

Abstract
The success of nifurtimox-eflornithine combination therapy (NECT) for the treatment of human African trypanosomiasis (HAT) has renewed interest in the potential of nitro drugs as chemotherapeutics. In order to study the implications of the more widespread use of nitro drugs against these parasites, we examined the in vivo and in vitro resistance potentials of nifurtimox and fexinidazole and its metabolites. Following selection in vitro by exposure to increasing concentrations of nifurtimox, Trypanosoma brucei brucei nifurtimox-resistant clones designated NfxR1 and NfxR2 were generated. Both cell lines were found to be 8-fold less sensitive to nifurtimox than parental cells and demonstrated cross-resistance to a number of other nitro drugs, most notably the clinical trial candidate fexinidazole (∼27-fold more resistant than parental cells). Studies of mice confirmed that the generation of nifurtimox resistance in these parasites did not compromise virulence, and NfxR1 remained resistant to both nifurtimox and fexinidazole in vivo. In the case of fexinidazole, drug metabolism and pharmacokinetic studies indicate that the parent drug is rapidly metabolized to the sulfoxide and sulfone form of this compound. These metabolites retained trypanocidal activity but were less effective in nifurtimox-resistant lines. Significantly, trypanosomes selected for resistance to fexinidazole were 10-fold more resistant to nifurtimox than parental cells. This reciprocal cross-resistance has important implications for the therapeutic use of nifurtimox in a clinical setting and highlights a potential danger in the use of fexinidazole as a monotherapy.
The protozoan parasites Trypanosoma brucei gambiense and T. b. rhodesiense are the causative agents of human African trypanosomiasis (HAT), commonly known as sleeping sickness. This epidemic disease was largely under control in the 1960s but has reemerged as a major threat due to limited financial resources for the maintenance of control programs as well as population displacement due to political conflict and famine (23). Currently, the World Health Organization estimates that about 400,000 people are infected with HAT, resulting in an annual death toll of around 50,000 (42), although this may be an overestimate (36). In the absence of an effective vaccine, treatment is dependent solely upon a small repertoire of drugs which suffer from a number of problems, including severe toxic side effects (12) and acquired drug resistance (2). In particular, treatment of the second stage of the disease, where parasites invade the central nervous system, has proven to be especially problematic. Two drugs are available: the arsenical melarsoprol and the ornithine decarboxylase inhibitor eflornithine (difluoromethylornithine [DFMO]). Melarsoprol is extremely toxic, with death in 3 to 6% of cases and treatment failures as high as 30% in certain areas. Although better tolerated than melarsoprol, eflornithine is not effective against T. b. rhodesiense infections, which account for 10% of all cases of sleeping sickness. The treatment regimen for T. b. gambiense is prolonged, requiring a total of 56 slow infusions over a 14-day period. As a result, melarsoprol often remains the treatment of choice in regions where health care provision is limited.
One important development in the treatment of T. b. gambiense infections is the introduction of nifurtimox-eflornithine combination therapy (NECT) (32). Nifurtimox, a drug used in the treatment of Chagas' disease, has previously been given on compassionate grounds for treatment of melarsoprol-refractory HAT. However, its efficacy as a monotherapy is low, and prolonged treatment is limited by severe toxicity (4, 5). Patients given NECT, consisting of oral nifurtimox over 10 days with DFMO infusions for 7 days, were found to fair just as well as those given the DFMO monotherapy, with cure rates of around 97%. The reduced frequency and duration of DFMO infusions in NECT is seen as highly advantageous in terms of cost, logistics, and human resources in areas of poverty, leading to its inclusion on the Model Lists of Essential Medicines of the World Health Organization.
The success of NECT has come at a time of renewed interest in nitroheterocyclic compounds for the treatment of infectious disease. In particular, PA-824 is being assessed for the treatment of tuberculosis (3, 15, 29), while nitazoxanide is currently in phase II clinical trials for the treatment of hepatitis C (15). The increased awareness of the antimicrobial potential of these compounds has also led to the renaissance of fexinidazole (Hoe 239) (41) as a potential chemotherapeutic for late-stage HAT. In 1983, Jennings and Urquhart reported that this compound, given in combination with suramin, effectively cured chronic T. brucei infections in mice (20). Some 26 years later, fexinidazole entered phase II clinical trials as a monotherapy for use against both early- and late-stage African sleeping sickness (8; report available from http://www.dndi.org/).
It is well established that naturally occurring strains of Trypanosoma cruzi can be inherently resistant to nifurtimox (28), but it is not known whether a similar situation might occur in the African trypanosome. However, clinical isolates of T. b. gambiense from Sudan and West and Central Africa show a 10-fold range of sensitivities to nifurtimox (25, 26). It was also recently demonstrated that laboratory-generated nifurtimox-resistant T. cruzi cell lines become resistant to another nitro drug, benznidazole (39). To determine whether this may also be the case for T. brucei, we have undertaken similar studies. Here, we examine the in vivo and in vitro resistance potentials of nifurtimox and fexinidazole and its metabolites in bloodstream trypanosomes. The potential for cross-resistance to other nitro compounds is also addressed.
MATERIALS AND METHODS
Cell lines and culture conditions.
