

Abstract
ExsA is a member of the AraC family of transcriptional activators and is required for expression of the Pseudomonas aeruginosa type III secretion system (T3SS). ExsA-dependent promoters consist of two binding sites for monomeric ExsA located approximately 50 bp upstream of the transcription start sites. Binding to both sites is required for recruitment of σ70-RNA polymerase (RNAP) to the promoter. ExsA-dependent promoters also contain putative −35 hexamers that closely match the σ70 consensus but are atypically spaced 21 or 22 bp from the −10 hexamer. Because several nucleotides located within the putative −35 region are required for ExsA binding, it is unclear whether the putative −35 region makes an additional contribution to transcription initiation. In the present study we demonstrate that the putative −35 hexamer is dispensable for ExsA-independent transcription from the PexsC promoter and that deletion of σ70 region 4.2, which contacts the −35 hexamer, has no effect on ExsA-independent transcription from PexsC. Region 4.2 of σ70, however, is required for ExsA-dependent activation of the PexsC and PexsD promoters. Genetic data suggest that ExsA directly contacts region 4.2 of σ70, and several amino acids were found to contribute to the interaction. In vitro transcription assays demonstrate that an extended −10 element located in the PexsC promoter is important for overall promoter activity. Our collective data suggest a model in which ExsA compensates for the lack of a −35 hexamer by interacting with region 4.2 of σ70 to recruit RNAP to the promoter.
Pseudomonas aeruginosa is an opportunistic human pathogen that causes a variety of acute and chronic infections in immunocompromised individuals (52, 53). A primary determinant of P. aeruginosa virulence is a type III secretion system (T3SS) (24, 70). The T3SS consists of a macromolecular conduit through which effector toxins are translocated into eukaryotic host cells (70). The translocated toxins promote tissue destruction and evasion of the host immune response (3, 55, 69). Mutants lacking a functional T3SS have reduced virulence in both tissue culture and animal infection models (2, 33).
The central regulator of T3SS gene expression is ExsA (25, 67, 68). ExsA directly binds to 10 different promoters and activates transcription of the core genes required for assembly and function of the T3SS (12, 64). ExsA belongs to the family of AraC/XylS transcriptional regulators. AraC family members generally consist of an amino-terminal ligand interaction domain and two carboxy-terminal helix-turn-helix DNA-binding motifs (14). AraC regulators are often classified by the type of ligand that regulates their activity. ExsA belongs to a small subset of AraC regulators that respond to protein ligands and control T3SS gene expression (50). Representatives of this subfamily are also found in Vibrio parahaemolyticus (ExsA), Shigella flexneri (MxiE), and Salmonella enterica (InvF) (17, 25, 42, 71). ExsA-dependent transcription in P. aeruginosa is antagonized by ExsD, which functions as an antiactivator by inhibiting the DNA-binding activity and self-association properties of ExsA (13, 43, 61). Similarly, the ExsA homolog in Vibrio parahaemolyticus is required for T3SS1 gene expression, and the ExsD homolog is thought to antagonize ExsA activity (71). Transcriptional activation by S. flexneri MxiE is also antagonized by a protein ligand (OspD1), but the inhibitory mechanism has not been established (48). In contrast, transcriptional activation by the S. enterica regulator InvF is positively regulated by the SicA coactivator through a direct binding interaction (18). S. flexneri IpgC, which copurifies with MxiE, may also function as a coactivator (49). In summary, modulation of activator function by protein ligands can occur in a positive or negative fashion and may be a common theme for AraC family members that regulate T3SS gene expression.
ExsA-dependent promoters in P. aeruginosa consist of two adjacent binding sites for monomeric ExsA. Binding site 1 completely overlaps a putative σ70-RNA polymerase (RNAP) −35 recognition hexamer, while binding site 2 extends upstream and includes an adenine-rich region (12). ExsA binds via a monomer assembly pathway in which ExsA bound to site 1 recruits a second ExsA monomer to binding site 2 (12, 14). Like most AraC family members, ExsA is dependent on σ70 for transcriptional activation (64), and ExsA-dependent promoters contain apparent σ70-RNAP hexamers that closely resemble the P. aeruginosa consensus sequences (TTGACA and TATAAT for the −35 and −10 sites, respectively) (12, 34). The placement of the −10 hexamers and transcription start sites has been established for several ExsA-dependent promoters by potassium permanganate footprinting experiments and 5′ rapid amplification of cDNA ends (RACE) mapping, respectively (64, 67). These experiments indicated that σ70-dependent transcription originates from the same start sites in the presence and absence of ExsA (64). The apparent −35 and −10 hexamers of ExsA-dependent promoters are spaced 4 to 5 bp farther apart than the 17 bp typical of most σ70-dependent promoters. Increased spacing has not been reported for any AraC family regulators but is seen for Spo0A, a transcriptional activator of the sporulation regulon in Bacillus subtilis (45). Spo0A activates promoters with extended spacing (20 to 22 bp) between near-consensus σA-RNAP (the σ70 homolog) −35 and −10 hexamers (8, 39, 57). The current model suggests that Spo0A activates transcription by repositioning RNAP prebound to the −35 site such that σA region 2 can interact with the −10 hexamer to initiate open complex formation (39). Despite the apparent similarity to the extended spacing of Spo0A-dependent promoters, genetic and biochemical experiments suggest an entirely different mechanism for ExsA-dependent activation. A kinetic analysis of abortive transcript production from the PexsC and PexsD promoters reveals that the primary function of ExsA is to recruit σ70-RNAP to promoter DNA (64). Additionally, ExsA-dependent promoters in which the spacing between the −35 and −10 hexamers has been reduced to 16 or 17 bp are severely reduced in expression. These data suggest that ExsA, unlike Spo0A, does not function by compensating for increased promoter spacing and raises the question as to whether the −35-like elements of ExsA-dependent promoters represent actual σ70-RNAP contact points.
Transcriptional activators typically promote transcription through specific contacts with the α and/or σ70 subunit of RNAP (41). The specific RNAP contacts made by these proteins are thought to depend largely on the location of the activator promoter-binding site relative to the −35 hexamer. Class I promoters usually contain an activator DNA-binding site located ≥20 bp upstream of the −35 hexamer (22). The available data suggest that activation of a class I promoter is typically mediated by specific contacts between the activator protein and the carboxy-terminal domain of the RNAP α subunit (α-CTD) (22). In contrast, class II promoters usually contain an activator DNA-binding site positioned in close proximity to or overlapping the −35 hexamer. Activation of a class II promoter is thought to occur by contacts with the σ70 subunit or both the σ70 and α subunits of RNAP (51, 56). Activation by AraC family members often involves interactions with region 4.2 of σ70 RNAP. This region contains a DNA-binding domain that recognizes the −35 hexamer. The carboxy-terminal end of region 4.2 also interacts with a diverse group of class II transcriptional activators (20). For example the AraC family regulators RhaR and RhaS, which are involved in the metabolism of rhamnose, contact several amino acids in region 4.2 of σ70, and this interaction is required for transcriptional activation (7, 66).
In this study we characterized the interaction between ExsA and the σ70 subunit. Our data indicate that ExsA functions as a class II transcriptional activator at the PexsC and PexsD promoters, does not require the α subunit of RNAP, and instead contacts several amino acids in region 4.2 of σ70. We also provide evidence that the −35-like element of the PexsC promoter is not an authentic RNAP recognition hexamer for ExsA-independent or -dependent transcription and demonstrate that ExsA-independent transcription at the PexsC promoter requires an extended −10 promoter element.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The bacterial strains and plasmids used in this study are summarized in Table 1. Escherichia coli strains were maintained on LB agar plates containing the following antibiotics/chemicals as necessary: gentamicin (15 μg/ml), ampicillin (50 or 100 μg/ml), tetracycline (10 μg/ml), kanamycin (50 μg/ml), and indole-3-acrylic acid (0.5 mM). P. aeruginosa strains were maintained on Vogel-Bonner minimal medium (65) with antibiotics as indicated (carbenicillin [300 μg/ml] and tetracycline [50 μg/ml]). For the LuxR experiments bacteria were grown in the presence of 200 nM N-(3-oxo-hexanoyl)-l homoserine lactone (Sigma, St. Louis, MO). To assay for ExsA-dependent gene expression in the presence of mutant and wild-type RNAP subunits, E. coli strains from LB agar plates grown overnight were inoculated into 10 ml of LB to an optical density at 600 nm (OD600) of 0.1 and grown with vigorous aeration at 30°C until the OD600 reached 0.6. β-Galactosidase assays were performed as previously described (19), and the reported values are the averages from at least three independent experiments.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristics or purpose | Reference or source |
|---|---|---|
| Pseudomonas aeruginosa strain PA103 | Wild-type parental strain | 26 |
| Escherichia coli strains | ||
| DH5α | recA cloning strain | 29 |
| GS162 | Wild-type strain carrying ΔlacU169 | 58 |
| SA1751 | Thermoinducible Int expression from the cryptic prophage for minicircle recombination | 16 |
| GA2071 | rpoD suppression strain | 40 |
| BL21(DE3) Tuner | Protein purification | Novagen |
| BW25141 | Maintenance of pir-dependent plasmids | 28 |
| Plasmids | ||
| pREiiα | rpoA expression vector | 9 |
| pGS490 | rpoA expression vector with a stop codon at 239 | 36 |
| pJN105 | Arabinose-inducible expression vector | 47 |
| pUY30 | Arabinose-inducible expression vector | 62 |
| pMini-CTX-lacZ | Vector for single-copy integration of lacZ reporters onto the P. aeruginosa chromosomal attB site | 31 |
| pMCTX-PlacUV5mut-lacZ | Transcriptional fusion of the PlacUV5mut-lacZ promoter to lacZ | This study |
| p2UY21-exsA | Plasmid that constitutively expresses exsA | This study |
| p2UY21-luxR | Plasmid that constitutively expresses luxR | This study |
| pMU102 | luxR expression vector | 63 |
| pAH125 | Vector for single-copy integration of lacZ reporters onto the E. coli λ attachment site | 28 |
| pluxI-lacZ | Translational fusion of the PluxI promoter to lacZ | 63 |
| pAH125-PluxI-lacZ | Translational fusion of the PluxI promoter to lacZ | This study |
| pAH125-PexsC-lacZ | Transcriptional fusion of the PexsC promoter to lacZ | This study |
| pAH125-PexsD-lacZ | Transcriptional fusion of the PexsD promoter to lacZ | This study |
| pAH125-PexoT-lacZ | Transcriptional fusion of the PexoT promoter to lacZ | This study |
| pGEX-rpoD and its derivatives | Plasmid that constitutively expresses rpoD or one of 16 alanine point mutations | 40 |
| pGEX-rpoD(K593A,R596A,R599A) | rpoD expression plasmid carrying the K593A, R596A, and R599A mutations | This study |
| pET-23b | Protein expression vector that includes a carboxy-terminal His6 tag | Novagen |
| pET23-rpoDHis and its derivatives | RpoD expression vector with a carboxy-terminal His6 tag | This study |
| pET-24a | Protein expression vector that includes a carboxy-terminal His6 tag | Novagen |
| pET24-rpoAHis | CTDHis6-tagged RpoA expression vector | This study |
| pET24-rpoB | Untagged RpoB expression vector | This study |
| pET24-rpoC | Untagged RpoC expression vector | This study |
| pOM90 | In vitro transcription template | 54 |
| pOM90-PexsC | In vitro transcription template containing the PexsC promoter | This study |
| pOM90-PexsD | In vitro transcription template containing the PexsD promoter | 64 |
| pSA508-PexsC and its derivatives | PexsC template vector yielding minicircle pMCPexsC | 64; this study |
| pTRCHIS-b | Source of Ptrc promoter | Invitrogen |
| pOM90-Ptrc(250) | In vitro transcription template containing the Ptrc promoter | This study |
| pOM90-Ptrc(180) | In vitro transcription template containing the Ptrc promoter | This study |
| pOM90-PRE# | In vitro transcription template containing the PRE# promoter | This study |
| pET23-rpoD (1-574) | RpoD expression vector lacking region 4.2 | This study |
Plasmid construction and promoter mutagenesis.
