

Abstract
The publication of the complete genome sequence for Mycobacterium tuberculosis H37Rv in 1998 has had a great impact on the research community. Nonetheless, it is suspected that genetic differences have arisen in stocks of H37Rv that are maintained in different laboratories. In order to assess the consistency of the genome sequences among H37Rv strains in use and the extent to which they have diverged from the original strain sequenced, we carried out whole-genome sequencing on six strains of H37Rv from different laboratories. Polymorphisms at 73 sites were observed, which were shared among the lab strains, though 72 of these were also shared with H37Ra and are likely to be due to sequencing errors in the original H37Rv reference sequence. An updated H37Rv genome sequence should be valuable to the tuberculosis research community as well as the broader microbial research community. In addition, several polymorphisms unique to individual strains and several shared polymorphisms were identified and shown to be consistent with the known provenance of these strains. Aside from nucleotide substitutions and insertion/deletions, multiple IS6110 transposition events were observed, supporting the theory that they play a significant role in plasticity of the M. tuberculosis genome. This genome-wide catalog of genetic differences can help explain any phenotypic differences that might be found, including a frameshift mutation in the mycocerosic acid synthase gene which causes two of the strains to be deficient in biosynthesis of the surface glycolipid phthiocerol dimycocerosate (PDIM). The resequencing of these six lab strains represents a fortuitous “in vitro evolution” experiment that demonstrates how the M. tuberculosis genome continues to evolve even in a controlled environment.
Publication of the whole genome sequence of the H37Rv strain of Mycobacterium tuberculosis by Stewart Cole and colleagues in 1998 provided a breakthrough in tuberculosis (TB) research (8), leading to insights into the biology, metabolism, and evolution of this infectious pathogen. Large protein families related to fatty acid and polyketide biosynthesis, regulation (e.g., sigma factors and two-component sensor systems), drug efflux pumps and transporters, and the PE_PGRS proteins (a large duplicated family unique to the M. tuberculosis group of mycobacteria) were identified. In addition, transposons, prophage-like elements, and other repetitive and/or mobile genetic elements were identified (18). This genomic information has played an essential role in interpreting gene expression studies, modeling persistence, and identifying essential proteins as putative targets for drug discovery. However, to date the functions of only half of the genes (1,756/4,066) have been determined or predicted, and the rest remain annotated as “hypothetical proteins” (6).
The H37Rv strain was initially selected for sequencing because it is a widely used laboratory strain that has retained its virulence. H37Rv was initially derived from a clinical isolate, H37, obtained from a patient with pulmonary tuberculosis in 1905. H37Rv falls in the T clade (5) and single-nucleotide polymorphism (SNP) cluster group SCG-6b (12). The virulence of H37Rv can be demonstrated in a number of animal models. For example, SCID mice infected with H37Rv typically have a mean time to death of 30 to 35 days, depending on the dose and route of inoculation (13).
An avirulent strain, H37Ra, was also derived from H37 by culturing on solid egg medium and selecting for resistance to lysis (42). The strain was found not to cause disease in guinea pigs (43) or in mice (27). It has a colony morphology (smooth) different from that of H37Rv (rough) and several other phenotypic differences (14, 29). The H37Rv (ATCC 25618) and H37Ra (ATCC 25177) strains are maintained at the Trudeau Institute in New York (3), although unfortunately, the original H37 clinical isolate has been lost. Strain ATCC 27294 (TMC 102) is also frequently used as a representative of H37Rv in studies and treated equivalently in the literature. ATCC 25618 and ATCC 27294 were both isolated from the same patient in different years, and both are fully drug susceptible.
