

Abstract
We recently reported that (2R,3R,4R,5R)-2-(4-amino-pyrrolo[2,3-d]pyrimidin-7-yl)-3-ethynyl-5-hydroxy-methyl-tetrahydro-furan-3,4-diol is a potent inhibitor of dengue virus (DENV), with 50% effective concentration (EC50) and cytotoxic concentration (CC50) values of 0.7 μM and >100 μM, respectively. Here we describe the synthesis, structure-activity relationship, and antiviral characterization of the inhibitor. In an AG129 mouse model, a single-dose treatment of DENV-infected mice with the compound suppressed peak viremia and completely prevented death. Mode-of-action analysis using a DENV replicon indicated that the compound blocks viral RNA synthesis. Recombinant adenosine kinase could convert the compound to a monophosphate form. Suppression of host adenosine kinase, using a specific inhibitor (iodotubercidin) or small interfering RNA (siRNA), abolished or reduced the compound's antiviral activity in cell culture. Studies of rats showed that 14C-labeled compound was converted to mono-, di-, and triphosphate metabolites in vivo. Collectively, the results suggest that this adenosine inhibitor is phosphorylated to an active (triphosphate) form which functions as a chain terminator for viral RNA synthesis.
The family Flaviviridae consists of three genera, Flavivirus, Pestivirus, and Hepacivirus. Many viruses from the genus Flavivirus are arthropod-borne and cause significant human diseases. The four serotypes of dengue virus (DENV) alone pose a health threat to 2.5 billion people worldwide, leading to 50 to 100 million human infections and 500,000 cases of dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS) each year (11). Besides DENV, other flaviviruses, such as West Nile virus (WNV), Japanese encephalitis virus (JEV), yellow fever virus (YFV), and tick-borne encephalitis virus (TBEV), also cause frequent outbreaks throughout the world. No clinically approved antiviral therapy is currently available for treatment of flavivirus infections. Human vaccines are available only for YFV, JEV, and TBEV. The development of a successful DENV vaccine has been challenging due to the facts that (i) the vaccine should simultaneously induce a long-lasting protection against all four DENV serotypes and (ii) an incompletely immunized individual may be sensitized to the life-threatening DHF or DSS. The challenge associated with the development of a vaccine underlines the importance of antiviral development for DENV and other flaviviruses.
The flavivirus genome is a single-strand RNA of plus-sense polarity. The single open reading frame from the 11-kb viral genome encodes three structural proteins (capsid [C], premembrane [prM], and envelope [E]) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5). Nonstructural proteins are responsible for viral replication. Of those, NS3 and NS5 have enzymatic activities. The N-terminal domain of NS3 (with NS2B as a cofactor) functions as a serine protease, and the C-terminal domain acts as an RNA helicase, an RNA triphosphatase, and an NTPase (10, 34, 35). The N-terminal domain of NS5 serves as a methyltransferase, and the C-terminal domain functions as an RNA-dependent RNA polymerase (RdRp) (1, 8, 27, 32). Both viral and host proteins are potential targets for antiviral development (22).
Nucleoside inhibitors represent the largest class of antiviral drugs. These inhibitors act as chain terminators during viral DNA (retroviruses and DNA viruses) or RNA (hepatitis C virus [HCV]) synthesis (7). Upon entering cells, most nucleoside inhibitors need to be converted into their corresponding triphosphates by viral or host kinases. The 5′-triphosphate metabolites compete with the natural substrate in the viral DNA or RNA polymerization reaction (4). Nucleoside analogs with ribose 2′-C-methyl (3, 5) or 4′-C-azido modifications (13, 14) are potent inhibitors of HCV, a virus closely related to the flaviviruses. Interestingly, the 7-deaza-2′-C-methyl-adenosine also inhibits DENV in cell culture and in mouse models (29). Although these nucleoside analogs possess an intact 3′-OH at the ribose moiety, the 2′-C-methyl and 4′-C-azido modifications may impose steric hindrance for subsequent chain elongation. We recently found that 7-deaza-2′-C-acetylene-adenosine (Fig. 1, compound 1) is a potent inhibitor of flaviviruses (36); however, a more detailed characterization is needed to further understand the antiviral activity of this compound.
FIG. 1.

Nucleoside flavivirus-specific inhibitor general structure A and compounds 1 to 4.
Here we report the synthesis, structure-activity relationship (SAR), and biological characterization of a number of nucleosides with the general structure A which led to the identification of compound 1 (Fig. 1). The results of biochemical experiments showed that compound 1 could be converted to the monophosphate by a recombinant human adenosine kinase. Suppression of the adenosine kinase expression by small interfering RNAs (siRNAs) or inhibition of the kinase activity by iodotubercidin decreased the cellular activity of compound 1. A 14C-labeled compound 1 was converted to mono-, di-, and triphosphate derivatives in rats. Furthermore, we show that compound 1 inhibits the viral RNA synthesis of DENV-1. Remarkably, a single-dose treatment of DENV-infected mice with compound 1 suppressed viremia and fully protected the infected mice from death.
MATERIALS AND METHODS
Antiviral assays.
Four types of antiviral assays were performed. (i) A cell-based flavivirus immunodetection (CFI) assay (33) was used to study SAR. Briefly, 2 × 104 A549 cells were seeded per well of a 96-well plate. On the following day, the cells were infected with DENV-2 at a multiplicity of infection (MOI) of 0.3 for 1 h. During the 1-h incubation, the compound was added to the viral inoculums. The culture fluid was then replenished with fresh medium containing the compound. On day 2 p.i., the cells were fixed with methanol and viral E protein detected by enzyme-linked immunosorbent assay (ELISA). The ELISA used mouse monoclonal antibody 4G2 and goat anti-mouse IgG conjugated with horseradish peroxidase as the primary and secondary antibodies, respectively. The 50% effective concentrations (EC50s) were calculated by nonlinear regression analysis using software Prism (Graphpad) or ActivityBase (ID Business Solutions). (ii) A viral titer reduction assay was performed to test the antiviral activities of compound 1 and other compounds (36). (iii) A luciferase-reporting replicon of DENV-2 was used to estimate the effects of the compound on viral translation and RNA synthesis (26). (iv) HEK-293 cells harboring the DENV-2 luciferase replicon (21) were used to examine the effect of siRNAs (targeting the human adenosine kinase or deoxycytidine kinase) on the efficacy of compound 1. The procedures for the second to fourth antiviral assays described above were described in detail in the references cited. The cytotoxic concentration (CC50) values were estimated by measuring the intracellular level of ATP (CellTiter-Glo luminescent cell viability assay; Promega) as previously described (33).
