

Abstract
In the absence of a vaccine, there is an urgent need for the development of safe and effective topical microbicides to prevent the sexual transmission of human immunodeficiency virus type 1 (HIV-1). In this study, we proposed to develop a novel class of microbicides using syndecan as the antiviral agent. Specifically, we generated a soluble syndecan-Fc hybrid molecule by fusing the ectodomain of syndecan-1 to the Fc domain of a human IgG. We then tested the syndecan-Fc hybrid molecule for various in vitro microbicidal anti-HIV-1 properties. Remarkably, the syndecan-Fc hybrid molecule possesses multiple attractive microbicidal properties: (i) it blocks HIV-1 infection of primary targets including T cells, macrophages, and dendritic cells (DC); (ii) it exhibits a broad range of antiviral activity against primary HIV-1 isolates, multidrug resistant HIV-1 isolates, HIV-2, and simian immunodeficiency virus (SIV); (iii) it prevents transmigration of HIV-1 through human primary genital epithelial cells; (iv) it prevents HIV-1 transfer from dendritic cells to CD4+ T cells; (v) it is potent when added 2 h prior to addition of HIV-1 to target cells; (vi) it is potent at a low pH; (vii) it blocks HIV-1 infectivity when diluted in genital fluids; and (viii) it prevents herpes simplex virus infection. The heparan sulfate chains of the syndecan-Fc hybrid molecule are absolutely required for HIV-1 neutralization. Several lines of evidence suggest that the highly conserved Arg298 in the V3 region of gp120 serves as the locus for the syndecan-Fc hybrid molecule neutralization. In conclusion, this study suggests that the syndecan-Fc hybrid molecule represents the prototype of a new generation of microbicidal agents that may have promise for HIV-1 prevention.
The dominant cell surface heparan sulfate proteoglycans (HSPG) (25, 29, 30, 33) are syndecans, which are transmembrane receptors highly expressed on adherent cells (macrophages and epithelial and endothelial cells) but poorly expressed on suspension cells (T cells) (2, 3, 4, 10, 35). Their ectodomain bears three linear heparan sulfate (HS) chains, which are composed of a repetition of a sulfated disaccharide motif (1). The sulfation pattern of HSs dictates the ligand specificity of syndecans (1). HSPG, including syndecans, serve as receptors for human deficiency virus type-1 (HIV-1) (16), herpes simplex virus (HSV) (7), human papillomavirus (HPV) (13, 37), and human T-lymphotropic virus type 1 (HTLV-1) (19, 20). Pretreatment of target cells such as macrophages with heparinase, an enzyme that removes HS moieties from syndecans, significantly reduces HIV-1 infectivity (35). Although syndecans do not alleviate the requirement for CD4 and chemokine receptors for viral entry (35), these in cis attachment receptors amplify HIV-1 infection by promoting viral adsorption to the surface of permissive cells. Syndecans also serve as in trans receptors for HIV-1 (2, 16). HIV-1 binds syndecans richly expressed on the endothelium and remains infectious for a week, whereas cell-free virus loses its infectivity after a single day (2). Moreover, HIV-1 attached onto the endothelium via syndecans represents an in trans source of infection for circulating T cells (2). Primary HIV-1, HIV-2, and simian immunodeficiency virus (SIV) isolates produced from peripheral blood mononuclear cells (PBMCs) exploit syndecans (2). Furthermore, syndecans on microvascular endothelial cells play a significant role in cell-free HIV-1 transmigration through the blood-brain barrier (3). Thus, HIV-1 has maximized its utilization of syndecans in the body.
A single conserved arginine (Arg298) in the V3 region of gp120 governs HIV-1 binding to syndecans (11). An amine group on the side chain of this residue is absolutely required for syndecan utilization by HIV-1 (11). HIV-1 binds syndecans via a 6-O sulfation (11) within the HS chains, demonstrating that this binding is not the result of random interactions between basic residues and negative charges but the result of specific contacts between gp120 and a well-defined sulfation in syndecans. Surprisingly, the Arg298 in gp120 that mediates HIV-1 binding to syndecans also mediates HIV-1 binding to CCR5 (42), suggesting that HIV-1 recognizes similar motifs on syndecans and CCR5 (11). Supporting this hypothesis, the 6-O sulfation recognized by HIV-1 on syndecans mimics the sulfated tyrosines recognized by HIV-1 in the N terminus of CCR5 (11). The finding that CCR5 and syndecans are exploited by HIV-1 via a single determinant echoes the mechanisms by which chemokines utilize these two disparate receptors and suggests that the gp120/chemokine mimicry may represent a common strategy in microbial pathogenesis.
More recent work suggests that syndecans play a critical role in HIV-1 transmission (4). HIV-1 transmission includes transmigration of HIV-1 through the genital epithelium and subsequent capture and transfer of infectious particles from dendritic cells (DC) and/or Langerhans cells (LC) to T cells (31, 34, 38). Importantly, human cervical and vaginal mucosal epithelia richly express syndecans in vivo (4). HS chain removal by heparinase treatment prevents HIV-1 transcytosis through primary human cervical and vaginal mucosal epithelia (4). The Arg298 V3 HIV-1 mutant, which cannot interact with syndecans (11), fails to transcytose (4). Thus, syndecans facilitate HIV-1 epithelial transmigration in vitro. More recently, we identified syndecan-3 as an HIV-1 receptor on DC (12). Syndecan-3 stabilizes the captured virus, enhances DC infection in cis, and promotes transfer to T cells (12). Removal of the HS from the cell surface by heparinase or by silencing syndecan-3 by small interfering RNA (siRNA) partially inhibited HIV-1 transmission by immature DC (12), whereas neutralizing both syndecan-3 and DC-SIGN abrogated HIV-1 capture and subsequent transmission (12). Our findings that syndecans modulate both HIV-1 epithelial transmigration and HIV-1 transfer from DC to CD4+ T cells suggest that syndecans represent new targets for the development of topical microbicides.
Nearly 40 million people currently live with HIV-1/AIDS; the majority of them reside in developing countries. It has been estimated that there are over 14,000 new HIV-1 infections per day in adults aged 15 to 50 years, with 50% of the newly infected being women aged 15 to 24 years (21, 22). In the absence of a vaccine, there is thus an urgent need for the development of safe and effective topical microbicides to prevent the sexual transmission of HIV-1 (14, 26). Topical microbicides are currently defined as vaginally or rectally applied products that prevent male-to-female or female-to-male transmission of HIV-1 infectious particles (39).
In this study, we proposed to develop a novel class of microbicides using syndecan as the antiviral agent. Specifically, we generated a soluble syndecan-Fc hybrid molecule by fusing the ectodomain of syndecan-1 to the Fc domain of a human IgG. We then tested this syndecan-Fc hybrid molecule for its in vitro microbicidal properties. Remarkably, the syndecan-Fc hybrid molecule possesses multiple attractive microbicidal properties: (i) it blocks HIV-1 infection of primary targets including T cells, macrophages, and DC; (ii) it exhibits a broad range of antiviral activity against primary HIV-1 isolates, multidrug-resistant HIV-1 isolates, HIV-1, and SIV; (iii) it prevents transmigration of HIV-1 through human primary genital epithelial cells (PGEC); (iv) it prevents HIV-1 transfer from DC to CD4+ T cells; (v) it is potent when added 2 h prior to addition of HIV-1 to target cells; (vi) it is potent at a low pH; (vii) it blocks HIV-1 infectivity when diluted in genital fluids; and (viii) it inhibits HSV infection. Thus, this syndecan-Fc hybrid molecule represents the prototype of a new generation of microbicidal agents that may have promise for HIV-1 prevention.
MATERIALS AND METHODS
Viruses.
293T cells were transfected with proviral plasmid (9 μg) and vesicular stomatitis virus G (VSV-G) envelope plasmid (1 μg). At day 2, VSV-G pseudotyped viruses (5 μg of p24) were harvested and used to acutely infect Jurkat cells (2 × 106 cells). Two days after infection, viruses were harvested, and p24 content was measured by enzyme-linked immunosorbent assay (ELISA; PerkinElmer, Waltham, MA). The following proviral constructs were used: wild-type pNL4.3 (X4); pNL4.3-BaL (R5), in which the wild-type NL4.3 envelope was switched for the R5 BaL envelope; pNL4.3ΔEnv, which lacks gp160; and pNL4.3-eGFP (X4) and pNL4.3-BaL-eGFP (R5), which encode the enhanced green fluorescent protein (eGFP) gene instead of the nef gene (6). Primary HIV-1, drug-resistant HIV-1, HIV-2, and SIV were obtained through the National Institutes of Health (NIH) AIDS Research and Reference Reagent Program and amplified in activated human PBMCs (5 × 106 cells) as described previously (5). The following HIV-1 group M viruses were used: (i) 92RW021, a subtype A Env, subtype A, R5 virus (contributed by WHO Network for HIV Isolation and Characterization); (ii) 92UG029, a subtype A Gag, subtype A Env, X4 (syncytium-inducing [SI]) virus (WHO Network for HIV Isolation and Characterization); (iii) 92TH026, a subtype B Gag, subtype B Env, R5 (non-syncytium-inducing [NSI]) virus (the UNAIDS Network for HIV Isolation and Characterization); (iv) 92HT599, a subtype B Env, X4 (SI) virus (Neal Halsey and the Division of AIDS [DAIDS], NIAID); (v) 93IN101, a subtype C Env, R5 (NSI) virus (Robert Bollinger and the UNAIDS Network for HIV Isolation and Characterization and the DAIDS, NIAID); (vi) 98IN017, a subtype C Env, X4 virus (Robert Bollinger and the UNAIDS Network for HIV Isolation and Characterization and the DAIDS, NIAID; courtesy of the NIBSC Center for AIDS Reagents, United Kingdom); (vii) 92UG005, a subtype D Gag, subtype D Env, R5 virus (UNAIDS Network for HIV Isolation and Characterization and the DAIDS, NIAID); (viii) 92UG024, a subtype D Gag, subtype D Env, X4 (SI) virus (UNAIDS Network for HIV Isolation and Characterization and the DAIDS, NIAID); (ix) 92TH006, a subtype E Env, R5 (NSI) virus (UNAIDS Network for HIV Isolation and Characterization and the DAIDS, NIAID); (x) 93TH053, a subtype E Env, X4 virus (UNAIDS Network for HIV Isolation and Characterization and the DAIDS, NIAID); (xi) 93BR029, a subtype F Gag, subtype F Env, R5 (NSI) virus (UNAIDS Network for HIV Isolation and Characterization and the DAIDS, NIAID); (xii) 93BR020, a subtype F Gag, subtype F Env subtype, R5 and X4 (SI) virus (UNAIDS Network for HIV Isolation and Characterization and the DAIDS, NIAID); (xiii) RU132, a subtype G Env, R5 virus (A. Bobkov, D. I. Ivanovsky Institute of Virology, Moscow, Russia; supplied by Jonathon Weber, Imperial College, London, United Kingdom); (xiv) Jv1083, a subtype G Env, R5 virus (A. Abimiku); and (xv) JR-CSF, a laboratory-adapted R5 (NSI) virus (I. Chen). The following drug-resistant viruses were used: HIV-174V/MT-2, a reverse transcriptase (RT) inhibitor-resistant X4 virus (HXB2; contributed by B. Larder, courtesy of the MRC AIDS Directed Program) (abbreviated RT74V); HIV-1LAI-M184V (lamivudine [3TC]-resistant), an RT inhibitor-resistant X4 virus (LAI; J. Mellors and R. Schinazi) (RTM184V); HIV-1 M46I/L63P/V82T/I84V, a protease (PR) inhibitor-resistant R5X4 virus (E. Emini) (PRM46I/L63P/V82T/I84V); and pNL4-3 gp41 (36G) V38A/N42D, a fusion inhibitor (T20)-resistant X4 virus (Trimeris, Inc.) (T20V38A/N42D). The following additional retroviruses were used: SIVmac251/43H (contributed by R. Desrosiers), SIVsyk1.2 (V. Hirsch and P. Johnson), HIV-2CDC310342 (M. Rayfield for S. Wiktor), and HIV-27312A (F. Gao and B. Hahn).