Plasmid construction is summarized in Table 2, and the primers used are listed in Table 3.
TABLE 2.
Construction of plasmids used in this study
| Figure | Product | Primer pair | Cloning vector |
|---|---|---|---|
| 1B | p2UY21-LuxR | 44122038-44122037 | p2UY21 |
| 1, 2 | pAH125-PexsC | 39530603-49188917 | pAH125 |
| 2 | pGEX-rpoD (K593A, R596A, R599A) | 48669731-48669730 | pGEX-rpoD |
| 3-6 | RpoDHis | 43812190-43812191 | pET-23b |
| 3 | RpoD (K597A) | 46001014-46001013 | pET23RpoDHis |
| 3 | RpoD (R603A) | 47437714-47437715 | pET23RpoDHis |
| 3 | RpoD (D616A) | 47437713-47437712 | pET23RpoDHis |
| 3 | RpoD (K597A,R603A) | 46001014-46001013 | pET23RpoDHis (R603A) |
| 3 | RpoD(K597A, R600A, R603A) | 48432036-48432035 | pET23RpoDHis (K597A,R603) |
| 3-6 | RpoAHisCTD | 46775590-46775589 | pET-24a |
| 3-6 | RpoB | 46775588-46775587 | pET-24a |
| 3-6 | RpoC | 47100507-46775585 | pET-24a |
| 3 | pOM90-PexsC | 35048925-35048926 | pOM90 |
| 4 | PexsC (G41T) | 43648443-43648442 | pSA508-PexsC |
| 4 | PexsC (T40A) | 48552525-48552524 | pSA508-PexsC |
| 4 | PexsC (G39C) | 48552527-48552526 | pSA508-PexsC |
| 4 | PexsC (A38T) | 43648441-43648440 | pSA508-PexsC |
| 4 | PexsC (C37G) | 43648439-43648438 | pSA508-PexsC |
| 4 | PexsC (A36T) | 48552529-48552528 | pSA508-PexsC |
| 4 | PexsC (A33G) | 43648437-43648436 | pSA508-PexsC |
| 4-6 | PexsC (TG) | 48552531-48552530 | pSA508-PexsC |
| 4-6 | PexsC (T8G) | 43579324-43579323 | pSA508-PexsC |
| 6 | pOM90-Ptrc(250) | 25444818-25444816 | pOM90 |
| 6 | pOM90-Ptrc(179) | 25444818-25444814 | pOM90 |
| 6 | pOM90-PRE# | 48495914-48495913 | pOM90 |
| 6 | RpoD (1-574) | 43812190-48495915 | pET-23b |
TABLE 3.
. Primers used in this study
| Primer identification no. | Sequence |
|---|---|
| 44122038 | CATGGCCATATGAAAAACATAAATGCCGAC |
| 44122037 | CATGGCGAGCTCTTAATTTTTAAAGTATGG |
| 39530603 | GCGACGCGGTACCATGAAGGACGTCCTGCAGCTCATCC |
| 49188917 | TGATGAATTCGCCTCCTAAAGCTCAGCGCATGC |
| 48669731 | CAGATCGAAGCGGCGGCGCTGGCCAAACTGGCTCACCCGAGCCGT |
| 48669730 | ACGGCTCGGGTGAGCCAGTTTGGCCAGCGCCGCCGCTTCGATCTG |
| 43812190 | CCGAGCCATATGTCCGGAAAAGCGCAA |
| 43812191 | GGCAGGAAGCTTCTCGTCGAGGAAGGAGCG |
| 46001014 | CAGATCGAAGCCGCGGCGTTGCGCAAG |
| 46001013 | CTTGCGCAACGCCGCGGCTTCGATCTG |
| 47437714 | TCGCGACGGATGGGCCAGCTTGCGCAA |
| 47437715 | TTGCGCAAGCTGGCCCATCCGTCGCGA |
| 47437713 | CGCTCCTTCCTCGCCGAGAAGCTTGCG |
| 47437712 | CGCAAGCTTCTCGGCGAGGAAGGAGCG |
| 48432036 | GCCGCGGCGTTGGCCAAGCTGGCCCAT |
| 48432035 | ATGGGCCAGCTTGGCCAACGCCGCGGC |
| 46775590 | GCCACCCATATGCAGAGTTCGGTAAATGAGTT |
| 46775589 | GCCTACGCGGCCGCGGCGGCAGTGGCCTTGTCGTCTTTCTTA |
| 46775588 | GCCACCCATATGGCTTACTCATACACTGAGAAAAAACG |
| 46775587 | GCCTACGCGGCCGCGGCTTATTCGGTTTCCAGTTCGATGTCG |
| 47100507 | GCCACCCATATGAAAGACTTGCTTAATCTGTTGAA |
| 46775585 | GCCTACGCGGCCGCGGCTTAGTTACCGCTCGAGTTCAGCGCTT |
| 35048925 | ATACTGGAATTCTGCGGTTCCCCCCC |
| 35048926 | ACGAATGAATTCCCACATCGGCCTCCAGCAAC |
| 43648443 | AAGAAAAGTCTCTCATTGACAAAAGCGATGC |
| 43648442 | GCATCGCTTTTGTCAATGAGAGACTTTTCTT |
| 48552525 | AAAGTCTCTCAGAGACAAAAGCGAG |
| 48552524 | CTCGCTTTTGTCTCTGAGAGACTTT |
| 48552527 | AAGTCTCTCAGTCACAAAAGCGAGG |
| 48552526 | CCTCGCTTTTGTGACTGAGAGACTT |
| 43648441 | AAAAGTCTCTCAGTGTCAAAAGCGATGCATA |
| 43648440 | TATGCATCGCTTTTGACACTGAGAGACTTTT |
| 43648439 | AAAGTCTCTCAGTGAGAAAAGCGATGCATAG |
| 43648438 | CTATGCATCGCTTTTCTCACTGAGAGACTTT |
| 48552529 | TCTCTCAGTGACTAAAGCGAGGCAT |
| 48552528 | ATGCCTCGCTTTAGTCACTGAGAGA |
| 43648437 | TCTCTCAGTGACAAAGGCGATGCATAGCCCG |
| 43648436 | CGGGCTATGCATCGCCTTTGTCACTGAGAGA |
| 48552531 | GGCATAGCCCGGACCTAGCATGCGCT |
| 48552530 | AGCGCATGCTAGGTCCGGGCTATGCC |
| 43579324 | AGCCCGGTGCTAGCAGGCGCTGAGCTTTAGG |
| 43579323 | CCTAAAGCTCAGCGCCTGCTAGCACCGGGCT |
| 25444818 | CTGCGAATTCAACGGTTCTGGCAAATATTC |
| 25444816 | CCGCGAATTCGGTTTATTCCTCCTTATTTAATCG |
| 25444814 | CTATGAATTCGAGTGCCCACACAGATTTC |
| 48495914 | GATCCTCGTTGCGTTTGTTTGCACGAGCTCTATGTTATAATTTCCTAAGCTTG |
| 48495913 | AATTCAAGCTTAGGAAATTATAACATAGAGTCGTGCAAACAAACGCAACGAG |
| 48495915 | GGCAGGAAGCTTCGACTGCATGGTGGAGTC |
The PexsC-lacZ, PexsD-lacZ, and PexoT-lacZ transcriptional reporters were generated by PCR amplification of the promoters and cloning into the KpnI/EcoRI (PexsC/PexsD) or SalI/EcoRI (PexoT) sites of the λ integration plasmid pAH125 (28). The PluxI-lacZ translational fusion reporter was generated by cloning the AatII/EcoRI restriction fragment from plasmid pluxI-lacZ (63) into plasmid pAH125. The resulting plasmids were integrated at the λ attachment site of E. coli strains GS162 and/or GA2071 by an electroporation method as described previously (28).