Cloning, expression, and purification of human adenosine kinase.
The cDNA fragment of human adenosine kinase was PCR amplified from an IMAGE clone (Invitrogen) and inserted into pET28a at the NdeI and BamHI restriction sites. Deoxycytidine kinase was similarly cloned into the pET28a vector at the NheI and XhoI restriction sites. The Escherichia coli BL21 RIL strain containing the plasmid was cultured at 37°C to an optical density at 600 nm (OD600) of 0.4 to 0.5. The entire culture was cooled at 4°C for 1 h before isopropyl-β-d-thiogalactopyranoside (IPTG; 0.4 μM) was added to induce protein expression. After the cells were cultured at 18°C for another 16 h, the cell pellet was harvested for protein purification. The recombinant proteins, containing an N-terminal His6 tag, were purified through a nickel column. The protein was eluted using an imidazole gradient of 0 to 500 mM. Fractions containing the recombinant protein were pooled and further purified through a HiLoad 16/60 Superdex 75 gel filtration column (GE Healthcare). The purified recombinant protein was stored in aliquots at −80°C.
Transient DENV-2 replicon assay.
DENV-2 replicon RNA (10 μg) was electroporated into baby hamster kidney cells (BHK-21) cells (8 × 106) as described previously (30). The transfected cells were seeded into 12-well plates (3.2 × 105 cells per well) and immediately incubated with 1 μM and 3 μM compound 1. As a control, 0.9% dimethyl sulfoxide (DMSO) was added to the cells (because the compound stock was dissolved in DMSO and assayed at a final concentration of 0.9% DMSO). Luciferase activity was measured as reported previously (25) at time points indicated below. For viral protein analysis, the cells were lysed at 24 h and 35 h posttransfection (p.t.) and assayed for NS3 expression using Western blotting. The blotting used an NS3 antibody (1:5,000 dilution; generated in-house in rabbit) and a mouse monoclonal anti-γ-tubulin antibody (1:1,000 dilution; Sigma) as primary antibodies; horseradish peroxidase-labeled anti-rabbit and anti-mouse IgG (1:8,000 dilution), respectively, were used as secondary antibodies.
Adenosine kinase assay.
The adenosine kinase assay was performed in 20 mM Tris-HCl, pH 7.5, 20 mM potassium phosphate, 100 μM MgCl2, 10 μM ATP, 10 μCi [γ-33P]ATP, 100 nM recombinant adenosine kinase, and 100 μM different nucleosides or compound 1 in a total volume of 10 μl. After incubation at 23°C for 1 h, the reactions were stopped by adding 10 μl of stop buffer (25 mM Tris-HCl, pH 7.5, 75 mM NaCl, and 20 mM EDTA). The resulting mixture (l μl) was spotted onto a polyethyleneimine (PEI)-cellulose thin-layer chromatography (TLC) plate (Merck). The TLC plate was developed with solvent containing 0.4 M ammonium sulfate for 2 h. The plate was then dried and analyzed by autoradiograph.
siRNA transfection.
Two sets of siRNAs were purchased from Dharmacon to target human adenosine or deoxycytidine kinase. Each siRNA set contained a mixture of four siRNAs, designed to maximize the chance of hitting the target gene while minimizing hits of off-target genes. The siRNAs (10 pmol) were mixed with 0.15 μl RNAiMax (Invitrogen) in 20 μl of Opti-MEM medium (Gibco BRL); the mixture was incubated at 23°C for 20 min in a 96-well tissue culture plate (Nunc). HEK-293 cells (2 × 104 in 100 μl) containing DENV-2 luciferase replicons were then added to the wells. After the transfected cells were incubated at 37°C for 24 h, various concentrations of compound 1 were added to the cells. The cells were assayed for luciferase activity 3 days after treatment with the compound using a Renilla luciferase kit (Promega).
In vivo phosphorylation of compound 1.
Plasma or blood pellet samples were obtained at different time points after a single intravenous (5 mg/kg of body weight) or oral (25 mg/kg) dose of 14C-labeled compound 1 to male Han-Wistar rats. Samples were extracted with ice-cold acetonitrile at a ratio of 3:1 (acetonitrile/sample [vol/vol]) to precipitate proteins. The resulting samples were then mixed by sonication using a Covaris sonicator (S2 series, SonoLab single-sample preparation system) and centrifuged at 1,000 × g for 10 min. The supernatants were dried and reconstituted in 50 μl of solvent {10 mM ammonium formate with 0.1% formic acid/methanol [98:2 (vol/vol)]} for sample analysis. The blood pellet sample was reconstituted with 2 volumes of 10 mM ammonium formate buffer containing 0.1% formic acid (pH 2.5) and then subjected to the extraction method described above. The samples were analyzed by high-performance liquid chromatography (HPLC) with off-line radioactive detection to determine the metabolite profiles. Following HPLC separation, the structural identification of metabolites was carried out using a Finnigan hybrid LTQ linear ion trap/orbitrap or linear ion trap LTQ mass spectrometer (Thermo Electron).
RESULTS
Chemistry.
The synthesis of the compounds is described in detail in the supplemental material. Nucleoside analogs 2 to 4 were prepared as described in the literature (9, 12), and the synthesis of compounds 11 to 14 is summarized in schemes 1 to 3 in the supplemental material. Dess-Martin oxidation of α-d-1,3,5-tri-O-benzoyl-ribofuranose (compound 5) followed by reaction of the ketone compound 6 with ethynylmagnesium bromide or 1-propynyl magnesium bromide afforded the desired modification of ribose. Further benzoylation of the tertiary alcohol gave the intermediate compounds 7 and 8, respectively (scheme 1 in the supplemental material). The bases were then introduced using Vorbruggen methodology followed by aminolysis, providing access to nucleoside analogs 11 to 13 (9, 12). Reaction of 6-chloro-9H-purin-2-ylamine with compound 7 and hydrolysis of the resulting chloro intermediate with NaOH led to compound 14 (schemes 2 and 3 in the supplemental material).