Cells.
TZM-bl cells were obtained through the NIH AIDS Research and Reference Reagent Program. TZM-bl cells express CD4, CXCR, and CCR5, which render them susceptible to infection, and contain an integrated Escherichia coli lacZ gene driven by the HIV-1 long terminal repeat (LTR) (44). Upon infection, Tat production from the integrated provirus leads to activation of the lacZ reporter, resulting in synthesis of β-galactosidase in these cells. Infected cells are identified by enzymatic activity measurement at 48 h postinfection, allowing quantitation after a single round of infection, as described previously (44). Blood-derived CD4+ T lymphocytes, macrophages, and DC were isolated as described previously (5). Briefly, human PBMCs were purified from fresh blood by banding on Ficoll-Hypaque (30 min at 800 × g at 25°C) (GE Healthcare Bio-Sciences Corp., Piscataway, NJ). Primary human CD4+ T cells were purified from PBMCs by positive selection with anti-CD4 Dynabeads and subsequent release using Detachabead (Invitrogen Corp., Carlsbad, CA). Cells were cultured in RPMI medium 1640 (Invitrogen Corp., Carlsbad, CA) supplemented with 10% fetal calf serum (FCS; Thermo Fisher Scientific Inc., Waltham, MA), minimal essential medium (MEM) amino acids, l-glutamine, MEM vitamins, sodium pyruvate (Invitrogen Corp., Carlsbad, CA), and penicillin plus streptomycin (Mediatech, Inc., Manassas, VA) and were subsequently activated with bacterial superantigen staphylococcal enterotoxin B (SEB; 100 ng/ml) and mitomycin C-killed PBMCs from another donor (cell ratio of 10:1 PBMCs to CD4+ cells). Three days after stimulation, cells were split 1:2 in medium containing interleukin-2 ([IL-2] final concentration, 200 units/ml; NIH AIDS Research and Reference Reagent Program). Cultures were then split 1:2 every 2 days in IL-2 medium and infected with HIV-1 at 7 days after stimulation. For generating primary human macrophages, monocytes were purified from human PBMCs by negative selection (Invitrogen Corp., Carlsbad, CA) and activated and cultured at a cell concentration of 106 cells/ml in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FCS (Thermo Fisher Scientific Inc., Waltham, MA), MEM amino acids, l-glutamine, MEM vitamins, sodium pyruvate (Invitrogen Corp., Carlsbad, CA), penicillin (100 units/ml), streptomycin (100 mg/ml), and 50 ng/ml recombinant human granulocyte-macrophage colony-stimulating factor (GM-CSF) (R&D Systems Inc., Minneapolis, MN) and maintained at 37°C in a humidified atmosphere supplemented with 5% CO2. To obtain monocyte-derived macrophages, cells were allowed to adhere to plastic and cultured for 6 days to allow differentiation. Immature DC were cultured from monocytes in the presence of IL-4 and GM-CSF (500 and 800 units/ml, respectively). At day 7, the phenotype of the cultured DC was confirmed by flow cytometric analysis. PGEC were provided by B. Kahn of the Department of Obstetrics and Gynecology at Scripps Clinic, La Jolla, CA. By rotation of cotton swabs against the vaginal walls, several million cells were collected per individual. Cells were immediately placed in sterile phosphate-buffered saline (PBS), held at 4°C, and transported to the laboratory. After centrifugation (at 300 × g for 5 min), the cell pellet was digested in 1 mg/ml collagenase/dispase (Roche Diagnostics Corp., Roche Applied Science, Indianapolis, IN) containing 1 mg/ml DNase (Sigma-Aldrich, St. Louis, MO) and 0.15 mg/ml Na-p-tosyl-l-lysine chloromethyl ketone (Sigma-Aldrich, St. Louis, MO) for 1 h at 37°C. The digest was spun down (1,000 × g for 20 min) and resuspended in 250 mg/ml PBS-bovine serum albumin (BSA). After additional centrifugation, the pellet was resuspended in 5 mg/ml PBS-BSA and loaded onto a 50% Percoll gradient. PGEC were then isolated from contaminating cells by fluorescence-activated cell sorting (FACS) as described previously (4) and propagated into collagen type I-coated T-25 flasks in DMEM-F12 medium containing 10% FCS and epithelial cell growth supplement (100 μg/ml) (Sigma-Aldrich, St. Louis, MO). PGEC were purified using magnetic cell sorting (MACS) columns (Miltenyi Biotec Inc., Auburn, CA) and MACS beads (Miltenyi Biotec Inc., Auburn, CA) coated with antibodies directed against specific cytokeratin (CK) members. Specifically, the following antibodies were used: CK3-3E4 (Miltenyi Biotec Inc., Auburn, CA), which recognizes CK8, CK18, and CK19; the CK3-6H5 (Miltenyi Biotec Inc., Auburn, CA), which recognizes CK7 and CK8; LL025 (Novocastra Laboratories, Ltd., Newcastle, United Kingdom), which recognizes CK16; and SY5 (Novocastra Laboratories Ltd., Newcastle, United Kingdom), which recognizes involucrin. As a second source of PGEC, cells were obtained from BioWhittaker (customized request) (BioWhittaker, Inc., Walkersville, MD). PGEC were passaged fewer than three times before use to maintain their original features.
Genital fluids and seminal plasma.
To collect genital and vaginal fluid/mucus, swabs were kept in the cervix and in the posterior vaginal fornix for about 20 s. Each swab was placed in a test tube containing 0.5 ml of PBS. A swab absorbs approximately 150 μl of fluid when saturated, and the average dilution of vaginal/cervical sample in the buffer will be approximately 1:5. The specimens were centrifuged at 15,000 rpm at 4°C for 10 min, and supernatants were pooled and stored at −80°C until use. Specimens were obtained from Lee Biosolutions Inc., St. Louis, MO. To collect seminal plasma, ejaculates were centrifuged at 1,300 × g for 15 min at 4°C to separate spermatozoa from seminal plasma. The seminal plasma was further clarified by centrifugation at 10,000 × g for 15 min, and supernatant was collected and stored at −80°C for further use. Specimens were obtained from GeneWay, San Diego, CA.
Infections.
TZM-bl cells (44) (100,000 cells/ml) were exposed to HIV-1 (100 pg of p24) for 4 h together with or without Fc proteins and washed, and infection was measured 48 h after infection by β-galactosidase activity. For 50% and 90% inhibitory concentration (IC50 and IC90, respectively) measurements, TZM-bl cells were exposed to HIV-1 variants (primary isolates and drug-resistant viruses) or HIV-2 and SIV isolates at 37°C in the presence of increasing concentrations of syndecan-1-Fc or control Fc. Viruses and Fc proteins were washed away after 4 h. Infection was measured 48 h after infection by β-galactosidase activity. Results (triplicate) were expressed in concentration (μg/ml) of Fc proteins required to inhibit 50% or 90% of virus infectivity. IC50 and IC90 measurements presented were representative of three independent experiments. To examine the inhibitory effect of Fc proteins on HIV-1 infection of primary cells, DC, CD4+ T lymphocytes, or macrophages (0.1 × 106 cells) were exposed to virus (1 ng of p24) for 1 day in the presence or absence of Fc proteins, washed three times with medium, and cultured in a flat-bottom 96-well plate. Supernatants were collected after different days, and viral replication was monitored by p24 ELISA (PerkinElmer, Waltham, MA). For cell-to-cell infection, activated PBMCs (200,000 cells) were infected with JR-CSF (500 pg of p24) for 3 days, washed three times to remove cell-free virus, and added to TZM-bl cells in the presence of increasing concentrations of syndecan-1-Fc or control Fc (0.1 to 100 μg/ml) diluted in a pool (n = 6) of seminal plasma. Infection was scored as above.