Trypanosoma brucei bloodstream-form “single-marker” S427 (T7RPOL TETR NEO) and nifurtimox-resistant cell lines were cultured at 37°C in HMI9-T medium (16) supplemented with 2.5 μg ml−1 G418 to maintain expression of T7 RNA polymerase and the tetracycline repressor protein (17). Cultures were initiated with 1 × 105 cells ml−1 and subcultured when cell densities approached 1 × 106 to 2 × 106 cells ml−1.
In order to directly compare the effects of nifurtimox and other compounds on the growth of these cell lines, the 50% effective concentration (EC50) of each compound was determined as previously described (21). Experimental data were corrected for background cell density and are expressed as percentages of values for nontreated cells before being fitted to the following two-parameter equation using GraFit software:
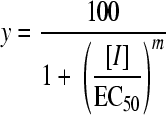 |
In this equation, [I] represents the inhibitor concentration and m is the slope factor. Experiments were repeated at least three times, and data are presented as the weighted means plus the weighted standard deviations.
Generation of nifurtimox- and fexinidazole-resistant T. brucei cell lines.
A nifurtimox-resistant T. brucei cell line was generated by subculturing bloodstream trypanosomes in the continuous presence of nifurtimox (a kind gift from Bayer Argentina). Parasites were exposed to stepwise-increased concentrations of drug, starting at a sublethal concentration of 1.5 μM, until they were routinely growing in 50 μM nifurtimox. After a total of 140 days in culture, resistant parasites were cloned in the absence of nifurtimox by limiting dilution, and the two clones (NfxR1 and NfxR2) demonstrating the highest levels of resistance to nifurtimox were selected for further study. A similar approach was used to generate a fexinidazole-resistant T. brucei cell line.
Nitro drug resistance in vivo.
Groups of five mice (NMRI) were infected with either wild-type (WT) bloodstream T. brucei or NfxR1 parasites by a single intraperitoneal injection of 104 cells in 0.2 ml of HMI9-T medium. Twenty-four hours following infection, WT- and NfxR1-infected mice were either dosed with nifurtimox (100 mg kg−1 of body weight) via an intraperitoneal injection or left untreated. The dosing of nifurtimox-treated mice was repeated in an identical manner for a further 3 days. Animals were inspected daily for clinical signs of infection, and wet smears of tail blood were examined microscopically as appropriate. Parasitemia was determined using a Neubauer hemocytometer, as previously described, and infected mice were monitored for 30 days (34). Mice with parasitemia that exceeded 108 cells ml−1 were humanely killed, since prior experience indicated that animals would succumb to an overwhelming infection by the following day. In addition, animals showing signs of acute drug toxicity were euthanized immediately.
The in vivo efficacy of fexinidazole was studied in WT- and NfxR1-infected mice in a manner similar to that described for nifurtimox. However, in this instance, fexinidazole was administered orally and at doses up to 200 mg kg−1. Parasitemia in fexinidazole-treated mice was monitored for 60 days.
Chemical synthesis of fexinidazole and derivatives.
Fexinidazole and fexinidazole sulfoxide were prepared in two steps from (1-methyl-5-nitro-1H-imidazol-2-yl)methanol (7), according to published procedures (18, 40). Fexinidazole sulfone was synthesized by reacting (1-methyl-5-nitro-1H-imidazol-2-yl)methanol and 4-(methylsulfonyl)phenol under Mitsunobu reaction conditions (37). PA-824 was prepared from (S)-2-nitro-6-(4-(trifluoromethoxy)benzyloxy)-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (Nowa Pharmaceuticals) as described by Orita et al. (30). The identities of all synthetic compounds were confirmed by 1H nuclear magnetic resonance (1H-NMR), 13C-NMR, high-resolution mass spectrometry, and, where appropriate, 19F-NMR. Compound purity was determined by liquid chromatography-mass spectrometry (LCMS), with all compounds found to be of >95% purity. Optical rotation measurements of the synthetic PA-824 were in close agreement with published values, confirming a high level of optical purity (30). Full experimental protocols and analytical data for fexinidazole sulfoxide, fexinidazole sulfone, and PA-824 are available from the authors upon request.
Determination of fexinidazole exposure in mice after acute oral dosing.
Fexinidazole (200 mg kg−1) was orally administered to NMRI mice. The dose solution was prepared on the day of dosing, and the vehicle was 10% (vol/vol) dimethyl sulfoxide (DMSO) in peanut oil. Blood samples (10 μl) were collected from the tail vein of each animal into Micronic tubes (Micronic BV) containing deionized water (20 μl) at 0.25, 0.5, 1, 2, 4, 6, and 8 h postdose and stored at −80°C until analyzed.