The synthesis of compounds with a substituent at the C-7 position of the base is depicted in scheme 4 in the supplemental material. Ketone compound 15 was prepared as described in the literature (2). A Grignard reaction with ethynylmagnesium bromide followed by protection of the tertiary alcohol by a 2,5-dichlorobenzyl group and deprotection of the acetonide furnished the diol compound 16. Protection of the diol with toluoylchloride followed by removal of the acetonide led to compound 17. Oxidation of compound 17 with periodic acid followed by treatment with triethylamine and BCl3 cleavage of the benzyl-protecting group gave the key intermediate compound 18. Epoxidation followed by treatment with the modified nucleobases in the presence of NaH yielded the corresponding protected nucleoside analogs 19a to 19c. Deprotection of the toluoyl-protecting groups followed by aminolysis provided the modified nucleoside analogs 1 and 20 to 22.
The required modified nucleobases for the above synthesis were prepared as shown in scheme 5 in the supplemental material. Commercially available 4-chloro-7H-pyrrolo[2,3-d] pyrimidine (compound 23) was converted to 4-chloro-5-fluoro-7H-pyrrolo[2,3-d]pyrimidine (compound 24) with Selectfluor. The nitrile derivative compound 26 was prepared from compound 23 in a two-step process using an iodination-cyanation sequence. Finally, compound 23 was converted to compound 27 with N-bromosuccinimide, followed by lithium exchange and quenching with methylchloroformate to give the required carboxylic acid methyl ester compound 28.
SAR analysis.
Nucleoside analogs with the general structure A (Fig. 1) are potent inhibitors of various members of the genus Flaviviridae (20). To explore the SAR for DENV inhibition, we synthesized a panel of adenosine nucleosides containing various 2′-C-alkyl substitutions (Table 1). For each compound, the antiviral activity was tested by quantifying viral E protein in a DENV-2 infection assay (CFI assay; see Materials and Methods for details); the cytotoxicity was monitored by measuring the intracellular level of ATP (33). The CFI assay consistently showed a signal-to-background ratio of 6- to 7-fold, with a Z′ factor of about 0.75; when studying DENV entry inhibitors, we previously showed that the EC50s derived from the CFI assay were comparable to the EC50s derived from the plaque reduction assays (33). The results (Table 1) showed that compound 2 with a 2′-C-methyl substitution inhibited DENV, confirming the results of the previous studies (20, 29). Substitution with 2′-C-ethyl (compound 3) and 2′-C-vinyl (compound 4) abolished the antiviral activity. In contrast, a 2′-C-acetylene substitution led to a potent nucleoside (compound 11) with an EC50 of 1.41 μM; however, the compound showed some cytotoxicity, with a CC50 of 40 μM. Further replacement of the 2′-C-acetylene proton with a methyl group (compound 15) led to a loss of activity. The results demonstrate that adenosine with a 2′-C-acetylene substitution is a potent inhibitor of DENV.
TABLE 1.
Results of 2′-C substitutions in the adenosine analogsa
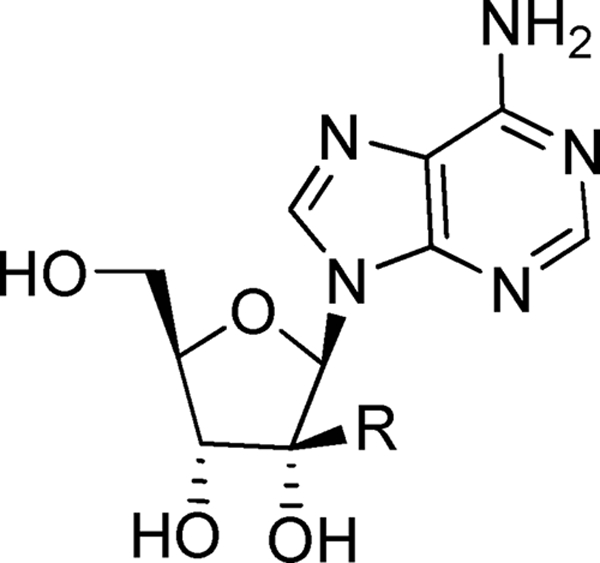 |
All values are the means of the results of at least three independent experiments.
Me, methyl; Et, ethyl.
The EC50s were determined by cellular flavivirus immunodetection (CFI) assay (see Materials and Methods for details).
The CC50s were estimated by measuring the intracellular level of ATP as previously described (33).
To examine whether other base types with a 2′-acetylene ribose would also inhibit DENV, we prepared a panel of 2′-C-acetylene derivatives containing cytosine (compound 13) and guanosine (compound 14) (Table 2). None of the base substitutions yielded active compounds. The results suggest that only adenosine with a 2′-C-acetylene ribose substitution is active against DENV.
TABLE 2.
Selectivity between adenine and other basesa
 |
All values are the means of the results of at least three independent experiments.
The EC50s were determined by cellular flavivirus immunodetection (CFI) assay.
The CC50s were estimated by measuring the intracellular level of ATP as previously described (33).
It was previously shown that adenosine with a ribose 2′-C-alkyl substitution could be deaminated by cellular adenosine deaminase to an inosine derivative (6) which was not active against DENV (data not shown). Since this metabolic pathway is also potentially relevant for compound 11, we replaced the adenine C-7 nitrogen with a carbon (compound 1; Table 3). This modification slightly improved the anti-DENV activity but significantly reduced the cytotoxicity (CC50 > 100 μM). We further explored the possible substitutions at the adenine C-7 position (Table 3). When the C-7 proton was replaced with F, the compound (compound 20) became slightly more potent (EC50, 0.4 μM) but more cytotoxic (CC50, 44 μM). CN (compound 21) and CONH2 substitutions (compound 22) led to less potent compounds, with EC50s of 3.1 μM and 2.0 μM, respectively. Overall, the SAR study identified 7-deaza-2′-C-acetylene-adenosine (compound 1) as a potent DENV inhibitor, which was selected for subsequent biological characterization.
TABLE 3.
Results of C-7 substitutions in 2′-acetylene-7-deaza-adenosinea
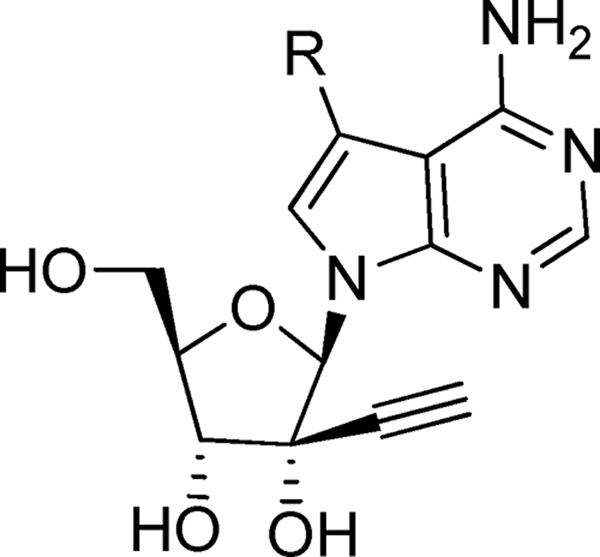 |
All results are the means of the results of at least three independent experiments.