Transmigration assay.
The HIV-1 transmigration assay using PGEC seeded onto polycarbonate membrane transwells was conducted as described previously (4). Briefly, PGEC were seeded onto the upper face of collagen I and fibronectin-coated 12-mm-diameter, 3-μm-pore-size polycarbonate membrane transwells at a density of 105 cells per well and were cultured until formation of tight junctions was achieved. The inserts were fed every 2 days. The monolayer on the filter effectively divides the well into an apical compartment and a basolateral compartment. The integrity of each cell monolayer was measured using an epithelial volt-ohm meter (Millipore Inc., Billerica, MA). To ensure the integrity of the PGEC barrier, we monitored the elevated transepithelial electrical resistance of each cell monolayer and measured the paracellular passage of the extracellular marker inulin by determination of the permeability coefficient as evaluated by a diffusion assay using [14C]carboxylated inulin (Sigma-Aldrich, St. Louis, MO) (molecular weight, 5,000) in the upper chamber, as we described previously (4). After verifying the integrity of the monolayer on the transwell filters, PGEC were exposed to HIV-1 (added to the upper chamber), and viral transcytosis was monitored by measuring amounts of particulate capsid in the basal chamber by p24 ELISA. We eliminated free capsid from particulate capsid by microcentrifuging medium collected from the basal chamber for 90 min at 4°C. The integrity of the monolayer of PGEC was preserved even 8 h after virus exposure (4). We verified by ELISA (horseradish peroxidase anti-human Fc antibody detection) that the Fc proteins did not cross the PGEC monolayer, indicating that the Fc fusion protein cannot act on transcytosed viruses. After particulate capsid measurement by p24 ELISA, infectivity of transcytosed viruses (1 ng of p24) was confirmed using TZM-bl indicator cells.
DC transmission assay.
Blood-derived immature DC were plated at 50,000 cells per well in 96-well v-bottom plates (BD Biosciences, San Jose, CA). Cells were incubated with NL4.3-eGFP (X4), NL4.3-BaL-eGFP (R5), or the single-round NL4.3ΔEnv-eGFP pseudotyped with NL4.3 gp160 (X4) (25 ng of p24) for 2 h at 37°C. Cells were washed three times with warm medium, and CCR5+ Jurkat T cells (100,000 cells) were added. Cells were cultured in a flat-bottom 96-well plate, harvested after 3 days, and fixed in 4% paraformaldehyde-PBS, and GFP expression was measured by FACS. The percentage of infected Jurkat T cells was selectively quantified by gating T cells using an anti-CD3 antibody as described previously (5).
Syndecan-1-Fc plasmid construction.
The ectodomain of human syndecan-1 was subcloned by PCR amplification. The amplified DNA product was digested with NotI and BamHI, gel purified, and ligated in frame with the Fc domain of human IgG1 in pCR3 as described previously (10). For amplification of the expression of Fc and syndecan-1-Fc protein, the glutamate synthetase (GS) amplification system was used as described previously (10). This system uses a vector that has the gene of interest and a GS gene, which allows selection and amplification in a glutamine-free environment in the presence of methionine sulfoximine, an inhibitor of the GS encoded by the vector and the CHO cells. For this purpose the Fc and syndecan-1-Fc fragments were subcloned under the control of the cytomegalovirus promoter in pRSC, a mammalian expression vector with two transcriptional units, as described previously (10). The GS gene was cloned by PCR amplification from a CHO cDNA library. The sense (5′-CCCACACCAAGCTTCTCGCCGCTC-3′) and antisense (5′-GATGAACTAGGAAAGCTTCAAGATCACTCAAAG-3′) primers, including HindIII sites (underlined), were designed from the nucleotide sequence of the GS gene as described previously (10). The PCR product was digested with HindIII and cloned under the control of the Rous sarcoma virus promoter in pRSC. The Δ3HS-syndecan-1-Fc devoid of its three HS chains was created by site-directed mutagenesis by replacing serines 37, 45, and 47 with alanines.
Expression, amplification, and purification of the syndecan-1-Fc fusion protein.
For production of stable cell lines expressing Fc and syndecan-1-Fc, 106 CHO-K1 cells were transfected with 10 μg of pRSC-GS-Fc, pRSC-GS-syndecan-1-Fc, and pRSC-GS-Δ3HS syndecan-1-Fc. At 36 h posttransfection, cells were trypsinized, plated in 96-wells of a microtiter plate, and grown in glutamine-free DMEM containing 25 μM methionine sulfoximine. After 14 to 21 days, clones surviving the selection were transferred to T25 flasks and grown to confluence. Supernatants were then harvested and incubated with 15 μl of protein A-Trisacryl at 4°C for 12 h. After three washes in PBS, beads were eluted in 25 μl of sodium dodecyl sulfate (SDS) sample buffer and loaded on an SDS-4 to 16% polyacrylamide gel, and the presence of Fc and syndecan-1-Fc proteins was analyzed by Western blotting with antibodies directed against human IgG1 Fc (GE Healthcare Bio-Sciences Corp., Piscataway, NJ). Blots were revealed by an enhanced chemiluminescence procedure (Thermo Fisher Scientific, Rockford, IL). Recombinant Fc and syndecan-1-Fc proteins were batch purified by affinity chromatography over protein A as specified by the manufacturer (Thermo Fisher Scientific, Rockford, IL). The concentrations of purified Fc and syndecan-1-Fc recombinant proteins were determined using a bicinchoninic acid protein assay (Thermo Fisher Scientific, Rockford, IL). Syndecan-1-Fc was incubated with heparinase I, II, and III (0.3 mIU) (Sigma-Aldrich, St. Louis, MO) for 2 h at 37°C to remove HS chains from the core protein.
Syndecan-1-Fc ELISA.
Nunc MaxiSorb eight-well strip plates were coated with anti-syndecan-1 IgG (clone ID4) or control isotype IgG (10 μg/ml) for 16 h at 4°C. Wells were washed extensively and blocked in PBS-BSA (1 mg/ml) for 4 h at room temperature. Medium containing (or not) syndecan-1-Fc was added to wells overnight at 4°C. Wells were washed, and captured syndecan-1-Fc molecules were revealed using anti-human Fc-horseradish peroxidase-IgG (1 μg/ml). Wells were washed, and o-phenylenediamine dihydrochloride substrate was added. After color development, the reaction was stopped with 4N sulfuric acid, and wells (triplicate) were read on a plate reader at 490 nm.
Cytotoxicity.
Cells were plated in clear-bottom 96-well plates (BD Biosciences, San Jose, CA). Cells were serially diluted from 55,000 cells to 25 cells in 100 μl of complete DMEM or RPMI medium. Fifteen microliters of the CellQuanti-MTTTM reagent (Gentaur, Kampenhout, Belgium) was added, and cells were incubated for 4 h at 37°C. Then, 100 μl of the solubilization solution was added, and the plate was shaken for 1 h at room temperature. The A570 was measured on a SpectraMax 384 Plus reader (Molecular Devices, Sunnyvale, CA). A linear relationship was observed between the A570 value and the cell number. The detection limit was estimated to be 950 cells from the blank control. To determine the cytotoxicity of Fc proteins on cells, 55,000 cells were plated per 80-μl well in clear-bottom 96-well plates. Cells were treated twice daily with 200 μg/ml of syndecan-1-Fc, Fc, or 0.01% saponin (Sigma-Aldrich, St. Louis, MO) for a week. No washes were performed to maintain a continuous exposure of cells to Fc proteins.
RESULTS
Construction, expression, amplification, and purification of the syndecan-Fc hybrid molecule.
The ectodomain of human syndecan-1 was subcloned in frame with the Fc domain of human IgG1. For amplification of the expression of Fc and syndecan-1-Fc, a GS amplification system was used that allows selection and amplification in CHO cells in a glutamine-free environment in the presence of methionine sulfoximine, an inhibitor of the GS encoded by the vector (10). For production of stable cell lines expressing Fc and syndecan-1-Fc, CHO cells were transfected with pRSC-GS-Fc and pRSC-GS-syndecan-1-Fc and grown in G-DMEM containing methionine sulfoximine. After 2 to 3 weeks of selection, clone supernatants were analyzed for Fc content by Western blotting using anti-Fc antibodies (Fig. 1; Fc and wild-type syndecan-1-Fc, lanes 1 and 2, respectively). Positive clones were then amplified for 2 to 3 additional weeks, and Fc and syndecan-1-Fc proteins were batch purified from 1 to 2 liters of cell supernatant by protein A affinity. The rationale for linking syndecan-1 to the Fc of human IgG1 was that the Fc moiety allowed efficient secretion of a syndecan molecule, which harbored heparan sulfate chains, into the extracellular medium and allowed subsequent affinity purification on a protein A column. Moreover, the Fc moiety was extremely useful to conduct ELISA, FACS analysis, and pulldown studies. Syndecan-1 was chosen because it is the most studied syndecan member and because it is highly expressed on genital epithelial cells (4).
FIG. 1.
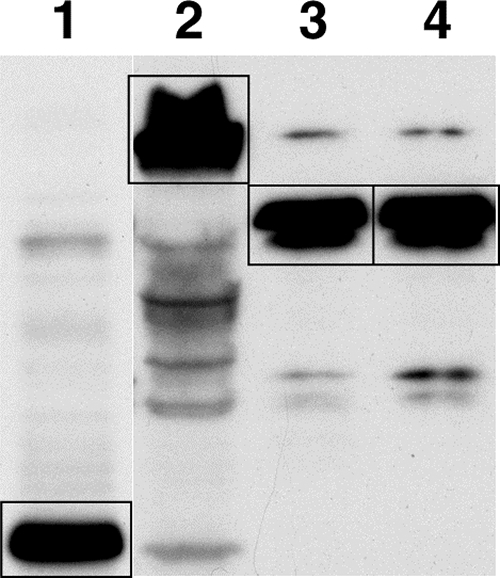
Western blot analysis of recombinant syndecan-Fc hybrid molecules. Protein A-purified Fc (1 μg) (lane 1), syndecan-1-Fc (1 μg) (lane 2), syndecan-1-Fc (1 μg) pretreated for 2 h at 37°C with a mixture of heparinases I, II and III (10 U) (lane 3), and the heparan sulfate chainless syndecan-1-Fc mutant called Δ3HS-syndecan-1-Fc (lane 4) (1 μg) were analyzed by Western blotting using anti-human Fc IgG (1/1,000 dilution). Fc and syndecan-1-Fc proteins (bands) are boxed.
