

Abstract
Meningococcal factor H-binding protein (fHbp) is a promising antigen that is part of two vaccines in clinical development. The protein specifically binds human complement factor H (fH), which downregulates complement activation on the bacterial surface and enables the organism to evade host defenses. In humans, the vaccine antigen forms a complex with fH, which may affect anti-fHbp antibody repertoire and decrease serum bactericidal activity by covering important fHbp epitopes. In a recent study, fHbp residues in contact with fH were identified from a crystal structure. Two fHbp glutamate residues that mediated ion-pair interactions with fH were replaced with alanine, and the resulting E218A/E239A mutant no longer bound the fH fragment. In the present study, we generated the E218A/E239A mutant recombinant protein and confirmed the lack of fH binding. By enzyme-linked immunosorbent assay (ELISA), the mutant fHbp showed similar respective concentration-dependent inhibition of binding of four bactericidal anti-fHbp monoclonal antibodies (MAbs) to fHbp, compared with inhibition by the soluble wild-type protein. In two mouse strains, the mutant fHbp elicited up to 4-fold-lower IgG anti-fHbp antibody titers and up to 20-fold-lower serum bactericidal titers than those elicited by the wild-type fHbp vaccine. Thus, although introduction of the two alanine substitutions to eliminate fH binding did not appear to destabilize the molecule globally, the mutations resulted in decreased immunogenicity in mouse models in which neither the mutant nor the wild-type control vaccine bound fH. These results cast doubt on the vaccine potential in humans of this mutant fHbp.
Neisseria meningitidis is a major cause of bacterial meningitis and sepsis worldwide. Conjugate vaccines based on the capsular polysaccharide are highly effective for prevention of disease caused by strains with capsular group A, C, W-135, or Y. Capsular group B strains, however, are responsible for about 35% of meningococcal disease burden in the United States (7, 15) and 80 to 90% in some European countries (14, 27). To date, there is no broadly effective vaccine for group B strains (12).
The group B polysaccharide is poorly immunogenic, which has been attributed to its chemical similarity to the polysialic acid moiety of the human neural cell adhesion molecule (NCAM) (8). There are also safety concerns for a vaccine that elicits antibody to the group B capsule. As a result, much effort has been devoted toward identifying noncapsular antigens as group B vaccine candidates (12, 21).
Among the most promising of the meningococcal protein antigens identified to date is factor H (fH)-binding protein (fHbp), which is a surface-exposed lipoprotein previously referred to as GNA1870 (19) or LP2086 (9). Recombinant fHbp vaccines elicited broad serum bactericidal antibodies in mice (9, 19). The protein binds human fH, which downregulates complement activation on the bacterial surface (17), and is critical for survival of the organism in human serum or blood (17, 24, 28). Binding to meningococci is specific for human fH (13, 25), which explains in part why N. meningitidis causes disease only in humans. In the absence of bound fH, the organism is readily killed by most nonhuman sera.
Factor H is present at high concentrations in human serum (∼500 μg/ml) (26). Consequently, when humans are immunized with fHbp, the vaccine antigen is expected to form a complex with fH. The presence of bound fH may cover important fHbp epitopes and adversely affect human anti-fHbp serum bactericidal antibody responses (24). The lack of fH binding in nonhuman animal models may explain why mice or rabbits immunized with recombinant fHbp appear to have broader serum bactericidal antibody responses (10, 16) than do immunized humans (22).
Recently, a crystal structure of fHbp bound to two short consensus repeat (SCR) domains of fH was described (24). The authors identified 20 amino acid residues of fHbp that interacted with fH. Two contact residues, which they designated glutamate (E) 283 and E304, mediated ion-pair interactions with fH and were conserved within the two previously described subfamilies of fHbps (9). Schneider et al. generated a site-specific mutant of fHbp (peptide ID 1 based on the classification system at http://Neisseria.org). The mutant contained two changes, E283 to alanine (A) and E304A. By surface plasmon resonance studies, the mutant protein did not bind with an fH fragment. Note that according to a more recent definition of the amino (N)-terminal amino acid sequence of the mature fHbp beginning with the lipidated cysteine residue (18), we refer to this mutant as E218A/E239A.
A mutant fHbp in which fH binding has been eliminated is a novel approach for improving the immunogenicity of an fHbp vaccine in humans. However, the effects of the two mutations on fHbp epitope expression, stability, or immunogenicity of the protein have not been investigated; thus, they are the objects of the present investigation.