The complete genome of H37Ra has been sequenced by Zheng et al. (48), who found 272 polymorphisms compared to the genome sequence determined by Cole et al. (8) for H37Rv. However, a subset of the polymorphic sites were found to match CDC1551, and upon resequencing of 85 such sites in H37Rv, 79 were determined to be errors in the H37Rv reference sequence. In addition, H37Ra has insertions of IS6110 at two novel sites and a loss of one, compared to the 16 sites in H37Rv. The 130 genuine H37Ra-specific polymorphisms found were divided into those in coding regions, those in upstream regulatory regions, and those in noncoding, nonregulatory intergenic regions in order to assess potential relevance to virulence. Polymorphisms in the promoter regions of sigC, nrdH (glutaredoxin-like electron transporter), and pabB (para-amino benzoate synthase), as well as nonsynonymous substitutions in mazG (regulator of stringent response), phoP (two-component sensor regulating biosynthesis of cell surface lipid antigens), pks12 (polyketide synthase involved in biosynthesis of mycoketides), and nrp (nonribosomal peptide synthetase potentially involved in phthiocerol dimycocerosate [PDIM] biosynthesis), were highlighted as possible causes of the loss of virulence. H37Ra does not synthesize a number of cell surface antigens, including sulfolipid-1, trehalose mycolates, and PDIM (7). The roles of mutations in phoP and sigM, both of which regulate expression of genes involved in biosynthesis of cell surface antigens, have been subsequently investigated, though neither seems to be singularly responsible for the avirulence of H37Ra (17, 35). Multiple mutations in PPE and PE_PGRS genes are also observed in H37Ra, and there has been speculation about the role of these genes in virulence (39). However, the RvD2 region (an 8-kb region present in H37Ra but deleted in H37Rv, including an IS6110 insertion element, mmpL14, and several hypothetical genes) is known not to be responsible for differences in virulence (25).
Because of its importance as a model strain used in laboratory studies, it is essential to determine how consistent different stocks of H37Rv in different laboratories are with the reference genome sequence and with each other. Different stocks could accumulate independent polymorphisms over time, and such inconsistencies could potentially make results of studies obtained with H37Rv cultures from different labs difficult to compare, particularly if they affect virulence, drug tolerance, metabolism, cell wall constitution, etc. Furthermore, sequencing errors in the original genome sequence are possible. In order to evaluate differences among currently used variants of H37Rv, we resequenced the complete genomes of six extant H37Rv strains (two samples of ATCC 25618 and four of ATCC 27294) using Illumina sequencing technology. We compared differences among them and differences from the reference sequences for H37Rv and H37Ra available from GenBank. The results of this study identify a common set of 73 polymorphisms shared among all six sequenced strains relative to the H37Rv reference strain. Most (72) of these are shared with H37Ra and likely correspond to sequencing errors in the original H37Rv genome sequence. However, there are several sites where additional polymorphisms are shared among a subset of strains, and several strains have a small number of unique polymorphisms. Furthermore, examination of insertion sites of the IS6110 transposable element reveals several changes that have occurred among these strains. These results illustrate the ongoing evolution of this strain and divergence from the sequenced reference strain of H37Rv and highlight the importance of understanding the genetic differences unique to the stock used in each laboratory.
MATERIALS AND METHODS
DNA sequencing.
Genomic DNA was extracted using the cetyltrimethylammonium bromide (CTAB)-lysozyme protocol described previously (26). The DNA library was constructed using a genomic DNA sample preparation kit from Illumina. The sample was first fragmented using a nebulization technique, and then the double-stranded DNA fragments comprised of 3′ or 5′ overhangs were converted into blunt ends, using T4 DNA polymerase and Klenow enzyme. Klenow exo− (lacking the 3′-to-5′ exonuclease) was used to add an A base to the 3′ end of the blunt phosphorylated DNA fragments so that the fragments could be ligated to the adaptors, which have a single T base overhang at their 3′ end. The ligated DNA was then size selected on a 2% agarose gel. DNA fragments of about 300 bp were excised from the preparative portion of the gel. DNA was then recovered using a Qiagen gel extraction kit and was PCR amplified to produce the final DNA library. Five picomoles of DNA from each strain was loaded onto a different lane of the sequencing chip (eight lanes total), and the clusters were generated on the cluster generation station of the GAII using the Illumina cluster generation kit. Bacteriophage φX174 DNA was used as a control. In the case of paired-end reads, distinct adaptors from Illumina were ligated to each end with PCR primers that allowed reading of each end as separate runs. The sequencing reaction was run for 36 cycles (tagging, imaging, and cleavage of one terminal base at a time), and four images of each tile on the chip were taken in different wavelengths for exciting each base-specific fluorophore. For paired-end reads, data were collected as two sets of matched 36-bp reads. Image analysis and base calling were done using the Illumina GA Pipeline software.
Comparative genome assembly.