The EC50s were determined by cellular flavivirus immunodetection (CFI) assay.
The CC50s were estimated by measuring the intracellular level of ATP as previously described (33).
Inhibition of DENV RNA synthesis.
We confirmed the antiviral activity of compound 1 using a viral titer reduction assay (36). Baby hamster kidney cells (BHK-21) were infected with DENV-2 at an MOI of 0.3; viral titers in culture fluid were measured at 48 h postinfection (p.i.). Treatment of the infected cells with compound 1 suppressed viral yield in a dose-responsive manner, with an EC50 of 0.7 μM; incubation of the culture with 6 μM compound reduced the viral titer by >160-fold (Fig. 2 A). Changes in MOI from 0.01 to 1 did not significantly affect the EC50s of compound 1 (data not shown). A cell viability assay showed that the compound was not cytotoxic up to 100 μM, the highest concentration tested (Fig. 2B).
FIG. 2.

Inhibition of DENV RNA synthesis by compound 1. (A) Results of viral titer reduction assay. BHK-21 cells were infected with DENV-2 (MOI of 0.3) and immediately treated with compound 1 at the indicated concentrations. Viral titers in culture fluid at 48 h p.i. were quantified by plaque assays. Average results and standard deviations (n = 3) are presented. (B) Results of cytotoxicity assay. BHK-21 cells were incubated with compound 1 for 48 h. A CellTiter-Glo luminescent assay was used to measure the intracellular level of ATP (to indicate cell viability) according to the manufacturer's protocol (Promega). Cell viability is presented as the percentage of luminescence derived from the compound-treated cells compared with the luminescence from the mock-treated cells. Average results and standard deviations (n = 3) are presented. (C) Results of transient DENV-2 replicon assay. A DENV-2 replicon containing a luciferase reporter was used to measure the effects of the compound on viral translation and RNA synthesis. The effects of compound 1 (1 μM and 3 μM) on viral translation and RNA synthesis were examined by measuring the luciferase signals at 2 to 6 h p.t. and at ≥24 h p.t., respectively. Average results and standard deviations (n = 3) are presented. (D) Results of Western blotting of viral NS3 protein. The expression levels of viral NS3 protein from the cells at 24 h and 35 h p.t., described for panel C, were analyzed using Western blotting. Host protein γ-tubulin was used as loading control (see Materials and Methods for details).
Next, we used a DENV-2 luciferase-reporting replicon to analyze the mechanism of inhibition. Transfection of BHK-21 cells with the replicon revealed two luciferase peaks. The first peak occurred at 2 to 4 h p.t., indicative of input replicon translation; the second peak occurred at >24 h p.t., indicative of RNA synthesis (16, 26). Treatment of the replicon-transfected cells with compound 1 did not affect the luciferase signals at 2 to 4 h but significantly suppressed the luciferase activities at 24 h and 35 h p.t. (Fig. 2C). Western blotting results showed that less viral NS3 protein was expressed when the transfected cells were treated with the compound (Fig. 2D). Collectively, the results demonstrate that the compound inhibits DENV through suppression of viral RNA synthesis. This conclusion is further supported by our previous results showing that the triphosphate form of compound 1 could directly inhibit RNA synthesis in a recombinant RdRp assay (36).
Phosphorylation of compound 1 by recombinant adenosine kinase.
To exhibit antiviral activity, compound 1 should be converted to the triphosphate form. Adenosine kinase is postulated to be the first enzyme in this intracellular sequence, converting compound 1 to a monophosphate form (18). To test this hypothesis, we cloned and expressed the human adenosine kinase using an E. coli system. The recombinant adenosine kinase, with an N-terminal His6 tag, was purified through a Ni2+ column, followed by gel filtration chromatography (Fig. 3 A). A sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis showed a protein of the expected size of 39 kDa in fractions 5 to 10 from the gel filtration column; the protein purity was >90%. The recombinant protein phosphorylated both compound 1 and adenosine to their monophosphate forms, although the phosphorylation of compound 1 was less efficient than that of the natural substrate adenosine, as judged by the intensities of their monophosphorylated derivatives analyzed on a thin-layer chromatograph (TLC) (Fig. 3B). The adenosine kinase did not phosphorylate uridine, guanosine, or cytidine. Since deoxycytidine kinase was reported to be a promiscuous kinase capable of phosphorylating both purine and pyrimidine nucleosides (28), we also prepared recombinant human deoxycytidine kinase. However, the recombinant deoxycytidine kinase did not phosphorylate compound 1 (data not shown). The results suggest that adenosine kinase is responsible for the phosphorylation of compound 1.
FIG. 3.

Conversion of compound 1 to its monophosphate form by recombinant adenosine kinase. (A) Purified recombinant human adenosine kinase. See Materials and Methods for cloning, expression, and purification of the adenosine kinase. Fractions from the gel filtration column (the final purification step) were analyzed on a 10% SDS-PAGE gel. The recombinant adenosine kinase is indicated by an arrow. (B) Thin-layer chromatography analysis of monophosphate nucleosides. Compound 1, adenosine, uridine, guanosine, and cytidine were incubated with [γ-33P]ATP in the presence of adenosine kinase. One microliter of the reaction mixture was spotted on the original and analyzed on a TLC plate. The plate was then dried and exposed to a film. Cold markers, ATP, AMP, and compound 1 monophosphate (indicated as NMP on the side of the TLC image), were analyzed on the TLC plate and detected under UV light.
Effects of adenosine kinase on antiviral activity in cell culture.