The constitutive ExsA expression plasmid p2UY21 was created through the following series of subcloning steps. ExsA expression plasmid pEB102 was created by PCR amplifying the exsA gene from P. aeruginosa strain PA103 using NdeI/SacI-containing primers and cloning the resulting fragment into plasmid pUY30 (62). Plasmid p2UY21 was created by cloning the 210-bp ApoI fragment from plasmid pMCTX-PlacUV5mut-lacZ (described below) into the MfeI/EcoRI sites of plasmid pEB102. Plasmid pMCTX-PlacUV5mut-lacZ was created by annealing complementary oligonucleotides (5′-AGCTTAGGCTTATCACTTTATGCTTCCGGCTCGTATAATGTGTG-3′ and 5′-AATTCACACATTATACGAGCCGGAAGCATAAAGTGATAAGCCTA-3′) and cloning the resulting fragment into the HindIII-EcoRI sites of plasmid pMini-CTX-lacZ (5). Constitutive LuxR expression plasmid p2UY21-luxR was created by cloning the NdeI-SacI fragment from pMU102 (63) into plasmid p2UY21. The pGEXrpoD(K593A,R596A,R599A) triple mutant σ70 expression plasmid and PexsC promoter point mutant transcription templates were generated by QuikChange site-directed mutagenesis (Stratagene).
The carboxy-terminal hexahistidine-tagged α subunit expression vector pET24-rpoAHis was created by PCR amplifying the rpoA gene from P. aeruginosa strain PA103 lacking its native stop codon by using NdeI-NotI-containing primers and cloning the resulting fragment into pET-24a (Novagen). The P. aeruginosa β and β′ subunit expression vectors (pET24-rpoB and pET24-rpoC, respectively) were created by PCR amplification of rpoB or rpoC from P. aeruginosa strain PA103 by using NdeI-NotI containing primers and cloning the resulting fragment into pET-24a. The carboxy-terminal hexahistidine-tagged σ70 expression vector (pET23-rpoDHis) was created by PCR amplification of the rpoD gene lacking its native stop codon from P. aeruginosa strain PA103 using primers incorporating NdeI-HindIII restriction sites and cloning the resulting fragment into pET-23b (Novagen). Point mutations in rpoD were introduced by QuikChange site-directed mutagenesis (Stratagene).
Purification of P. aeruginosa RNAP core enzyme, σ70 and holoenzyme reconstitution.
ExsA was purified as previously described under native conditions as an amino-terminal decahistidine-tagged fusion protein (12). Individual P. aeruginosa RNAP subunits were purified as described previously (60) with modifications. E. coli Tuner(DE3) carrying pET24-rpoAHis was grown at 37°C in 50 ml of Luria broth containing 50 μg/ml kanamycin to an OD600 of 0.7, at which time IPTG (isopropyl-β-d-thiogalactopyranoside) (1 mM) was added and the culture was incubated for an additional 3 h at 37°C. Bacteria were harvested by centrifugation and suspended in 4 ml of buffer A (20 mM Tris-HCl [pH 7.9], 500 mM NaCl, and 5 mM imidazole). Cells were lysed via sonication on ice, and unbroken cells were removed by centrifugation (15 min, 16,000 × g, 4°C). Solid ammonium sulfate (60% of saturation) was added, and samples were allowed to precipitate for 15 min at 4°C with agitation. The precipitate was collected by centrifugation (20 min, 16,000 × g, 4°C) and resuspended in 10 ml of buffer B (20 mM Tris-HCl [pH 7.9], 6 M guanidine HCl, and 500 mM NaCl) containing 5 mM imidazole. Prior to Ni2+ affinity chromatography (see below), the material was subjected to ultracentrifugation (30 min, 100,000 × g, 4°C) to remove particulates.
E. coli Tuner(DE3) carrying pET23-rpoDHis was grown at 37°C in 200 ml LB containing 200 μg/ml ampicillin to an OD600 of 0.5, at which time IPTG (1 mM) was added and the culture was incubated for an additional 3 h at 37°C. Bacteria were harvested by centrifugation and suspended in 5 ml buffer B containing 5 mM imidazole. Cells were lysed via sonication on ice, and unbroken cells were removed by centrifugation (15 min, 38,000 × g, 4°C).
The α and σ subunits were denatured and solubilized with guanidine as described above and purified under denaturing conditions by Ni2+ affinity chromatography. Lysates were applied to a 1-ml HisTrap column (GE Healthcare) previously equilibrated with buffer B containing 5 mM imidazole, washed with 10 ml buffer B containing 30 mM imidazole, and developed with a 10-ml linear imidazole gradient (30 to 500 mM) in buffer B. The elution peaks were established by SDS-PAGE. The purified α subunit was stored on ice for immediate use in core RNAP reconstitution. Purified σ70 was dialyzed overnight against buffer E (50 mM Tris-HCl [pH 7.9], 200 mM KCl, 10 mM MgCl2, 10 μM ZnCl2, 1 mM EDTA, 5 mM 2-mercaptoethanol, and 20% [vol/vol] glycerol) at 4°C, subjected to ultracentrifugation (30 min, 100,000 × g, 4°C), and stored in 50% glycerol at −20°C.
The β and β′ RNAP subunits were purified from E. coli inclusion bodies. E. coli Tuner(DE3) carrying either pET24-rpoB or pET24-rpoC was grown at 37°C in 1 liter of LB containing 50 μg/ml kanamycin to an OD600 of 0.5, at which time IPTG (1 mM) was added and the culture was incubated for an additional 3 h at 37°C. Bacteria were harvested by centrifugation and suspended in 16 ml of buffer C (40 mM Tris-HCl [pH 7.9], 300 mM KCl, 10 mM EDTA, 1 mM dithiothreitol [DTT], and 1× protease inhibitor cocktail [Roche]) containing 0.2 mg/ml lysozyme and 0.2% (wt/vol) sodium deoxycholate. The bacteria were incubated on ice for 20 min and lysed by sonication. Inclusion bodies were collected by centrifugation (30 min, 38,000 × g, 4°C) and washed with 16 ml buffer C containing 0.2% n-octyl-β-d-glucoside. Inclusion bodies were sonicated and centrifuged as described above, followed by a final wash with 16 ml buffer C. Washed inclusion bodies were solubilized in 2 ml of buffer D (50 mM Tris-HCl [pH 7.9], 6 M guanidine-HCl, 10 mM MgCl2, 10 μM ZnCl2, 1 mM EDTA, 10 mM DTT, and 10% [vol/vol] glycerol) and incubated at 25°C for 10 min. The resulting material was subjected to ultracentrifugation (30 min, 100,000 × g, 4°C), and the soluble fraction was stored on ice for immediate use in core enzyme reconstitution.
RNAP core enzyme was reconstituted by mixing 0.3 mg purified α subunit, 1.5 mg purified β subunit, and 3 mg β′ subunit in buffer D (2 ml) and dialyzing twice against 500 ml of buffer E at 4°C with constant stirring. The resulting material was subjected to ultracentrifugation (30 min, 100,000 × g, 4°C), and the soluble fraction was applied to a 1-ml HiTrap heparin HP column (GE Healthcare) equilibrated with buffer E. The column was washed with 10 ml buffer E containing 0.4 M KCl and developed with a 10-ml linear KCl gradient (0.4 to 2 M) in buffer E. The elution peaks were analyzed by SDS-PAGE, and pure fractions containing stoichiometric core RNAP (α2ββ′) were dialyzed against 1 liter buffer E containing 50% glycerol and stored at −20°C.
RNAP holoenzyme was reconstituted by mixing core RNAP (500 nM) and σ70 (1 μM) in 35 μl 1× transcription buffer (40 mM Tris-HCl [pH 7.5], 50 mM KCl, 10 mM MgCl2, 1 mM DTT, 0.1% Tween 20, and 0.5 mg/ml bovine serum albumin [BSA]) for 30 min at 25°C. The resulting holoenzyme (1 μl) was then used in a 20-μl transcription reaction mixture.
In vitro transcription assays.
Supercoiled transcription templates containing the PexsC and PexsD promoters were described previously (14, 64). The pOM90-PexsC template was generated by PCR amplifying the PexsC promoter (nucleotides [nt] −207 to +192 relative to the transcriptional start site) and cloning as an EcoRI fragment into pOM90 (54). The resulting template contains a fusion of the PexsC promoter to the rpoC transcriptional terminator (rpoCter) on pOM90 and directs synthesis of a 261-nt transcript. The pOM90-Ptrc180 and pOM90-Ptrc250 templates were generated by PCR amplifying the Ptrc promoter (nucleotides −61 to +109/179 relative to the transcriptional start site) from pTRCHIS-b (Invitrogen) and cloning as an EcoRI fragment into pOM90. The pOM90-Ptrc180 and pOM90-Ptrc250 templates fuse the Ptrc promoter to rpoCter, resulting in 180- and 250-base transcripts, respectively. Finally, the pOM90-PRE# template was generated by annealing complementary oligonucleotides, and the resulting BamHI-EcoRI fragment was cloned into pOM90. The pOM90-PRE# template fuses the synthetic PRE# promoter to the rrnB T1 terminator and results in a 135-base transcript.
Single-round transcription assays (20-μl final volume) were performed by incubating ExsAHis (35 nM) with transcription templates (2 nM) at 25°C in 1× transcription buffer (40 mM Tris-HCl [pH 7.5], 50 mM KCl, 10 mM MgCl2, 1 mM DTT, 0.1% Tween 20, and 0.5 mg/ml BSA) containing the initiating nucleotides ATP and GTP (0.75 mM). After 10 min, 25 nM reconstituted P. aeruginosa RNAP holoenzyme was added, and open complexes were allowed to form for 1 min at 25°C in the presence of ExsAHis or for 20 min at 25°C in the absence of ExsAHis. Elongation was allowed to proceed by the addition of the remaining nucleotides (0.25 mM ATP/GTP/CTP, 0.75 mM UTP, and 2.5 μCi [α-32P]CTP) in 1× transcription buffer containing heparin (final concentration, 50 μg/ml). Reactions were stopped after 5 min at 25°C by the addition of 20 μl stop buffer (98% formamide, 20 mM EDTA, 0.05% bromophenol blue, and 0.05% xylene cyanol). Samples were heated at 95°C for 5 min and immediately incubated on ice before electrophoresis on 5% denaturing polyacrylamide gels.