MATERIALS AND METHODS
Materials.
Chemical reagents and alkaline phosphatase (AP)-conjugated secondary antibodies were purchased from Sigma-Aldrich, St. Louis, MO, unless specified otherwise.
Site-specific mutagenesis and protein purification.
The pET21b-derived plasmid encoded fHbp ID 1, which is a member of subfamily B or variant group 1 according to the Wyeth (9) and Novartis (19) classifications, respectively, or modular group I according to our recently described classification based on five variable segments, each derived from one of two genetic lineages (3, 20). Site-specific mutagenesis was performed using the QuikChange II kit (Stratagene, La Jolla, CA). The oligonucleotide primers 5′-CTTTACAACCAAGCCGCGAAAGGCAGTTACTC and 5′-GGCAGCGCGGCAGTGAAAACCGTAAAC (mutated codons underlined) and their reverse complementary sequences were used to introduce the E218A and E239A substitutions sequentially. The mutant plasmid clones were confirmed by DNA sequencing. The wild-type (WT) and E218A/E239A mutant fHbp plasmids were transformed into the Escherichia coli strain BL21(DE3) (Novagen, Madison, WI), and the recombinant proteins were expressed and purified by immobilized metal ion chromatography as described previously (2). The protein concentrations were determined by UV absorbance at 280 nm using a molar extinction coefficient of 8,940 M−1 cm−1. The purified proteins were dialyzed overnight against phosphate-buffered saline (PBS; Roche Applied Science, Indianapolis, IN) containing 3% (wt/vol) sucrose and stored frozen in aliquots at −30°C.
Binding of fH to recombinant fHbp.
Binding of fH to fHbp was measured by enzyme-linked immunosorbent assay (ELISA) (5). In brief, purified recombinant fHbp (2 μg/ml in PBS) was added to the wells of a microtiter plate and incubated overnight at 4°C. The plates were washed three times with PBS, 1% bovine serum albumin (BSA), 0.1% Tween 20, 0.01% NaN3, pH 7.4, and blocked with PBS, 1% BSA, 0.01% NaN3, pH 7.4, for 1 h at room temperature (RT). Purified human fH (Complement Technology, Tyler, TX) in PBS, 1% BSA, 0.1% Tween 20, 0.01% sodium azide, pH 7.4, was added (serial 5-fold dilutions starting at 25 μg/ml), and the plate was incubated for 2 h at RT. After washing, sheep anti-human fH (1:2,000 dilution; Lifespan Biosciences, Seattle, WA) was added and the plate was incubated for 1 h at 37°C. AP-conjugated donkey anti-sheep IgG (1:5,000 dilution) was added, and the plate was incubated for 1 h at RT. Phosphatase substrate (1 mg/ml p-nitrophenyl phosphate) diluted in 50 mM NaH2CO3, 1 mM MgCl2, pH 9.8, was added, and the optical density (OD) was read at 405 nm after 30 min of incubation at RT.
Anti-fHbp MAb binding and inhibition of binding by ELISA.
The ability of soluble mutant or wild-type recombinant fHbp to inhibit binding of murine anti-fHbp monoclonal antibodies (MAbs) to solid-phase wild-type fHbp was determined by ELISA. The four bactericidal MAbs tested, JAR 1, 4, and 5 (4, 5, 29) and MAb 502 (11, 23), were described previously. For testing inhibition of binding, the wells of the microtiter plate were incubated with wild-type (WT) fHbp and washed and blocked as described above. Fifty microliters of serial 5-fold dilutions of soluble wild-type or mutant fHbp was added to the wells along with equal volumes of anti-fHbp MAbs. The final concentrations of MAbs in the reaction mixtures were 0.5 μg/ml (JAR 1 and JAR 4) or 0.1 μg/ml (JAR 5 and MAb 502). After incubation at 37°C for 1 h, the wells were washed, and bound MAb was detected with AP-conjugated goat anti-mouse IgG (Fc specific, 1:5,000 dilution) and phosphatase substrate as described above.
DSC.