We took two approaches to demonstrate the biological relevance of the cellular adenosine kinase to the antiviral activity of compound 1. If the adenosine kinase is essential for the antiviral activity, inactivation of the kinase activity (using an inhibitor, first approach) or suppression of the kinase expression (using siRNAs, second approach) should reduce the potency of the compound. For the first approach, DENV-2-infected human alveolar basal epithelial cells (A549) were treated with compound 1 in the presence or absence of iodotubercidin, a known inhibitor of adenosine kinase (17). As shown in Fig. 4 A, the addition of iodotubercidin (0.2 μM) completely abolished the antiviral activity of compound 1 (up to 20 μM, the highest concentration tested). No cytotoxicity of iodotubercidin was detected at the concentrations tested (data not shown). For the second approach, a set of four siRNAs (Fig. 4B) targeting different regions of the human adenosine kinase mRNA were cotransfected into human embryonic kidney cells (HEK-293) harboring the DENV-2 luciferase replicon (21). The replicon cells were then treated with various concentrations of compound 1. As shown in Fig. 4B, transfection of the siRNAs targeting the adenosine kinase shifted the EC50 from 0.79 μM to 9.2 μM, resulting in an 11-fold decrease in antiviral efficacy. The adenosine kinase siRNAs did not completely abolish the antiviral activity of compound 1, possibly due to residual expression of adenosine kinase. As a negative control, cotransfection of a set of four siRNAs targeting the deoxycytidine kinase did not affect the potency of compound 1. No observable cytotoxicity was observed when cells were transfected with the siRNAs alone (data not shown). These results demonstrate that adenosine kinase plays an important role in activating the antiviral activity of compound 1 in cell culture.
FIG. 4.

Effect of adenosine kinase on antiviral activity. (A) Effect of iodotubercidin on antiviral efficacy. A CFI antiviral assay (see Materials and Methods for details) was performed using compound 1 in the presence or absence of iodotubercidin (0.2 μM). The antiviral activity is presented as the percent inhibition. (B) Effect of siRNAs on antiviral activity. HEK-293 cells harboring the DENV-2 luciferase replicon were cotransfected with a set of four siRNAs targeting human adenosine kinase or deoxycytidine kinase. The target sequences of each interfering RNA are shown (top panel). The cells were treated with compound 1 at the indicated concentrations at 24 h p.t. and assayed for luciferase activities at 72 h after treatment with the compound. The antiviral activity is presented as the percent luciferase activity. The averages of the results of two independent experiments are shown.
Conversion of compound 1 to mono-, di-, and triphosphate forms in rats.
We examined the phosphorylation profile of compound 1 in rats using a 14C-labeled compound. The compound was 14C labeled at the C-2 position of the adenine base. After the rats were dosed with 5 mg/kg of compound 1 intravenously or with 25 mg/kg orally, nucleoside metabolites were extracted from plasma and blood cells, respectively. Fast-protein liquid chromatography (FPLC) analysis of these extracts showed that only the parent compound was detected in the plasma (Fig. 5 A), whereas the mono-, di-, and triphosphate derivatives, together with the parent compound, were detected in the blood cells (Fig. 5B). The results demonstrate that compound 1 can be converted into the corresponding triphosphate form in vivo.
FIG. 5.

Conversion of compound 1 to mono-, di-, and triphosphate derivatives in vivo. (A) Analysis of metabolite in rat plasma. The 14C-labeled compound 1 was either intravenously dosed at 5 mg/kg or orally dosed at 25 mg/kg in rats. Plasma was collected from 4 to 24 h after dosing. Metabolites of compound 1 were extracted from the pooled plasma and analyzed using FPLC (see Materials and Methods for details). (B) Analysis of metabolites in blood cells. Total blood cells were harvested at 1 to 12 h after dosing. The cell pellets collected at different time points were pooled together, and compound metabolites extracted. The extracts were then subjected to HPLC analysis.
Single-dose efficacy of compound 1 in dengue mouse models.
We recently reported the potent antiviral activity of compound 1 in repeat-dose regimens in DENV-infected mice (36). However, the single-dose efficacy of compound 1 remains to be determined in vivo. We examined this possibility in both DENV viremia and lethal mouse models. For the viremia model, AG129 mice (defective in alpha/beta interferon [IFN-α/β] and IFN-γ receptors) were intraperitoneally infected with DENV-2 (strain TSVO1) and treated with a single oral dose of compound 1 (from 25 to 300 mg/kg) at 12 h postinfection. The animals were sacrificed at the peak of viremia on day 3 p.i., and plasma viremia was quantified by plaque assay. As shown in Fig. 6 A, the viremia level was reduced in a dose-dependent manner from 25 to 75 mg/kg, achieving a >10-fold reduction after a single-dose treatment of 75 mg/kg. Higher dosing at 150 and 300 mg/kg did not further lower the viremia, indicating that the uptake of compound 1 reached saturation at 75 mg/kg. For comparison, we also included the results from the optimal multiple-dose regimen (twice daily dosing at 25 mg/kg per dose for 3 days starting immediately after infection, equating to a total exposure of 150 mg/kg).
FIG. 6.

A single-dose efficacy of compound 1 was seen in mouse DENV models. (A) Results for a mouse model of viremia (29). AG129 mice were intraperitoneally inoculated with 2 × 106 PFU of DENV-2 (strain TSV01). The mice (8 animals per group) were orally dosed once with the indicated amount of compound 1 at 12 h p.i. The peak viremia on day 3 p.i. was measured by plaque assay. An asterisk indicates that the difference between the compound-treated and the vehicle-treated group is statistically significant (P value < 0.05 using Student's t test). Error bars show standard deviations. (B) Results for lethal mouse model. AG129 mice (6 mice per group) were intravenously inoculated with 3 × 107 PFU of DENV-2 strain D2S10 (31), treated with a single dose of 75 mg/kg of compound 1 at 12 h p.i., and monitored for mortality. The detailed protocols for the mouse experiments were reported previously (36). The difference between the compound-treated and the vehicle-treated groups is statistically significant at a P value of <0.01.
Next, we used a lethal mouse model to determine the antiviral effect of a single-dose treatment on disease severity. AG129 mice were intravenously infected with a lethal dose (3 × 107 PFU) of the mouse-adapted DENV-2 strain D2S10 (31) and treated with a single 75-mg/kg dose of compound 1 at 12 h postinfection. The single-dose treatment provided full protection against the lethal virus challenge (Fig. 6B). The single-dose efficacy is similar to the results when mice were treated with a multiple-dose regimen (twice-daily dosing at 10 mg/kg per dose for 3 days, which was previously shown to be the lowest dose to give full protection [36]). Collectively, the results demonstrated that a single-dose treatment of DENV-infected mice with compound 1 suppressed peak viremia and completely protected the infected mice from death.
