54.1.49. [DOI] [PubMed] [Google Scholar]
- 31.Shelmet JJ, Reichard GA, Skutches CL, Hoeldtke RD, Owen OE, Boden G. Ethanol causes acute inhibition of carbohydrate, fat, and protein oxidation and insulin resistance. J Clin Invest. 1988;81:1137–1145. doi: 10.1172/JCI113428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frayn KN. Physiological regulation of macronutrient balance. Int J Obes Relat Metab Disord. 1995;19:S4–S10. [PubMed] [Google Scholar]
- 33.Knowler WC, Pettitt DJ, Saad MF, Charles MA, Nelson RG, Howard BV, et al. Obesity in the Pima Indians: its magnitude and relationship with diabetes. Am J Clin Nutr. 1991;53:1543S–1551S. doi: 10.1093/ajcn/53.6.1543S. [DOI] [PubMed] [Google Scholar]
- 34.Ravussin E, Lillioja S, Knowler WC, Christin L, Freymond D, Abbott WG, et al. Reduced rate of energy expenditure as a risk factor for body-weight gain. N Engl J Med. 1988;318:467–472. doi: 10.1056/NEJM198802253180802. [DOI] [PubMed] [Google Scholar]
- 35.Tataranni PA, Harper IT, Snitker S, Del Parigi A, Vozarova B, Bunt J, et al. Body weight gain in free-living Pima Indians: effect of energy intake vs expenditure. Int J Obes Relat Metab Disord. 2003;27:1578–1583. doi: 10.1038/sj.ijo.0802469. [DOI] [PubMed] [Google Scholar]
- 36.Astrup A, Gotzsche PC, van de Werken K, Ranneries C, Toubro S, Raben A, et al. Meta-analysis of resting metabolic rate in formerly obese subjects. Am J Clin Nutr. 1999;69:1117–1122. doi: 10.1093/ajcn/69.6.1117. [DOI] [PubMed] [Google Scholar]
- 37.Amatruda JM, Statt MC, Welle SL. Total and resting energy expenditure in obese women reduced to ideal body weight. J Clin Invest. 1993;92:1236–1242. doi: 10.1172/JCI116695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weinsier RL, Nelson KM, Hensrud DD, Darnell BE, Hunter GR, Schutz Y. Metabolic predictors of obesity. Contribution of resting energy expenditure, thermic effect of food, and fuel utilization to four-year weight gain of post-obese and never-obese women. J Clin Invest. 1995;95:980–985. doi: 10.1172/JCI117807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zurlo F, Ferraro RT, Fontvielle AM, Rising R, Bogardus C, Ravussin E. Spontaneous physical activity and obesity: crosssectional and longitudinal studies in Pima Indians. Am J Physiol. 1992;95:E296–E300. doi: 10.1152/ajpendo.1992.263.2.E296. [DOI] [PubMed] [Google Scholar]
- 40.Snitker S, Tataranni PA, Ravussin E. Spontaneous physical activity in a respiratory chamber is correlated to habitual physical activity. Int J Obes Relat Metab Disord. 2001;25:1481–1486. doi: 10.1038/sj.ijo.0801746. [DOI] [PubMed] [Google Scholar]
- 41.Levine JA, Eberhardt NL, Jensen MD. Role of nonexercise activity thermogenesis in resistance to fat gain in humans. Science. 1999;283:212–214. doi: 10.1126/science.283.5399.212. [DOI] [PubMed] [Google Scholar]
- 42.Johannsen DI, Ravussin E. Spontaneous physical activity: relationship between fidgeting and body weight control. Curr Opin Endocrinol Diabetes Obes. 2008;15:409–415. doi: 10.1097/MED.0b013e32830b10bb. [DOI] [PubMed] [Google Scholar]