RESULTS
The carboxy-terminal domain of the RNAP α subunit is not required for ExsA-dependent transcriptional activation.
Since ExsA activates transcription primarily through recruitment of RNAP (64) and many transcriptional activators that recruit do so by contacting the carboxy-terminal domain of the RNAP α subunit (α-CTD), we tested the hypothesis that ExsA uses a similar mechanism. Previous studies have shown that ExsA activates transcription in vitro to similar levels using RNAP from either P. aeruginosa or E. coli (64). To demonstrate that ExsA can activate transcription from the PexsC, PexsD, and PexoT promoters in E. coli, ExsA was expressed from a plasmid (p2UY21-exsA) under the transcriptional control of a constitutive α-CTD-independent promoter. ExsA-dependent transcription was measured from transcriptional reporters consisting of ExsA-dependent promoters (PexsC, PexsD, and PexoT) fused to lacZ and integrated at the E. coli λ phage attachment site. Significant ExsA-dependent activation of all three promoters was observed relative to a control plasmid (Fig. 1 A), demonstrating that ExsA is sufficient to activate transcription from PexsC, PexsD, and PexoT, as was previously shown for PexsC in E. coli (61).
FIG. 1.
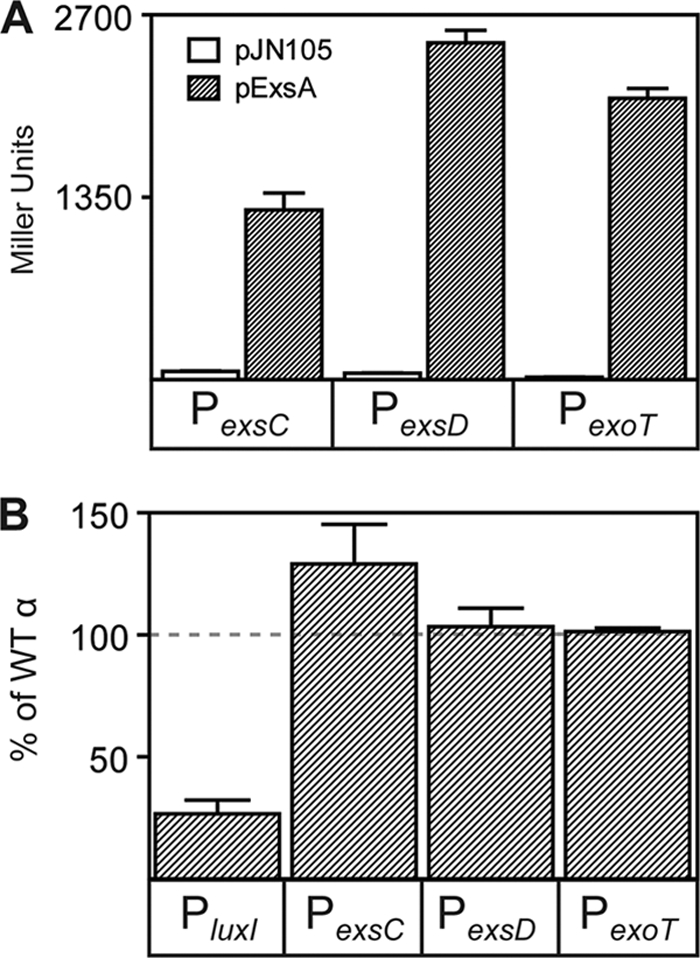
The RNAP α-CTD is not required for ExsA-dependent activation of transcription. (A) E. coli strain GS162 carrying the indicated transcriptional reporters (PexsC-lacZ, PexsD-lacZ, or PexoT-lacZ) was transformed with a vector control (pJN105) or a constitutive ExsA expression plasmid (p2UY21, labeled pExsA in the figure). The resulting strains were grown in LB to an OD600 of 0.6 and assayed for β-galactosidase activity (reported in Miller units). (B) E. coli GS162 carrying a PluxI-lacZ reporter and a LuxR expression plasmid (p2UY21-luxR) and the reporter strains from panel A were transformed with a plasmid expressing the native α or αΔCTD subunit. The resulting strains were grown in LB to an OD600 of 0.6 and assayed for β-galactosidase activity. The reporter activities obtained in cells expressing αΔCTD were normalized to the same strain expressing native (WT) α and reported as the percentage of native activity. The results represent the averages for three independent experiments, and error bars represent the standard errors of the means.
To determine the role of the α-CTD, we used an established E. coli assay in which the native α subunit (α-wt) or α lacking the C-terminal 239 amino acids (α-ΔCTD) was expressed from a plasmid such that its cellular concentration exceeded that of native α subunit expressed from the chromosome. This approach was necessary because deletion of the α subunit CTD is lethal in E. coli (37). ExsA-dependent transcription following overexpression of α-ΔCTD was plotted as a percentage of the activation observed with overexpressed α-wt. As a control, we also measured LuxR-dependent activation of a PluxI-lacZ transcriptional fusion (1). LuxR is an activator known to require the α-CTD (59). ExsA-dependent activation of the PexsC-lacZ, PexsD-lacZ, and PexoT-lacZ reporters in the presence of α-ΔCTD was ≥100% of that seen with α-wt, indicating that ExsA does not require the α-CTD for transcriptional activation (Fig. 1B). Curiously, activation from the PexsC promoter in the presence of α-ΔCTD was 125% of the wild-type value, suggesting that the α-CTD might have an inhibitory function at this promoter. In contrast, activation of the PluxI-lacZ reporter was reduced to ∼33% of the wild-type value in the presence of α-ΔCTD.
ExsA-dependent transcription in E. coli is dependent on specific amino acids within region 2 of σ70 domain 4.
A number of class II transcriptional activators interact with a basic amino acid region (region 2) of σ70 domain 4. Since the ExsA-binding sites overlap a near-consensus −35 RNAP recognition hexamer, we hypothesized that ExsA recruits RNAP through an interaction with region 4.2 of the σ70 subunit. Lonetto et al. generated an rpoD plasmid expression library containing alanine point mutations in 16 nonessential positions of region 4.2 to test for activator-specific defects in gene expression (40). Those experiments were performed in E. coli strain GA2071 where expression of chromosomal rpoD is tightly repressed. To measure ExsA activity, the PexsC-lacZ and PexsD-lacZ transcriptional reporters were introduced at the λ phage attachment site of E. coli strain GA2071, and the resulting strains were transformed with a plasmid expressing wild-type RpoD or the RpoD point mutants. Given the tendency for reversion of rpoD point mutants to the native sequence (40), the integrity of each expression plasmid was verified by nucleotide sequence analysis after introduction into strain GA2071. ExsA was constitutively expressed from plasmid p2UY21. Since the RpoD mutants in this library do not affect activator-independent transcription, ExsA expression levels were similar in each of the tested strains (Fig. 2A) (40). ExsA-dependent expression from the PexsC and PexsD reporters in the presence of RpoD mutants was plotted relative to that in the presence of wild-type RpoD (Fig. 2B and C). The most drastic effect on ExsA-dependent activation of the PexsC-lacZ reporter resulted with the K593A, R596A, and R599A substitutions, which exhibited 50%, 29%, and 25% of the activity seen with native RpoD, respectively (Fig. 2B). Similarly, PexsD-lacZ reporter activity was also impaired by the K593A, R596A, and R599A substitutions to 42%, 67%, and 36% of the native RpoD levels (Fig. 2C).
FIG. 2.
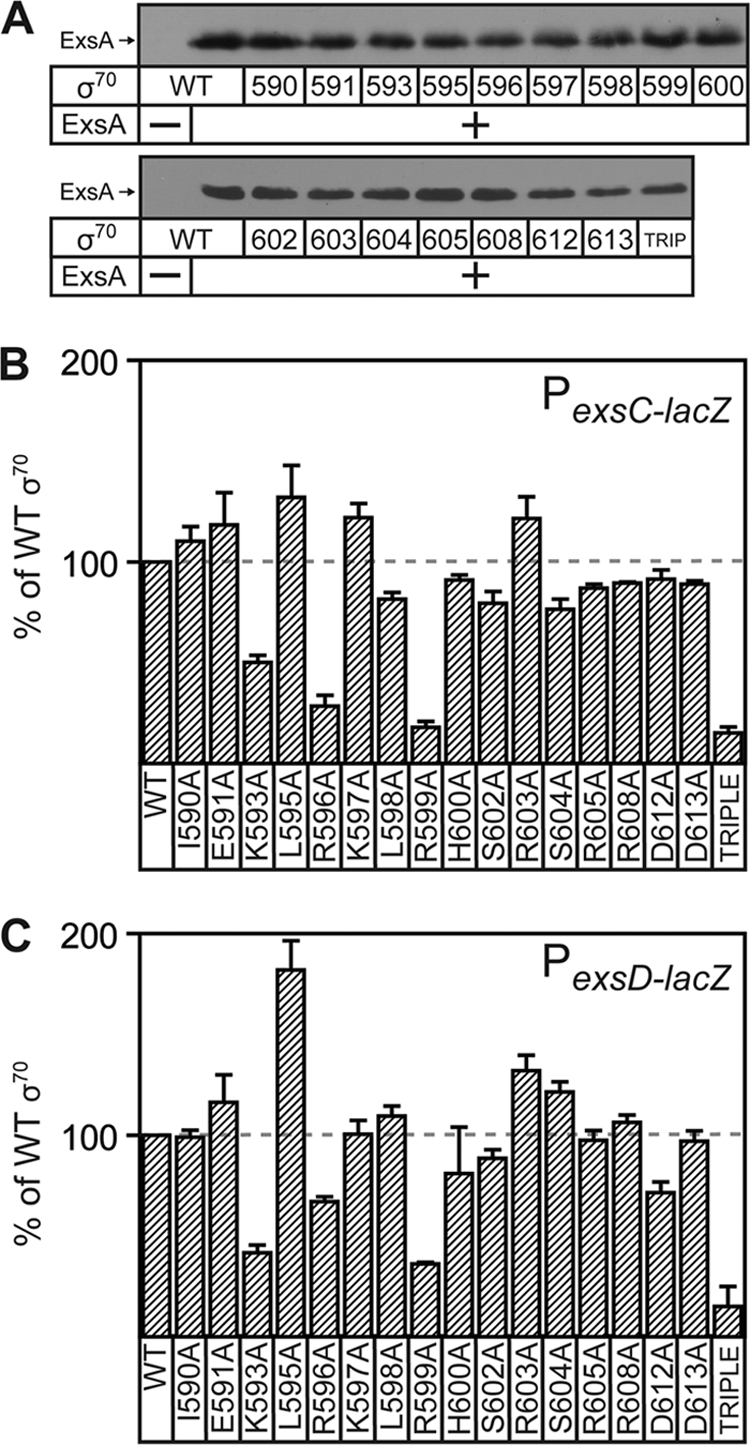
ExsA-dependent transcription requires several amino acids in region 4.2 of E. coli σ70. (A) ExsA immunoblots demonstrating that steady-state expression levels are similar in each of the strains used below. (B and C) E. coli strain GA2071 (tightly suppressed for native σ70 expression) carrying the PexsC-lacZ (B) or PexsD-lacZ (C) transcriptional reporter and the p2UY21 ExsA expression plasmid was transformed with a wild-type σ70 expression plasmid or an σ70 expression plasmid carrying the indicated mutations in region 4.2. The resulting strains were grown in LB to an OD600 of 0.6 and assayed for β-galactosidase activity. The reported values (percentage of activity in the presence of wild-type σ70 subunit) are the averages from three independent experiments, and error bars represent the standard errors of the means.