Extraction of blood samples was performed by a method based on protein precipitation using acetonitrile and a new chemical entity from our own compound collection as an internal standard. Levels of fexinidazole and two major metabolites (sulfone and sulfoxide) in these extracted samples were determined by ultraperformance liquid chromatography (UPLC)-tandem mass spectrometry (MS-MS) on a Quattro Premier XE mass spectrometer using positive electrospray ionization (Waters, United Kingdom) in multiple-reaction-monitoring mode. Samples (5 μl) were injected using an Acquity UPLC system (Waters, United Kingdom) onto a 2.1- by 50-mm Acquity UPLC 1.7-μm bridged ethyl hybrid (BEH) C18 column (Waters, United Kingdom). The column was maintained at 45°C, and samples were maintained at 4°C. Samples were eluted with a flow rate of 0.6 ml min−1. Mobile phase A consisted of Milli-Q water with formic acid (0.1%), and mobile phase B consisted of acetonitrile with formic acid (0.1%). The gradient profile started with 5% B for 0.1 min and then increased linearly over 1.1 min to 95% B. The flow remained for 0.1 min at 95% B and then dropped linearly over 1.1 min to starting conditions, 5% B, until the end of the run at 2.4 min. The entire flow was directed from the column through a photodiode array detector scanning between 210 and 400 nm, with a resolution of 1.2 nm, and then sequentially onto the mass spectrometer, where flow entered the instrument between 0.3 min and 2.4 min via the built-in switching valve (Rheodyne, CA). Calibration curves were constructed from values from blood to cover at least 3 orders of magnitude for fexinidazole (i.e., 1 to 1,000 ng ml−1) and approximately 2 orders of magnitude for the sulfone and sulfoxide metabolites (i.e., 200 to 15,000 ng ml−1).
RESULTS
Generation of a nifurtimox-resistant T. brucei cell line.
To establish the ease with which resistance to nifurtimox could be generated in vitro, bloodstream trypanosomes (strain 427) were cultured in the continuous presence of nifurtimox. Parasites were exposed to stepwise-increased concentrations of drug, starting at a sublethal concentration of 1.5 μM, until they were continuously growing in 50 μM nifurtimox (Fig. 1 A). In the presence of drug, the doubling time of nifurtimox-resistant parasites was 12.8 h, compared to 7.3 h for WT cells in the absence of drug. After a total of 140 days in culture, resistant parasites were cloned in the absence of nifurtimox by limiting dilution. The relative sensitivities of these cloned cell lines to nifurtimox were established and compared to that of WT parasites. Cloned lines NfxR1 and NfxR2 were found to be 8-fold less sensitive to nifurtimox than parental WT cells, with EC50s of 20.1 ± 0.9 and 20.3 ± 1.3 μM, respectively (Fig. 1B and Table 1). Nifurtimox resistance in NfxR1 and NfxR2 was found to be stable over 30 passages (60 days) in culture in the absence of drug (data not shown).
FIG. 1.

Nifurtimox resistance in vitro. (A) Schematic representation of the generation of a nifurtimox-resistant cell line in T. brucei. Each passage of cells in culture (open circle) and loss of a culture due to drug toxicity (skull and crossbones) are indicated. (B) EC50s were determined for nifurtimox (circles) and pentamidine (squares) against WT (open symbols) and NfxR1 (closed symbols) cells, respectively. The curves are the nonlinear fits of data using a two-parameter EC50 equation provided by GraFit (see Materials and Methods). EC50s of 0.95 ± 0.02 and 2.6 ± 0.1 nM were determined for pentamidine against WT and NfxR1 cells, respectively, while values of 2.4 ± 0.1 and 20.1 ± 0.9 μM were determined for nifurtimox. Data are the weighted means ± standard deviations of at least triplicate measurements.
TABLE 1.
Sensitivities of wild-type and nifurtimox-resistant clones of T. brucei to standard drugs and experimental nitro compoundsa
 |
The results are the weighted means and standard deviations from at least three independent experiments. The values in parentheses are Hill slopes, i.e., the weighted means from at least three independent experiments. The standard deviation was less than 20% for all values. Resistance factors (RF) were determined as the ratio of the EC50 of the resistant cells to that of the wild-type cells. ND, not determined.
P < 0.001 by Student's t test compared to wild-type cells.
Generation of nifurtimox resistance in these cells did not greatly alter their sensitivity to drugs from other chemical classes, with the diamidine pentamidine and the fluorinated amino acid eflornithine having similar potencies against NfxR and parental wild-type cells (Fig. 1B and Table 1). However, NfxR1 and NfxR2 did demonstrate cross-resistance to a number of other nitro compounds, including nitrofurazone, previously used for the treatment of late-stage sleeping sickness (1). The anticancer nitrobenzene prodrug CB1954 (22), identified in a phenotypic screen in our laboratory as one of the most potent growth inhibitors of WT parasites, also showed marked cross-resistance in the NfxR cell lines. Most strikingly, the highest levels of resistance were observed against the nitroimidazole fexinidazole and its putative metabolites (sulfoxide and sulfone) (41). NfxR1 and NfxR2 were found to be 27- and 29-fold less sensitive to fexinidazole than wild-type parasites, respectively, and about 15-fold less sensitive to its sulfoxide and sulfone metabolites. As previously demonstrated in T. cruzi, nifurtimox resistance in bloodstream T. brucei also resulted in a similar level of resistance to benznidazole (5-fold higher than that of wild-type parasites). Notably, NfxR cells were not cross-resistant to the antitubercular compound PA-824, which is considered to have a mode of action different from those of other nitro drugs (27, 35). Metronidazole, a drug used to treat microaerophilic bacterial and protozoal infections, had no effect on the growth of either wild-type or NfxR parasites at the highest concentration tested (500 μM).
Nitro drug resistance in vivo.