The 36-bp reads that were generated for each strain were mapped against H37Rv as a reference sequence (accession on. NC_000962, downloaded from NCBI) via ungapped alignments allowing up to two mismatches. For reads that mapped to multiple locations, one was chosen at random. For paired-end data, mapping locations of each read were restricted to sites within 300 bp of mapping locations of its partner. The base calls were made by a multiplying the base probabilities of the individual bases covering each site (extracted from Q-values output by the Pipeline software, encoding uncertainty about spot interpretation during image analysis) and taking the base with the highest combined likelihood. Apparent differences (at sites where the consensus base from overlapping reads differed from the expected base in the reference sequence), along with sites where coverage was low (<5×) or observed bases were heterogeneous (majority base < 70%), were identified, and local contig building was applied. Contigs were built by a best-first-search algorithm that starts with a read matching ∼100 bp upstream, contains a sequence of reads with perfect overlaps of at least 25 bp, and ends in a read matching ∼100 bp downstream. Alignment of the consensus sequence of the contig to the corresponding region in the reference genome revealed whether SNPs or insertions/deletions (indels) were present. These polymorphisms were compiled for each strain and used to modify the H37Rv sequence into edited genome sequences for each strain. Finally, all the reads for each strain were remapped against the corresponding intermediate sequence, and the base calls at each site were recalculated as described above. Whole-genome alignments for comparative analyses were generated using MUMMER v 3.20 (24).
RESULTS
Samples of six H37Rv strains obtained from separate laboratories were sequenced using an Illumina GenomeAnalyzerII (Table 1). All strains were sequenced with a read length of 36 bp, though two were sequenced in single-end mode and four in paired-end mode (where reads are sequenced from both ends of ∼250-bp fragments, providing additional localization constraints). The depth of coverage ranged between 29.4× (H37RvHA) and 151.3× (H37RvJO). The genomes were sequenced to >97.97% completion (defined as sites covered by at least one read), with the remaining sites with zero depth of coverage largely confined to the highly GC-rich coding regions of PE_PGRS genes, where sequencing is not as efficient. The spoligotypes of the strains were determined by matching sequencing data (reads) to the oligonucleotide sequences of the 43 spacers in the direct-repeat (DR) region (20). While H37RvLP, H37RvJO, and H37RvCO have spoligotype patterns that match the general pattern for the H37Rv reference sequence (octal representation, 777777477760771; missing spacers 20 to 21 and 33 to 36), the other three strains (H37RvAE, H37RvMA, and H37RvHA) are missing an additional spacer, number 26. The distinct spoligotypes suggest a clustering of the strains into subgroups based on phylogenetic relationships (see below).
TABLE 1.
H37Rv strains sequenced in this study
| Strain | ATCC no. | Source | Spoligotypea | Sequencing | Coverage (×) | Completion (%) |
|---|---|---|---|---|---|---|
| H37RvAE | 27294 | William Jacobs (Albert Einstein College of Medicine) | 777777475760771 | Single end | 63.3 | 99.98 |
| H37RvMA | 27294 | Chris Sassetti (University of Massachusetts) | 777777475760771 | Paired end | 62.3 | 99.22 |
| H37RvCO | 27294 | Karen Dobos (Colorado State University) | 777777477760771 | Paired end | 79.2 | 98.77 |
| H37RvHA | 27294 | Sarah Fortune (Harvard University) | 777777475760771 | Single end | 29.4 | 99.37 |
| H37RvLP | 25618 | Tanya Parish (IDRI, Seattle, WA) | 777777477760771 | Paired end | 151.3 | 99.00 |
| H37RvJO | 25618 | Valerie Mizrahi (Johannesburg, South Africa) | 777777477760771 | Paired end | 49.7 | 98.78 |
Spoligotype inferred from matching sequence data (36-bp reads) to spacer oligonucleotides. For comparison, the spoligotype of the H37Rv reference strain is 777777477760771.
There are 72 polymorphisms that are shared in common among all six H37Rv variants plus H37Ra, in comparison to the H37Rv reference sequence. These include 57 single-nucleotide polymorphisms (SNPs) and 15 insertions/deletions (indels) 1 to 3 bp in length, although polymorphisms in PE_PGRS genes were excluded (due to ambiguity caused by low coverage). Among the SNPs, there are 6 in noncoding regions, 21 synonymous mutations, and 30 nonsynonymous mutations. Rv0197 and pstA1 show truncations (mutations to a stop codon mid-open reading frame [ORF]), and pks3 and Rv1783 show mutations in their stop codons to extend their ORFs. There are eight indels in coding regions, causing frameshift mutations in Rv0197, PPE7, Rv0907, Rv1046c, Rv1575, Rv2251, Rv3655c, and sigM. Of the 15 mutations in noncoding regions, 6 are within putative regulatory regions (<100 bp upstream of coding regions). It should also be noted that there are two local clusters of SNPs (12 within a 137-bp span in PPE9 and 14 within an 80-bp span in PPE47) that are not included in this analysis.