Differential scanning calorimetry (DSC) experiments were performed using purified fHbp that had been dialyzed overnight against PBS and adjusted to a concentration of 0.5 mg/ml. The PBS used for dialysis was the reference solution. A VP-DSC microcalorimeter (MicroCal, Northampton, MA) was employed using a scan rate of 60°C/h and the middle feedback setting. The data were analyzed using Origin software v. 5.0 (MicroCal), which was used to subtract baseline scans, normalize the data to the protein concentration, and fit the data to two unfolding transitions. The normalized data were exported, and the thermograms were plotted using Prism 5.0b (GraphPad, La Jolla, CA).
Mouse immunization.
The immunogenicity of the wild-type and mutant fHbps was evaluated in experiments conducted with two different mouse strains, CD-1 (outbred) and BALB/c (inbred) mice, and performed at Children's Hospital Oakland Research Institute (CHORI) or the University of Massachusetts Medical Center, respectively, using protocols approved by the Institutional Animal Care and Use committees. Purified, recombinant fHbp was dialyzed against 10 mM histidine-HCl, pH 6.5, 150 mM NaCl, 3% (wt/vol) sucrose (>1,000-fold excess) overnight. One day prior to the immunization, fHbp was adsorbed with aluminum hydroxide [Al(OH)3] as the adjuvant. Each 200-μl dose contained 20 μg fHbp and 600 μg Al(OH)3. The final concentrations of the buffer were 10 mM histidine-HCl, pH 6.5, and 150 mM NaCl. The CD-1 and BALB/c mouse studies used three intraperitoneal (i.p.) injections of vaccine given at 3-week intervals. After the results were obtained, a second study was done with CD-1 mice using one injection of vaccine to confirm the lower immunogenicity of the mutant vaccine observed in the first studies. In all three studies, blood samples were obtained 3 weeks after the last dose.
Measurement of anti-fHbp IgG responses.
Anti-fHbp IgG responses were measured by ELISA using the recombinant wild-type fHbp as the antigen on the plate and serial 5-fold dilutions of serum starting at 1:100. Bound IgG antibody was detected with AP-conjugated goat anti-mouse IgG and phosphatase substrate as described above. Anti-fHbp ELISA titers were expressed as reciprocal mean log10 titers ± 2 standard errors (SE). The mean and SE were calculated from log10-transformed values using Prism (GraphPad Software, La Jolla, CA).
Complement-dependent serum bactericidal antibody activity.
The bactericidal assay was performed as described elsewhere using log-phase organisms of group B strain H44/76 (strain designation B: P1.7,16: F3-3: ST-32 [cc32]), which were grown in broth culture as described previously (1). The complement source was serum from a nonimmune healthy adult human with normal hemolytic complement activity. The serum was passed over a protein G Sepharose column (HiTrap Protein G HP; GE Healthcare, Piscataway, NJ) to remove IgG antibodies prior to use in the assay. Geometric mean titers (GMTs) and log10-transformed mean titers and SE were calculated using Prism (GraphPad Software).
RESULTS
We constructed the mutant of fHbp, E218A/E239A, which previously was reported not to bind with a fragment of human fH (24). By SDS-PAGE, the respective molecular mass and purity of the mutant protein were similar to those of WT fHbp vaccine (Fig. 1). As expected, only the wild-type protein bound purified, intact fH (Fig. 2).
FIG. 1.
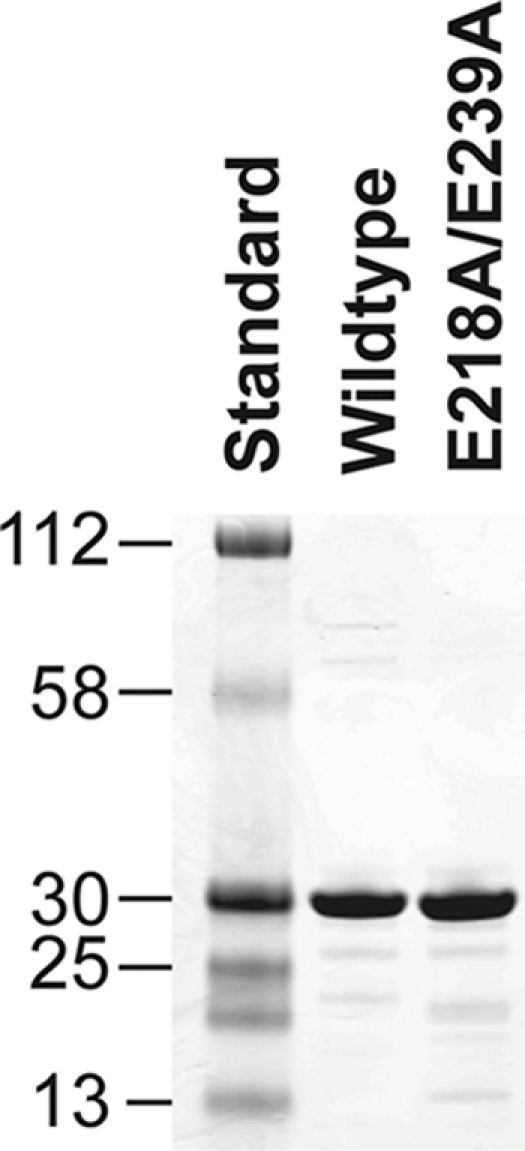
Molecular mass and purity of WT and mutant fHbps. Purified proteins (5 μg each) were separated by SDS-PAGE and visualized by Coomassie blue staining. Lane 1, Kaleidoscope broad-range molecular mass standards (Bio-Rad, Richmond, CA); lane 2, WT fHbp (peptide ID 1); lane 3, E218A/E239A mutant fHbp. The masses of the standards (in kDa) are shown on the left.