To confirm the biological relevance of nifurtimox resistance in NfxR clones, groups of mice were inoculated with either WT or NfxR1 parasites. Previous studies had established that the WT strain has a 50% effective dose (ED50) of 36.7 mg kg−1 when administered once daily for 4 days (m = 3.7; ED99, 128 mg kg−1 [recalculated from reference 13]). Twenty-four hours following infection, WT- and NfxR1-infected mice were either dosed with nifurtimox (100 mg kg−1) via an intraperitoneal injection or left untreated. The dosing of nifurtimox-treated mice was repeated in an identical manner for a further 3 days. Infections were monitored over a 30-day period, and survival curves of each infection are shown in Fig. 2. Mice infected with WT cells succumbed to infection between days 5 and 7, and this was also the case for mice infected with NfxR1, confirming that nifurtimox resistance does not compromise virulence in these bloodstream trypanosomes. Nifurtimox treatment ensured that WT-infected mice remained free from parasitemia for the entire 30 days of the study. However, one mouse from this group died due to drug toxicity, and this was also the case in the NfxR1-infected group, indicating that 100 mg kg−1 may exceed the maximum tolerable dose of nifurtimox in these animals. Most significantly, nifurtimox treatment of NfxR1-infected mice did little to impede the growth of these parasites, and lethal levels of parasitemia were reached within 6 days. Collectively, these results confirm that NfxR1 parasites retain their nifurtimox-resistant phenotype in vivo.
FIG. 2.

Nifurtimox resistance in vivo. Groups of 5 mice were infected with either WT or NfxR1 cells (1 × 104 parasites). Twenty-four hours following infection, WT- and NfxR1-infected mice were either dosed with nifurtimox (100 mg kg−1) via an intraperitoneal injection or left untreated. Data are presented in the form of a Kaplan-Meier survival plot. Animals that succumbed to drug toxicity rather than parasitemia are annotated (skull and crossbones). WT, pink; WT treated with nifurtimox, blue; NfxR1, green; NfxR1 treated with nifurtimox, red.
To determine if nifurtimox-resistant parasites were cross-resistant to the clinical trial candidate fexinidazole in vivo, WT- and NfxR1-infected mice were treated orally with up to 200 mg kg−1 fexinidazole once a day for 4 days. In general, fexinidazole seemed to be well tolerated in mice, and there were no signs of the toxicity issues previously seen following intraperitoneal administration of nifurtimox. Treatment of WT-infected mice with fexinidazole at 50 mg kg−1 affected a cure in 100% of these animals, with an established ED50 value of 21 ± 0.3 mg kg−1 (m = 7.9) (Fig. 3). In keeping with our in vitro studies, NfxR1 parasites in vivo were found to have a relative resistance to fexinidazole that was 8.5-fold higher than that of WT parasites (ED50, ∼171 mg kg−1). At the highest dose of the drug (200 mg kg−1), only 2 out of 5 mice were aparasitemic following treatment. The other three mice showed relapsing peaks of parasitemia after treatment, with one apparently being self-cured, since no further signs of infection were observed from days 18 to 60, when the experiment was terminated.
FIG. 3.

Fexinidazole resistance in vivo. Groups of 5 mice were infected with either WT cells (open circles) or NfxR1 cells (closed circles) (1 × 104 parasites). Twenty-four hours following infection, WT- and NfxR1-infected mice were dosed orally with fexinidazole (25 to 200 mg kg−1). Cured mice were determined to be those which survived to 60 days following inoculation.
Pharmacokinetic properties of fexinidazole.
In early studies regarding the potential of fexinidazole as a treatment for HAT, Winkelmann and Raether (41) proposed that the sulfur atom in this compound is almost certainly metabolized in vivo to sulfoxide and/or sulfone groups (see the structures in Table 1). Analysis of blood samples from mice orally dosed with fexinidazole (200 mg kg−1) confirmed that this was indeed the case (Fig. 4). Fifteen minutes after oral dosing, levels of fexinidazole in the blood were found to peak at 961 ng ml−1, before declining steadily over time. The sulfoxide metabolite accumulated to considerably higher levels in the blood than the parent compound, reaching 40,000 ng ml−1 2 h following dosing. Fexinidazole sulfone, the final metabolite to appear in the blood, continued to accumulate throughout the 8 h of the study, reaching a concentration of 55,000 ng ml−1.
FIG. 4.

Blood exposure of fexinidazole and its metabolites. Levels of fexinidazole and two major metabolites (sulfone and sulfoxide) were monitored in blood samples collected from mice at defined intervals, following oral dosing of fexinidazole (200 mg kg−1). The EC99 values of fexinidazole and its sulfoxide and sulfone metabolites, established in vitro, are annotated as solid, dotted, and dashed lines, respectively. Symbols: closed squares, fexinidazole; open circles, sulfoxide; closed circles, sulfone. Data are the means of triplicate measurements.
Comparison of the maximum achievable total blood levels of fexinidazole with the previously established in vitro EC99 (4,900 ng ml−1) suggests that the unmetabolized form of this drug is unlikely to be the major trypanocidal agent in vivo. In contrast, total blood levels of both the sulfoxide and sulfone metabolites comfortably exceeded their respective EC99 levels for several hours following dosing. Additional studies established that the fraction unbound (fu) of fexinidazole and its sulfoxide and sulfone metabolites in mouse plasma is high (fu, 0.147, 0.858, and 0.728, respectively), indicating that the free-drug metabolite levels are also well in excess of the parasite EC99. Collectively, these findings suggest that fexinidazole acts as a prodrug, which is oxidized in vivo to these more therapeutically relevant species.