There is only one site where there is a polymorphism shared only among the five H37Rv variant strains but not the H37Rv or H37Ra reference strains: A459399C in a noncoding region, at −84 bp upstream of Rv0383c. This SNP appears to be unique to the H37Rv lab strains as a group. H37Ra has a 55-bp deletion in this region.
Many of these sites are consistent with errors discovered in the H37Rv reference sequence upon resequencing by Zheng et al. (48). Of 48 successfully resequenced sites with apparent SNPs in the H37Ra genome that matched CDC1551 but not H37Rv (excluding SNPs in PE_PGRS genes, PPE9, and PPE47), they found only 3 sites with genuine differences, and the rest turned out to be errors in the H37Rv sequence. Our results agree exactly with the resequencing, in that all of the 45 errors appear as shared SNPs among our H37Rv lab strains shown in Table 2, and the 3 genuine SNPs between H37Rv and H37Ra were not found in the H37Rv lab strains (position 754186, G to A; position 1077312, G to A; and position 4100975, C to T [these remain unique to H37Ra]). Nine of the 15 indels in our study were also resequenced by Zheng et al. (48), and all were confirmed as errors in the H37Rv sequence, including the frameshift in amino acid (aa) 160 of sigM (shortening the ORF from 222 aa in H37Rv to 196 aa in H37Ra).
TABLE 2.
Shared polymorphisms among all five H37Rv lab strains and H37Ra, relative to H37Rva
| Rv no. | Gene | Coordinateb | Mutation | Amino acid substitutionc | Comment |
|---|---|---|---|---|---|
| Rv0012 | 14785 | T → C | C233R | ||
| Rv0050 | ponA1 | 55553 | C → T | P631S | |
| Rv0064 | 69989 | G → A | G457D | ||
| Rv0082 | 90144 | A → G | Q74R | ||
| Rv0083 | 91071 | T → C | I224I | ||
| Rv0101 | nrp | 116000 | T → G | V2000V | |
| NC | 131177 | +G | −73 bp upstream of Rv0108c | ||
| Rv0197 | 234477 | T → G | Y749* | Reduces length by 14 aa | |
| Rv0197 | 234497 | +GT | |||
| Rv0204c | 242299 | C → G | V306L | ||
| Rv0323c | 390828 | T → C | S142G | ||
| Rv0354c | PPE7 | 424323 | +C | ||
| Rv0355c | PPE8 | 426909 | A → C | W2591G | |
| Rv0382c | pyrE | 458282 | A → G | Y33Y | |
| Rv0425c | ctpH | 511518 | T → G | I1268I | |
| Rv0442c | PPE10 | 532097 | T → C | K40E | |
| Rv0461 | 552085 | A → G | Q20Q | ||
| Rv0473 | 563577 | A → G | K5R | ||
| Rv0682 | rpsL | 781922 | A → G | K121K | |
| Rv0890c | 990001 | G → C | P866A | ||
| Rv0907 | 1010207 | +G | |||
| Rv0919 | 1025106 | T → C | F141F | ||
| Rv0930 | pstA1 | 1037911 | C → T | R305* | Reduces length by 4 aa |
| Rv1046c | 1168718 | +T | |||
| Rv1121 | zwf1 | 1244700 | T → C | L332L | |
| NC | 1313338 | A → G | −39 bp upstream of Rv1179c | ||
| NC | 1313339 | +C | −40 bp upstream of Rv1179c | ||
| Rv1180 | pks3 | 1315191 | A → C | *489Y | Fuses ORF with pks4 |
| Rv1181 | pks4 | 1315884 | G → A | A217A | |
| Rv1185c | fadD21 | 1327402 | T → C | E37E | |
| Rv1188 | 1331696 | A → C | R226R | ||
| Rv1266c | pknH | 1414021 | C → T | R607Q | |
| Rv1297 | rho | 1453608 | T → C | G135G | |
| NC | 1471659 | C → T | |||
| Rv1575 | 1780588 | +G | In aa A220 | ||
| Rv1677 | dsbF | 1901816 | A → G | Q23Q | |
| Rv1783 | 2020563 | A → T | *463L | Fuses ORF with Rv1784 | |
| Rv1808 | PPE32 | 2050913 | A → G | E331E | |
| Rv1809 | PPE33 | 2051746 | T → C | A155A | |
| Rv1815 | 2057774 | A → T | I83F | ||
| NC | 2167489 | T → C | |||
| Rv1925 | fadD31 | 2177654 | A → C | M190L | |
| NC | 2207592 | +C | |||
| Rv1979c | 2221796 | C → T | V457I | ||
| NC | 2251999 | A → G | −3 bp upstream from otsB1 | ||
| Rv2037c | 2282787 | C → T | C312Y | ||
| Rv2048c | pks12 | 2297976 | G → A | S3004L | |
| Rv2101 | helZ | 2361623 | A → C | M462L | |