FIG. 2.

Binding of human fH to purified recombinant fHbp as measured by ELISA. Filled circles, WT fHbp; open squares, E218A/E239A mutant fHbp.
By ELISA, the wild-type and mutant fHbps were indistinguishable in their respective concentration-dependent binding with four bactericidal anti-fHbp MAbs (data not shown). These MAbs included MAb 502 and JAR 1, which served as probes for epitopes on the carboxyl (C)-terminal domain of the protein (23). The remaining two MAbs recognized an epitope including residues 25 to 27 and 57 to 59 of the N-terminal domain (JAR 4) (4) or amino acid residues 121 and 122 of the N-terminal domain (JAR 5) (5). By ELISA, the WT and mutant proteins also showed similar respective concentration-dependent inhibition of binding of each of these MAbs to fHbp (Fig. 3). Thus, introduction of the two amino acid substitutions in the mutant protein did not alter expression of the epitopes recognized by these four anti-fHbp MAbs.
FIG. 3.

Inhibition of binding of anti-fHbp MAbs to recombinant WT fHbp by soluble WT or mutant fHbp as measured by ELISA. Filled circles, WT fHbp inhibitor; open squares, E218A/E239A mutant fHbp inhibitor. Data are shown for the anti-fHbp MAbs MAb 502 (A), JAR 1 (B), JAR 4 (C), and JAR 5 (D).
As a further test of the structural integrity of the E218A/E239A mutant protein, we determined the thermal stability by differential scanning calorimetry (DSC). The wild-type protein unfolded with two transitions, with midpoints (Tm) at 69.0 and 84.1°C (Fig. 4, solid line). The second transition occurred at a temperature similar to that (82.0°C) which was observed previously for a C-terminal fragment of fHbp (18). Therefore, the two transitions likely corresponded to thermal unfolding of the respective N- and C-terminal domains revealed in the solution structures (6, 18). The E218A/E239A mutant protein also unfolded with two transitions, centered at 69.1 and 82.5°C (dashed line). The first transition was indistinguishable from that of the wild-type protein, and the second transition with the mutant protein was slightly lower than that with the WT protein (ΔTm = −1.6°C). The slightly lower temperature for the second transition of the mutant was consistent with the locations of both amino acid substitutions in the C-terminal domain.
FIG. 4.

Thermal stability of WT and mutant fHbps as measured by differential scanning calorimetry. The excess heat capacity (Cp) versus temperature is plotted for WT (solid line) and E218A/E239A mutant (dashed line) fHbp. The lower and higher temperature transitions correspond to the unfolding of the N- and C-terminal domains, respectively.
Immunogenicity of wild-type and E218A/E239A mutant fHbps.
As described in Materials and Methods, we conducted three mouse immunogenicity studies with the wild-type and E218A/E239A mutant fHbps. In study 1, CD-1 mice were given three doses of the proteins adsorbed with aluminum hydroxide [Al(OH)3]. Study 2 was similar to study 1 except that in study 2 the CD-1 mice received only one dose of vaccine. In study 3, we used BALB/c mice that were immunized with three doses of either the WT or mutant protein. In all three studies there were trends for lower reciprocal geometric mean anti-fHbp IgG antibody titers in the mice given the mutant protein (Fig. 5). The respective differences were significant in studies 2 (1/GMT of 1.8 × 103 versus 4.4 × 103, P = 0.02) and 3 (2 × 104 versus 9 × 104; P = 0.007), where individual sera were assayed instead of serum pools, which were assayed in study 1.
