

Summary
Objective
The pathogenicity of anti-human fVIII monoclonal antibodies (MAbs) was tested in a murine bleeding model.
Methods
MAbs were injected into the tail veins of hemophilia A mice to a peak plasma concentration of 60 nM, followed by injection of human B domain-deleted factor VIII (fVIII) at 180 U/kg, producing peak plasma concentrations of ∼2 nM. At 2 hours, blood loss following a 4 mm tail snip was measured. The following MAbs were tested: 1) 4A4, a type I anti-A2 fVIII inhibitor, 2) I54 and 1B5, classical type I anti-C2 inhibitors, 3) 2-77 and B45, non-classical type II anti-C2 inhibitors, and 4) 2-117, a non-classical anti-C2 MAb with inhibitory activity less than 0.4 Bethesda Units/mg IgG.
Results
All MAbs except 2-117 produced similar amounts of blood loss that were significantly greater than control mice injected with fVIII alone. Increasing the dose of fVIII to 360 U/kg overcame the bleeding diathesis produced by the type II MAbs 2-77 and B45, but not the type I antibodies, 4A4, I54, and 1B5. These results were consistent with the in vitro Bethesda assay in which 4A4 completely inhibited both 1 U/ml and 3 U/ml fVIII, while there was 40% residual activity at saturating concentrations of 2-77 at either concentration of fVIII.
Conclusions
For patients with an inhibitor response dominated by non-classical anti-C2 antibodies both the in vivo and in vitro results suggest that treatment with high-dose fVIII rather than bypassing agents may be warranted.
Keywords: Hemophilia A, Factor VIII, Inhibitor, Antibody
Introduction
Approximately 30% of patients with hemophilia A develop detectable anti-factor VIII (fVIII) antibodies in response to infusions of fVIII [1, 2, 3, 4]. The immune response to fVIII currently is the most significant complication in the management of patients with hemophilia A. In addition, autoimmune antibodies to fVIII can develop in non-hemophiliacs, producing acquired hemophilia A, which frequently produces life- or limb-threatening bleeding.
FVIII contains a domain sequence designated A1-A2-B-ap-A3-C1-C2. It circulates as an A1-A2-B/ap-A3-C1-C2 heterodimer bound to von Willebrand factor (VWF). During the activation of fVIII by thrombin, the B domain and the light chain activation peptide, ap, are released, and cleavage between the A1 and A2 domains occurs, producing an A1/A2/A3-C1-C2 heterotrimer [5]. Factor Xa also activates fVIII and catalyzes these cleavages along with additional cleavages in the A1 and A2 domains and a slow cleavage in the A3 domain. Activated fVIII (fVIIIa) functions as a cofactor for factor IXa during intrinsic pathway factor X activation on phospholipid membrane surfaces. Thus, fVIII inhibitors potentially can act by interfering with the activation of fVIII, decreasing the binding of fVIII to VWF, or inhibiting fVIIIa function. FVIII inhibitors can either inhibit fVIII completely or incompletely at saturating concentrations, corresponding to type I and type II behavior, respectively [6].
In either congenital or acquired hemophilia A, the majority of inhibitory antibodies are directed at either the 40-kDa A2 or the 15-kDa C2 domains of fVIII [7]. Congenital hemophilia patients typically have a polyclonal response consisting of antibodies to both the A2 and C2 domain, whereas acquired hemophilia patients typically have a more limited B cell epitope response and either have anti-A2 or anti-C2 antibodies but not both [7]. Inhibitor plasmas from acquired hemophilia patients often have type II characteristics with incomplete fVIII activity at saturating concentrations of antibodies. Despite this incomplete inhibition of fVIII many acquired hemophilia patients have a severe bleeding diathesis. Initially, anti-C2 antibodies were identified that inhibit the binding of fVIII to phospholipid membranes [8]. However, it has been reported that the C2 domain also contributes to the binding of fVIII to VWF, thrombin and factor Xa [9, 10, 11]. Phospholipid membranes and VWF bind to at least partially overlapping sites on fVIII, because their binding to fVIII is mutually exclusive [12, 13, 14]. Consistent with this observation, anti-C2 antibodies have been identified that inhibit the binding of fVIII to both phospholipid membranes and VWF [15, 16, 17]. However, these sites are not identical because the ap region makes a major contribution to the interaction of fVIII with VWF, but not phospholipid [18, 19]. Additionally, although most antibodies that inhibit phospholipid binding also inhibit VWF binding, differential inhibition has been observed with some antibodies [20, 21]. Because VWF is not necessary for the procoagulant function of fVIII per se, antibodies that solely inhibit the binding of fVIII to VWF might not have inhibitory activity in in vitro coagulation assays. However, they could be pathogenic by decreasing the circulatory lifetime of fVIII, which decreases when it is not bound to VWF. Additionally, murine anti-C2 monoclonal antibodies (MAbs) [10, 22] and anti-C2 antibodies in polyclonal patient plasmas have been identified that interfere with the activation of fVIII by thrombin or factor Xa [9, 22].