Fexinidazole-resistant T. brucei lines.
To determine whether fexinidazole-resistant cell lines would show reciprocal cross-resistance to nifurtimox, fexinidazole-resistant parasites were generated in a manner identical to that previously described for nifurtimox. The results of a representative drug sensitivity experiment are shown for cloned line FxR1 in Fig. 5. Overall, the cloned lines (FxR1 and FxR2) were found to be 10-fold less sensitive to fexinidazole than parental WT cells, with weighted mean EC50 (and Hill slope) values from 4 independent experiments of 16.9 ± 0.8 (1.5) and 16.0 ± 1.2 μM (1.2), respectively. Significantly, FxR1 and FxR2 demonstrated considerable cross-resistance to nifurtimox and were found to be 9.5- and 9.3-fold less sensitive than WT parasites, respectively (3 independent experiments). Notably, the Hill slopes for nifurtimox are considerably steeper than for fexinidazole in both sensitive and resistant cell lines. Thus, although the EC50 for fexinidazole in WT cells is 2.4-fold lower than that of nifurtimox (Table 1), the resulting MICs (defined as the EC99 calculated from the EC50 and Hill slope) are roughly equivalent (15.2 and 17.7 μM for nifurtimox and fexinidazole, respectively). In contrast, the mean EC50s of the two FxR cell lines for fexinidazole and nifurtimox are equivalent (16.4 versus 18.8 μM), whereas the calculated mean MIC values are 4.3-fold higher for fexinidazole than for nifurtimox (510 versus 120 μM for fexinidazole and nifurtimox, respectively).
FIG. 5.

Cross-resistance to nifurtimox in FxR cell lines. EC50s were determined for fexinidazole (circles) and nifurtimox (squares) against WT (open symbols) and FxR1 (closed symbols) cells, respectively. The curves are the nonlinear fits of data using a two-parameter EC50 equation provided by GraFit (see Materials and Methods). In this experiment, the EC50 (and Hill slope) values for fexinidazole are 1.6 ± 0.1 μM (1.6) and 17.8 ± 1.5 μM (1.4) for WT and FxR1 cells, respectively, and for nifurtimox are 1.8 ± 0.1 μM (3.1) and 18.4 ± 0.6 μM (3.9), respectively.
DISCUSSION
The resistance potential of nitro drugs such as nifurtimox and fexinidazole has yet to be adequately studied in the African trypanosome. With ongoing phase II clinical trials of fexinidazole as a treatment for HAT and the inclusion of NECT on the Model Lists of Essential Medicines of the World Health Organization, the need for such studies has become more pressing. T. b. brucei is a suitable model for T. b. gambiense, since it is more amenable to culture in vitro and in vivo and displays similar sensitivities to nifurtimox, eflornithine, and pentamidine (25, 26). In the current study, we have confirmed that resistance to nifurtimox can be generated relatively easily in bloodstream trypanosomes in vitro and that these nifurtimox-resistant cell lines are cross-resistant to a number of other nitro drugs. Most significantly, NfxR parasites are highly cross-resistant to fexinidazole and its active metabolites (13- to 29-fold more resistant than WT parasites). Conversely, our fexinidazole-resistant lines of T. brucei are cross-resistant to nifurtimox (10-fold more resistant than WT parasites). Should this occur in a clinical setting, widespread resistance to nifurtimox would preclude the use of fexinidazole and vice versa. In addition, based on pharmacological data from human subjects (31), it can be calculated that, with an oral-dose schedule of 15 mg kg−1 of nifurtimox every 8 h for 10 days for HAT patients (32), the total serum concentration will reach approximately 3 μM. Thus, if this concentration is reflected in the central nervous system, then this might account for the partial efficacy of nifurtimox as a monotherapy (4, 5) since the mean EC50 in vitro for clinical isolates of T. b. gambiense ranges from 0.26 to 2.6 μM (25, 26). This 10-fold range of inherent susceptibility to nifurtimox underlines the need for future clinical trials with fexinidazole to include testing of T. b. gambiense field isolates for their intrinsic sensitivity to this drug.
It is not known whether the success of NECT in the treatment of the Gambian form of sleeping sickness is due to the additive or synergistic toxic effects of nifurtimox and eflornithine. However, eflornithine treatment lowers the intracellular concentrations of the unique antioxidant trypanothione in African trypanosomes (14) and nifurtimox may increase oxidant stress as discussed below.