FIG. 5.

IgG Anti-fHbp antibody responses (1/GMT) of mice immunized with WT or mutant fHbp adsorbed with aluminum hydroxide. In study 1, CD-1 mice were immunized with three doses of recombinant WT or mutant fHbp; in study 2, CD-1 mice were immunized with one dose of WT or mutant fHbp; in study 3, BALB/c mice were immunized with three doses of WT or mutant fHbp. Shaded bars, WT fHbp; open bars, E218A/E239A mutant fHbp. In study 1, the sera were pooled (3 pools per vaccine group, each pool from sera of 3 to 4 mice). In studies 2 and 3, individual sera were assayed (n = 6 to 8 mice per vaccine group). *, P = 0.02; **, P = 0.007. 95% CI, 95% confidence interval.
Table 1 summarizes the respective serum bactericidal antibody responses measured against group B strain H44/76, which expressed an fHbp variant that matched the vaccine antigen. In all three studies, the mice immunized with the mutant protein showed lower responses than the controls immunized with the WT protein. The differences were significant in studies 2 and 3, in which individual sera from the mice were tested (1/GMT of 6 versus 18 after the one dose of vaccine used in study 2, P = 0.03, and 48 versus 1,986 after three doses used in study 3, P < 0.03) (Table 1).
TABLE 1.
Complement-mediated serum bactericidal antibody responses of mice immunized with fHbp vaccines
| Studya | Mouse strain | No. of doses | fHbp vaccine |
|||
|---|---|---|---|---|---|---|
| WT |
E218A/E239A mutant |
|||||
| 1/mean log10 titer ± 2 SE | 1/GMT | 1/mean log10 titer ± 2 SE | 1/GMT | |||
| 1 | CD-1 | 3 | 2.63 ± 0.54 | 427 | 2.31 ± 0.25 | 206 |
| 2 | CD-1 | 1 | 1.26 ± 0.36 | 18b | 0.80 ± 0.14 | 6b |
| 3 | BALB/c | 3 | 3.30 ± 0.30 | 1,986b | 1.68 ± 0.60 | 48b |
In study 1, the sera were pooled (3 pools per vaccine group, each pool from sera of 3 to 4 mice). The respective GMTs between vaccine groups were not significantly different. In studies 2 and 3, individual sera were assayed (n = 6 to 8 mice per vaccine group) and the respective differences were significant for both studies. Study 2 also used different lots of WT and mutant fHbp vaccines than those used in study 1 or 3.
P was <0.05 for comparison of the respective WT and mutant vaccine groups in each study.
DISCUSSION
The effect of binding of fH to fHbp-based vaccines on their immunogenicity in humans is unknown. A mutant fHbp molecule that does not bind fH may have increased immunogenicity by exposing fHbp epitopes that ordinarily are not available when humans are immunized with the WT fHbp antigen, which binds human fH. As a first step in developing such a vaccine, we prepared the previously described mutant E218A/E239A, which does not bind to fH, and tested its immunogenicity in mice. Because mouse fH was not expected to bind to either the WT or mutant fHbp, our hypothesis was that the mutant fHbp would be similar in immunogenicity to the WT fHbp. Our most important finding was that, contrary to this hypothesis, the mutant fHbp vaccine elicited lower IgG anti-fHbp antibody titers and lower serum bactericidal titers than did the corresponding WT fHbp vaccine. The lower immunogenicity of the mutant fHbp vaccine in mice in the absence of fH binding suggested that the introduction of the two alanine substitutions, while abolishing binding to human fH, also adversely affected expression of epitopes that were important for eliciting serum bactericidal antibody responses. The largest decrease in immunogenicity of the mutant protein was observed in BALB/c mice, but similar trends for lower immunogenicity of the mutant protein were observed in two studies of a second mouse strain (CD-1). The decrease was statistically significant in the second study of CD-1 mice when individual mouse sera were assayed instead of serum pools. To decrease the possibility that the results from the first immunogenicity study resulted from a poor preparation of the purified proteins, the second CD-1 study also used new lots of freshly prepared recombinant protein vaccines. Note that the studies that we performed did not address directly whether a mutant protein that did not bind to human fH would have increased immunogenicity in the presence of human fH. To address this question would require immunogenicity studies of humans or studies with a transgenic animal model that expresses human fH.