We have characterized the diversity of a large panel of anti-C2 MAbs that were produced in a murine hemophilia A immunogenicity model [20]. Five groups of structural epitopes were defined based on patterns of overlapping epitopes. Group A, AB and B antibodies correspond to classical anti-C2 antibodies that inhibit the binding of fVIII and fVIIIa to phospholipid and VWF. Group BC antibodies are the most frequent and are type II inhibitors with inhibitory titers usually greater than 10,000 Bethesda units per mg IgG. These antibodies inhibit the activation of fVIII by thrombin and factor Xa in the presence and absence of VWF. Group C antibodies, which are rare, are represented by the well known commercial MAb, ESH8, which blocks the release of VWF from fVIII following thrombin activation [22].
We recently reported that non-classical Group BC/C antibodies are present in the plasmas of most human fVIII inhibitor patients [23]. Group BC antibodies have inhibitory titers on an equimolar basis that are usually at least 10-fold higher than classical anti-C2 antibodies. However, at saturating concentrations they produce residual fVIII levels of 20 – 40%. Because fVIII levels in this range in the absence of inhibitory antibodies are sufficient for normal hemostasis, the question is raised whether non-classical anti-C2 antibodies are pathogenic. In this study, we compared the pathogenicity of type I and type II anti-C2 MAbs, along with an anti-A2 inhibitor, in an in vivo bleeding model.
Methods
Materials
Pooled citrated normal plasma (FACT) and fVIII deficient plasma were obtained from George King Biomedical (Overland Park, KS). Isoflurane (Hospira, Lake Forest, IL) and 0.9% sterile saline (Hospira, Lake Forest, IL) were obtained from the Emory University Hospital Pharmacy. All other materials were reagent grade or are described in the cited literature.
Recombinant fVIII
A recombinant B domain deleted (BDD) human fVIII construct, HSQ, was expressed from a baby hamster kidney-derived cell line as previously described [24, 25]. HSQ was purified from conditioned serum-free cell culture media using a two-step ion-exchange chromatography procedure as previously described [26]. Fractions were analyzed by one-stage coagulation assay as described previously [27], absorbance at 280 nm, and sodium dodecyl sulfate polyacrylamide gel electrophoresis. The purity of the IgGs was judged to be greater than 90% by SDS-PAGE. The amino acid content was used to estimate the molar extinction coefficient and then the absorbance at 280 nm was used to estimate the concentration of the fVIII protein[28]. This concentration was used to calculate the specific activity in a one-stage coagulation assay.
Hemophilia A mice
Exon 16-disrupted (E16) hemophilia A mice in a C57BL/6 background were originally obtained from Dr. Leon Hoyer (American Red Cross, Holland Lab) and a breeding colony was established [29, 30]. In these experiments 8-12 week old male or female mice were used.
Anti-fVIII MAbs from anti-fVIII hybridomas
Murine anti-fVIII C2 domain MAbs were isolated as described previously [20]. IgG concentrations were calculated using an extinction coefficient at 280 nm of 1.37 (mg/ml)-1 cm-1. 4A4 is a type I anti-A2 inhibitor that blocks the activation of factor X by the intrinsic pathway factor X activation complex and has an inhibitory titer of 40,000 Bethesda units (BU)/mg IgG. I54 is a classical type I Group A anti-C2 inhibitor that has a specific inhibitory titer of 1300 BU/mg IgG. 1B5 is a classical type I Group B anti-C2 inhibitor that has a specific inhibitory titer of 930 BU/mg IgG. 2-77 and B45 are non-classical type II Group BC anti-C2 inhibitors that block the activation of fVIII, produce a residual fVIII level of 30-40% at saturating concentrations and have inhibitory titers of 25,000 and 21,000 BU/mg IgG, respectively. 2-117 is a non-classical Group C anti-C2 MAb with inhibitory activity less than 0.4 BU/mg IgG (Table 1). All inhibitory titers were calculated using the Bethesda assay with buffered pooled normal plasma as the human fVIII source [20, 31].
Table 1.
Characteristics of anti-fVIII MAbs
| MAb | Domain Specificity | Type | Inhibitor Titer (BU/mg IgG) |
|---|---|---|---|
| 4A4 | A2 | I | 40,000 |
| I54 | C2-Group A | I | 1300 |
| 1B5 | C2-Group B | I | 930 |
| 2-77 | C2-Group BC | II | 25,000 |
| B45 | C2-Group BC | II | 21,000 |
| 2-117 | C2-Group C | - | <0.4 |
In vivo bleeding model
After determination of body weight, hemophilia A mice were placed on a heating pad for approximately 5 minutes to dilate the tail veins and then were injected with 140 μl of saline or 140 μl of 0.5 mg/kg MAb diluted in saline. At 15 min after the initial injection, mice were injected with 60 μl of 180 or 360 U/kg (∼0.02 – 0.04 mg/kg) BDD human fVIII in sterile saline. The peak molar concentration of MAb in vivo was approximately ten-fold higher than the peak concentration of the higher dose of fVIII. At 110 min after the initial injection, anesthesia was induced with 3% isoflurane at a flow of 1000 ml/min using a RC2 Rodent Circuit Controller (VetEquip®, Pleasanton, CA) and then decreased to 2% isoflurane at a flow rate of 500ml/min. Tails were placed into a 15 ml conical tube containing 13 ml of normal saline at 37°C. At 120 min 4mm of the distal tail was transected using a scalpel. The tail then was placed into a new 15 ml conical tube filled containing 13 ml of saline at 37°C. Following the tail-snip, the anesthesia was reduced to 1.5% isoflurane and a flow rate of 500 ml/min which was maintained for the remainder of the experiment. The amount of blood loss into the tube in 40 minutes or at the time of death from hemorrhage was recorded as mg of blood loss per g of body weight.