The effects observed from the single amino acid substitutions were modest (2- to 3-fold) and likely reflect the fact that each of the individual positions represents only a portion of the ExsA-σ70 interaction site. We predicted that ExsA-dependent transcription might result from synergistic interactions with each of the three amino acid positions. This proved to be true, as the activity of the PexsC and PexsD reporters in the presence of a triple RpoD mutant (K593A, R596A, R599A) was only 15% of the wild-type activity in both cases (Fig. 2B and C). We did note that strain GA2071 expressing the RpoD triple mutant exhibited a 2-fold growth defect yet had ExsA levels equivalent to those of GA2071 expressing native RpoD (Fig. 2A).
ExsA-dependent transcription in vitro is dependent on P. aeruginosa σ70 region 4.2.
To further characterize the role of σ70 region 4.2, the mutations from E. coli rpoD (K593A, R596A, and R599A) that affect ExsA-dependent transcription in vivo were introduced into P. aeruginosa rpoD. Native and mutant forms of P. aeruginosa RpoD were expressed in E. coli and purified under denaturing conditions by Ni2+ affinity chromatography. Core RNAP was generated by expressing the P. aeruginosa α, β, and β′ subunits in E. coli, purifying the individual purified components (Fig. 3A), and reconstituting σ-saturated RNAP holoenzyme with either native RpoD, RpoD-K593A, RpoD-R596A, RpoD-R599A, or the triple RpoD mutant. RNAP holoenzyme activity was normalized between the different RpoD-reconstituted polymerases by comparing the production of single-round in vitro transcription products from the Ptrc promoter. Transcription from the Ptrc promoter was not affected by the K593A, K596A, or K599A mutation in region 4.2 of σ70 (40).
FIG. 3.

ExsA-dependent in vitro transcription is dependent on region 4.2 of σ70 from P. aeruginosa. (A) Silver-stained SDS-polyacrylamide gel of purified and reconstituted core polymerase subunits α, β, and β′ (lane 1), native σ70 (lane 2); σ70 carrying the K597A, R596A, and R599A amino acid substitutions (lane 3); and σ70 lacking region 4.2 (lane 4). (B and C) Single-round in vitro transcription assays. ExsAHis (35 nM) was incubated with 2 nM supercoiled PexsC or PexsD promoter template (pOM90-PexsC or pOM90-PexsD) at 25°C in the presence of rATP and rGTP. After 10 min, P. aeruginosa core RNAP, σ70-RNAP, or σ70 (K597A/R596A/R599A)-RNAP was added (25 nM each; the activity of σ-saturated enzymes was normalized with Ptrc), and the reaction mixture was incubated for 1 min at 25°C. Heparin and substrate nucleotides (including 2.5 μCi [α-32P]CTP) were immediately added, and the reaction mixture was incubated for 5 min at 25°C. Reactions were terminated, and the resulting products were electrophoresed on a 5% denaturing polyacrylamide-urea gel and subjected to phosphorimaging. The ExsA-dependent terminated transcripts (261 nt) from the PexsC or PexsD promoter and the runoff transcripts (250 or 180 nt) from the Ptrc promoter are indicated.
Reconstituted RNAP holoenzymes were then assayed for ExsA-dependent transcription in vitro using supercoiled plasmid templates containing the PexsC and PexsD promoters fused to the rpoCter terminator. Each of the templates generates a 261-nucleotide, terminated transcript. As expected, terminated transcripts were not observed with core RNAP alone. We initially tested the individual σ70 mutants (K597A, R600A, and R603A) for ExsA-dependent activation of the PexsC or PexsD promoters but found that none had an activation defect greater than 50% of native RpoD (data not shown). This result was not surprising given that a similar observation was made when testing the individual σ70 mutants for activation in E. coli (Fig. 2B and C). In contrast, the triple RpoD mutant produced far less exsC and exsD transcripts than did native RpoD (Fig. 3B and C). These combined data indicate that σ70 region 4.2 is required for ExsA-dependent activation of the PexsC and PexsD promoters both in vivo and in vitro.
The near-consensus −35 sequence at the PexsC promoter is not required for ExsA-independent transcription.
We previously demonstrated that the PexsC promoter has low basal activity in the absence of ExsA (64). To determine whether the putative −35 sequence is required for ExsA-independent promoter activity, we generated PexsC transcription templates containing point mutations at each of the −35 nucleotide positions. Each of the nucleotide substitutions, with the exception of G41T, was divergent from the σ70 consensus (Fig. 4A). The mutant promoters were assayed for ExsA-independent transcript levels and compared to the native PexsC promoter and to a negative control containing a single point mutation (T8G) in the established −10 Pribnow box (Fig. 4A). To account for subtle differences in template concentration and purity, the PexsC transcripts were normalized to a constitutive transcript generated from a promoter located on the plasmid backbone (64). Whereas the negative control (T8G) lacking a functional −10 hexamer exhibited a 50-fold decrease in transcription compared to PexsC (Fig. 4), the remaining point mutants had little (less than 2-fold) or no effect on ExsA-independent transcription (Fig. 4B and C). These data indicate that the putative −35 hexamer is not important for ExsA-independent transcription at the PexsC promoter.
FIG. 4.

The near-consensus −35 hexamer in the PexsC promoter is not required for ExsA-independent transcription. (A) Diagram showing the mutant PexsC promoter derivatives used in this experiment. The −35, extended −10, and −10 elements are boxed, and the individual point mutations are in bold. (B) Single-round in vitro transcription assays showing ExsA-independent transcription from PexsC derivatives containing −35 (G41T, T40A, G39C, A38T, C37G, A36T, and A33G), extended −10 (TG), and −10 (T8G) point mutations. Reactions were performed as described in the legend to Fig. 3, except open complexes were allowed to form for 20 min in the absence of ExsA. (C) Quantification of the in vitro transcription data shown in panel B. The amount of exsC transcript produced in each experiment was normalized to an ExsA-independent transcript (64) produced from a weak promoter on the minicircle backbone. The reported values are the averages from three independent experiments, and error bars represent the standard errors of the means.
Although a near-consensus, but improperly spaced, −35 sequence is present at the PexsC promoter, it is possible that a weak, unrecognizable −35 hexamer with a poor match to the σ70 consensus is present and optimally spaced (16/17 bp) from the −10 hexamer. Potential −35 hexamers spaced at either 16 or 17 bp would have the sequence AAAGCG or AAAAGC, respectively (matches to consensus are underlined). To test this hypothesis, we constructed a single point mutation in the PexsC promoter (A33G) such that the potential −35 hexamer spaced 16 bp (AAGGCG) from the −10 hexamer more closely resembles the −35 consensus sequence and the potential −35 hexamer spaced at 17 bp (AAAGGC) would be a weaker match to the consensus. The A33G mutation had no significant effect (<2-fold) compared to native PexsC. These combined data suggest that the putative −35 sequence is not important for ExsA-independent transcription.
The PexsC promoter sequence located immediately upstream of the −10 box resembles an extended −10 promoter (Fig. 4A). Extended −10 promoters contain the sequence TGxTATAAT and can function in either the presence or absence of a −35 hexamer (4, 44). To determine whether the PexsC promoter contains an extended −10 element, we mutated the consensus TG sequence to AC (here referred to as PexsC-TG). As expected, the mutant PexsC-TG promoter had a significant reduction in ExsA-independent transcription (5-fold) compared to the native PexsC promoter (Fig. 4B and C). These combined data suggest that the PexsC promoter lacks a −35 hexamer and that an extended −10 element may provide basal promoter activity.
The extended −10 element is important for ExsA-independent and -dependent PexsC promoter activity.
Since ExsA-independent activity of the PexsC promoter requires an apparent extended −10 sequence, we asked whether ExsA-dependent activation had a similar requirement using in vitro transcription assays. PexsC-TG promoter activity was reduced 3-fold in the presence of ExsA, demonstrating that the extended −10 element affects PexsC to similar extents in the presence and absence of ExsA (Fig. 5A and B). In contrast, the T8G mutation ablates both ExsA-dependent and -independent promoter activity. Note that ExsA-independent transcripts were not observed under these conditions due to the short RNAP incubation time (1 min) required to detect ExsA-dependent open complex formation in the linear range (Fig. 5A and data not shown). To rule out the trivial explanation that the DNA-binding activity of ExsA is affected by the TG mutation, we employed electrophoretic mobility shift assays (EMSAs) and found no significant difference in the binding affinity of ExsA for the PexsC-TG and native PexsC promoters or in formation of shift complexes 1 and 2 (Fig. 5C).