Based upon inhibition of MAb binding, there was no evidence of decreased expression of epitopes on the mutant protein recognized by any of the four murine anti-fHbp MAbs tested (Fig. 3). In previous studies, all four of these MAbs were bactericidal with human and/or rabbit complement (11, 29). Although none of the epitopes recognized by these anti-fHbp MAbs appeared to be affected by introduction of the two alanine substitutions in our mutant fHbp vaccine, conceivably, replacement of the two negatively charged residues with alanine affected the electrostatics and/or conformation of the protein surface and resulted in decreased expression of other important epitopes. By calorimetry, there was evidence of slight destabilization of the mutant protein; however, we did not consider this destabilization sufficient to affect the thermal stability or proteolytic susceptibility of the mutant under physiological conditions.
Collectively, the data indicated that introduction of the two mutations in the C-terminal domain of fHbp resulted in loss of vaccine immunogenicity in mouse models in which neither the WT or mutant protein bound fH. A previous report identified residues 101 to 255, which contained the E218A/E239A mutations, as the region of fHbp important for eliciting bactericidal antibody responses (11). Thus, it may be more feasible to engineer immunogenic fHbp mutants that do not bind fH by using amino acid substitutions in the N-terminal domain (residues 1 to 134).
Acknowledgments
This work was supported, in part, by Public Health Service grants R01 AI 046464 and AI 082263 (to D.M.G.), AI 054544 (to S.R.), and AI 070955 (to P.T.B.) from the National Institute of Allergy and Infectious Diseases, NIH. J.S was supported, in part, by NIH grant AI 032725. The work at Children's Hospital Oakland Research Institute was performed in a facility funded by Research Facilities Improvement Program grant number C06 RR 016226 from the National Center for Research Resources, NIH.
Emily Braga, Ryan Palapaz, and Tracy Wong, Children's Hospital Oakland Research Institute, provided expert technical assistance. MAb 502 was a generous gift of Marzia Giuliani and Mariagrazia Pizza, Novartis Vaccines and Diagnostics, Siena, Italy.
Footnotes
Published ahead of print on 2 June 2010.
REFERENCES
- 1.Beernink, P. T., D. A. Caugant, J. A. Welsch, O. Koeberling, and D. M. Granoff. 2009. Meningococcal factor H-binding protein variants expressed by epidemic capsular group A, W-135, and X strains from Africa. J. Infect. Dis. 199:1360-1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beernink, P. T., and D. M. Granoff. 2008. Bactericidal antibody responses induced by meningococcal recombinant chimeric factor H-binding protein vaccines. Infect. Immun. 76:2568-2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beernink, P. T., and D. M. Granoff. 2009. The modular architecture of meningococcal factor H-binding protein. Microbiology 155:2873-2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beernink, P. T., C. Lopasso, A. Angiolillo, F. Felici, and D. Granoff. 2009. A region of the N-terminal domain of meningococcal factor H-binding protein that elicits bactericidal antibody across antigenic variant groups. Mol. Immunol. 46:1647-1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beernink, P. T., J. A. Welsch, M. Bar-Lev, O. Koeberling, M. Comanducci, and D. M. Granoff. 2008. Fine antigenic specificity and cooperative bactericidal activity of monoclonal antibodies directed at the meningococcal vaccine candidate, factor H-binding protein. Infect. Immun. 76:4232-4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cantini, F., D. Veggi, S. Dragonetti, S. Savino, M. Scarselli, G. Romagnoli, M. Pizza, L. Banci, and R. Rappuoli. 2009. Solution structure of the factor H-binding protein, a survival factor and protective antigen of Neisseria meningitidis. J. Biol. Chem. 284:9022-9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohn, A. C., J. R. MacNeil, L. H. Harrison, C. Hatcher, J. Theodore, M. Schmidt, T. Pondo, K. E. Arnold, J. Baumbach, N. Bennett, A. S. Craig, M. Farley, K. Gershman, S. Petit, R. Lynfield, A. Reingold, W. Schaffner, K. A. Shutt, E. R. Zell, L. W. Mayer, T. Clark, D. Stephens, and N. E. Messonnier. 2010. Changes in Neisseria meningitidis disease epidemiology in the United States, 1998-2007: implications for prevention of meningococcal disease. Clin. Infect. Dis. 50:184-191. [DOI] [PubMed] [Google Scholar]