Nitroheterocyclic compounds, such as nifurtimox, are believed to function as prodrugs requiring enzyme-mediated reduction by nitroreductases (NTRs) to generate cytotoxic species that cause damage to DNA, lipids, and proteins. Depending on the organism and the nature of the nitro drug, reduction can occur via sequential one-electron or two-electron processes (6, 9, 10). In one-electron processes, the first intermediate formed is the nitro anion radical (RNO2−), which, under aerobic conditions, is rapidly oxidized back to the parent drug, forming a superoxide anion (O2−). Superoxide can dismute to form hydrogen peroxide (H2O2) and dioxygen (O2). If the cell's capacity to remove O2− and H2O2 is exceeded, then the highly reactive hydroxyl radical can be formed via the transition metal-catalyzed Fenton reaction (19). In addition, RNO2− can be further reduced to form the reactive and toxic nitroso species, particularly under anaerobic conditions. Curiously, the nitrofuran nifurtimox readily undergoes futile redox cycling in T. cruzi, whereas the nitroimidazole benznidazole does not (see the reviews in references 9 and 10). Whether the lower potency and lower resistance factor of benznidazole compared with nifurtimox are related to a different mode of action in T. brucei needs to be investigated. In two-electron processes, reduction is catalyzed by type I NTRs (6), enzymes which are insensitive to futile redox recycling by oxygen and utilize flavin mononucleotide as a cofactor, directly forming toxic species, such as the nitroso intermediate, or, following a second round of reduction, the hydroxylamine species (33). Recent studies of T. cruzi and T. brucei have identified a trypanosomal type I NTR which is capable of metabolizing a number of nitroheterocyclic drugs, including nifurtimox (39). Wilkinson and colleagues reported that modulation of NTR levels within these parasites directly affected their sensitivity to nitro compounds, with reduced levels of the enzyme leading to nitro drug resistance. These findings not only confirm a crucial role for NTR in drug activation in the trypanosomes but also suggest depletion of this enzyme as one possible mechanism of nitro drug resistance. Reliance on a single enzyme for prodrug activation could leave these drugs particularly vulnerable to the emergence of drug resistance, a hypothesis that seems to be supported by the ease with which nitro drug resistance was generated in NfxR and FxR cell lines. NTR activity is too low to be measured in cell lysates, and our attempts to generate a specific antiserum to estimate NTR protein levels have proved unsuccessful. However, we have eliminated the possibility that point mutations in T. brucei NTR are responsible for resistance. Furthermore, deletion of one copy of the NTR gene in this diploid parasite increases resistance to nifurtimox only by a factor of 2- to 3-fold (39; A. Y. Sokolova et al., unpublished data). Characterization of the molecular basis of nitro drug resistance in these cell lines will be the focus of our subsequent studies.
Notably, many of the structurally diverse nitroheterocyclic compounds tested in this study were found to have similar potencies in inhibiting cell growth and display similar resistance factors to nifurtimox (Table 1, compounds 1 to 5 and 9), suggesting similar mechanisms of cell killing. The most striking exceptions were the antituberculosis drug PA-824, which showed no cross-resistance, and metronidazole, which was inactive. PA-824, a bicyclic nitroimidazole, is reduced in Mycobacterium tuberculosis by an unusual deazaflavin-dependent nitroreductase (35), while in Trichomonas vaginalis, metronidazole is reduced either by the pyruvate-ferredoxin oxidoreductase system (11) or thioredoxin reductase (24), none of which are present in T. brucei. The inability of these compounds to efficiently kill wild-type parasites combined with the lack of resistance to PA-824 in NfxR cell lines supports the hypothesis that NTR is intimately involved in the mode of action of nitro drugs in T. brucei.
With the pitifully small armory of drugs available for the treatment of HAT, it is essential that effective policies are in place to prevent the development of resistance to new and existing antitrypanosomal drugs. One such strategy involves the use of drugs in combination, with the rationale that the likelihood of developing resistance to a single agent is relatively high, but the likelihood of developing resistance to two compounds is much lower (38). The use of drug combinations to circumvent resistance has been successfully employed with antimalarials (38), and ideally, partner drugs should be found for new antitrypanosomal drugs prior to widespread clinical use. The introduction of NECT represents the first widely used combination therapy for HAT and could serve as a template for the introduction of subsequent therapies. In light of the findings of this study, it is imperative that fexinidazole should be used clinically only with a partner drug, such as eflornithine. Failure to do so may have dire consequences for the future use of nitro drugs as effective treatments for HAT.
Acknowledgments
This work was supported by grants from the Wellcome Trust to A.H.F. (WT079838, WT077705, and WT083481) and a studentship from the BBSRC to A.Y.S.
We thank Laste Stojanovski and Frederick Simeons for assistance with the in vivo drug susceptibility testing and Robert Kime and Suzanne Norval for analysis of drug metabolites.
Footnotes
Published ahead of print on 3 May 2010.
The authors have paid a fee to allow immediate free access to this article.
REFERENCES
- 1.Apted, F. I. C. 1980. Present status of chemotherapy and chemoprophylaxis of human trypanosomiasis in the eastern hemisphere. Pharmacol. Ther. 11:391-413. [DOI] [PubMed] [Google Scholar]
- 2.Barrett, M. P., and A. H. Fairlamb. 1999. The biochemical basis of arsenical-diamidine cross-resistance in African trypanosomes. Parasitol. Today 15:136-140. [DOI] [PubMed] [Google Scholar]
- 3.Barry, C. E., H. I. M. Boshoff, and C. S. Dowd. 2004. Prospects for clinical introduction of nitroimidazole antibiotics for the treatment of tuberculosis. Curr. Pharm. Des. 10:3239-3262. [DOI] [PubMed] [Google Scholar]
- 4.Bisser, S., F. X. N′Siesi, V. Lejon, P. M. Preux, S. Van Nieuwenhove, C. M. M. Bilenge, and P. Buscher. 2007. Equivalence trial of melarsoprol and nifurtimox monotherapy and combination therapy for the treatment of second-stage Trypanosoma brucei gambiense sleeping sickness. J. Infect. Dis. 195:322-329. [DOI] [PubMed] [Google Scholar]
- 5.Bouteille, B., O. Oukem, S. Bisser, and M. Dumas. 2003. Treatment perspectives for human African trypanosomiasis. Fundam. Clin. Pharmacol. 17:171-181. [DOI] [PubMed] [Google Scholar]
- 6.Bryant, D. W., D. R. McCalla, M. Leeksma, and P. Laneuville. 1981. Type I nitroreductases of Escherichia coli. Can. J. Microbiol. 27:81-86. [DOI] [PubMed] [Google Scholar]
- 7.Chauviere, G., B. Bouteille, B. Enanga, C. de Albuquerque, S. L. Croft, M. Dumas, and J. Perie. 2003. Synthesis and biological activity of nitro heterocycles analogous to megazol, a trypanocidal lead. J. Med. Chem. 46:427-440. [DOI] [PubMed] [Google Scholar]
- 8.DNDi. 2009. Annual report 2008-2009. Delivering innovation and building a robust pipeline. Drugs for Neglected Diseases Initiative, Geneva, Switzerland.