| Rv2205c | 2470149 | T → C | E105E | ||
| NC | 2505919 | A → G | |||
| NC | 2523208 | +CCG | |||
| Rv2251 | 2525727 | −G | In aa E55 | ||
| NC | 2718852 | T → G | −44 bp upstream of nadD | ||
| Rv2450c | rpfE | 2751804 | C → T | R126Q | |
| Rv2495c | pdhC | 2809621 | T → C | T107A | |
| Rv2614A | 2943411 | T → C | L12L | ||
| Rv2627c | 2954439 | T → C | R104G | ||
| Rv2680 | 2996194 | T → A | V30V | ||
| Rv2695 | 3012293 | A → G | T126T | ||
| Rv2896c | 3205978 | A → C | S153A | ||
| Rv2932 | ppsB | 3254365 | T → C | L1098L | |
| Rv3011c | gatA | 3370177 | T → G | M420L | |
| Rv3144c | PPE52 | 3510642 | T → C | S226G | |
| NC | 3580637 | −T | −1 bp upstream from lipV | ||
| NC | 3590687 | +C | |||
| Rv3331 | sugI | 3718357 | C → T | P423L | |
| NC | 3862474 | −A | −84 bp upstream from rplM | ||
| Rv3479 | 3896340 | T → G | L174R | ||
| Rv3655c | 4095002 | −G | |||
| Rv3704c | gshA | 4147070 | A → G | L373L | |
| Rv3911 | sigM | 4400663 | −C | In aa R160 | |
| Rv3919c | gidB | 4407904 | G → A | S100F |
Amino acid translations for mutations in coding regions are given. Mutations in noncoding regions (NC) were checked to see if they were within 100 bp of the translational start site of a nearby coding region, and if so, the gene and distance are indicated.
Relative to the H37Rv reference sequence.
*, stop codon.
Polymorphisms unique to certain strains or shared among a subset of strains were also identified, potentially representing differences among the stock cultures (Table 3). None of the differences were shared with H37Ra, which matched H37Rv at all these sites. Six polymorphisms were shared among H37RvAE, H37RvMA, H37RvHA, and H37RvCO, which are ATCC 27294 specific. One of these is a −CCG deletion in ponA1 (a predicted penicillin-binding transpeptidase/transglycosylase gene) corresponding to Pro630, which is an apparent reduction in copy number of a CCGn tandem-repeat region from six copies in H37Rv and H37Ra to five copies. Four polymorphisms are shared by only H37RvAE, H37RvMA, and H37RvHA, including three SNPs (in lprO [silent], Rv2604c, and a noncoding region) and +CAC in Rv2553c (a hypothetical protein). The similarity of these strains is consistent with the spoligotyping results, suggesting that these three strains form a derived subgroup. Three unique mutations are observed in H37RvAE (in Rv1063c [silent], fadD31, and ppsA). The mutation H620N in fadD31, encoding an acyl coenzyme A (acyl-CoA) ligase, is outside the boundaries of the AMP-binding domain (spanning residues 70 to 553 according to Pfam [http://pfam.sanger.ac.uk]) and thus is unlikely to affect function. The mutation in ppsA is a nonsense mutation (W1294*). ppsA encodes a multidomain polyketide synthase involved in PDIM biosynthesis, specifically the polymerization of the phthiocerol component. The mutation occurs near the C terminus of the protein product (total length, 1,876 amino acids) and would truncate the terminal acyl carrier protein (ACP) domain and thus presumably abrogate the function of ppsA. It is most likely that this mutation is a one-off mutation (specific to the sample sequenced) that occurred between passaging and is not propagated in the parental strain, as this strain is regularly passaged in mice and retains virulence and PDIM production (W. R. Jacobs, Jr., unpublished results). Supporting this, we did not observe the ppsA W1294* mutation in several isogenic mutants that we sequenced. Mutations in genes involved in PDIM biosynthesis are frequently observed in strains maintained in the laboratory without the selective pressure to maintain virulence (11), reinforcing the value of regular passaging in mice.
TABLE 3.