FIG. 5.

The extended −10 element within the PexsC promoter is important for overall promoter activity independent of ExsA function. (A and B) Single-round in vitro transcription assays and quantification of the corresponding transcripts from the PexsC, PexsC-TG, and PexsCT8G promoters. Experiments were performed as described in the legend to Fig. 3, allowing 1 min for open complex formation in both the absence and presence of ExsA. The reported values (arbitrary densitometry units) are the averages from three independent experiments, and error bars represent the standard errors of the mean. (C) Electrophoretic mobility shift assays (EMSAs) of the PexsC and PexsC-TG promoter probes. Specific (SP) and nonspecific (Non-SP) probes (0.25 nM each) were incubated in the absence of ExsAHis (−) or with increasing concentrations of ExsAHis (1 to 36 nM; 2-fold dilutions) for 15 min, followed by electrophoresis and phosphorimaging. ExsAHis-dependent shift products 1 and 2 are indicated.
Region 4.2 of σ70 is required for ExsA-dependent but not ExsA-independent transcription.
Region 4.2 of σ70 recognizes the −35 hexamer and is essential for recognition of most bacterial promoters (15). Region 4.2 is also a common target for AraC family transcriptional activators. We have provided evidence that ExsA interacts with this region and that the putative −35 sequence is not a determinant for RNAP recruitment at the PexsC promoter. Based on these data, we hypothesized that the PexsC extended −10 element compensates for the lack of a functional −35 hexamer. To test this idea, we employed in vitro transcription assays utilizing RNAP holoenzyme reconstituted with σ70 lacking the carboxy-terminal 43 amino acids, σ70Δ4.2, which encompasses region 4.2. Whereas deletion of region 4.2 renders promoters that are dependent upon −35 hexamers nonfunctional, the same deletion has little effect on transcription initiation and elongation from extended −10 promoters (38). The following promoters were used as controls for this experiment: (i) Ptrc, which contains a strong −35 hexamer and requires region 4.2 of σ70, and (ii) PRE#, a synthetic promoter which lacks a −35 hexamer and does not require region 4.2 but is dependent upon an extended −10 element (10, 38) (Fig. 6A). Although σ70Δ4.2 has slightly reduced affinity for core RNAP enzyme (38), holoenzyme reconstituted with σ70Δ4.2 (here referred to as RNAP-σΔ4.2) and native RNAP holoenzyme generated similar levels of transcript from the PRE# promoter (Fig. 6B). In contrast, RNAP-σ70Δ4.2 generated significantly less transcript from the Ptrc promoter than did RNAP-σ70 (Fig. 6B). Consistent with our hypothesis that the PexsC promoter lacks a functional −35 hexamer, RNAP-σ70Δ4.2 and RNAP-σ70 generated similar levels of PexsC transcript in the absence of ExsA (Fig. 6B). In addition, the PexsC-TG and PexsCT8G mutants were essentially devoid of RNAP-σ70Δ4.2-dependent activity. Finally, we tested whether ExsA-dependent transcripts were produced from the PexsC promoter using RNAP-σ70Δ4.2. Although ExsA-dependent transcription was drastically reduced with RNAP-σ70Δ4.2, a detectable transcript was made when reactions were allowed to proceed for 1 min for open complex formation. These same conditions do not support the detection of transcription in the absence of ExsA using native RNAP-σ70 holoenzyme (Fig. 5A). It is unclear whether the weak ExsA-dependent transcription in the absence of region 4.2 represents additional contacts between ExsA and σ70 outside region 4.2 or additional contacts between ExsA and other RNAP subunits.
FIG. 6.

Region 4.2 of σ70 is not required for ExsA-independent transcription of the PexsC promoter. (A) Diagram of transcription templates used in this experiment. The −35 regions (underlined), extended −10 elements (boxed), −10 elements (boxed), and point mutations (bold) are indicated. (B) Single-round in vitro transcription assays were performed with σ70 and σ70Δ4.2 reconstituted RNAP holoenzymes normalized for specific activity using the PRE# extended −10 promoter (lanes 3 and 4). Reactions were performed as described in the legend to Fig. 3, and open complexes were allowed to form for 1 min (lanes 1 to 4, 9, and 10) or 20 min (lanes 5 to 8) as indicated.
DISCUSSION
In the present study we find that recruitment of RNAP by ExsA does not require the CTD of the RNAP α subunit, a common target for AraC family regulators. Although these studies were performed with E. coli we believe the findings would be identical for P. aeruginosa. Data supporting this claim include the following: (i) ExsA activates transcription from T3SS promoters in vitro to similar extents with RNAP (normalized for specific activity using an α-CTD-independent promoter) from either E. coli or P. aeruginosa (64). (ii) the carboxy-terminal 90 amino acids of the α subunits from E. coli and P. aeruginosa share 86% identity, and (iii) heterologous activators known to require the α-CTD, including LuxR from Vibrio fischeri (used in this study), can efficiently activate E. coli RNAP (59). For these reasons, we believe that the involvement of the α-CTD in ExsA-dependent activation would have been detected in our experiments.
Interestingly, ExsA-dependent transcription from the PexsC promoter was slightly elevated (125%) following expression of α-ΔCTD compared to the full-length α subunit (Fig. 1B). A possible explanation for this finding is that the α-CTD may bind the PexsC promoter and antagonize ExsA function. In this scenario, the α-CTD-PexsC promoter interaction might sterically hinder the DNA-binding activity of ExsA or its ability to contact RNA polymerase. We did not test whether ExsA interacts with the amino-terminal domain (NTD), since Egan et al. have shown that an extremely diverse group of AraC family members do not require this domain for transcriptional activation (23).
Using a plasmid-based mutant rpoD expression library, we found that ExsA requires the K593, R596, and R599 amino acids of σ70 for full activation of the PexsC and PexsD promoters (Fig. 2). These specific residues are some of the most frequently observed contact points for AraC family members and unrelated transcriptional regulators (20, 40). In fact, an alignment of ExsA with the AraC family members RhaS and MelR reveals a conserved aspartate residue known to interact with R599 of σ70 (27, 66). Whether this aspartate or other conserved positions are important for the interaction of ExsA with σ70 will be the subject of future studies. Although ExsA-dependent activation defects of greater than 2-fold were not routinely observed with a single point mutation in rpoD, expression of the chromosomal rpoD gene is only suppressed in these experiments, and leaky expression of rpoD may result in higher levels of ExsA-dependent activation that would bias the data toward transcriptional activation defects smaller than those observed. Furthermore, the literature suggests that RNAP-activator interaction regions most likely consist of several amino acid contacts (40). Consistent with this, we find that the σ70 triple mutant (K593A, R596A, R599A) showed a cumulative 6-fold effect on ExsA-dependent transcription in vitro and in vivo (Fig. 2 and 3). It is possible that ExsA may interact with amino acids in region 4.2 that we did not test, other regions in σ70, and/or different RNAP subunits. The 16 amino acids in the mutant rpoD expression library were selected because these positions are reported to have little effect on activator-independent transcription (40). Some amino acids in region 4.2 were omitted from this library because alanine substitution resulted in unstable protein or because they are required for interaction with the −35 hexamer (40). It is therefore possible that other amino acids are also important for the interaction with ExsA. Finally, the finding that a σ70 derivative lacking region 4.2 (σΔ4.2) is still capable of weak ExsA-dependent activation supports the hypothesis that ExsA interacts with several regions of σ70 and/or multiple RNAP subunits (Fig. 6B).
Although the mechanism of transcription activation is known for only a small number of AraC family activators, most activate transcription by facilitating both closed and open complex formation (11). Activators that facilitate both closed and open complex formation do so by contacting the α-CTD and σ70 region 4.2, respectively (11). It is therefore curious that ExsA requires region 4.2 of σ70 and functions primarily to recruit RNAP. A possible explanation for this finding is that the ExsA-σ70 region 4.2 interaction affects the rate of isomerization to an open complex. This explanation seems unlikely, however, as disruption of the ExsA-σ70 region 4.2 interaction results in at least a 5-fold defect in activation, whereas ExsA is known to only marginally affect (2-fold) the rate of isomerization to an open complex. We believe a more likely explanation is that ExsA interaction with σ70 region 4.2 results primarily in the recruitment of RNAP. This is in contrast to the reported activity of the well-characterized cI protein of phage lambda, which increases the isomerization rate at the PRM promoter by contacting σ70 region 4.2 (21, 30). The cI example is somewhat paradoxical, since it has been well established that in the absence of a transcriptional activator, σ70 region 4.2 normally interacts directly with DNA at the −35 position to facilitate the initial binding of RNAP to the promoter (15). In fact, the observation that ExsA recruits RNAP through contacts with σ70 region 4.2 seems to better support the known function of region 4.2. We believe the most likely explanation for these discrepancies is that protein-protein interactions with σ70 region 4.2 can affect both closed and open complex formation. In support of this claim, a single point mutation (R596H) in σ70 region 4.2 changes the mechanism of cI activation to an enhancement of closed complex formation while having almost no effect on the rate of isomerization to an open complex (21). This finding may indicate that the specific contacts between transcriptional activators and σ70 region 4.2 do not determine whether closed or open complex formation is enhanced. In fact, Dove et al. have suggested that the promoter sequence and location of the activator-binding site may play the most important part in determining the mechanism of transcriptional activation by an activator (21). Further studies analyzing the structure of activator-RNAP complexes are needed to address this curious observation.