- 9.Docampo, R., and S. N. J. Moreno. 1984. Free radical metabolites in the mode of action of chemotherapeutic agents and phagocytic cells on Trypanosoma cruzi. Rev. Infect. Dis. 6:223-238. [DOI] [PubMed] [Google Scholar]
- 10.Docampo, R., and S. N. J. Moreno. 1986. Free-radical metabolism of antiparasitic agents. Fed. Proc. 45:2471-2476. [PubMed] [Google Scholar]
- 11.Dunne, R. L., L. A. Dunn, P. Upcroft, P. J. O'Donoghue, and J. A. Upcroft. 2003. Drug resistance in the sexually transmitted protozoan Trichomonas vaginalis. Cell Res. 13:239-249. [DOI] [PubMed] [Google Scholar]
- 12.Fairlamb, A. H. 2003. Chemotherapy of human African trypanosomiasis: current and future prospects. Trends Parasitol. 19:488-494. [DOI] [PubMed] [Google Scholar]
- 13.Fairlamb, A. H., N. S. Carter, M. Cunningham, and K. Smith. 1992. Characterisation of melarsen-resistant Trypanosoma brucei brucei with respect to cross-resistance to other drugs and trypanothione metabolism. Mol. Biochem. Parasitol. 53:213-222. [DOI] [PubMed] [Google Scholar]
- 14.Fairlamb, A. H., G. B. Henderson, C. J. Bacchi, and A. Cerami. 1987. In vivo effects of difluoromethylornithine on trypanothione and polyamine levels in bloodstream forms of Trypanosoma brucei. Mol. Biochem. Parasitol. 24:185-191. [DOI] [PubMed] [Google Scholar]
- 15.Ginsberg, A. M., M. W. Laurenzi, D. J. Rouse, K. D. Whitney, and M. K. Spigelman. 2009. Safety, tolerability, and pharmacokinetics of PA-824 in healthy subjects. Antimicrob. Agents Chemother. 53:3720-3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greig, N., S. Wyllie, S. Patterson, and A. H. Fairlamb. 2009. A comparative study of methylglyoxal metabolism in trypanosomatids. FEBS J. 276:376-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirumi, H., and K. Hirumi. 1989. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 75:985-989. [PubMed] [Google Scholar]
- 18.Hoff, D. R., and D. W. Henry. January 1967. Nitroimidazoles. U.S. patent 3,299,090.
- 19.Imlay, J. A. 2003. Pathways of oxidative damage. Annu. Rev. Microbiol. 57:395-418. [DOI] [PubMed] [Google Scholar]
- 20.Jennings, F. W., and G. M. Urquhart. 1983. The use of the 2 substituted 5-nitroimidazole, fexinidazole (Hoe 239) in the treatment of chronic T. brucei infections in mice. Z. Parasitenkd. 69:577-581. [DOI] [PubMed] [Google Scholar]
- 21.Jones, D. C., A. Ariza, W. H. Chow, S. L. Oza, and A. H. Fairlamb. 2010. Comparative structural, kinetic and inhibitor studies of Trypanosoma brucei trypanothione reductase with T. cruzi. Mol. Biochem. Parasitol. 169:12-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knox, R. J., P. J. Burke, S. Chen, and D. J. Kerr. 2003. CB 1954: from the Walker tumor to NQO2 and VDEPT. Curr. Pharm. Des. 9:2091-2104. [DOI] [PubMed] [Google Scholar]
- 23.Legros, D., G. Ollivier, M. Gastellu-Etchegorry, C. Paquet, C. Burri, J. Jannin, and P. Buscher. 2002. Treatment of human African trypanosomiasis—present situation and needs for research and development. Lancet Infect. Dis. 2:437-440. [DOI] [PubMed] [Google Scholar]
- 24.Leitsch, D., D. Kolarich, M. Binder, J. Stadlmann, F. Altmann, and M. Duchêne. 2009. Trichomonas vaginalis: metronidazole and other nitroimidazole drugs are reduced by the flavin enzyme thioredoxin reductase and disrupt the cellular redox system. Implications for nitroimidazole toxicity and resistance. Mol. Microbiol. 72:518-536. [DOI] [PubMed] [Google Scholar]
- 25.Likeufack, A. C. L., R. Brun, A. Fomena, and P. Truc. 2006. Comparison of the in vitro drug sensitivity of Trypanosoma brucei gambiense strains from West and Central Africa isolated in the periods 1960-1995 and 1999-2004. Acta Trop. 100:11-16. [DOI] [PubMed] [Google Scholar]
- 26.Maina, N., K. J. Maina, P. Maser, and R. Brun. 2007. Genotypic and phenotypic characterization of Trypanosoma brucei gambiense isolates from Ibba, South Sudan, an area of high melarsoprol treatment failure rate. Acta Trop. 104:84-90. [DOI] [PubMed] [Google Scholar]