- 43.Spraul M, Ravussin E, Fontvieille AM, Rising R, Larson DE, Anderson EA. Reduced sympathetic nervous activity. A potential mechanism predisposing to body weight gain. J Clin Invest. 1993;92:1730–1735. doi: 10.1172/JCI116760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwartz RS, Jaeger LF, Veith RC. Effect of clonidine on the thermic effect of feeding in humans. Am J Physiol. 1988;254:R90–R94. doi: 10.1152/ajpregu.1988.254.1.R90. [DOI] [PubMed] [Google Scholar]
- 45.Christin L, O’Connell M, Bogardus C, Danforth E, Jr, Ravussin E. Norepinephrine turnover and energy expenditure in Pima Indian and white men. Metabolism. 1993;42:723–729. doi: 10.1016/0026-0495(93)90239-k. [DOI] [PubMed] [Google Scholar]
- 46.Snitker S, Tataranni PA, Ravussin E. Respiratory quotient is inversely associated with muscle sympathetic nerve activity. J Clin Endocrinol Metab. 1998;83:3977–3979. doi: 10.1210/jcem.83.11.5265. [DOI] [PubMed] [Google Scholar]
- 47.Tataranni PA, Young JB, Bogardus C, Ravussin E. A low sympathoadrenal activity is associated with body weight gain and development of central adiposity in Pima Indian men. Obes Res. 1997;5:341–347. doi: 10.1002/j.1550-8528.1997.tb00562.x. [DOI] [PubMed] [Google Scholar]
- 48.Astrup A. The sympathetic nervous system as a target for intervention in obesity. Int J Obes Relat Metab Disord. 1995;19(Suppl 7):S24–S28. [PubMed] [Google Scholar]
- 49.McNeill G, Bruce AC, Ralph A, James WP. Inter-individual differences in fasting nutrient oxidation and the influence of diet composition. Int J Obes. 1988;12:455–463. [PubMed] [Google Scholar]
- 50.Zurlo F, Lillioja S, Esposito-Del Puente A, Nyomba BL, Raz I, Saad MF, et al. Low ratio of fat to carbohydrate oxidation as predictor of weight gain: study of 24-h RQ. Am J Physiol. 1990;259:E650–E657. doi: 10.1152/ajpendo.1990.259.5.E650. [DOI] [PubMed] [Google Scholar]
- 51.Toubro S, Sorensen TI, Hindsberger C, Christensen NJ, Astrup A. Twenty-four-hour respiratory quotient: the role of diet and familial resemblance. J Clin Endocrinol Metab. 1998;83:2758–2764. doi: 10.1210/jcem.83.8.5044. [DOI] [PubMed] [Google Scholar]
- 52.Seidell JC, Muller DC, Sorkin JD, Andres R. Fasting respiratory exchange ratio and resting metabolic rate as predictors of weight gain: the Baltimore Longitudinal Study on Aging. Int J Obes Relat Metab Disord. 1992;16:667–674. [PubMed] [Google Scholar]
- 53.Astrup A, Buemann B, Christensen NJ, Toubro S. Failure to increase lipid oxidation in response to increasing dietary fat content in formerly obese women. Am J Physiol. 1994;266:E592–E599. doi: 10.1152/ajpendo.1994.266.4.E592. [DOI] [PubMed] [Google Scholar]
- 54.Larson DE, Ferraro RT, Robertson DS, Ravussin E. Energy metabolism in weight-stable postobese individuals. Am J Clin Nutr. 1995;62:735–739. doi: 10.1093/ajcn/62.4.735. [DOI] [PubMed] [Google Scholar]
- 55.Froidevaux F, Schutz Y, Christin L, Jequier E. Energy expenditure in obese women before and during weight loss, after refeeding, and in the weight-relapse period. Am J Clin Nutr. 1993;57:35–42. doi: 10.1093/ajcn/57.1.35. [DOI] [PubMed] [Google Scholar]