We provide evidence that the putative −35 hexamer in the PexsC promoter is not sufficient for ExsA-independent expression. This is consistent with a previous study demonstrating that the −35 hexamer from PexsD, although a close match to the σ70 consensus, is also not used as an RNAP recognition site (12, 64). To further characterize the role of the PexsC −35 region, we generated point mutations at every position in the −35 site, and the resulting mutations had no significant effect (<2-fold) on ExsA-independent transcription, while control mutations in the −10 hexamer resulted in undetectable levels of transcript (Fig. 4). An explanation for this result is that an authentic −35 hexamer is located at a more favorable position (16 or 17 bp relative to the −10 sequence) but has few matches to the consensus sequence. We tested this hypothesis by creating a single point mutation in PexsC (A33G), which should significantly increase or decrease ExsA-independent activation if the −35 hexamer is positioned 16 or 17 bp from the −10 hexamer, respectively (46). No significant effect was observed with this mutant, suggesting that a −35 hexamer is not required for ExsA-independent transcription at the PexsC promoter. Unfortunately, we were unable to assess the role of the −35 hexamer with respect to ExsA-dependent transcription, since mutations in the −35 region are known to disrupt ExsA binding to site 1 (12).
Consistent with the hypothesis that a −35 hexamer is not required for ExsA-independent transcription from the PexsC promoter, we identified a putative extended −10 element (38). A point mutation within this element resulted in a significant reduction in both ExsA-dependent and ExsA-independent transcription (Fig. 4 and 5). EMSA experiments demonstrated that the extended −10 mutation had no effect on ExsA binding to the promoter (Fig. 5C). These data indicate that the PexsC promoter contains an extended −10 promoter that might partially compensate for the lack of a functional −35 hexamer. Since exsA expression is autoregulated through the PexsC promoter, it is tempting to speculate that the extended −10 element is important in maintaining a basal level of the exsCEBA transcript. The fact that the extended −10 element is required for maximal PexsC promoter activity, however, prevented us from directly testing this hypothesis. Nevertheless, 5′ RACE promoter-mapping experiments demonstrate that exsCEBA transcript is detectable in an exsA mutant (64), suggesting that PexsC exhibits some level of basal activity. We propose a model in which ExsA recruits RNA polymerase to an extended −10 promoter (PexsC) by contacting σ70 region 4.2. Interestingly, the residual transcription from PexsC seen with σ70Δ4.2-RNAP was shown to be ExsA dependent (compare Fig. 5A and 6B), further suggesting that an additional region of σ70 or perhaps an RNAP subunit other than α and σ70 may be involved in ExsA-dependent transcriptional activation, as has been suggested for other AraC regulators (6, 23, 32, 35).
Acknowledgments
This study was supported by the National Institutes of Health (grant RO1-AI055042-07).
Footnotes
Published ahead of print on 7 May 2010.
REFERENCES
- 1.Antunes, L. C., A. L. Schaefer, R. B. Ferreira, N. Qin, A. M. Stevens, E. G. Ruby, and E. P. Greenberg. 2007. Transcriptome analysis of the Vibrio fischeri LuxR-LuxI regulon. J. Bacteriol. 189:8387-8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Apodaca, G., M. Bomsel, R. Lindstedt, J. Engel, D. Frank, K. E. Mostov, and J. Wiener-Kronish. 1995. Characterization of Pseudomonas aeruginosa-induced MDCK cell injury: glycosylation-defective host cells are resistant to bacterial killing. Infect. Immun. 63:1541-1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barbieri, J. T., and J. Sun. 2004. Pseudomonas aeruginosa ExoS and ExoT. Rev. Physiol. Biochem. Pharmacol. 152:79-92. [DOI] [PubMed] [Google Scholar]
- 4.Barne, K. A., J. A. Bown, S. J. Busby, and S. D. Minchin. 1997. Region 2.5 of the Escherichia coli RNA polymerase sigma70 subunit is responsible for the recognition of the extended −10′ motif at promoters. EMBO J. 16:4034-4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Becher, A., and H. P. Schweizer. 2000. Integration-proficient Pseudomonas aeruginosa vectors for isolation of single-copy chromosomal lacZ and lux gene fusions. Biotechniques 29:948-950, 952. [DOI] [PubMed] [Google Scholar]
- 6.Bhende, P. M., and S. M. Egan. 1999. Amino acid-DNA contacts by RhaS: an AraC family transcription activator. J. Bacteriol. 181:5185-5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhende, P. M., and S. M. Egan. 2000. Genetic evidence that transcription activation by RhaS involves specific amino acid contacts with sigma 70. J. Bacteriol. 182:4959-4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bird, T. H., J. K. Grimsley, J. A. Hoch, and G. B. Spiegelman. 1996. The Bacillus subtilis response regulator Spo0A stimulates transcription of the spoIIG operon through modification of RNA polymerase promoter complexes. J. Mol. Biol. 256:436-448. [DOI] [PubMed] [Google Scholar]
- 9.Blatter, E. E., W. Ross, H. Tang, R. L. Gourse, and R. H. Ebright. 1994. Domain organization of RNA polymerase alpha subunit: C-terminal 85 amino acids constitute a domain capable of dimerization and DNA binding. Cell 78:889-896. [DOI] [PubMed] [Google Scholar]
- 10.Brosius, J., M. Erfle, and J. Storella. 1985. Spacing of the −10 and −35 regions in the tac promoter. Effect on its in vivo activity. J. Biol. Chem. 260:3539-3541. [PubMed] [Google Scholar]
- 11.Browning, D. F., and S. J. Busby. 2004. The regulation of bacterial transcription initiation. Nat. Rev. Microbiol. 2:57-65. [DOI] [PubMed] [Google Scholar]
- 12.Brutinel, E. D., C. A. Vakulskas, K. M. Brady, and T. L. Yahr. 2008. Characterization of ExsA and of ExsA-dependent promoters required for expression of the Pseudomonas aeruginosa type III secretion system. Mol. Microbiol. 68:657-671. [DOI] [PubMed] [Google Scholar]
- 13.Brutinel, E. D., C. A. Vakulskas, and T. L. Yahr. 2010. ExsD inhibits expression of the Pseudomonas aeruginosa type III secretion system by disrupting ExsA self-association and DNA binding activity. J. Bacteriol. 192:1479-1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brutinel, E. D., C. A. Vakulskas, and T. L. Yahr. 2009. Functional domains of ExsA, the transcriptional activator of the Pseudomonas aeruginosa type III secretion system. J. Bacteriol. 191:3811-3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campbell, E. A., O. Muzzin, M. Chlenov, J. L. Sun, C. A. Olson, O. Weinman, M. L. Trester-Zedlitz, and S. A. Darst. 2002. Structure of the bacterial RNA polymerase promoter specificity sigma subunit. Mol. Cell 9:527-539. [DOI] [PubMed] [Google Scholar]
- 16.Choy, H. E., and S. Adhya. 1993. RNA polymerase idling and clearance in gal promoters: use of supercoiled minicircle DNA template made in vivo. Proc. Natl. Acad. Sci. U. S. A. 90:472-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Darwin, K. H., and V. L. Miller. 1999. InvF is required for expression of genes encoding proteins secreted by the SPI1 type III secretion apparatus in Salmonella typhimurium. J. Bacteriol. 181:4949-4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Darwin, K. H., and V. L. Miller. 2001. Type III secretion chaperone-dependent regulation: activation of virulence genes by SicA and InvF in Salmonella typhimurium. EMBO J. 20:1850-1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dasgupta, N., G. L. Lykken, M. C. Wolfgang, and T. L. Yahr. 2004. A novel anti-anti-activator mechanism regulates expression of the Pseudomonas aeruginosa type III secretion system. Mol. Microbiol. 53:297-308. [DOI] [PubMed] [Google Scholar]
- 20.Dove, S. L., S. A. Darst, and A. Hochschild. 2003. Region 4 of sigma as a target for transcription regulation. Mol. Microbiol. 48:863-874. [DOI] [PubMed] [Google Scholar]
- 21.Dove, S. L., F. W. Huang, and A. Hochschild. 2000. Mechanism for a transcriptional activator that works at the isomerization step. Proc. Natl. Acad. Sci. U. S. A. 97:13215-13220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ebright, R. H. 1993. Transcription activation at class I CAP-dependent promoters. Mol. Microbiol. 8:797-802. [DOI] [PubMed] [Google Scholar]
- 23.Egan, S. M., A. J. Pease, J. Lang, X. Li, V. Rao, W. K. Gillette, R. Ruiz, J. L. Ramos, and R. E. Wolf, Jr. 2000. Transcription activation by a variety of AraC/XylS family activators does not depend on the class II-specific activation determinant in the N-terminal domain of the RNA polymerase alpha subunit. J. Bacteriol. 182:7075-7077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frank, D. W. 1997. The exoenzyme S regulon of Pseudomonas aeruginosa. Mol. Microbiol. 26:621-629. [DOI] [PubMed] [Google Scholar]
- 25.Frank, D. W., and B. H. Iglewski. 1991. Cloning and sequence analysis of a trans-regulatory locus required for exoenzyme S synthesis in Pseudomonas aeruginosa. J. Bacteriol. 173:6460-6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frank, D. W., G. Nair, and H. P. Schweizer. 1994. Construction and characterization of chromosomal insertional mutations of the Pseudomonas aeruginosa exoenzyme S trans-regulatory locus. Infect. Immun. 62:554-563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grainger, D. C., C. L. Webster, T. A. Belyaeva, E. I. Hyde, and S. J. Busby. 2004. Transcription activation at the Escherichia coli melAB promoter: interactions of MelR with its DNA target site and with domain 4 of the RNA polymerase sigma subunit. Mol. Microbiol. 51:1297-1309. [DOI] [PubMed] [Google Scholar]
- 28.Haldimann, A., and B. L. Wanner. 2001. Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria. J. Bacteriol. 183:6384-6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hanahan, D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557-580. [DOI] [PubMed] [Google Scholar]