Polymorphic sites containing mutations unique to one lab strain or shared among a subset (excluding polymorphisms in PPE and PE_PGRS proteins)
| Mutation type and positiona | Mutation in strainb: |
|||||
|---|---|---|---|---|---|---|
| H37RvCO | H37RvAE | H37RvMA | H37RvHA | H37RvLP | H37RvJO | |
| Mutations shared by all H37Rv variants but not found in H37Ra | ||||||
| 459399/NCc | A → C | A → C | A → C | A → C | A → C | A → C |
| Mutations shared by cluster 1 | ||||||
| Rv0050/ponA1 | −CCG | −CCG | −CCG | −CCG | ||
| Rv0543c | A81A | A81A | A81A | A81A | ||
| Rv0861c/ercc3 | A410A | A410A | A410A | A410A | ||
| Rv1520 | Y200Y | Y200Y | Y200Y | Y200Y | ||
| Rv1771 | Q291R | Q291R | Q291R | Q291R | ||
| Rv1907c | V158A | V158A | V158A | V158A | ||
| Mutations shared by cluster 2 | ||||||
| Rv0179c/lprO | L239L | L239L | L239L | |||
| 986204/NC | C → A | C → A | C → A | |||
| Rv2553cd | +CAC(1) | +CAC(1) | +CAC(1) | |||
| Rv2604c | D151E | D151E | D151E | |||
| Mutations unique to H37RvAE | ||||||
| Rv1063c | R83R | |||||
| Rv1925/fadD31 | H620N | |||||
| Rv2931/ppsA | W1294* | |||||
| Mutations shared by cluster 3 | ||||||
| Rv0480c | R9H | R9H | ||||
| Rv0538 | P402S | P402S | ||||
| Rv0573c | L275L | L275L | ||||
| Rv2940c/mase | +GC | +GC | ||||
| Rv3785 | −17 bp | −17 bp | ||||
| Mutations unique to H37RvLP | ||||||
| Rv0282 | Y275H | |||||
| Rv0573c | F252V | |||||
| Rv1949c | G236G | |||||
| Rv2946c/pks1 | D1463E | |||||
| Rv3645 | V393A | |||||
| Mutations unique to H37RvJO | ||||||
| Rv0282 | D138G | |||||
| 1108537/NCf | A → G | |||||
| Rv2542 | A348T | |||||
NC, mutation is in a noncoding region.
*, mutation to a stop codon.
−84 bp upstream of Rv0383c; H37Ra has a −55 bp deletion here.
In amino acid 53 out of 417 in Rv2553c (hypothetical protein).
In amino acid 829 out of 2,111 in mas (mycocerosic acid synthase).
−33 bp upstream of Rv0991c.
The two ATCC 25618 samples, H37RvLP and H37RvJO, share five mutations, including a frameshift mutation +GC in mas (encoding mycocerosic acid synthase, which is also involved in PDIM biosynthesis) in amino acid 829 out of 2,111. This mutation is known to disrupt the function of mas, as H37RvJO has been shown to be PDIM deficient (21). However, both H37RvLP and H37RvJO are fully virulent in mice (21, 33). This would appear to contradict reports that PDIM deficiency correlates with reduced virulence (10). One possible explanation for this discrepancy could be a difference in genetic backgrounds, e.g., a compensating mutation that allows these strains to retain virulence despite loss of PDIM. There are three mutations shared between H37RvLP and H37RvJO (other than the frameshift in mas and a synonymous mutation in Rv0573c): R9H in Rv0480c, P402S in Rv0538, and a 17-bp deletion in Rv3785. Rv0480c encodes a putative nitrilase, but the mutation occurs in the N terminus, which is on the surface of the protein, not the active site, based on the crystal structure of the yeast homolog (Protein Data Bank [PDB] accession no. 1F89). Rv3785 is a hypothetical protein of unknown function. However, Rv0538 has previously been recognized as an immunogenic protein (38). Rv0538 is a 548-amino acid membrane protein of unknown function containing Pro/Thr repeats, and hence it is also called proline-threonine repetitive protein (PTRP). In a study aimed at identifying antigenic proteins for potential diagnostic development, antibodies to Rv0538 were found with high frequency in HIV-negative, TB-positive patients (38). Peptide epitope mapping identified four 20-amino-acid regions that each bound antibodies in sera of >50% of patients with active infections, and Pro402 is located directly in the middle of the third region, PT40. Though the connection between Rv0538 and virulence is still unresolved, this study directly links the site of the P402S SNP in Rv0538 shared by H37RvLP and H37RvJO to stimulation of the immune response of the host and therefore could be part of the explanation for how these strains retain their virulence despite the loss of PDIM production.