Anti-HIV-1 spectrum of the syndecan-Fc hybrid molecule.
We first tested the ability of the syndecan-Fc hybrid molecule to block the infection of CD4+ CXCR4+ CCR5+ HeLa TZM-bl reporter (β-galactosidase) cells by three distinct categories of viruses. The first category includes HIV-1 isolates representative of various HIV-1 subtypes with different coreceptor usage, either CCR5 (R5 viruses) or CXCR4 (X4 viruses). The second category includes HIV-1 isolates that are resistant to reverse transcriptase (RT), protease (PR), or fusion (T20) inhibitors. The third category is other retroviruses such as HIV-2 and SIV isolates. Viruses were added to TZM-bl cells together with Fc or syndecan-1-Fc for 4 h; cells were washed, and infection was measured 48 h after infection as β-galactosidase activity. Importantly, we found that syndecan-1-Fc, but not Fc, efficiently blocks infection of all HIV-1, HIV-2, and SIV isolates (Table 1).
TABLE 1.
Syndecan-1-Fc exhibits a broad anti-HIV spectrum
| Virus and/or isolate | Clade | Coreceptor usage | Syndecan-1-Fc activity (μg/ml) |
Fc control activity (μg/ml) |
||
|---|---|---|---|---|---|---|
| IC50 | IC90 | IC50 | IC90 | |||
| HIV-1 group M isolatesa | ||||||
| 92RW021 | A | R5 | 1.9 ± 0.06 | 6.1 ± 0.2 | >100 | >100 |
| 92UG029 | A | X4 | 2.2 ± 0.08 | 8.2 ± 0.07 | >100 | >100 |
| 92TH026 | B | R5 | 0.3 ± 0.02 | 4.9 ± 0.11 | >100 | >100 |
| 92HT599 | B | X4 | 2.1 ± 0.11 | 7.7 ± 0.23 | >100 | >100 |
| 93IN101 | C | R5 | 1.2 ± 0.04 | 4.6 ± 0.14 | >100 | >100 |
| 98IN017 | C | X4 | 1.6 ± 0.1 | 3.6 ± 0.06 | >100 | >100 |
| 92UG005 | D | R5 | 1.9 ± 0.03 | 9.3 ± 0.14 | >100 | >100 |
| 92UG024 | D | X4 | 0.8 ± 0.05 | 7.8 ± 0.06 | >100 | >100 |
| 92TH006 | E | R5 | 2.4 ± 0.12 | 4.5 ± 0.23 | >100 | >100 |
| 93TH053 | E | X4 | 2.3 ± 0.11 | 7.4 ± 0.19 | >100 | >100 |
| 93BR029 | F | R5 | 2.1 ± 0.06 | 6.3 ± 0.07 | >100 | >100 |
| 93BR020 | F | X4 | 1.0 ± 0.02 | 4.9 ± 0.25 | >100 | >100 |
| RU132 | G | R5 | 0.4 ± 0.06 | 3.3 ± 0.03 | >100 | >100 |
| Jv1083 | G | R5 | 2.4 +/− 0.13 | 7.2 ± 0.17 | >100 | >100 |
| Drug-resistant HIV-1 virusesa | ||||||
| RT74V | 2.4 ± 0.14 | 9.1 ± 0.24 | >100 | >100 | ||
| RTM184V | 0.8 ± 0.03 | 5.4 ± 0.07 | >100 | >100 | ||
| PRM46I/L63P/V82T/I84V | 0.5 ± 0.01 | 3.9 ± 0.08 | >100 | >100 | ||
| PRG48V/L90M | 1.1 ± 0.03 | 7.4 ± 0.13 | >100 | >100 | ||
| T20V38A/N42D | 0.6 ± 0.07 | 5.6 ± 0.04 | >100 | >100 | ||
| T20N425 | 1.4 ± 0.1 | 8.3 ± 0.5 | >100 | >100 | ||
| Other retroviruses | ||||||
| SIVmac251 43H | 1.7 ± 0.04 | 6.9 ± 0.3 | >100 | >100 | ||
| SIVsyk1.2 | 1.3 ± 0.02 | 5.4 ± 0.16 | >100 | >100 | ||
| HIV-2CDC310342 | 2.3 ± 0.11 | 8.6 ± 0.26 | >100 | >100 | ||
| HIV-27312A | 2.9 ± 0.04 | 9.2 ± 0.05 | >100 | >100 | ||
Representative of various HIV-1 subtypes with different coreceptor usage.
See Materials and Methods for a description of the viruses.
The syndecan-Fc hybrid molecule blocks HIV-1 infection of CD4+ T lymphocytes, macrophages, and DC.
An ideal microbicide should inhibit HIV-1 infection of primary in vivo targets of HIV-1. We thus asked whether syndecan-1-Fc could block HIV-1 infection in human primary CD4+ T lymphocytes, macrophages, and DC. Cells were exposed to HIV-1 (R5 JR-CSF) together with syndecan-1-Fc or Fc control. Twenty-four hours postinfection, cells were washed, and viral replication was monitored by measuring the amount of HIV-1 capsid protein present in the cell culture supernatant by p24 ELISA every 2 days for up to 10 to 14 days thereafter. Remarkably, syndecan-1-Fc, but not Fc, blocked HIV-1 infection of all three in vivo cell targets of HIV-1 (Fig. 2A). We conducted similar inhibitory experiments using increasing concentrations of Fc or syndecan-1-Fc rather than a fixed concentration of Fc proteins. IC50 and IC90 values are presented in Table 2. Importantly, syndecan-1-Fc exhibited no cytotoxic effect on target cells (Fig. 2B). We used the natural detergent saponin as positive control for cytotoxicity (Fig. 2B). These results suggest that the syndecan-Fc hybrid molecule has the potential to specifically prevent infection of T cells, macrophages, and DC during the early steps of HIV-1 transmission.
FIG. 2.

The syndecan-Fc hybrid molecule blocks HIV-1 infection of primary in vivo cell targets. (A) Blood-derived CD4+ T lymphocytes, macrophages, or DC (0.5 × 106 cells) were exposed to JR-CSF (1 ng of p24) and syndecan-1-Fc or control Fc (5 μg/ml) for 1 day at 37°C and washed. The amount of virus released was monitored by measuring amounts of capsid protein in cell supernatants by p24 ELISA. The points are median values, and error bars represent standard errors of triplicates. The P values were calculated using a one-sided t test assuming equal variances. d, day. (B) Cells were serially diluted from 55,000 cells to 25 cells in 100 μl of complete medium. Fifteen microliters of the CellQuanti-MTTTM reagent was added, and cells were incubated for 4 h at 37°C. Then, 100 μl of the solubilization solution was added, and the plate was shaken for 1 h at room temperature. The A570 was measured on a SpectraMax 384 Plus reader. To determine the cytotoxicity of Fc proteins, cells (55,000) were treated twice daily with 200 μg/ml of syndecan-1-Fc, Fc, or 0.01% saponin for a week. No washes were performed to maintain a continuous exposure of cells to Fc proteins. OD, optical density.
TABLE 2.
Syndecan-Fc hybrid inhibition of HIV-1 JR-CSF infection of CD4+ T lymphocytes, macrophages, and DC
| Target cell type and donor | Syndecan-1-Fc activity (μg/ml) |
Fc control activity (μg/ml) |
||
|---|---|---|---|---|
| IC50 | IC90 | IC50 | IC90 | |
| CD4+ T lymphocytes | ||||
| Donor 1 | 1.1 ± 0.03 | 3.2 ± 0.14 | >100 | >100 |
| Donor 2 | 0.8 ± 0.04 | 2.5 ± 0.13 | >100 | >100 |
| Macrophages | ||||
| Donor 1 | 1.5 ± 0.02 | 4.6 ± 0.21 | >100 | >100 |
| Donor 2 | 0.9 ± 0.03 | 3.1 ± 0.13 | >100 | >100 |
| DC | ||||
| Donor 1 | 1.6 ± 0.1 | 4.2 ± 0.18 | >100 | >100 |
| Donor 2 | 1.2 ± 0.04 | 3 ± 0.15 | >100 | >100 |
Antiviral mechanism of action of the syndecan-Fc hybrid molecule.
After demonstrating the broad potency of the syndecan-Fc hybrid molecule at blocking HIV-1 infection of its primary in vivo targets, we examined its mechanisms of antiviral action. Since we along with others previously reported that cell surface HSPG including syndecans facilitate the initial adsorption of the virus onto target cells (16, 25, 29, 30, 33), we asked whether syndecan-1-Fc interferes with HIV-1 adsorption onto CD4+ T lymphocytes and macrophages. To address this issue, HIV-1 (JR-CSF) together with Fc or syndecan-1-Fc was added to CD4+ T lymphocytes and macrophages. After 2 h at 4°C, cells were washed and lysed, and amounts of attached virus were quantified by p24 ELISA. Importantly, the syndecan-Fc hybrid molecule, but not Fc, profoundly reduced HIV-1 adsorption onto target cells (Fig. 3A). After demonstrating that syndecan-1-Fc blocks HIV-1 adsorption, we asked whether it also interferes with the subsequent entry step: the CD4/coreceptor-mediated internalization. To address this issue, plates containing cells and viruses were first centrifuged at 2,000 × g (spinoculation) for 3 h at 4°C to avoid any internalization. The spinoculation step artificially forces the initial attachment of the virus onto the cells (28). Cells were washed twice to remove unbound virus and then placed at 37°C for 4 h to permit internalization in the presence of Fc or syndecan-1-Fc. Cells were then trypsinized and lysed. Amounts of internalized virus were quantified by measuring amounts of capsid in cell lysates by p24 ELISA. We found that syndecan-1-Fc, but not Fc, significantly prevented HIV-1 internalization into CD4+ T lymphocytes and macrophages (Fig. 3B). Together these data suggest that the syndecan-Fc hybrid molecule blocks HIV-1 infection by interfering with HIV-1 entry.