- 30.Hawley, D. K., and W. R. McClure. 1982. Mechanism of activation of transcription initiation from the lambda PRM promoter. J. Mol. Biol. 157:493-525. [DOI] [PubMed] [Google Scholar]
- 31.Hoang, T. T., A. J. Kutchma, A. Becher, and H. P. Schweizer. 2000. Integration-proficient plasmids for Pseudomonas aeruginosa: site-specific integration and use for engineering of reporter and expression strains. Plasmid 43:59-72. [DOI] [PubMed] [Google Scholar]
- 32.Holcroft, C. C., and S. M. Egan. 2000. Roles of cyclic AMP receptor protein and the carboxyl-terminal domain of the alpha subunit in transcription activation of the Escherichia coli rhaBAD operon. J. Bacteriol. 182:3529-3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holder, I. A., A. N. Neely, and D. W. Frank. 2001. Type III secretion/intoxication system important in virulence of Pseudomonas aeruginosa infections in burns. Burns 27:129-130. [DOI] [PubMed] [Google Scholar]
- 34.Hovey, A. K., and D. W. Frank. 1995. Analyses of the DNA-binding and transcriptional activation properties of ExsA, the transcriptional activator of the Pseudomonas aeruginosa exoenzyme S regulon. J. Bacteriol. 177:4427-4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jair, K. W., R. G. Martin, J. L. Rosner, N. Fujita, A. Ishihama, and R. E. Wolf, Jr. 1995. Purification and regulatory properties of MarA protein, a transcriptional activator of Escherichia coli multiple antibiotic and superoxide resistance promoters. J. Bacteriol. 177:7100-7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jourdan, A. D., and G. V. Stauffer. 1999. GcvA-mediated activation of gcvT-lacZ expression involves the carboxy-terminal domain of the alpha subunit of RNA polymerase. FEMS Microbiol. Lett. 181:307-312. [DOI] [PubMed] [Google Scholar]
- 37.Klein-Marcuschamer, D., C. N. Santos, H. Yu, and G. Stephanopoulos. 2009. Mutagenesis of the bacterial RNA polymerase alpha subunit for improvement of complex phenotypes. Appl. Environ. Microbiol. 75:2705-2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumar, A., R. A. Malloch, N. Fujita, D. A. Smillie, A. Ishihama, and R. S. Hayward. 1993. The minus 35-recognition region of Escherichia coli sigma 70 is inessential for initiation of transcription at an “extended minus 10” promoter. J. Mol. Biol. 232:406-418. [DOI] [PubMed] [Google Scholar]
- 39.Kumar, A., and C. P. Moran, Jr. 2008. Promoter activation by repositioning of RNA polymerase. J. Bacteriol. 190:3110-3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lonetto, M. A., V. Rhodius, K. Lamberg, P. Kiley, S. Busby, and C. Gross. 1998. Identification of a contact site for different transcription activators in region 4 of the Escherichia coli RNA polymerase sigma70 subunit. J. Mol. Biol. 284:1353-1365. [DOI] [PubMed] [Google Scholar]
- 41.Martin, R. G., and J. L. Rosner. 2001. The AraC transcriptional activators. Curr. Opin. Microbiol. 4:132-137. [DOI] [PubMed] [Google Scholar]
- 42.Mavris, M., A. L. Page, R. Tournebize, B. Demers, P. Sansonetti, and C. Parsot. 2002. Regulation of transcription by the activity of the Shigella flexneri type III secretion apparatus. Mol. Microbiol. 43:1543-1553. [DOI] [PubMed] [Google Scholar]
- 43.McCaw, M. L., G. L. Lykken, P. K. Singh, and T. L. Yahr. 2002. ExsD is a negative regulator of the Pseudomonas aeruginosa type III secretion regulon. Mol. Microbiol. 46:1123-1133. [DOI] [PubMed] [Google Scholar]
- 44.Mitchell, J. E., D. Zheng, S. J. Busby, and S. D. Minchin. 2003. Identification and analysis of ′extended −10′ promoters in Escherichia coli. Nucleic Acids Res. 31:4689-4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Molle, V., M. Fujita, S. T. Jensen, P. Eichenberger, J. E. Gonzalez-Pastor, J. S. Liu, and R. Losick. 2003. The Spo0A regulon of Bacillus subtilis. Mol. Microbiol. 50:1683-1701. [DOI] [PubMed] [Google Scholar]
- 46.Moyle, H., C. Waldburger, and M. M. Susskind. 1991. Hierarchies of base pair preferences in the P22 ant promoter. J. Bacteriol. 173:1944-1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Newman, J. R., and C. Fuqua. 1999. Broad-host-range expression vectors that carry the l-arabinose-inducible Escherichia coli araBAD promoter and the araC regulator. Gene 227:197-203. [DOI] [PubMed] [Google Scholar]
- 48.Parsot, C., E. Ageron, C. Penno, M. Mavris, K. Jamoussi, H. d'Hauteville, P. Sansonetti, and B. Demers. 2005. A secreted anti-activator, OspD1, and its chaperone, Spa15, are involved in the control of transcription by the type III secretion apparatus activity in Shigella flexneri. Mol. Microbiol. 56:1627-1635. [DOI] [PubMed] [Google Scholar]
- 49.Pilonieta, M. C., and G. P. Munson. 2008. The chaperone IpgC copurifies with the virulence regulator MxiE. J. Bacteriol. 190:2249-2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Plano, G. V. 2004. Modulation of AraC family member activity by protein ligands. Mol. Microbiol. 54:287-290. [DOI] [PubMed] [Google Scholar]
- 51.Rhodius, V. A., D. M. West, C. L. Webster, S. J. Busby, and N. J. Savery. 1997. Transcription activation at class II CRP-dependent promoters: the role of different activating regions. Nucleic Acids Res. 25:326-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Richards, M. J., J. R. Edwards, D. H. Culver, and R. P. Gaynes. 2000. Nosocomial infections in combined medical-surgical intensive care units in the United States. Infect. Control Hosp. Epidemiol. 21:510-515. [DOI] [PubMed] [Google Scholar]
- 53.Richards, M. J., J. R. Edwards, D. H. Culver, and R. P. Gaynes. 1999. Nosocomial infections in medical intensive care units in the United States. National Nosocomial Infections Surveillance System. Crit. Care Med. 27:887-892. [DOI] [PubMed] [Google Scholar]
- 54.Richet, E., and L. Sogaard-Andersen. 1994. CRP induces the repositioning of MalT at the Escherichia coli malKp promoter primarily through DNA bending. EMBO J. 13:4558-4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sato, H., and D. W. Frank. 2004. ExoU is a potent intracellular phospholipase. Mol. Microbiol. 53:1279-1290. [DOI] [PubMed] [Google Scholar]
- 56.Savery, N. J., G. S. Lloyd, M. Kainz, T. Gaal, W. Ross, R. H. Ebright, R. L. Gourse, and S. J. Busby. 1998. Transcription activation at class II CRP-dependent promoters: identification of determinants in the C-terminal domain of the RNA polymerase alpha subunit. EMBO J. 17:3439-3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Seredick, S. D., and G. B. Spiegelman. 2004. The Bacillus subtilis response regulator Spo0A stimulates sigmaA-dependent transcription prior to the major energetic barrier. J. Biol. Chem. 279:17397-17403. [DOI] [PubMed] [Google Scholar]
- 58.Stauffer, G. V., M. D. Plamann, and L. T. Stauffer. 1981. Construction and expression of hybrid plasmids containing the Escherichia coli glyA genes. Gene 14:63-72. [DOI] [PubMed] [Google Scholar]
- 59.Stevens, A. M., N. Fujita, A. Ishihama, and E. P. Greenberg. 1999. Involvement of the RNA polymerase alpha-subunit C-terminal domain in LuxR-dependent activation of the Vibrio fischeri luminescence genes. J. Bacteriol. 181:4704-4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang, H., Y. Kim, K. Severinov, A. Goldfarb, and R. H. Ebright. 1996. Escherichia coli RNA polymerase holoenzyme: rapid reconstitution from recombinant alpha, beta, beta′, and sigma subunits. Methods Enzymol. 273:130-134. [DOI] [PubMed] [Google Scholar]
- 61.Thibault, J., E. Faudry, C. Ebel, I. Attree, and S. Elsen. 2009. Anti-activator ExsD forms a 1:1 complex with ExsA to inhibit transcription of type III secretion operons. J. Biol. Chem. 284:15762-15770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Urbanowski, M. L., E. D. Brutinel, and T. L. Yahr. 2007. Translocation of ExsE into Chinese hamster ovary cells is required for transcriptional induction of the Pseudomonas aeruginosa type III secretion system. Infect. Immun. 75:4432-4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Urbanowski, M. L., C. P. Lostroh, and E. P. Greenberg. 2004. Reversible acyl-homoserine lactone binding to purified Vibrio fischeri LuxR protein. J. Bacteriol. 186:631-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vakulskas, C. A., K. M. Brady, and T. L. Yahr. 2009. Mechanism of transcriptional activation by Pseudomonas aeruginosa ExsA. J. Bacteriol. 191:6654-6664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vogel, H. J., and D. M. Bonner. 1956. Acetylornithinase of Escherichia coli: partial purification and some properties. J. Biol. Chem. 218:97-106. [PubMed] [Google Scholar]
- 66.Wickstrum, J. R., and S. M. Egan. 2004. Amino acid contacts between sigma 70 domain 4 and the transcription activators RhaS and RhaR. J. Bacteriol. 186:6277-6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yahr, T. L., and D. W. Frank. 1994. Transcriptional organization of the trans-regulatory locus which controls exoenzyme S synthesis in Pseudomonas aeruginosa. J. Bacteriol. 176:3832-3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yahr, T. L., A. K. Hovey, S. M. Kulich, and D. W. Frank. 1995. Transcriptional analysis of the Pseudomonas aeruginosa exoenzyme S structural gene. J. Bacteriol. 177:1169-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yahr, T. L., A. J. Vallis, M. K. Hancock, J. T. Barbieri, and D. W. Frank. 1998. ExoY, an adenylate cyclase secreted by the Pseudomonas aeruginosa type III system. Proc. Natl. Acad. Sci. U. S. A. 95:13899-13904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yahr, T. L., and M. C. Wolfgang. 2006. Transcriptional regulation of the Pseudomonas aeruginosa type III secretion system. Mol. Microbiol. 62:631-640. [DOI] [PubMed] [Google Scholar]
- 71.Zhou, X., D. H. Shah, M. E. Konkel, and D. R. Call. 2008. Type III secretion system 1 genes in Vibrio parahaemolyticus are positively regulated by ExsA and negatively regulated by ExsD. Mol. Microbiol. 69:747-764. [DOI] [PMC free article] [PubMed] [Google Scholar]