- 27.Manjunatha, U., H. I. Boshoff, and C. E. Barry. 2009. The mechanism of action of PA-824: novel insights from transcriptional profiling. Commun. Integr. Biol. 2:215-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murta, S. M., R. T. Gazzinelli, Z. Brener, and A. J. Romanha. 1998. Molecular characterization of susceptible and naturally resistant strains of Trypanosoma cruzi to benznidazole and nifurtimox. Mol. Biochem. Parasitol. 93:203-214. [DOI] [PubMed] [Google Scholar]
- 29.Nuermberger, E., S. Tyagi, R. Tasneen, K. N. Williams, D. Almeida, I. Rosenthal, and J. H. Grosset. 2008. Powerful bactericidal and sterilizing activity of a regimen containing PA-824, moxifloxacin, and pyrazinamide in a murine model of tuberculosis. Antimicrob. Agents Chemother. 52:1522-1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orita, A., K. Miwa, G. Uehara, and J. Otera. 2007. Integration of solventless reaction in a multi-step process: application to an efficient synthesis of PA-824. Adv. Synth. Catal. 349:2136-2144. [Google Scholar]
- 31.Paulos, C., J. Paredes, I. Vasquez, S. Thambo, A. Arancibia, and G. Gonzalez-Martin. 1989. Pharmacokinetics of a nitrofuran compound, nifurtimox, in healthy volunteers. Int. J. Clin. Pharmacol. 27:454-457. [PubMed] [Google Scholar]
- 32.Priotto, G., S. Kasparian, W. Mutombo, D. Ngouama, S. Ghorashian, U. Arnold, S. Ghabri, E. Baudin, V. Buard, S. Kazadi-Kyanza, M. Ilunga, W. Mutangala, G. Pohlig, C. Schmid, U. Karunakara, E. Torreele, and V. Kande. 2009. Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: a multicentre, randomised, phase III, non-inferiority trial. Lancet 374:56-64. [DOI] [PubMed] [Google Scholar]
- 33.Race, P. R., A. L. Lovering, R. M. Green, A. Ossor, S. A. White, P. F. Searle, C. J. Wrighton, and E. I. Hyde. 2005. Structural and mechanistic studies of Escherichia coli nitroreductase with the antibiotic nitrofurazone. Reversed binding orientations in different redox states of the enzyme. J. Biol. Chem. 280:13256-13264. [DOI] [PubMed] [Google Scholar]
- 34.Sienkiewicz, N., S. Jaroslawski, S. Wyllie, and A. H. Fairlamb. 2008. Chemical and genetic validation of dihydrofolate reductase-thymidylate synthase as a drug target in African trypanosomes. Mol. Microbiol. 69:520-533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singh, R., U. Manjunatha, H. I. M. Boshoff, Y. H. Ha, P. Niyomrattanakit, R. Ledwidge, C. S. Dowd, I. Y. Lee, P. Kim, L. Zhang, S. H. Kang, T. H. Keller, J. Jiricek, and C. E. I. Barry. 2008. PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science 322:1392-1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stuart, K. D., R. Brun, S. L. Croft, A. H. Fairlamb, R. E. Gurtler, J. H. McKerrow, S. Reed, and R. L. Tarleton. 2008. Kinetoplastids: related protozoan pathogens, different diseases. J. Clin. Invest. 118:1301-1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Swamy, K. C. K., N. N. B. Kumar, E. Balaraman, and K. V. P. P. Kumar. 2009. Mitsunobu and related reactions: advances and applications. Chem. Rev. 109:2551-2651. [DOI] [PubMed] [Google Scholar]
- 38.White, N. J., and P. L. Olliaro. 1996. Strategies for the prevention of antimalarial drug resistance: rationale for combination chemotherapy for malaria. Parasitol. Today 12:399-401. [DOI] [PubMed] [Google Scholar]
- 39.Wilkinson, S. R., M. C. Taylor, D. Horn, J. M. Kelly, and I. Cheeseman. 2008. A mechanism for cross-resistance to nifurtimox and benznidazole in trypanosomes. Proc. Natl. Acad. Sci. U. S. A. 105:5022-5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Winkelmann, E., and W. Raether. August 1977. 1-Methyl-2-(phenyl-oxymethyl)-5-nitro-imidazoles and process for their manufacture. U.S. patent 4,042,705.
- 41.Winkelmann, E., and W. Raether. 1978. Chemotherapeutically active nitro compounds. 4. 5-Nitroimidazoles (part III). Arzneimittelforschung 28:739-749. [PubMed] [Google Scholar]
- 42.World Health Organization. 2004. The world health report 2004: changing history. World Health Organization, Geneva, Switzerland.