FIG. 3.

The syndecan-Fc hybrid molecule blocks HIV-1 infection by inhibiting viral entry. CD4+ T lymphocytes (500,000 cells) or macrophages (150,000 cells) were exposed to 5 ng of p24 of JR-CSF in the presence or absence of syndecan-1-Fc or control Fc (5 μg/ml). To measure virus adsorption (A), cells were exposed to virus for 2 h at 4°C, washed twice with PBS, and lysed (not trypsinized), and amounts of capsid in cell lysate were quantified by p24 ELISA. To measure virus internalization (B), plates containing cells and viruses were first centrifuged at 2,000 × g (spinoculation) for 3 h at 4°C. Cells were washed twice to remove unbound virus and then placed at 37°C for 4 h in the presence of Fc or syndecan-1-Fc (5 μg/ml). Cells were then trypsinized and lysed. Amounts of internalized virus were quantified by measuring amounts of capsid in cell lysates by p24 ELISA. Results are expressed as the percentage of internalization relative to maximal adsorption, and internalization in the absence of Fc proteins is arbitrarily fixed at 100. The points are median values (triplicate) and error bars represent standard errors of triplicates. The P values were calculated using a one-sided t test assuming equal variances.
Since we previously obtained evidence that HIV-1-syndecan interactions are mediated via contacts between the Arg298 of gp120 and the 6-O sulfation of HS chains of syndecans (11), we asked whether syndecan-1-Fc mediates its inhibitory effect via its HS chains. To test this hypothesis, syndecan-1-Fc was pretreated with heparinase, which removes HS chains from the core syndecan protein (1) (Fig. 1, lane 3), and added to CD4+ T-lymphocytes together with HIV-1 (JR-CSF). Viral replication was monitored over a period of 10 days by measuring amounts of capsid in cell culture supernatant by p24 ELISA. In contrast to untreated syndecan-1-Fc, heparinase-treated syndecan-1-Fc, like Fc, did not influence viral replication in CD4+ T lymphocytes (Fig. 4A). This finding suggests that the syndecan-Fc hybrid molecule mediates its inhibitory effect via its sulfated heparan chains.
FIG. 4.

HS chain removal abrogates the anti-HIV activity of the syndecan-Fc hybrid molecule. (A) Blood-derived CD4+ T lymphocytes (0.5 × 106 cells) were exposed to JR-CSF (1 ng of p24) and untreated syndecan-1-Fc, heparinase-treated syndecan-1-Fc, or control Fc (5 μg/ml) for 1 day at 37°C and washed. The amount of virus released was monitored by measuring amounts of p24 capsid protein in cell supernatants by ELISA. (B) The same experiment as in panel A was performed except that the Δ3HS-syndecan-1-Fc, which is devoid of its HS chains after mutagenesis of its three glycosaminoglycan attachment sites, was tested for anti-HIV-1 activity. The points are median values, and error bars represent standard errors of triplicates. The P values were calculated using a one-sided t test assuming equal variances.
A distinct structural feature of the four syndecans (syndecan-1 to -4) is a conserved region of three glycosaminoglycan attachment sites near the N terminus of their core proteins (1, 23, 45). These sites predominantly initiate HS chains (1, 23, 45). To determine whether HS chains are important for the inhibitory effect of syndecan-1-Fc, we generated Δ3HS-syndecan-1-Fc, which is devoid of its three HS chains as a result of oligonucleotide-directed mutagenesis. Specifically, serine residues at position 37, 45, and 47 were changed to alanines, thereby preventing the addition of HS chains at those sites (Fig. 1, lane 4). Importantly, we found that in contrast to wild-type syndecan-1-Fc, Δ3HS-syndecan-1-Fc, like Fc, fails to block HIV-1 infection of CD4+ T lymphocytes (Fig. 4B). This further demonstrates that the HS chains of the syndecan-Fc hybrid molecule are responsible for the neutralization of HIV-1, likely by targeting the highly conserved Arg298 within gp120 (11).
Antiviral effect of the syndecan-Fc hybrid molecule before, during, and after virus exposure.
To determine the likely duration of antiviral activity of the syndecan-Fc hybrid molecule, we asked whether syndecan-1-Fc inhibits HIV-1 infection when added to cells for various lengths of time before or after the addition of virus. Initially, we added syndecan-1-Fc or Fc to TZM-bl cells at 1, 2, or 4 h before adding HIV-1 (R5 JR-CSF) (1 ng of p24) or together with the virus (time zero), and it was maintained in the culture until infection was monitored 48 h later. Syndecan-1-Fc significantly inhibited infection when it was added together with the virus (time zero) and up to 2 h before the addition of the virus (Fig. 5A), indicating that the syndecan-Fc hybrid molecule is highly potent in vitro at 37°C for 4 h. Next, we asked whether syndecan-1-Fc can inhibit HIV-1 infection when it was added after the virus is added to target cells. Syndecan-1-Fc does not significantly block HIV-1 infection when it is added to cells after virus inoculation (Fig. 5A, posttreatment 1, 2, or 4 h) (). These data suggest that the syndecan-Fc hybrid molecule is potent when added before virus exposure.
FIG. 5.

Antiviral effect of the syndecan-Fc hybrid molecule before, during, or after virus exposure. (A) Syndecan-1-Fc or control Fc (5 μg/ml) was added to TZM-bl cells 1, 2, or 4 h before (negative values on the y axis) or after (positive values on the y axis) the addition of HIV-1 (R5 JR-CSF) (1 ng of p24) or together (time zero) with the virus. Infection was measured 48 h after infection as β-galactosidase activity. (B) Activated PBMCs (200,000 cells) were infected with JR-CSF (500 pg of p24) for 3 days, washed three times to remove cell-free virus, and added to TZM-bl cells in the presence of increasing concentrations of syndecan-1-Fc or control Fc (0.1 to 100 μg/ml) diluted in a pool (n = 6) of seminal plasma. Infection was scored as above. The points are median values, and error bars represent standard errors of triplicates. The P values were calculated using a one-sided t test assuming equal variances.
Antiviral effect of the syndecan-Fc hybrid molecule on HIV-1 cell-to-cell transmission.
Since HIV-1 can be transmitted either as a cell-free or as cell-associated virus, we examined the effect of syndecan-1-Fc on cell-to-cell transmission. To address this issue, we added HIV-1-infected PBMCs to target TZM-bl reporter cells in the presence of semen and increasing concentrations of syndecan-1-Fc. Importantly, we found that syndecan-1-Fc significantly inhibits the infection of TZM-bl cells by infected PBMCs (Fig. 5B). It is likely that syndecan-1-Fc, by binding to the V3 region of gp120 on the surface of infected PBMCs, interferes with cell-to-cell fusion. We found that higher concentrations (100 μg/ml) of syndecan-1-Fc were necessary to inhibit cell-to-cell infection than cell-free infection (Fig. 5B). This is likely due to the fact that cell-to-cell HIV-1 infection is more efficacious than cell-free infection (36).
The syndecan-Fc hybrid molecule blocks HIV-1 transmission in vitro.
HIV-1 is thought to be sexually transmitted by genital epithelium transmigration followed by DC- and/or LC-mediated capture and transmission to CD4+ T cells. We first examined the antiviral effect of syndecan-1-Fc on HIV-1 epithelial transmigration. To address this issue, we developed an in vitro transwell chamber assay that mimics HIV-1 transcytosis through PGEC (4). HIV-1 was added to the apical surface of PGEC in the upper chamber of a transwell system for 8 h at 37°C, and the amount of transcytosed virus was quantified by p24 ELISA in the lower chamber medium in contact with the basal PGEC surface. Syndecan-1-Fc or control Fc was added to the apical surface of the PGEC monolayer 30 min before the virus was added. We chose to add Fc-syndecan to the transwell 30 min before the addition of the virus to simplify the design of the experiment since we found that adding the fusion protein 1 h before adding the virus to cells or at the same time similarly inhibited HIV-1 infection (Fig. 5). In the absence of Fc proteins, HIV-1 efficiently transmigrated through PGEC, in contrast to a virus lacking gp160 (Fig. 6A), suggesting that the viral glycoprotein is absolutely required for transcytosis and that the PGEC layer does not allow nonspecific transmigration of viruses. Amounts of transcytosed viruses were quantified by measuring amounts of particulate capsid in the basal chamber. Remarkably, syndecan-1-Fc, but not Fc, prevents HIV-1 transmigration through PGEC (Fig. 6A). We obtained similar results when we applied syndecan-1-Fc together with virus addition (data not shown). Importantly, syndecan-1-Fc exhibited no cytotoxic effect on PGEC (Fig. 6C).
FIG. 6.

The syndecan-Fc hybrid molecule inhibits both HIV-1 transmigration and HIV-1 transfer from DC to T cells. (A) Pseudotyped NL4.3ΔEnv/gp160 R5 Env virus (25 ng of p24) or Gp160-deficient NL4.3ΔEnv virus (25 ng of p24) was added to the apical surface of PGEC. Syndecan-1-Fc or control Fc (5 μg/ml) was added to PGEC 30 min before the virus was added. Amounts of transcytosed viruses in the basal chamber were quantified by p24 ELISA. Results are expressed as a percentage of p24 of the original inoculum. The points are median values, and error bars represent standard errors of triplicates. (B) DC (100,000 cells) were incubated for 2 h at 37°C with wild-type NL4.3-eGFP (X4 virus) and NL4.3-BaL-eGFP (R5 virus) viruses or with the pseudotyped NL4.3ΔEnv-eGFP/gp160 X4 Env virus (25 ng of p24). Syndecan-1-Fc or control Fc (5 μg/ml) was added 2 h later. DC were washed 2 h after the Fc proteins were added, Jurkat T cells (100,000 cells) were added for 3 days, and the percentage of infected Jurkat T cells was analyzed. The percentage of HIV-infected Jurkat T cells (gated by using an anti-CD3 antibody) in DC-T-cell coculture was analyzed by GFP expression on day 5. Error bars represent standard errors of duplicates. (C) The experiment is as described in the legend of Fig. 2B.