In vivo recovery of fVIII
In some experiments, at 120 min after the fVIII injection mice were euthanized with carbon dioxide instead of a tail-snipping and a cardiac puncture was performed using a 22-gauge needle. Between 0.5-1.0 mL of blood was collected and anticoagulated with 3.8% (w/v) sodium citrate at a ratio of 1 volume to 9 volumes blood. Plasma was obtained by centrifugation and assayed for fVIII activity using the one-stage coagulation assay [27] in which BDD human fVIII diluted into E16 mouse plasma was the source of fVIII for the standard curve.
FVIII inhibitor assays
FVIII inhibitor titers were measured using the Bethesda assay [31] with the modifications previously described [32]. Recombinant BDD human fVIII was added to human fVIII deficient plasma at 1 U/ml and 3 U/ml and used as the source of fVIII activity. One Bethesda unit (BU) per ml is defined as the dilution of inhibitor that produces 50% inhibition of fVIII activity. At least 8 MAb concentrations were tested in duplicate and the resulting inhibition curve was fitted using the 4-parameter logistic equation to estimate the concentration of MAb producing 50% inhibition.
FVIII inhibitor titers were also measured with the Coatest® SP FVIII kit (Chromogenix, Lexington, MA). Briefly, 25 μl of buffered normal pooled human plasma was incubated for 2 hours at 37°C with 25 μl of MAb diluted in 0.15 M NaCl, 20 mM HEPES, 0.05% (w/v) Tween 80, pH 7.4 (HBS/Tween) or with 25 μl of HBS/Tween as the control. A standard curve based on serial dilutions of pooled normal plasma was used. FVIII levels were measured in triplicate for each sample using the methods of the Coatest® SP FVIII kit.
Statistics
Comparisons between groups were made using the Mann-Whitney U test and SigmaStat version 3.5 (Systat, Port Richmond, CA). A P value of less than 0.05 was considered statistically significant.
Results
Non-classical anti-C2 MAbs behave differently in one-stage and chromogenic fVIII assays
Inhibitory activity of MAbs toward BDD human fVIII was determined using both the one-stage coagulation assay as well as a chromogenic assay. The type I anti-A2 MAb, 4A4, produced 50% inhibition of fVIII activity at similar concentrations in both assays: 0.1 μg/ml and 0.05 μg/ml in the Bethesda and chromogenic assays, respectively (Fig. 1). Similarly, BO2C11, a classical type I Group AB anti-C2 antibody [15, 20] produced 50% inhibition of fVIII activity at similar concentrations in both assays, 0.05 μg/ml and 0.1 μg/ml in the Bethesda and chromogenic assays, respectively (data not shown). In contrast, the non-classical type II MAb 2-77 produced 50% inhibition at 0.2 μg/ml in the Bethesda assay but did not produce 50% inhibition at concentrations as high as 0.8 mg/ml in the chromogenic assay. Increasing the concentration of fVIII to 3 U/ml in the one-stage assay shifted the 4A4 and 2-77 inhibition curves to the right as expected given the increased amount of antibody necessary to produce inhibition. At both concentrations of fVIII, there was 40% residual fVIII activity at saturating concentrations of 2-77.
Fig. 1. In vitro inhibition of fVIII by MAbs 4A4 and 2-77.
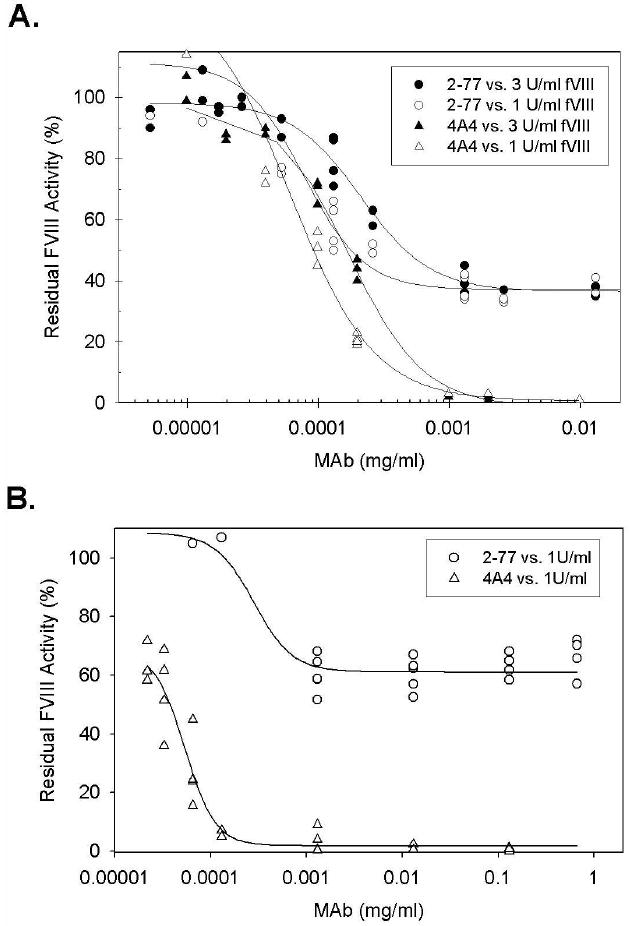
FVIII was reconstituted into hemophilia A plasma at the indicated concentrations and the inhibition of fVIII by MAbs 4A4 (triangles) or 2-77 (circles) was measured by (A) one-stage coagulation assay and (B) chromogenic assay as described in Methods. The curves represent 4-parameter logistic fits to the data.