- 56.Stock MJ. Gluttony and thermogenesis revisited. Int J Obes Relat Metab Disord. 1999;23:1105–1117. doi: 10.1038/sj.ijo.0801108. [DOI] [PubMed] [Google Scholar]
- 57.Bouchard C, Tremblay A, Despres JP, Nadeau A, Lupien PJ, Theriault G, et al. The response to long-term overfeeding in identical twins. N Engl J Med. 1990;322:1477–1482. doi: 10.1056/NEJM199005243222101. [DOI] [PubMed] [Google Scholar]
- 58.Neumann R. Experimentelle Beitrage zur lehre von dem taglichen Nahrungsbedarf des Menschen unter besonderer Berucksichtigung der notwendigen Eiweissmenge. Arch Hyg. 1902;45:1–87. [Google Scholar]
- 59.Miller DS, Mumford P, Stock MJ. Gluttony. 2. Thermogenesis in overeating man. Am J Clin Nutr. 1967;20:1223–1229. doi: 10.1093/ajcn/20.11.1223. [DOI] [PubMed] [Google Scholar]
- 60.Sims EA, Danforth E, Jr, Horton ES, Bray GA, Glennon JA, Salans LB. Endocrine and metabolic effects of experimental obesity in man. Recent Prog Horm Res. 1973;29:457–496. doi: 10.1016/b978-0-12-571129-6.50016-6. [DOI] [PubMed] [Google Scholar]
- 61.Befroy DE, Peterson KF, Dufour S, Mason GF, Rothman DL, Shulman GI. Increased substrate oxidation and mitochondrial uncoupling in skeletal muscle of endurance-trained individuals. PNAS. 2008;105:16701–16706. doi: 10.1073/pnas.0808889105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lebon V, Dufour S, Petersen KF, Ren J, Jucker BM, Slezak LA, et al. Effect of triiodothyronine on mitochondrial energy coupling in human skeletal muscle. J Clin Invest. 2001;108:733–737. doi: 10.1172/JCI11775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jucker BM, Dufour S, Ren J, Cao X, Previs SF, Underhill B, et al. Assessment of mitochondrial energy coupling in vivo by 13C/31P NMR. Proc Natl Acad Sci USA. 2000;97:6880–6884. doi: 10.1073/pnas.120131997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fernstrom M, Bakkman L, Tonkonogi M, Shabalina IG, Rozh-destvenskaya Z, Mattsson CM, et al. Reduced efficiency, but increased fat oxidation, in mitochondria from human skeletal muscle after 24-h ultraendurance exercise. J Appl Physiol. 2007;102:1844–1849. doi: 10.1152/japplphysiol.01173.2006. [DOI] [PubMed] [Google Scholar]
- 65.Rosenbaum M, Vandenborne K, Goldsmith R, Simoneau JA, Heymsfield S, Joanisse DR, et al. Effects of experimental weight perturbation on skeletal muscle work efficiency in human subjects. Am J Physiol Regul Integr Comp Physiol. 2003;285:R183–R192. doi: 10.1152/ajpregu.00474.2002. [DOI] [PubMed] [Google Scholar]
- 66.Coll AP, Farooqi IS, O’Rahilly S. The hormonal control of food intake. Cell. 2007;129:251–262. doi: 10.1016/j.cell.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Russek M. Participation of hepatic glucoreceptors in the control of intake of food. Nature. 1963;197:79–80. doi: 10.1038/197079b0. [DOI] [PubMed] [Google Scholar]
- 68.Flatt JP. The difference in the storage capacities for carbohydrate and for fat, and its implications in the regulation of body weight. Ann NY Acad Sci. 1987;499:104–123. doi: 10.1111/j.1749-6632.1987.tb36202.x. [DOI] [PubMed] [Google Scholar]
- 69.Hardie DG, Scott JW, Pan DA, Hudson ER. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003;546:113–120. doi: 10.1016/s0014-5793(03)00560-x. [DOI] [PubMed] [Google Scholar]