- 8.Finne, J., M. Leinonen, and P. H. Makela. 1983. Antigenic similarities between brain components and bacteria causing meningitis. Implications for vaccine development and pathogenesis. Lancet ii:355-357. [DOI] [PubMed] [Google Scholar]
- 9.Fletcher, L. D., L. Bernfield, V. Barniak, J. E. Farley, A. Howell, M. Knauf, P. Ooi, R. P. Smith, P. Weise, M. Wetherell, X. Xie, R. Zagursky, Y. Zhang, and G. W. Zlotnick. 2004. Vaccine potential of the Neisseria meningitidis 2086 lipoprotein. Infect. Immun. 72:2088-2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giuliani, M. M., J. Adu-Bobie, M. Comanducci, B. Arico, S. Savino, L. Santini, B. Brunelli, S. Bambini, A. Biolchi, B. Capecchi, E. Cartocci, L. Ciucchi, F. Di Marcello, F. Ferlicca, B. Galli, E. Luzzi, V. Masignani, D. Serruto, D. Veggi, M. Contorni, M. Morandi, A. Bartalesi, V. Cinotti, D. Mannucci, F. Titta, E. Ovidi, J. A. Welsch, D. Granoff, R. Rappuoli, and M. Pizza. 2006. A universal vaccine for serogroup B meningococcus. Proc. Natl. Acad. Sci. U. S. A. 103:10834-10839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giuliani, M. M., L. Santini, B. Brunelli, A. Biolchi, B. Arico, F. Di Marcello, E. Cartocci, M. Comanducci, V. Masignani, L. Lozzi, S. Savino, M. Scarselli, R. Rappuoli, and M. Pizza. 2005. The region comprising amino acids 100 to 255 of Neisseria meningitidis lipoprotein GNA 1870 elicits bactericidal antibodies. Infect. Immun. 73:1151-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Granoff, D. M. 2010. Review of meningococcal group B vaccines. Clin. Infect. Dis. 50(Suppl. 2):S54-S65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Granoff, D. M., J. A. Welsch, and S. Ram. 2009. Binding of complement factor H (fH) to Neisseria meningitidis is specific for human fH and inhibits complement activation by rat and rabbit sera. Infect. Immun. 77:764-769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gray, S. J., C. L. Trotter, M. E. Ramsay, M. Guiver, A. J. Fox, R. Borrow, R. H. Mallard, and E. B. Kaczmarski. 2006. Epidemiology of meningococcal disease in England and Wales 1993/94 to 2003/04: contribution and experiences of the Meningococcal Reference Unit. J. Med. Microbiol. 55:887-896. [DOI] [PubMed] [Google Scholar]
- 15.Kaplan, S. L., G. E. Schutze, J. A. Leake, W. J. Barson, N. B. Halasa, C. L. Byington, C. R. Woods, T. Q. Tan, J. A. Hoffman, E. R. Wald, K. M. Edwards, and E. O. Mason, Jr. 2006. Multicenter surveillance of invasive meningococcal infections in children. Pediatrics 118:e979-e984. [DOI] [PubMed] [Google Scholar]
- 16.Koeberling, O., A. Seubert, and D. M. Granoff. 2008. Bactericidal antibody responses elicited by a meningococcal outer membrane vesicle vaccine with overexpressed factor H-binding protein and genetically attenuated endotoxin. J. Infect. Dis. 198:262-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Madico, G., J. A. Welsch, L. A. Lewis, A. McNaughton, D. H. Perlman, C. E. Costello, J. Ngampasutadol, U. Vogel, D. M. Granoff, and S. Ram. 2006. The meningococcal vaccine candidate GNA1870 binds the complement regulatory protein factor H and enhances serum resistance. J. Immunol. 