Non-classical anti-C2 antibodies are pathogenic in an in vivo bleeding model
An in vivo bleeding model was established in which blood loss following a 4 mm tail-snip was used to determine the bleeding phenotype in hemophilia A mice injected with BDD human fVIII and various anti-fVIII MAbs. A total of 135 hemophilia A mice aged 8-12 weeks received intravenous injections of MAb, followed 15 minutes later by intravenous injection of fVIII or saline control. Nine mice were excluded from the analysis secondary to missed injections or 4 mm tail-snips falling over a scarred portion of the tail or a large kink in the tail. There were between 8 and 14 mice in each group. Mean blood loss in saline control mice was 32.2 mg/g body weight (Fig. 2). In contrast, 9 of 10 mice that received no MAb and 180 U/kg fVIII bled less than 5 mg/g body weight. There was a significant decrease in bleeding between the mice that received fVIII compared to the saline control group (P = 0.003).
Fig. 2. Bleeding produced by anti-fVIII MAbs in a murine tail snip model.
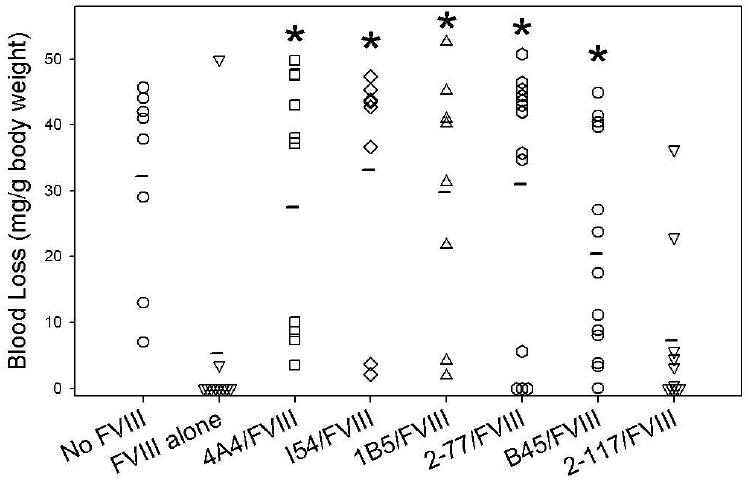
Blood loss in hemophilia A mice following tail snipping was measured in the presence and absence of injections of 180 U/kg (0.02 mg/kg) BDD human fVIII and 0.5 mg/kg murine anti-human fVIII MAbs as described in Methods. The asterisk indicates MAbs with significantly more bleeding than control mice treated with fVIII but no MAb.
Once the amount of fVIII needed to correct the bleeding phenotype of the hemophilia A mice was established, saturating concentrations of MAbs were injected, followed by injection of fVIII (Fig. 2). MAb 4A4 was pathogenic, producing a mean blood loss of 27.5 mg/g body weight that was significantly higher than mice that received fVIII and no MAb (P = 0.003)), but not significantly different than the saline controls that received no fVIII (P = 0.74) The classical type I anti-C2 inhibitors, I54 and 1B5, also were pathogenic, producing mean blood losses of 33.2 and 29.8 mg/g body weight (P = 0.005 and P = 0.003, respectively), but not significantly different than the saline controls (P = 0.72 for both groups) The non-classical type II anti-C2 inhibitors, 2-77 and B45, also were pathogenic, producing mean blood losses of 31.0 and 20.5 mg/g body weight (P = 0.01 and P = 0.004, respectively), which was not significantly different than saline control mice (P = 0.86 and P = 0.11, respectively). The non-classical Group C MAb, 2-117, was not pathogenic, producing a mean blood loss of 7.3 mg/g body weight (P = 0.14). Thus, all MAbs except 2-117 significantly increased blood loss compared to mice injected only with fVIII.
The pathogenicity of non-classical anti-C2 antibodies is overcome with higher doses of fVIII
A higher dose (360 U/kg) of fVIII was given to hemophilia A mice in an attempt to overcome the pathogenicity of MAbs which caused significant bleeding at a lower dose of 180 U/kg. The peak plasma concentrations of MAbs remained over 10-fold higher than the plasma concentration of fVIII at this dose. The higher fVIII dose failed to correct the bleeding produced by the anti-A2 MAb 4A4 or the classical anti-C2 MAb I54. The classical MAb 1B5 had a slight decrease in blood loss from 29.8 to 25.3 mg/g body weight but this was not statistically significant (P = 0.7) (Fig. 3). In contrast, the bleeding produced by the non-classical anti-C2 inhibitors 2-77 and B45 was significantly less at the higher dose of fVIII (P = 0.04 and P = 0.004, respectively).