Several of the mutations observed among these six H37Rv strains (7 out of 20 in coding regions) are silent and thus unlikely to cause phenotypic differences. However, the functional relevance of the remaining mutations, many of which are conservative (e.g., D151E in Rv2604c and V393A in Rv3645), is unknown. The SNP in pks1 in H37RvLP probably does not cause a phenotype, since it is part of a disrupted polyketide synthase gene that produces phenolglycolipid (PGL) in other strains of M. tuberculosis but which has been split into two ORFs (pks15 and pks1) by a frameshift mutation in H37Rv and thus is already nonfunctional (9). Of the three SNPs that occur in noncoding regions, two are in putative regulatory regions near the transcriptional start sites of operons. The closest one is an A-to-G substitution 33 bp upstream of Rv0991c (encoding a hypothetical protein). Of four indels observed, two are in frame (−3 bp in ponA1 and +3 bp in Rv2553c), and two cause frameshifts (in mas [discussed above] and Rv3785, encoding another hypothetical protein whose function is presumably disrupted in H37RvLP and H37RvJO).
The polymorphisms shared among certain strains suggest that they can be clustered by similarity. A phylogenetic tree was constructed using maximum parsimony (dnapars in PHYLIP 3.66) based on the polymorphic sites list in Table 3 (Fig. 1), augmented with genuine differences between H37Rv and H37Ra not attributed to sequencing errors (48). As expected, this phylogeny suggests that H37RvLP and H37RvJO diverged from H37RvCO, H37RvAE, H37RvMA, and H37RvHA, forming two distinct clusters, consistent with their typing as ATCC 25618 and ATCC 27294, respectively. There are five ATCC 25618-specific polymorphisms and six ATCC 27294-specific polymorphisms distinguishing the highest-level branches. Subsequently, H37RvAE, H37RvMA, and H37RvHA diverged from H37RvCO as a subcluster. Several of the strains subsequently accumulated a small number of independent mutations (three in H37RvAE, five in H37RvLP, and three in H37RvJO). The number of polymorphisms in each strain is small compared to the number of differences between the H37Rv and H37Ra (30 SNPs, excluding those in PE_PGRS genes). Note that H37Rv is on a shallow branch because all of the sequencing errors have been filtered out, leaving only one site (position 459399, A to C) where it differs from all the other strains.
FIG. 1.
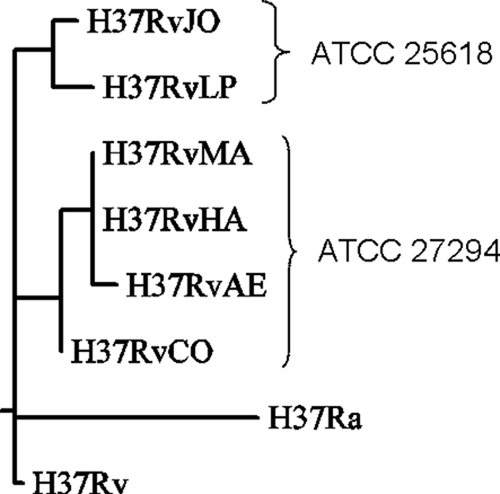
Maximum-parsimony tree of relationships among strains. Branch lengths represent number of nucleotide differences (total tree length = 57).
In addition to the small number of substitutions and insertions/deletions distinguishing these variant strains of H37Rv, there are also several differences in insertion sites of the IS6110 transposable element. All six strains have the same 16 copies of IS6110 as in the H37Rv reference strain, with three exceptions. First, H37RvLP and H37RvJO have lost IS6110 12, located at coordinate 3.55 Mb. Second, H37RvJO has a novel insertion site at coordinate 3.49 Mb, disrupting Rv3128c, encoding a hypothetical protein of unknown function. This site occurs in a 556-bp region called NTF-1, in which Beijing strains of M. tuberculosis also have one or two unique IS6110 insertions (23, 34). Third, H37RvCO contains a novel IS6110 insertion site in plcD 617 bp upstream from the site in H37Rv. In addition, H37RvAE, H37RvMA, and H37RvHA appear to have only one copy in this region and lack the intervening portion of plcD. This can be explained by a series of three IS6110 insertion/loss events (Fig. 2). H37Ra shows an insertion in the coding region of plcD, along with an additional one downstream, creating an adjacent pair and spanning several genes in between (including mmpL14). It was postulated that the IS pair may have been ancestral and that the 8-kb region, including the upstream IS element, was lost in H37Rv (25). It appears that after this event, a new insertion of IS6110 occurred even further upstream in plcD in a progenitor of four of the six H37Rv lab strains, resulting in the pattern observed in H37RvCO. Subsequently, this IS and the intervening portion of plcD in H37RvAE, H37RvMA, and H37RvHA were deleted, probably through homologous recombination with the IS6110 element downstream. Given the frequency of insertion events in this region, it is likely that plcD represents a “hot spot” for IS6110 insertion sites (37, 45). However, these insertion/deletion events probably do not have any functional relevance, since they are in a gene that is already disrupted.