We then examined the capacity of the syndecan-Fc hybrid molecule to prevent DC-mediated transmission of HIV-1. We took advantage of a replication-defective virus (NL4.3ΔEnv-eGFP), which does not encode Env but which has been pseudotyped with HIV-1 Env (6). Pseudotyped viruses can infect cells because they contain Env. However, cells that have been infected by pseudotyped viruses cannot produce infectious viruses because de novo viruses do not encode Env. Thus, the use of pseudotyped viruses allowed the analysis of the effect of the syndecan-Fc hybrid molecule on the transmission of infectious particles from DC to T cells independently of DC infection. DC were incubated with wild-type NL4.3-eGFP and NL4.3-BaL-eGFP or pseudotyped NL4.3ΔEnv-eGFP/gp160 Env viruses. Two hours later, at which time the entry of the virus into DC is completed (18), syndecan-1-Fc or control Fc was added. After 2 h, DC were washed to remove both free virus and Fc proteins. To measure DC-T-cell transmission, Jurkat T cells were added for 3 days, and the percentage of infected T cells (gated with an anti-CD3 antibody) was analyzed by FACS. Only pseudotyped viruses that have been rapidly transferred from DC to T cells through the virological synapse (independently of DC infection) can infect T cells. Indeed, pseudotyped viruses that infect DC can no longer infect T cells because they do not encode Env. Because DC were washed before T cells were added, the T-cell infection by the pseudotyped virus observed in Fig. 6B could arise only from pseudotyped particles that were transferred from DC to T cells. Importantly, syndecan-1-Fc added to DC prevents subsequent T-cell infection with the pseudotyped virus (Fig. 6B). We obtained similar results when syndecan-1-Fc was added to DC 1 h before virus was added (data not shown). This finding suggests that syndecan-1-Fc neutralizes HIV-1 either before DC infection or during DC transfer to T cells. Thus, syndecan-1-Fc interferes with HIV-1 transmission at multiple levels: genital epithelial transmigration, DC infection or transfer to T cells, and T cell infection.
Antiviral activities of the syndecan-Fc hybrid molecule are preserved after low pH and genital fluid exposure.
An ideal microbicide should maintain its potency in the genital environment. We thus examined the inhibitory effect of the syndecan-Fc hybrid molecule under conditions that mimic the genital environment, such as exposure to low pH and genital fluids. We first examined the antiviral effect of the syndecan-Fc hybrid molecule at various pHs. Syndecan-1-Fc and Fc were incubated at pH 2 to 9 and tested for their antiviral activities. As expected, syndecan-1-Fc treated at a neutral pH (pH 7) neutralizes HIV-1. Importantly, syndecan-1-Fc maintained its potency in a basic (pH 8) or acidic (pH 4) environment (Fig. 7A), suggesting that antiviral activities of the syndecan-Fc hybrid molecule are preserved at low pH. We then examined the antiviral effect of the syndecan-Fc hybrid molecule in seminal plasma, cervical fluid, and vaginal mucus. Seminal plasma was separated from sperm by centrifugation at 500 × g for 10 min. The pH was determined before and after the seminal plasma was mixed with syndecan-1-Fc or Fc diluted in PBS. We found that the original pH of seminal plasma is approximately 8.0 to 8.05 (n = 8). Efficacy of syndecan-1-Fc against HIV-1 in the presence of seminal plasma was conducted as follows. Fifty microliters of serially 2-fold diluted syndecan-1-Fc in DMEM was mixed with 50 μl of JR-CSF and 50 μl of either seminal plasma or DMEM. The mixtures were added to TZM-bl indicator cells, and infection was scored. Similar studies were conducted with cervical fluid and vaginal mucus. Importantly, syndecan-1-Fc maintains its potent antiviral effect when diluted in each of these genital fluids (Fig. 7B). Together, these features are important with regard to the future use of the syndecan-Fc hybrid molecule as a microbicide.
FIG. 7.

The syndecan-Fc hybrid molecule is still active after low pH and genital fluid exposure. (A) TZM-bl cells (20,000 cells) were exposed to JR-CSF (100 pg of p24). Syndecan-1-Fc or control Fc was treated for 8 h at various pHs and added to cells (final concentration of 5 μg/ml) together with virus. Infection was measured 48 h postinfection by β-galactosidase activity. (B) Syndecan-1-Fc or control Fc was diluted in medium, a pool (n = 6) of seminal plasma, a pool of cervical fluid (n = 8), and a pool (n = 4) of vaginal mucus. The dilutions were then added to TZM-bl cells together with HIV-1 (100 pg of p24). Infection was measured 48 h postinfection by β-galactosidase activity. Results are expressed as relative light units. The points are median values, and error bars represent standard errors of triplicates. The P values were calculated using a one-sided t test assuming equal variances.
The syndecan-Fc hybrid molecule blocks HSV infection.
Since genital herpes increases transmission of HIV-1 (17), an ideal microbicide should inhibit HIV-1 as well as HSV infection. We thus investigated whether the syndecan-Fc hybrid molecule could also block HSV infection. To address this issue, Vero cells were exposed to HSV-1-GFP or HSV-2-GFP (multiplicity of infection [MOI] of 0.1) in the presence of syndecan-1-Fc or Fc. After 2 days, HSV infection was scored by FACS by measuring the percentage of GFP-positive (GFP+) Vero cells. Importantly, we found that syndecan-1-Fc, but not Fc, inhibits HSV-1 and HSV-2 infection (Fig. 8A). It is likely that syndecan-1-Fc blocks HSV infection by interfering with the interaction between HSV glycoproteins and entry receptors on Vero cells. Since HSV exploits HS proteoglycans to enhance its infection (7), it is likely that syndecan-1-Fc blocks HSV infection by interfering with the attachment of HSV to cell surface HS expressed on Vero cells (i.e., syndecans). Supporting this hypothesis, HS removal by enzymatic (heparinase treatment) or genetic (Δ3HS-syndecan-1-Fc) manipulation abrogates the anti-HSV effect of the syndecan-Fc hybrid molecule (Fig. 8A). As above (Fig. 2B, 5B and 6B), syndecan-1-Fc exhibited no cytotoxic effect on Vero cells (Fig. 8B). Dual neutralization of HIV-1 and HSV infection represents an additional microbicidal property for the syndecan-Fc hybrid molecule.
FIG. 8.

The syndecan-Fc hybrid molecule blocks HSV infection. Vero cells (250,000 cells) were exposed to HSV-1-GFP or HSV-2-GFP (MOI of 0.1) together with control Fc, syndecan-1-Fc, heparinase-treated syndecan-1-Fc, or Δ3HS syndecan-1-Fc lacking HS chains (5 μg/ml). After 2 days, Vero cell infection was scored by FACS by quantifying the percentage of GFP+ cells. Data are expressed in percentage of GFP+ cells. The points are median values, and error bars represent standard errors of triplicates. The P values were calculated using a one-sided t test assuming equal variances. (B) The experiment is as described in the legend of Fig. 2B.
DISCUSSION
In this study, we report that a syndecan-Fc hybrid molecule has potent antiviral activity against HIV-1. Interestingly, many antiviral properties of syndecan-1-Fc satisfy important preconditions for the use of syndecan-1-Fc as a potential microbicide. Syndecan-1-Fc (i) blocks HIV-1 infection of primary targets including T cells, macrophages and DC; (ii) exhibits a broad range of antiviral activity against primary HIV-1 isolates, multidrug resistant HIV-1 isolates, HIV-2, and SIV; (iii) prevents transmigration of HIV-1 through primary human genital epithelial cells; (iv) prevents HIV-1 transfer from DC to T cells; (v) is potent when added 2 h prior to addition of HIV-1 to cells; (vi) is potent at a low pH; (vii) efficiently blocks HIV-1 infectivity when diluted in genital fluids; and (viii) blocks HSV infection. Thus, this syndecan-Fc hybrid molecule represents the prototype of a new generation of microbicidal agents that may have promise for HIV-1 prevention.
We previously showed that HIV-1 binds to cell surface syndecans via a highly conserved residue (Arg298) within the V3 region of gp120 (11). Interestingly, the same residue is critical for gp120 binding to the CCR5 chemokine receptor (42). It is thus likely that the syndecan-Fc hybrid molecule blocks HIV-1 infection by interacting with Arg298 and, therefore, by interfering with both viral adsorption and coreceptor-mediated entry. Since compounds that interrupt gp120-syndecan interactions have never been exploited as microbicides, they represent a novel class of microbicides and thus differ from existing tools. One advantage of using compounds that target gp120-syndecan interactions as microbicides is that all primary R5, X4, and R5X4 HIV-1 as well as HIV-2 isolates bind syndecans. Thus, these compounds will have a broader impact than, for example, RANTES derivatives, which neutralize R5 viruses only (24). Since we found that compounds that block gp120-syndecan interactions also block gp120-CCR5 interactions (11), their dual inhibitory effects represent another advantage of using them as microbicides. Supporting this notion, we found that the syndecan-Fc hybrid molecule blocks a broad spectrum of X4, R5, and R5X4 viruses. Although it is likely that in vivo syndecan-1-Fc-resistant HIV-1 variants should arise, we were unsuccessful at developing such resistant variants in vitro. Nevertheless, it is important to emphasize that the “perfect” microbicide will likely be composed of several anti-HIV-1 agents that target viral proteins other than gp120 (i.e., RT) and therefore should neutralize emerging syndecan-1-Fc-resistant HIV-1 variants. Another advantage of using a syndecan-Fc hybrid molecule as a microbicide is that many sexually transmitted pathogens also exploit syndecans for host colonization, such as HSV (7), HPV (13, 37), and Neisseria gonorrhoeae (15, 41). Supporting this hypothesis, we found that the syndecan-Fc hybrid molecule efficiently blocks HSV infection (Fig. 8).