Fig. 3. Antibody-dependent blood loss in hemophilia A mice as a function of fVIII dose.
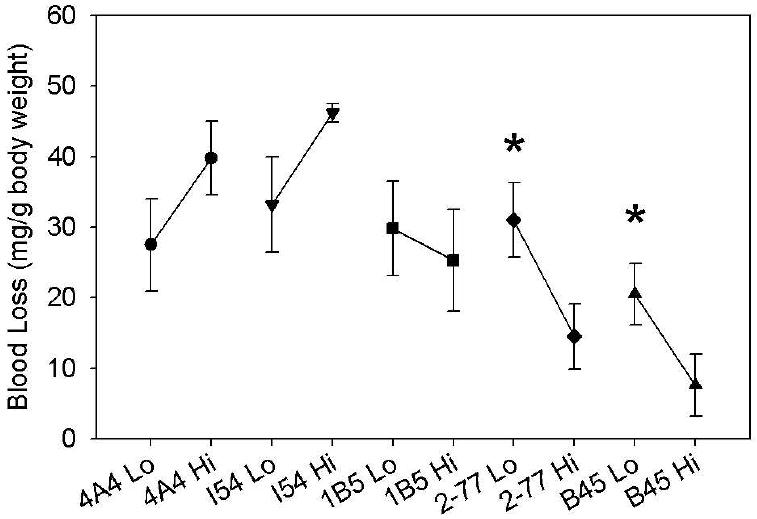
Blood loss in hemophilia A mice following tail snipping was measured after injections of 180 U/kg (“Lo”) or 360 U/kg (“Hi”) BDD human fVIII and 0.5 mg/kg murine anti-human fVIII MAbs as described in Methods. The asterisk indicates a significant difference in bleeding between groups.
FVIII recovery correlates with bleeding phenotype
Residual fVIII levels were evaluated for hemophilia A mice injected with saturating concentrations of MAb followed by fVIII at doses ranging from 0-360 U/kg (Fig. 4). 4A4 completely inhibited fVIII throughout the dose range. In contrast, there was a dose-dependent increase in fVIII recovery in mice treated with the non-classical inhibitors, 2-77 and B45.
Fig. 4. In vivo recovery of fVIII in the presence of saturating concentrations of MAb.
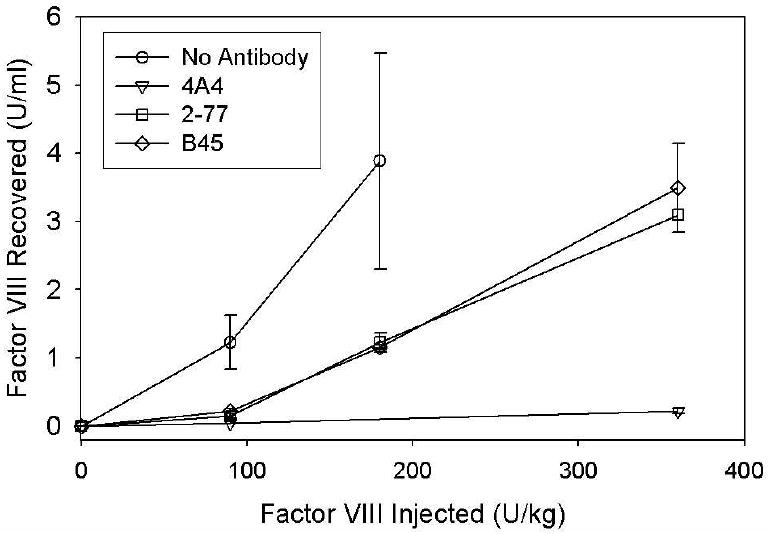
Blood was obtained by cardiac puncture 120 min after injection of BDD human fVIII to the indicated peak plasma concentrations and injection of 0.5 mg/kg of MAb4A4 (triangles), 2-77 (squares), B45 (diamonds) or saline control (circles). FVIII recovery was determined one-stage coagulation assay as described in Methods. Error bars represent SEM (n = 2-10 per group).
Discussion
In this study, we found that type II non-classical anti-C2 antibodies, as well as type I anti-A2 and classical anti-C2 inhibitors, are pathogenic in a murine tail snip model (Fig. 2). Non-classical antibodies produce similar amounts of blood loss as type I inhibitors despite the fact that substantial residual fVIII activity remains at saturating antibody concentrations. For example, there is 40% residual fVIII activity at saturating concentrations of 2-77 at nominal fVIII concentrations of 1 or 3 U/ml in the one-stage coagulation assay (Fig. 1A). An anti-C1 antibody, MAb-LE2E9, with similar in vitro characteristics has also been described[33]. This behavior is consistent with that of acquired hemophilia patients who have type II inhibitors and a bleeding diathesis in the presence of substantial fVIII activity. Our recent discovery of the presence of non-classical Group BC/C anti-C2 antibodies in the plasmas of most inhibitor patients [23] suggests that these antibodies may be a dominant population in many patients with acquired hemophilia.