DISCUSSION
A number of strategies have been pursued to develop flavivirus inhibitors (22). Since nucleoside analogs have been used successfully as therapy for other viral diseases, we took this approach to develop DENV inhibitors. Our chemistry efforts led to the discovery of compound 1 as a potent inhibitor of DENV. We recently showed that this compound was active in vivo; a multiple-dose treatment suppressed peak viremia, reduced cytokine elevation, and completely protected the infected mice from death (36). In this study, we demonstrated that a single-dose treatment of DENV-infected mice with compound 1 could also suppress peak viremia by >10-fold and completely protected the infected mice from death. To our knowledge, compound 1 represents the first small-molecule inhibitor that shows a potent in vivo efficacy against a flavivirus after a single-dose treatment.
Our results strongly indicate that compound 1 exerts its antiviral activity through RNA chain termination. This conclusion is supported by three lines of evidences. First, using a DENV replicon, we showed that the compound suppressed viral RNA synthesis; it did not inhibit viral translation (Fig. 2). Second, we previously showed that the triphosphate form of compound 1 was able to directly inhibit RNA elongation catalyzed by a recombinant DENV NS5 (36). Third, cocrystallization of DENV RdRp in complex with the triphosphate form of compound 1 showed that the triphosphate moiety was located at the active site of the RdRp (S. S. Yeong, T. L. Yap, Y.-L. Chen, D. Luo, P. Niyomrattanakit, H. Dong, C. C. Lim, P.-Y. Shi, and J. Lescar, unpublished results). The results also lead to the inference that upon entering cells, the compound is phosphorylated to its triphosphate form before it can be utilized by viral RdRp to terminate RNA synthesis. This notion is further supported by the results showing that after oral dosing of rats with compound 1, the compound was converted to the mono-, di-, and triphosphate forms in blood cells (Fig. 5).
The nucleoside-based approach for antiviral development faces several challenges. Because multiple cellular enzymes are required to convert a nucleoside (inactive) into its triphosphate form (active), the antiviral efficacy is a combined outcome derived from multiple enzymatic reactions. Besides the intrinsic potency against the viral polymerase, the efficiency of kinase-mediated phosphorylation may contribute significantly to the antiviral outcome. An inactive nucleoside may be the result of an inefficient conversion to the triphosphate form. For this reason, nucleoside inhibitors are often discovered serendipitously. To determine the intrinsic potency against viral polymerase, one needs to synthesize the triphosphate compound and test it directly in a viral polymerase assay. Different cell lines may have different expression levels of nucleoside kinases, leading to discrepant EC50s of the same compound (15). It is therefore useful to evaluate nucleoside inhibitors in more than one clinically relevant cell line.
Since the conversion of a nucleotide to its monophosphate form is often the rate-limiting step in triphosphorylation (19), we examined the monophosphorylation of compound 1. Recombinant human adenosine kinase could efficiently convert the compound to its monophosphate form (Fig. 3). Inhibition of adenosine kinase using a specific inhibitor (iodotubercidin) or siRNAs abolished or reduced the compound's antiviral activity (Fig. 4). In contrast, recombinant deoxycytidine kinase did not phosphorylate compound 1; treatment of cells with siRNAs targeting the deoxycytidine kinase did not affect the potency of compound 1. These results indicate that adenosine kinase may be responsible for intracellular monophosphorylation of the compound. For nucleosides that could not be efficiently converted to their monophosphate forms, one could synthesize the monophosphate or phosphonate prodrugs. The phosphoramidate approach was successfully used to convert inactive nucleoside analogs to potent inhibitors of HCV (23, 24). Other examples of nucleotide phosphonates are adefovir disoproxil and tenofovir disoproxil, which are in clinical use for treatment of hepatitis B virus and HIV, respectively.
Another challenge for the development of nucleoside antivirals is the unpredictable toxicity. This was experienced during the development of compound 1. The compound did not show any negative signs in in vitro assays, including a mitochondrial toxicity assay, the Ames test for genotoxicity, the hERG channel test for cardiovascular toxicity, and a micronucleus assay for mutagenicity, as well as various receptor, ion channel, and kinase assays. However, when rats (10 mg/kg/day) and dogs (1 mg/kg/day) were dosed with the compound daily for 2 weeks, a no-observed-adverse-effect level (NOAEL) could not be achieved; NOAEL was achieved when rats were orally dosed with compound 1 at 50 mg/kg/day for 1 week (36). Experiments are ongoing to determine the nature of the toxicity of compound 1.
Two major issues remain to be addressed for further development of compound 1. One issue is related to the in vivo efficacy. Although the AG129 mouse model could be used to estimate the antiviral efficacy (29), caution should be taken when interpreting the results from such experiments. Since the AG129 mouse lacks IFN-α/β and IFN-γ receptors, the mouse is defective in an innate immune response that plays a critical role in host defense when encountering a pathogen. Thus, this model may underestimate the real efficacy of the compound. Another issue is related to the length of therapeutic treatment versus the 2-week NOAEL result. Dengue is an acute disease with a fever duration of less than a week (11), so the length of treatment is expected to be less than a week. This is in contrast to the treatment of chronic diseases which require long-term treatment (such as HCV and HIV). The difference between the acute and chronic diseases should be considered during preclinical development of DENV inhibitors. Notably, the above-described issues are related to each other. An improved animal model that allows a better estimation of in vivo potency will be critical to set up an appropriate level for a preclinical toxicity study.
This study has extended our previous finding that an adenosine nucleoside is a potent inhibitor of DENV. A single-dose treatment with the compound completely protected the DENV-infected mice from death. Upon entering cells, the compound is likely converted to its monophosphate form by adenosine kinase and further metabolized to its active triphosphate forms. The compound functions as a chain terminator to inhibit viral RNA synthesis.
Supplementary Material
Acknowledgments
We thank Julien Lescar for help and discussion during the course of the project.
Footnotes
Published ahead of print on 10 May 2010.