One cannot exclude the possibility that syndecan-1-Fc has adverse effects in the vaginal environment after its topical administration. One can envision that the hybrid molecule, by capturing specific cytokines or chemokines via its HS chains, enhances the suppression of immune activation and increases the inflammation state mediated by HIV-1 (9). Similarly, syndecan-1-Fc may play a detrimental role during HIV-1 transmission by capturing and neutralizing cationic antimicrobial polypeptides (i.e., defensins), which are the principal effector molecules of mucosal innate immunity against microbes and viruses such as HIV-1 (8). However, we do not anticipate that syndecan-Fc (60 to 90 kDa), which is extremely soluble and does not make aggregates even at high concentrations, would cause discomfort to humans or animals. Indeed, the ectodomain of syndecans, which we fused to an IgG1 Fc portion, is constitutively and constantly shed by human cells without affecting cell viability (1). The extracellular domain of syndecan-1 is proteolytically cleaved at a juxtamembrane site by a tissue inhibitor of metalloprotease-3-sensitive metalloproteinases in response to a variety of physiological stimulators in a process known as shedding (43). Shedding converts syndecan-1 from a membrane-bound coreceptor into a soluble effector capable of binding the same ligands. Thus, the nature of syndecan-1-Fc (ectodomain of existing human receptors fused to human antibody fragments) significantly diminishes the risk of adverse effects.
Another disadvantage of using syndecan-1-Fc as a topical microbicide is that its antiviral effect wanes in 4 h (Fig. 5). Interestingly, we found by FACS analysis that syndecan-Fc binds efficiently to HeLa cells (data not shown). It is likely that syndecan-Fc binds to HeLa cells by binding either to Fc receptors expressed on HeLa cells or to cytokines, chemokines, or growth factors bound onto the surface of HeLa cells. An adequate formulation of syndecan-1-Fc allowing a slow release of the fusion protein should, hopefully, partially alleviate this obstacle. Since syndecan-1-Fc loses its antiviral effect when added to cells 1 h after virus exposure, it would therefore need to be applied shortly before coitus and would not have long-lasting effects like other potential microbicides.
A series of microbicides has been developed (i.e., sulfated and sulfonated polymers) that most likely work similarly to syndecan-1-Fc. Importantly, a majority of these microbicidal candidates (carrageenan [the active ingredient in Carraguard], cellulose sulfate, PRO-2000, and dextran sulfate) failed to provide protection in humans (27, 40). It has been proposed that this failure arises from a limited in vitro potency of these sulfated compounds against R5 viruses, the most commonly transmitted strains of HIV-1 (27, 40). Moreover, evidence emerged that cellulose sulfate enhances HIV-1 infection in vitro, particularly of R5 viruses (27, 40). Thus, although syndecan-1-Fc efficiently blocks both X4 and R5 viruses and does not promote HIV-1 infection, it is still possible that the topical administration of syndecan-1-Fc in vivo would not provide any protection. Importantly, syndecan-1-Fc is currently being tested in a vaginal HIV-1 transmission model in macaque. Indeed, we believe that any microbicide candidate should demonstrate proof of both safety and efficacy in a macaque model before being considered for human trials. Evidently, success in a macaque model would not guarantee that syndecan-1-Fc will protect women, but a failure to protect monkeys would be considered a serious sign that it lacks potency.
Another concern with regard to the future development of syndecan-1-Fc as a microbicide is that it exhibits a potent antiviral activity only when used at a μg/ml range (Tables 1 and 2). To avoid having to use too high a concentration of syndecan-1-Fc in vivo, we propose to optimize the potency of the Fc hybrid molecule. We previously reported that the degree of sulfation of heparan sulfates directly correlates with their potential to neutralize HIV-1 (3). Specifically, we showed that the removal of the 6-O sulfation dramatically decreases the anti-HIV-1 effect of heparan sulfate analogs, whereas oversulfation significantly increases their inhibitory effect (3). Thus, with regard to future optimization of syndecan-1-Fc, we will examine whether manipulating the degree of sulfation of syndecan-1-Fc would improve its potency and therefore would diminish the amounts of syndecan-1-Fc needed for protection.
Protein microbicides such as syndecan-1-Fc could represent promising alternatives to the current generation of small-molecule drugs. However, producing and distributing such proteins in key areas of HIV endemicity are difficult and expensive because of the lack of a fermentor and a cold chain. Interestingly, many therapeutic proteins have been expressed in plants, including a large number of vaccine candidates and various recombinant antibody formats (32). The most important benefits of using plants such as maize seeds over fermentor-based systems include the enhanced stability of recombinant proteins accumulating in the endosperm, which means that a cold chain is not necessary for product distribution (32), as well as the economy of large-scale production, which will make the product more affordable. In other words, if syndecan-1-Fc provides protection in a vaginal SHIV transmission model in macaques, the production of syndecan-1-Fc in plants should be considered.
In conclusion, this study demonstrates that syndecan-1-Fc possesses numerous microbicidal in vitro properties against HIV-1. Specifically, syndecan-1-Fc can block HIV-1 transmission at multiple levels: trans-epithelial migration, DC-mediated transfer to T cells, and cis infection of CD4+ T lymphocytes, macrophages, and DC. Future safety and efficacy in vivo studies using humanized mice and macaque vaginal simian-human immunodeficiency virus (SHIV) transmission models will determine whether syndecan-Fc hybrid molecules truly represent prototypes of a new generation of microbicidal agents against HIV-1.
Acknowledgments
We thank J. Kuhns for secretarial assistance. We thank C. Aiken for 293T and CHO-K1 cells, J. Kappes for TZM-bl cells, M. Emerman for CCR5+ Jurkat T cells, and B. Kahn for providing us with primary genital epithelial cells. We thank D. Trono for the VSV-G plasmid and C. Aiken for NL4.3 pseudovirus plasmids. We thank G. David for providing the human syndecan-1 plasmid as well as the ID4 anti-syndecan-1 IgG. We thank P. Desai for providing us with HSV-1-GFP. We thank L. de Witte and T. Geijtenbeek for providing us with HSV-2-GFP as well as purified DC. We thank A. de Parseval, J. Elder and the Protein Expression and Proteomics Core of the University of California, San Diego, Center for AIDS Research, for providing us with control adhesin proteins as well as plasmids necessary for the cloning and expression of the syndecan-1-Fc fusion protein.
We acknowledge financial support from U.S. Public Health Service grants AI071952 and AI076005 (P.A.G.).
This is publication 20446 from the Department of Immunology and Microbial Science, The Scripps Research Institute, La Jolla, CA.
Footnotes
Published ahead of print on 3 May 2010.