The Bethesda assay, as originally described [31], is based on the one-stage coagulation assay and is a sensitive and specific test in the diagnosis of clinically significant fVIII inhibitors. The chromogenic fVIII assay also is used in some clinical laboratories to test for fVIII inhibitors and frequently produces results that are discordant with respect to the one-stage assay (Meeks, S.L and Lollar, P., unpublished observations). In the present study, MAb 2-77 produced significant bleeding despite having an undetectable inhibitor titer in the chromogenic inhibitor assay, although it is a high-titer inhibitor in the one-stage assay (Fig. 1B). These results indicate that the chromogenic assay is not a suitable replacement for the one-stage assay in the measurement of fVIII inhibitors.
Although the Bethesda assay is useful for the detection of clinically significant fVIII inhibitors, the inhibitor titer and the residual fVIII activity in the plasma are not necessarily good predictors of clinical severity[34]. For example, fVIII levels and inhibitor titers at presentation were not predictive of the severity of bleeding events in a recent observational study of patients with acquired hemophilia in the United Kingdom [35]. In that study, the median fVIII level was 4% in those with a fatal bleeding event compared to 3% in patients who did not require treatment for their bleeding symptoms. Initial inhibitory titers were similar in both groups with medians of 7.2 and 7 BU/ml, respectively. Interestingly, in our bleeding model the non-classical MAbs produced a bleeding phenotype at a fVIII dose of 180 U/kg despite a recovery of ∼1U/ml fVIII at the time of cardiac puncture. The bleeding phenotype was corrected at a fVIII dose of 360 U/kg, which corresponds to a recovery of ∼3U/ml of fVIII. Thus, saturating concentrations of non-classical MAbs evidently decrease fVIII function in vivo more than in vitro.
Dosing formulas have been developed to estimate the amount of fVIII needed to saturate a low titer inhibitor and obtain a clinical response to fVIII. One example is total fVIII infused is equal to the dose needed to saturate the inhibitor (plasma volume in ml × inhibitor titer) plus the dose of fVIII desired to treat the bleeding episode[36]. However, it has also been reported that patients with high-titer inhibitors have had a clinical response to high doses of fVIII despite being given significantly less fVIII than would be needed to bind all circulating anti-fVIII antibody [37]. These examples underscore the need for the investigation into the correlation between in vitro and in vivo assessments of fVIII activity and inhibitors.
The pathogenic mechanism of action of non-classical anti-C2 antibodies is not known. MAb 2-77 inhibits the activation of fVIII by thrombin or factor Xa and has little, if any, ability to inhibit activated fVIII [20]. During the one-stage coagulation assay, fVIII initially is activated by thrombin or factor Xa relatively slowly until sufficient thrombin or factor Xa are formed to produce rapid feedback activation. In contrast, in the chromogenic assay, exogenous thrombin is added to rapidly activate fVIII. Thus, the one-stage assay is potentially more sensitive than the chromogenic assay to the presence of inhibitors that interfere with the activation of fVIII. One explanation for the pathogenicity of non-classical anti-C2 antibodies is that hemostasis in vivo may be even more sensitive to inhibition of fVIII activation than the one-stage coagulation assay.
An important finding in this study is that the pathogenicity of high-titer non-classical anti-C2 inhibitors can be overcome by increasing the dose of fVIII (Fig. 3). In contrast, classical type I anti-C2 inhibitors continued to produce bleeding at the higher dose of fVIII. Patients with congenital hemophilia often develop a polyclonal antibody response to fVIII that is dominated by type I inhibitors. In contrast, patients with acquired hemophilia often have type II inhibitors and a more limited epitope spectrum [7]. Historically, patients with high-titer inhibitors usually have been treated with bypassing agents, such as FEIBA™ or recombinant factor VIIa, or porcine fVIII. These agents are selected because of high anti-fVIII inhibitor titers or, in the case of patients with acquired hemophilia, bleeding in the presence of detectable fVIII levels. Our results suggest that high-dose fVIII should be considered more actively in patients with inhibitor responses dominated by non-classical anti-C2 antibodies, especially in those patients who respond poorly to FEIBA™ or recombinant factor VIIa. This requires further clinical investigation.
Acknowledgments
Supported by grants from the National Institutes of Health (HL082609 and HL40921) and Hemophilia of Georgia, Inc. (P.L.), and Hemophilia and Thrombosis Research Society and Hemophilia of Georgia (S.L.M.).