177:501-510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mascioni, A., B. E. Bentley, R. Camarda, D. A. Dilts, P. Fink, V. Gusarova, S. K. Hoiseth, J. Jacob, S. L. Lin, K. Malakian, L. K. McNeil, T. Mininni, F. Moy, E. Murphy, E. Novikova, S. Sigethy, Y. Wen, G. W. Zlotnick, and D. H. Tsao. 2009. Structural basis for the immunogenic properties of the meningococcal vaccine candidate LP2086. J. Biol. Chem. 284:8738-8746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Masignani, V., M. Comanducci, M. M. Giuliani, S. Bambini, J. Adu-Bobie, B. Arico, B. Brunelli, A. Pieri, L. Santini, S. Savino, D. Serruto, D. Litt, S. Kroll, J. A. Welsch, D. M. Granoff, R. Rappuoli, and M. Pizza. 2003. Vaccination against Neisseria meningitidis using three variants of the lipoprotein GNA1870. J. Exp. Med. 197:789-799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pajon, R., P. T. Beernink, L. H. Harrison, and D. M. Granoff. 2010. Frequency of factor H-binding protein modular groups and susceptibility to cross-reactive bactericidal activity in invasive meningococcal isolates. Vaccine 28:2122-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pizza, M., V. Scarlato, V. Masignani, M. M. Giuliani, B. Arico, M. Comanducci, G. T. Jennings, L. Baldi, E. Bartolini, B. Capecchi, C. L. Galeotti, E. Luzzi, R. Manetti, E. Marchetti, M. Mora, S. Nuti, G. Ratti, L. Santini, S. Savino, M. Scarselli, E. Storni, P. Zuo, M. Broeker, E. Hundt, B. Knapp, E. Blair, T. Mason, H. Tettelin, D. W. Hood, A. C. Jeffries, N. J. Saunders, D. M. Granoff, J. C. Venter, E. R. Moxon, G. Grandi, and R. Rappuoli. 2000. Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science 287:1816-1820. [DOI] [PubMed] [Google Scholar]
- 22.Plested, J. S., J. A. Welsch, and D. M. Granoff. 2009. Ex vivo model of meningococcal bacteremia using human blood for measuring vaccine-induced serum passive protective activity. Clin. Vaccine Immunol. 16:785-791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scarselli, M., F. Cantini, L. Santini, D. Veggi, S. Dragonetti, C. Donati, S. Savino, M. M. Giuliani, M. Comanducci, F. Di Marcello, G. Romagnoli, M. Pizza, L. Banci, and R. Rappuoli. 2009. Epitope mapping of a bactericidal monoclonal antibody against the factor H binding protein of Neisseria meningitidis. J. Mol. Biol. 386:97-108. [DOI] [PubMed] [Google Scholar]
- 24.Schneider, M. C., B. E. Prosser, J. J. Caesar, E. Kugelberg, S. Li, Q. Zhang, S. Quoraishi, J. E. Lovett, J. E. Deane, R. B. Sim, P. Roversi, S. Johnson, C. M. Tang, and S. M. Lea. 2009. Neisseria meningitidis recruits factor H using protein mimicry of host carbohydrates. Nature 458:890-893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shaughnessy, J., L. A. Lewis, H. Jarva, and S. Ram. 2009. Functional comparison of the binding of factor H short consensus repeat 6 (SCR 6) to factor H binding protein from Neisseria meningitidis and the binding of factor H SCR 18 to 20 to Neisseria gonorrhoeae porin. Infect. Immun. 77:2094-2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stoiber, H., C. Pinter, A. G. Siccardi, A. Clivio, and M. P. Dierich. 1996. Efficient destruction of human immunodeficiency virus in human serum by inhibiting the protective action of complement factor H and decay accelerating factor (DAF, CD55). J. Exp. Med. 183:307-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trotter, C. L., M. Chandra, R. Cano, A. Larrauri, M. E. Ramsay, C. Brehony, K. A. Jolley, M. C. Maiden, S. Heuberger, and M. Frosch. 2007. A surveillance network for meningococcal disease in Europe. FEMS Microbiol. Rev. 31:27-36. [DOI] [PubMed] [Google Scholar]
- 28.Welsch, J. A., S. Ram, O. Koeberling, and D. M. Granoff. 2008. Complement-dependent synergistic bactericidal activity of antibodies against factor H-binding protein, a sparsely distributed meningococcal vaccine antigen. J. Infect. Dis. 197:1053-1061. [DOI] [PubMed] [Google Scholar]
- 29.Welsch, J. A., R. Rossi, M. Comanducci, and D. M. Granoff. 2004. Protective activity of monoclonal antibodies to genome-derived neisserial antigen 1870, a Neisseria meningitidis candidate vaccine. J. Immunol. 172:5606-5615. [DOI] [PubMed] [Google Scholar]