- 70.Goldberg GR, Murgatroyd PR, McKenna AP, Heavey PM, Prentice AM. Dietary compensation in response to covert imposition of negative energy balance by removal of fat or carbohydrate. Br J Nutr. 1998;80:141–147. [PubMed] [Google Scholar]
- 71.Snitker S, Larson DE, Tataranni PA, Ravussin E. Ad libitum food intake in humans after manipulation of glycogen stores. Am J Clin Nutr. 1997;65:941–946. doi: 10.1093/ajcn/65.4.941. [DOI] [PubMed] [Google Scholar]
- 72.Shetty PS, Prentice AM, Goldberg GR, Murgatroyd PR, McKenna AP, Stubbs RJ, et al. Alterations in fuel selection and voluntary food intake in response to isoenergetic manipulation of glycogen stores in humans. Am J Clin Nutr. 1994;60:534–543. doi: 10.1093/ajcn/60.4.534. [DOI] [PubMed] [Google Scholar]
- 73.Stubbs RJ, Murgatroyd PR, Goldberg GR, Prentice AM. Carbohydrate balance and the regulation of day-to-day food intake in humans. Am J Clin Nutr. 1993;57:897–903. doi: 10.1093/ajcn/57.6.897. [DOI] [PubMed] [Google Scholar]
- 74.Pannacciulli N, Salbe AD, Ortega E, Venti CA, Bogardus C, Krakoff J. The 24-h carbohydrate oxidation rate in a human respiratory chamber predicts ad libitum food intake. Am J Clin Nutr. 2007;86:625–632. doi: 10.1093/ajcn/86.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Langhans W, Scharrer E. Metabolic control of eating. World Rev Nutr Diet. 1992;70:1–67. [PubMed] [Google Scholar]
- 76.Eckel RH, Hernandez TL, Bell ML, Weil KM, Shepard TY, Grunwald GK, et al. Carbohydrate balance predicts weight and fat gain in adults. Am J Clin Nutr. 2006;83:803–808. doi: 10.1093/ajcn/83.4.803. [DOI] [PubMed] [Google Scholar]
- 77.Galgani JE, Heilbronn LK, Azuma K, Kelley DE, Albu JB, Pi-Sunyer X, et al. Metabolic flexibility in response to glucose is not impaired in people with type 2 diabetes after controlling for glucose disposal rate. Diabetes. 2008;57:841–845. doi: 10.2337/db08-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ukropcova B, Sereda O, de Jonge L, Bogacka I, Nguyen T, Xie H, et al. Family history of diabetes links impaired substrate switching and reduced mitochondrial content in skeletal muscle. Diabetes. 2007;56:720–727. doi: 10.2337/db06-0521. [DOI] [PubMed] [Google Scholar]
- 79.Schrauwen P, Westerterp KR. The role of high-fat diets and physical activity in the regulation of body weight. Br J Nutr. 2000;84:417–427. doi: 10.1017/s0007114500001720. [DOI] [PubMed] [Google Scholar]
- 80.Schrauwen P, van Marken Lichtenbelt WD, Saris WH, Westerterp KR. Changes in fat oxidation in response to a high-fat diet. Am J Clin Nutr. 1997;66:276–282. doi: 10.1093/ajcn/66.2.276. [DOI] [PubMed] [Google Scholar]
- 81.Hill JO, Peters JC, Reed GW, Schlundt DG, Sharp T, Greene HL. Nutrient balance in humans: effects of diet composition. Am J Clin Nutr. 1991;54:10–17. doi: 10.1093/ajcn/54.1.10. [DOI] [PubMed] [Google Scholar]
- 82.Smith SR, de Jonge L, Zachwieja JJ, Roy H, Nguyen T, Rood JC, et al. Fat and carbohydrate balances during adaptation to a high-fat. Am J Clin Nutr. 2000;72:450–457. doi: 10.1093/ajcn/71.2.450. [DOI] [PubMed] [Google Scholar]