References
- 1.Bray GL, Gomperts ED, Courter S, Gruppo R, Gordon EM, Manco-Johnson M, et al. A multicenter study of recombinant factor VIII (recombinate): safety, efficacy, and inhibitor risk in previously untreated patients with hemophilia A. The Recombinate Study Group. Blood. 1994;83(9):2428–35. [PubMed] [Google Scholar]
- 2.Kreuz W, Ettingshausen CE, Zyschka A, Oldenburg J, Saguer IM, Ehrenforth S, et al. Inhibitor development in previously untreated patients with hemophilia A: a prospective long-term follow-up comparing plasma-derived and recombinant products. Semin Thromb Hemost. 2002;28(3):285–90. doi: 10.1055/s-2002-32664. [DOI] [PubMed] [Google Scholar]
- 3.Lusher JM, Arkin S, Abildgaard CF, Schwartz RS. Recombinant factor VIII for the treatment of previously untreated patients with hemophilia A. Safety, efficacy, and development of inhibitors. Kogenate Previously Untreated Patient Study Group. N Engl J Med. 1993;328(7):453–9. doi: 10.1056/NEJM199302183280701. [DOI] [PubMed] [Google Scholar]
- 4.Lusher JM, Lee CA, Kessler CM, Bedrosian CL. The safety and efficacy of B-domain deleted recombinant factor VIII concentrate in patients with severe haemophilia A. Haemophilia. 2003;9(1):38–49. doi: 10.1046/j.1365-2516.2003.00708.x. [DOI] [PubMed] [Google Scholar]
- 5.Lollar P, Parker CG. Subunit structure of thrombin-activated porcine factor VIII. Biochemistry. 1989;28(2):666–74. doi: 10.1021/bi00428a038. [DOI] [PubMed] [Google Scholar]
- 6.Gawryl MS, Hoyer LW. Inactivation of factor VIII coagulant activity by two different types of human antibodies. Blood. 1982;60(5):1103–9. [PubMed] [Google Scholar]
- 7.Prescott R, Nakai H, Saenko EL, Scharrer I, Nilsson IM, Humphries JE, et al. The inhibitor antibody response is more complex in hemophilia A patients than in most nonhemophiliacs with factor VIII autoantibodies. Recombinate and Kogenate Study Groups. Blood. 1997;89(10):3663–71. [PubMed] [Google Scholar]
- 8.Arai M, Scandella D, Hoyer LW. Molecular basis of factor VIII inhibition by human antibodies. Antibodies that bind to the factor VIII light chain prevent the interaction of factor VIII with phospholipid. J Clin Invest. 1989;83(6):1978–84. doi: 10.1172/JCI114107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nogami K, Shima M, Hosokawa K, Nagata M, Koide T, Saenko EL, et al. Factor VIII C2 domain contains the thrombin-binding site responsible for thrombin-catalyzed cleavage at Arg1689. J Biol Chem. 2000;275(33):25774–80. doi: 10.1074/jbc.M002007200. [DOI] [PubMed] [Google Scholar]
- 10.Nogami K, Shima M, Hosokawa K, Suzuki T, Koide T, Saenko EL, et al. Role of factor VIII C2 domain in factor VIII binding to factor Xa. J Biol Chem. 1999;274(43):31000–7. doi: 10.1074/jbc.274.43.31000. [DOI] [PubMed] [Google Scholar]
- 11.Saenko EL, Shima M, Rajalakshmi KJ, Scandella D. A role for the C2 domain of factor VIII in binding to von Willebrand factor. J Biol Chem. 1994;269(15):11601–5. [PubMed] [Google Scholar]
- 12.Andersson LO, Brown JE. Interaction of factor VIII-von Willebrand Factor with phospholipid vesicles. Biochem J. 1981;200(1):161–7. doi: 10.1042/bj2000161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lajmanovich A, Hudry-Clergeon G, Freyssinet JM, Marguerie G. Human factor VIII procoagulant activity and phospholipid interaction. Biochim Biophys Acta. 1981;678(1):132–6. doi: 10.1016/0304-4165(81)90056-8. [DOI] [PubMed] [Google Scholar]
- 14.Saenko EL, Scandella D. A mechanism for inhibition of factor VIII binding to phospholipid by von Willebrand factor. J Biol Chem. 1995;270(23):13826–33. doi: 10.1074/jbc.270.23.13826. [DOI] [PubMed] [Google Scholar]
- 15.Jacquemin MG, Desqueper BG, Benhida A, Vander Elst L, Hoylaerts MF, Bakkus M, et al. Mechanism and kinetics of factor VIII inactivation: study with an IgG4 monoclonal antibody derived from a hemophilia A patient with inhibitor. Blood. 1998;92(2):496–506. [PubMed] [Google Scholar]
- 16.Shima M, Nakai H, Scandella D, Tanaka I, Sawamoto Y, Kamisue S, et al. Common inhibitory effects of human anti-C2 domain inhibitor alloantibodies on factor VIII binding to von Willebrand factor. Br J Haematol. 1995;91(3):714–21. doi: 10.1111/j.1365-2141.1995.tb05374.x. [DOI] [PubMed] [Google Scholar]
- 17.Shima M, Scandella D, Yoshioka A, Nakai H, Tanaka I, Kamisue S, et al. A factor VIII neutralizing monoclonal antibody and a human inhibitor alloantibody recognizing epitopes in the C2 domain inhibit factor VIII binding to von Willebrand factor and to phosphatidylserine. Thromb Haemost. 1993;69(3):240–6. [PubMed] [Google Scholar]
- 18.Saenko E, Sarafanov A, Greco N, Shima M, Loster K, Schwinn H, et al. Use of surface plasmon resonance for studies of protein-protein and protein-phospholipid membrane interactions. Application to the binding of factor VIII to von Willebrand factor and to phosphatidylserine-containing membranes. J Chromatogr A. 1999;852(1):59–71. doi: 10.1016/s0021-9673(99)00491-4. [DOI] [PubMed] [Google Scholar]