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1.Ackermann, M., and R. Padmanabhan. 2001. De novo synthesis of RNA by the dengue virus RNA-dependent RNA polymerase exhibits temperature dependence at the initiation but not elongation phase. J. Biol. Chem. 276:39926-39937. [DOI] [PubMed] [Google Scholar]
- 2.Bio, M. M., F. Xu, M. Waters, J. M. Williams, K. A. Savary, C. J. Cowden, C. Yang, E. Buck, Z. J. Song, D. M. Tschaen, R. P. Volante, R. A. Reamer, and E. J. Grabowski. 2004. Practical synthesis of a potent hepatitis C virus RNA replication inhibitor. J. Org. Chem. 69:6257-6266. [DOI] [PubMed] [Google Scholar]
- 3.Carroll, S. S., S. Ludmerer, L. Handt, K. Koeplinger, N. R. Zhang, D. Graham, M. E. Davies, M. MacCoss, D. Hazuda, and D. B. Olsen. 2009. Robust antiviral efficacy upon administration of a nucleoside analog to hepatitis C virus-infected chimpanzees. Antimicrob. Agents Chemother. 53:926-934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carroll, S. S., and D. B. Olsen. 2006. Nucleoside analog inhibitors of hepatitis C virus replication. Infect. Disord. Drug Targets 6:17-29. [DOI] [PubMed] [Google Scholar]
- 5.Carroll, S. S., J. E. Tomassini, M. Bosserman, K. Getty, M. W. Stahlhut, A. B. Eldrup, B. Bhat, D. Hall, A. L. Simcoe, R. LaFemina, C. A. Rutkowski, B. Wolanski, Z. Yang, G. Migliaccio, R. De Francesco, L. C. Kuo, M. MacCoss, and D. B. Olsen. 2003. Inhibition of hepatitis C virus RNA replication by 2′-modified nucleoside analogs. J. Biol. Chem. 278:11979-11984. [DOI] [PubMed] [Google Scholar]
- 6.Cristalli, G., S. Costanzi, C. Lambertucci, G. Lupidi, S. Vittori, R. Volpini, and E. Camaioni. 2001. Adenosine deaminase: functional implications and different classes of inhibitors. Med. Res. Rev. 21:105-128. [DOI] [PubMed] [Google Scholar]
- 7.De Clercq, E., and J. Neyts. 2009. Antiviral agents acting as DNA or RNA chain terminators. Handb. Exp. Pharmacol. 2009(189):53-84. [DOI] [PubMed] [Google Scholar]
- 8.Egloff, M. P., D. Benarroch, B. Selisko, J. L. Romette, and B. Canard. 2002. An RNA cap (nucleoside-2′-O-)-methyltransferase in the flavivirus RNA polymerase NS5: crystal structure and functional characterization. EMBO J. 21:2757-2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eldrup, A. B., C. R. Allerson, C. F. Bennett, S. Bera, B. Bhat, N. Bhat, M. R. Bosserman, J. Brooks, C. Burlein, S. S. Carroll, P. D. Cook, K. L. Getty, M. MacCoss, D. R. McMasters, D. B. Olsen, T. P. Prakash, M. Prhavc, Q. Song, J. E. Tomassini, and J. Xia. 2004. Structure-activity relationship of purine ribonucleosides for inhibition of hepatitis C virus RNA-dependent RNA polymerase. J. Med. Chem. 47:2283-2295. [DOI] [PubMed] [Google Scholar]
- 10.Falgout, B., R. H. Miller, and C. J. Lai. 1993. Deletion analysis of dengue virus type 4 nonstructural protein NS2B: identification of a domain required for NS2B-NS3 protease activity. J. Virol. 67:2034-2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gubler, D., G. Kuno, and L. Markoff. 2007. Flaviviruses, p. 1153-1253. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 5th ed., vol. 1. Lippincott William & Wilkins, Philadelphia, PA. [Google Scholar]
- 12.Harry-O'kuru, R. E., J. M. Smith, and M. S. Wolfe. 1997. A short, flexible route toward 2′-C-branched ribonucleosides. J. Org. Chem. 62:1754-1759. [Google Scholar]
- 13.Klumpp, K., G. Kalayanov, H. Ma, S. Le Pogam, V. Leveque, W. R. Jiang, N. Inocencio, A. De Witte, S. Rajyaguru, E. Tai, S. Chanda, M. R. Irwin, C. Sund, A. Winqist, T. Maltseva, S. Eriksson, E. Usova, M. Smith, A. Alker, I. Najera, N. Cammack, J. A. Martin, N. G. Johansson, and D. B. Smith. 2008. 2′-Deoxy-4′-azido nucleoside analogs are highly potent inhibitors of hepatitis C virus replication despite the lack of 2′-alpha-hydroxyl groups. J. Biol. Chem. 283:2167-2175. [DOI] [PubMed] [Google Scholar]
- 14.Klumpp, K., V. Leveque, S. Le Pogam, H. Ma, W. R. Jiang, H. Kang, C. Granycome, M. Singer, C. Laxton, J. Q. Hang, K. Sarma, D. B. Smith, D. Heindl, C. J. Hobbs, J. H. Merrett, J. Symons, N. Cammack, J. A. Martin, R. Devos, and I. Najera. 2006. The novel nucleoside analog R1479 (4′-azidocytidine) is a potent inhibitor of NS5B-dependent RNA synthesis and hepatitis C virus replication in cell culture. J. Biol. Chem. 281:3793-3799. [DOI] [PubMed] [Google Scholar]
- 15.Leary, J. J., R. Wittrock, R. T. Sarisky, A. Weinberg, and M. J. Levin. 2002. Susceptibilities of herpes simplex viruses to penciclovir and acyclovir in eight cell lines. Antimicrob. Agents Chemother. 46:762-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lo, L., M. Tilgner, K. Bernard, and P.-Y. Shi. 2003. Functional analysis of mosquito-borne flavivirus conserved sequence elements within 3′ untranslated region of West Nile virus using a reporting replicon that differentiates between viral translation and RNA replication. J. Virol. 77:10004-10014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopez, J., A. Samtodrian, C. Campas, and J. Gil. 2003. 5-Aminoimidazole-4-carboxamide riboside induces apoptosis in Jurkat cells, but the AMP-activated protein kinase is not involved. Biochem. J. 370:1027-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maj, M. C., B. Singh, and R. S. Gupta. 2000. Structure-activity studies on mammalian adenosine kinase. Biochem. Biophys. Res. Commun. 275:386-393. [DOI] [PubMed] [Google Scholar]
- 19.McGuigan, C., D. Cahard, H. M. Sheeka, E. De Clercq, and J. Balzarini. 1996. Aryl phosphoramidate derivatives of d4T have improved anti-HIV efficacy in tissue culture and may act by the generation of a novel intracellular metabolite. J. Med. Chem. 39:1748-1753. [DOI] [PubMed] [Google Scholar]