REFERENCES
- 1.Bernfield, M., M. Gotte, P. W. Park, O. Reizes, M. L. Fitzgerald, J. Lincecum, and M. Zako. 1999. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 68:729-777. [DOI] [PubMed] [Google Scholar]
- 2.Bobardt, M. D., A. C. Saphire, H. C. Hung, X. Yu, B. Van der Schueren, Z. Zhang, G. David, and P. A. Gallay. 2003. Syndecan captures, protects, and transmits HIV-1 to T lymphocytes. Immunity 18:27-39. [DOI] [PubMed] [Google Scholar]
- 3.Bobardt, M. D., P. Salmon, L. Wang, J. D. Esko, D. Gabuzda, M. Fiala, D. Trono, B. van der Schueren, G. David, and P. A. Gallay. 2004. Contribution of proteoglycans to human immunodeficiency virus type 1 brain invasion. J. Virol. 78:6567-6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bobardt, M. D., U. Chatterji, S. Selvarajah, B. Van der Schueren, G. David, B. Kahn, and P. A. Gallay. 2007. Cell-free human immunodeficiency virus type 1 transcytosis through primary genital epithelial cells. J. Virol. 81:395-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bobardt, M. D., G. Cheng, L. de Witte, S. Selvarajah, U. Chatterji, B. E. Sanders-Beer, T. B. Geijtenbeek, F. V. Chisari, and P. A. Gallay. 2008. Hepatitis C virus NS5A anchor peptide disrupts human immunodeficiency virus. Proc. Natl. Acad. Sci. U. S. A. 105:5525-5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chatterji, U., M. D. Bobardt, R. Stanfield, R. G. Ptak, L. A. Pallansch, P. A. Ward, M. J. Jones, C. A. Stoddart, P. Scalfaro, J.-M. Dumont, K. Besseghir, B. Rosenwirth, and P. A. Gallay. 2005. Naturally occurring capsid substitutions render HIV-1 cyclophilin A independent in human cells and TRIM-cyclophilin-resistant in Owl monkey cells. J. Biol. Chem. 280:40293-40300. [DOI] [PubMed] [Google Scholar]
- 7.Cheshenko, N., W. Liu, L. M. Satlin, and B. C. Herold. 2007. Multiple receptor interactions trigger release of membrane and intracellular calcium stores critical for herpes simplex virus entry. Mol. Biol. Cell 18:3119-3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cole, A. M., and A. L. Cole. 2008. Antimicrobial polypeptides are key anti-HIV-1 effector molecules of cervicovaginal host defense. Am. J. Reprod. Immunol. 59:27-34. [DOI] [PubMed] [Google Scholar]
- 9.Deeks, S. G., and B. D. Walker. 2004. The immune response to AIDS virus infection: good, bad, or both? J. Clin. Invest. 113:808-810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Parseval, A., and J. H. Elder. 2001. Binding of recombinant feline immunodeficiency virus surface glycoprotein to feline cells: role of CXCR4, cell-surface heparans, and an unidentified non-CXCR4 receptor. J. Virol. 75:4528-4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Parseval, A., M. D. Bobardt, A. Chatterji, U. Chatterji, J. H. Elder, G. David, S. Zolla-Pazner, M. Farzan, T. H. Lee, and P. A. Gallay. 2005. A highly conserved arginine in gp120 governs HIV-1 binding to both syndecans and CCR5 via sulfated motifs. J. Biol. Chem. 280:39493-39504. [DOI] [PubMed] [Google Scholar]
- 12.de Witte, L., M. Bobardt, U. Chatterji, G. Degeest, G. David, T. B. Geijtenbeek, and P. Gallay. 2007. Syndecan-3 is a dendritic cell-specific attachment receptor for HIV-1. Proc. Natl. Acad. Sci. U. S. A. 104:19464-19469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Witte, L., Y. Zoughlami, B. Aengeneyndt, G. David, Y. van Kooyk, L. Gissmann, and T. B. Geijtenbeek. 2007. Binding of human papilloma virus L1 virus-like particles to dendritic cells is mediated through heparan sulfates and induces immune activation. Immunobiology 212:679-691. [DOI] [PubMed] [Google Scholar]
- 14.Dhawan, D., M. Keller, and M. E. Klotman. 2006. Topical microbicides: the time has come. AIDS Read. 16:144-148, 155-158, 161. [PubMed] [Google Scholar]
- 15.Freissler, E., A. Meyer auf der Heyde, G. David, T. F. Meyer, and C. Dehio. 2000. Syndecan-1 and syndecan-4 can mediate the invasion of OpaHSPG-expressing Neisseria gonorrhoeae into epithelial cells. Cell. Microbiol. 2:69-82. [DOI] [PubMed] [Google Scholar]
- 16.Gallay, P. 2004. Syndecans and HIV-1 pathogenesis. Microbes Infect. 6:617-622. [DOI] [PubMed] [Google Scholar]
- 17.Galvin, S. R., and M. S. Cohen. 2004. The role of sexually transmitted diseases in HIV-1 transmission. Nat. Rev. Microbiol. 2:33-42. [DOI] [PubMed] [Google Scholar]
- 18.Geijtenbeek, T. B., D. S. Kwon, R. Torensma, S. J. van Vliet, G. C. van Duijnhoven, J. Middel, I. L. Cornelissen, H. S. Nottet, V. N. KewalRamani, D. R. Littman, C. G. Figdor, and Y. van Kooyk. 2000. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 100:587-597. [DOI] [PubMed] [Google Scholar]
- 19.Jones, K. S., C. Petrow-Sadowski, D. C. Bertolette, Y. Huang, and F. W. Ruscetti. 2005. Heparan sulfate proteoglycans mediate attachment and entry of human T-cell leukemia virus type 1 virions into CD4+ T cells. J. Virol. 79:12692-12702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones, K. S., C. Petrow-Sadowski, Y. K. Huang, D. C. Bertolette, and F. W. Ruscetti. 2008. Cell-free HTLV-1 infects dendritic cells leading to transmission and transformation of CD4+ T cells. Nat. Med. 4:429-436. [DOI] [PubMed] [Google Scholar]
- 21.Klasse, P. J., R. J. Shattock, and J. P. Moore. 2006. Which topical microbicides for blocking HIV-1 transmission will work in the real world? PLoS Med. 3:e351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klasse, P. J., R. Shattock, and J. P. Moore. 2008. Antiretroviral drug-based microbicides to prevent HIV-1 sexual transmission. Annu. Rev. Med. 59:455-471. [DOI] [PubMed] [Google Scholar]
- 23.Kokenyesi, R., and M. Bernfield. 1994. Core protein structure and sequence determine the site and presence of heparan sulfate and chondroitin sulfate on syndecan-1. J. Biol. Chem. 269:12304-12309. [PubMed] [Google Scholar]
- 24.Lederman, M. M., R. S. Veazey, R. Offord, D. E. Mosier, J. Dufour, M. Mefford, M. Piatak, Jr., J. D. Lifson, J. R. Salkowitz, B. Rodriguez, A. Blauvelt, and O. Hartley. 2004. Prevention of vaginal SHIV-1 transmission in rhesus macaques through inhibition of CCR5. Science 306:485-487. [DOI] [PubMed] [Google Scholar]
- 25.Mondor, I., S. Ugolini, and Q. J. Sattentau. 1998. Human immunodeficiency virus type 1 attachment to HeLa CD4 cells is CD4 independent and gp120 dependent and requires cell surface heparans. J. Virol. 72:3623-3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moore, J. P., Topical microbicides become topical. 2005. N. Engl. J. Med. 352:298-300. [DOI] [PubMed] [Google Scholar]
- 27.Morris, G. C., and C. J. Lacey. 2010. Microbicides and HIV prevention: lessons from the past, looking to the future. Curr. Opin. Infect. Dis. 23:57-63. [DOI] [PubMed] [Google Scholar]
- 28.O'Doherty, U., W. J. Swiggard, and M. H. Malim. 2000. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J. Virol. 74:10074-10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohshiro, Y., T. Murakami, K. Matsuda, K. Nishioka, K. Yoshida, and N. Yamamoto. 1996. Role of cell surface glycosaminoglycans of human T cells in human immunodeficiency virus type-1 (HIV-1) infection. Microbiol. Immunol. 40:827-835. [DOI] [PubMed] [Google Scholar]
- 30.Patel, M., M. Yanagishita, G. Roderiquez, D. C. Bou-Habib, T. Oravecz, V. C. Hascall, and M. A. Norcross. 1993. Cell-surface heparan sulfate proteoglycan mediates HIV-1 infection of T-cell lines. AIDS Res. Hum. Retroviruses 9:167-174. [DOI] [PubMed] [Google Scholar]
- 31.Pope, M., and A. T. Haase. 2003. Transmission, acute HIV-1 infection and the quest for strategies to prevent infection. Nat. Med. 9:847-852. [DOI] [PubMed] [Google Scholar]
- 32.Ramessar, K., T. Rademacher, M. Sack, J. Stadlmann, D. Platis, G. Stiegler, N. Labrou, F. Altmann, J. Ma, E. Stöger, T. Capell, and P. Christou. 2008. Cost-effective production of a vaginal protein microbicide to prevent HIV transmission. Proc. Natl. Acad. Sci. U. S. A. 105:3727-3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roderiquez, G., T. Oravecz, M. Yanagishita, D. C. Bou-Habib, H. Mostowski, and M. A. Norcross. 1995. Mediation of human immunodeficiency virus type 1 binding by interaction of cell surface heparan sulfate proteoglycans with the V3 region of envelope gp120-gp41. J. Virol. 69:2233-2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Royce, R. A., A. Sena, W. Cates, Jr., and M. S. Cohen. 1997. Sexual transmission of HIV-1. N. Engl. J. Med. 336:1072-1078. [DOI] [PubMed] [Google Scholar]
- 35.Saphire, A. C., M. D. Bobardt, Z. Zhang, G. David, and P. A. Gallay. 2001. Syndecans serve as attachment receptors for human immunodeficiency virus type 1 on macrophages. J. Virol. 75:9187-9200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sato, H., J. Orenstein, D. Dimitrov, and M. Martin. 1992. Cell-to-cell spread of HIV-1 occurs within minutes and may not involve the participation of virus particles. Virology 186:712-724. [DOI] [PubMed] [Google Scholar]
- 37.Shafti-Keramat, S., A. Handisurya, E. Kriehuber, G. Meneguzzi, K. Slupetzky, and R. Kirnbauer. 2003. Different heparan sulfate proteoglycans serve as cellular receptors for human papillomaviruses. J. Virol. 77:13125-13135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shattock, R. J., and J. P. Moore. 2003. Inhibiting sexual transmission of HIV-1 infection. Nat. Rev. Microbiol. 1:25-34. [DOI] [PubMed] [Google Scholar]
- 39.Turpin, J. A. 2002. Considerations and development of topical microbicides to inhibit the sexual transmission of HIV. Expert Opin. Investig. Drugs 11:1077-1097. [DOI] [PubMed] [Google Scholar]
- 40.Van Damme, L., R. Govinden, F. M. Mirembe, F. Guédou, S. Solomon, M. L. Becker, B. S. Pradeep, A. K. Krishnan, M. Alary, B. Pande, G. Ramjee, J. Deese, T. Crucitti, and D. Taylor for the C. S. Study Group. 2008. Lack of effectiveness of cellulose sulfate gel for the prevention of vaginal HIV transmission. N. Engl. J. Med. 359:463-472. [DOI] [PubMed] [Google Scholar]
- 41.van Putten, J. P., and S. M. Paul. 1995. Binding of syndecan-like cell surface proteoglycan receptors is required for Neisseria gonorrhoeae entry into human mucosal cells. EMBO J. 14:2144-2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang, W. K., T. Dudek, Y. J. Zhao, H. G. Brumblay, M. Essex, and T. H. Lee. 1998. CCR5 coreceptor utilization involves a highly conserved arginine residue of HIV type 1 gp120. Proc. Natl. Acad. Sci. U. S. A. 95:5740-5745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang, Z., M. Götte, M. Bernfield, and O. Reizes. 2005. Constitutive and accelerated shedding of murine syndecan-1 is mediated by cleavage of its core protein at a specific juxtamembrane site. Biochemistry 44:12355-12361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wei, X., J. M. Decker, H. Liu, Z. Zhang, R. B. Arani, J. M. Kilby, M. S. Saag, X. Wu, G. M. Shaw, and J. C. Kappes. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 46:1896-1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang, L., and J. D. Esko. 1994. Amino acid determinants that drive heparan sulfate assembly in a proteoglycan. J. Biol. Chem. 269:19295-19299. [PubMed] [Google Scholar]