- 19.Saenko EL, Scandella D. The acidic region of the factor VIII light chain and the C2 domain together form the high affinity binding site for von willebrand factor. J Biol Chem. 1997;272(29):18007–14. doi: 10.1074/jbc.272.29.18007. [DOI] [PubMed] [Google Scholar]
- 20.Meeks SL, Healey JF, Parker ET, Barrow RT, Lollar P. Antihuman factor VIII C2 domain antibodies in hemophilia A mice recognize a functionally complex continuous spectrum of epitopes dominated by inhibitors of factor VIII activation. Blood. 2007;110(13):4234–42. doi: 10.1182/blood-2007-06-096842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scandella D, Gilbert GE, Shima M, Nakai H, Eagleson C, Felch M, et al. Some factor VIII inhibitor antibodies recognize a common epitope corresponding to C2 domain amino acids 2248 through 2312, which overlap a phospholipid-binding site. Blood. 1995;86(5):1811–9. [PubMed] [Google Scholar]
- 22.Saenko EL, Shima M, Gilbert GE, Scandella D. Slowed release of thrombin-cleaved factor VIII from von Willebrand factor by a monoclonal and a human antibody is a novel mechanism for factor VIII inhibition. J Biol Chem. 1996;271(44):27424–31. doi: 10.1074/jbc.271.44.27424. [DOI] [PubMed] [Google Scholar]
- 23.Meeks SL, Healey JF, Parker ET, Barrow RT, Lollar P. Nonclassical anti-C2 domain antibodies are present in patients with factor VIII inhibitors. Blood. 2008;112(4):1151–3. doi: 10.1182/blood-2008-01-132639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barrow RT, Healey JF, Gailani D, Scandella D, Lollar P. Reduction of the antigenicity of factor VIII toward complex inhibitory antibody plasmas using multiply-substituted hybrid human/porcine factor VIII molecules. Blood. 2000;95(2):564–8. [PubMed] [Google Scholar]
- 25.Healey JF, Barrow RT, Tamim HM, Lubin IM, Shima M, Scandella D, et al. Residues Glu2181-Val2243 contain a major determinant of the inhibitory epitope in the C2 domain of human factor VIII. Blood. 1998;92(10):3701–9. [PubMed] [Google Scholar]
- 26.Lollar P, Fay PJ, Fass DN. Factor VIII and factor VIIIa. Methods Enzymol. 1993;222:128–43. doi: 10.1016/0076-6879(93)22010-d. [DOI] [PubMed] [Google Scholar]
- 27.Doering CB, Healey JF, Parker ET, Barrow RT, Lollar P. High level expression of recombinant porcine coagulation factor VIII. J Biol Chem. 2002;277(41):38345–9. doi: 10.1074/jbc.M206959200. [DOI] [PubMed] [Google Scholar]
- 28.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4(11):2411–23. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bi L, Lawler AM, Antonarakis SE, High KA, Gearhart JD, Kazazian HH., Jr Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nat Genet. 1995;10(1):119–21. doi: 10.1038/ng0595-119. [DOI] [PubMed] [Google Scholar]
- 30.Bi L, Sarkar R, Naas T, Lawler AM, Pain J, Shumaker SL, et al. Further characterization of factor VIII-deficient mice created by gene targeting: RNA and protein studies. Blood. 1996;88(9):3446–50. [PubMed] [Google Scholar]
- 31.Kasper CK, Aledort L, Aronson D, Counts R, Edson JR, van Eys J, et al. Proceedings: A more uniform measurement of factor VIII inhibitors. Thromb Diath Haemorrh. 1975;34(2):612. [PubMed] [Google Scholar]
- 32.Barrow RT, Lollar P. Neutralization of antifactor VIII inhibitors by recombinant porcine factor VIII. J Thromb Haemost. 2006;4(10):2223–9. doi: 10.1111/j.1538-7836.2006.02135.x. [DOI] [PubMed] [Google Scholar]
- 33.Singh I, Smith A, Vanzieleghem B, Collen D, Burnand K, Saint-Remy JM, et al. Antithrombotic effects of controlled inhibition of factor VIII with a partially inhibitory human monoclonal antibody in a murine vena cava thrombosis model. Blood. 2002;99(9):3235–40. doi: 10.1182/blood.v99.9.3235. [DOI] [PubMed] [Google Scholar]
- 34.Hay CR. Acquired haemophilia. Baillieres Clin Haematol. 1998;11(2):287–303. doi: 10.1016/s0950-3536(98)80049-8. [DOI] [PubMed] [Google Scholar]
- 35.Collins PW, Hirsch S, Baglin TP, Dolan G, Hanley J, Makris M, et al. Acquired hemophilia A in the United Kingdom: a 2-year national surveillance study by the United Kingdom Haemophilia Centre Doctors' Organisation. Blood. 2007;109(5):1870–7. doi: 10.1182/blood-2006-06-029850. [DOI] [PubMed] [Google Scholar]
- 36.Negrier C. Inhibitors to factor VIII: treatment of acute bleeds. In: Lee CA, Berntorp EE, Hoots WK, editors. Textbook of Hemophilia. Blackwell Publishing Ltd.; 2005. pp. 80–85. [Google Scholar]
- 37.Luna-Zaizar H, Esparza-Flores MA, Lopez-Guido B, Aguilar-Lopez LB, Cortes Alvarez CR, Jaloma-Cruz AR. Kinetics of factor VIII:C inhibitors and treatment response in severe hemophilia A patients. Int J Lab Hematol. 2008 doi: 10.1111/j.1751-553X.2008.01099.x. [DOI] [PubMed] [Google Scholar]