- 20.Migliaccio, G., J. E. Tomassini, S. S. Carroll, L. Tomei, S. Altamura, B. Bhat, L. Bartholomew, M. R. Bosserman, A. Ceccacci, L. F. Colwell, R. Cortese, R. De Francesco, A. B. Eldrup, K. L. Getty, X. S. Hou, R. L. LaFemina, S. W. Ludmerer, M. MacCoss, D. R. McMasters, M. W. Stahlhut, D. B. Olsen, D. J. Hazuda, and O. A. Flores. 2003. Characterization of resistance to non-obligate chain-terminating ribonucleoside analogs that inhibit hepatitis C virus replication in vitro. J. Biol. Chem. 278:49164-49170. [DOI] [PubMed] [Google Scholar]
- 21.Ng, C. Y., F. Gu, W. Y. Phong, Y. L. Chen, S. P. Lim, A. Davidson, and S. G. Vasudevan. 2007. Construction and characterization of a stable subgenomic dengue virus type 2 replicon system for antiviral compound and siRNA testing. Antiviral Res. 76:222-231. [DOI] [PubMed] [Google Scholar]
- 22.Noble, C. G., Y. L. Chen, H. Dong, F. Gu, S. P. Lim, W. Schul, Q. Y. Wang, and P. Y. Shi. 2010. Strategies for development of dengue virus inhibitors. Antiviral Res. 85:450-462. [DOI] [PubMed] [Google Scholar]
- 23.Perrone, P., F. Daverio, R. Valente, S. Rajyaguru, J. A. Martin, V. Leveque, S. Le Pogam, I. Najera, K. Klumpp, D. B. Smith, and C. McGuigan. 2007. First example of phosphoramidate approach applied to a 4′-substituted purine nucleoside (4′-azidoadenosine): conversion of an inactive nucleoside to a submicromolar compound versus hepatitis C virus. J. Med. Chem. 50:5463-5470. [DOI] [PubMed] [Google Scholar]
- 24.Perrone, P., G. M. Luoni, M. R. Kelleher, F. Daverio, A. Angell, S. Mulready, C. Congiatu, S. Rajyaguru, J. A. Martin, V. Leveque, S. Le Pogam, I. Najera, K. Klumpp, D. B. Smith, and C. McGuigan. 2007. Application of the phosphoramidate ProTide approach to 4′-azidouridine confers sub-micromolar potency versus hepatitis C virus on an inactive nucleoside. J. Med. Chem. 50:1840-1849. [DOI] [PubMed] [Google Scholar]
- 25.Puig-Basagoiti, F., T. S. Deas, P. Ren, M. Tilgner, D. M. Ferguson, and P.-Y. Shi. 2005. High-throughput assays using luciferase-expressing replicon, virus-like particle, and full-length virus for West Nile virus drug discovery. Antimicrob. Agent. Chemother. 49:4980-4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Puig-Basagoiti, F., M. Tilgner, B. M. Forshey, S. M. Philpott, N. G. Espina, D. E. Wentworth, S. J. Goebel, P. S. Masters, B. Falgout, P. Ren, D. M. Ferguson, and P.-Y. Shi. 2006. Triaryl pyrazoline compound inhibits flavivirus RNA replication. Antimicrob. Agents Chemother. 50:1320-1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ray, D., A. Shah, M. Tilgner, Y. Guo, Y. Zhao, H. Dong, T. Deas, Y. Zhou, H. Li, and P.-Y. Shi. 2006. West Nile virus 5′-cap structure is formed by sequential guanine N-7 and ribose 2′-O methylations by nonstructural protein 5. J. Virol. 80:8362-8370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sabini, E., S. Hazra, S. Ort, M. Konrad, and A. Lavie. 2008. Structural basis for substrate promiscuity of dCK. J. Mol. Biol. 378:607-621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schul, W., W. Liu, H. Y. Xu, M. Flamand, and S. G. Vasudevan. 2007. A dengue fever viremia model in mice shows reduction in viral replication and suppression of the inflammatory response after treatment with antiviral drugs. J. Infect. Dis. 195:665-674. [DOI] [PubMed] [Google Scholar]
- 30.Shi, P. Y., M. Tilgner, M. K. Lo, K. A. Kent, and K. A. Bernard. 2002. Infectious cDNA clone of the epidemic West Nile virus from New York City. J. Virol. 76:5847-5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shresta, S., K. L. Sharar, D. M. Prigozhin, P. R. Beatty, and E. Harris. 2006. Murine model for dengue virus-induced lethal disease with increased vascular permeability. J. Virol. 80:10208-10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan, B. H., J. Fu, R. J. Sugrue, E. H. Yap, Y. C. Chan, and Y. H. Tan. 1996. Recombinant dengue type 1 virus NS5 protein expressed in Escherichia coli exhibits RNA-dependent RNA polymerase activity. Virology 216:317-325. [DOI] [PubMed] [Google Scholar]
- 33.Wang, Q. Y., S. J. Patel, E. Vangrevelinghe, H. Y. Xu, R. Rao, D. Jaber, W. Schul, F. Gu, O. Heudi, N. L. Ma, M. K. Poh, W. Y. Phong, T. H. Keller, E. Jacoby, and S. G. Vasudevan. 2009. A small-molecule dengue virus entry inhibitor. Antimicrob. Agents Chemother. 53:1823-1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wengler, G., and G. Wengler. 1991. The carboxy-terminal part of the NS 3 protein of the West Nile flavivirus can be isolated as a soluble protein after proteolytic cleavage and represents an RNA-stimulated NTPase. Virology 184:707-715. [DOI] [PubMed] [Google Scholar]
- 35.Wengler, G., and G. Wengler. 1993. The NS3 nonstructural protein of flaviviruses contains an RNA triphosphatase activity. Virology 197:265-273. [DOI] [PubMed] [Google Scholar]
- 36.Yin, Z., Y.-L. Chen, W. Schul, Q.-Y. Wang, F. Gu, J. Duraiswamy, R. R. Kondreddi, P. Niyomrattanakit, S. B. Lakshminarayana, A. Goh, H. Y. Xu, W. Liu, B. Liu, J. Y. H. Lim, C. Y. Ng, M. Qing, C. C. Lim, A. Yip, G. Wang, W. L. Chan, H. P. Tan, K. Lin, B. Zhang, G. Zou, K. A. Bernard, C. Garrett, K. Beltz, M. Dong, M. Weaver, H. He, A. Pichota, V. Dartois, T. H. Keller, and P.-Y. Shi. 2009. An adenosine nucleoside inhibitor of dengue virus. Proc. Natl. Acad. Sci. U. S. A. 106:20435-20439. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.