FIG. 2.

Proposed history of IS6110 insertion/deletion events in the region of plcD. “hypo” indicates coding regions annotated as encoding hypothetical proteins in the H37Ra genome.
DISCUSSION
The inferred phylogenetic relationships among the strains based on SNP analysis (Fig. 2) are consistent with the known provenance of these six strains. H37RvCO was obtained from the Trudeau Institute as ATCC 27294 (TMC 102) in 1969 for the laboratory at Colorado State University. H37RvAE in W. Jacob's lab was obtained from the original ATCC 27294 stock at the Trudeau Institute in the 1970s. Subsequently, strain H37RvHA was obtained from Barry Bloom's laboratory, which had obtained it from the Albert Einstein stock, and was transferred to S. Fortune's lab at Harvard in the late 1990s. H37RvMA was derived from this stock for C. Sassetti's group at the University of Massachusetts Medical School in 2000. Independently, H37RvLP (“London Pride”) was obtained by T. Parish as ATCC 25618 from the National Institute of Medical Research (NIMR) in London around 1994. This strain was then maintained at the London School of Hygiene & Tropical Medicine (LSHTM). H37RvJO was obtained from LSHTM in 1998. This history supports the main division between the clusters of strains based on SNP analysis (H37RvAE, H37RvSF, H37RvMA, and H37RvCO [three of which passed through the Jacobs lab] versus H37RvLP and H37RvJO [which passed through NIMR]). It can also be used to give an interpretation of the IS6110 insertion/deletion events in the plcD region. It is likely that the novel IS6110 insertion in plcD occurred early in the Trudeau H37Rv stock before H37RvCO was sampled and that it was subsequently lost by H37RvAE during maintenance in the Jacobs lab and inherited by the other strains derived from it (H37RvHA and H37RvMA). Three other shared SNPs and an indel are associated with this lineage, though Jacobs' stock has gone on to acquire several additional unique polymorphisms.
While H37Rv is widely regarded as a standard virulent reference strain for studies of tuberculosis, it has been recognized that genetic differences among stocks from different labs are possible. Furthermore, it has been suggested that there are sequencing errors in the H37Rv reference genome sequence, which can complicate the interpretation of polymorphisms observed in isogenic mutants. Thus, we undertook the sequencing of the complete genomes of six strains of H37Rv from distinct laboratories. Indeed, the lab strains were found to have 73 common differences from the H37Rv reference strain. However, nearly all of these are shared with the avirulent strain H37Ra, and upon resequencing of H37Rv by Zheng et al. (48), they appear to be errors in the original H37Rv sequence. Hence, apparent polymorphisms at these loci can be disregarded.
Nonetheless, there are several differences that were observed among the six H37Rv lab strains. Several strains share polymorphisms, dividing them into two distinct clusters with five and six common polymorphisms each. This primarily reflects the difference between the two culture types, ATCC 25618 and ATCC 27294. Both are frequently used as H37Rv reference strains in experimental studies and often are treated interchangeably. However, they are not always phenotypically identical (for example, ATCC 25618 has a slightly higher pyrazinamide MIC than ATCC 27294 [40]), and to date it has not been clear how they differ genetically. Our sequencing results show that there are a combined 11 differences between these two versions of H37Rv. In addition, several strains have a small number of unique mutations (three to five) that were acquired independently, making them distinct even within each cluster. Further genetic variations observed include two independent insertions and two deletions of IS6110 transposable elements, as well as the loss of an additional spacer in the direct-repeat (DR) region in one cluster of strains, leading to a change in spoligotype. It has been argued that the transposition of IS6110 insertion elements plays a significant role in the evolution of the M. tuberculosis genome (31), and this was observed even on the small scale of this study. For comparison, the rate of change among IS6110 insertions sites in vivo (among clinical isolates) has been estimated at 2.87% per site per year (36). The observed differences among the strains sequenced clearly demonstrate that the stocks of H37Rv in different laboratories are genetically distinct.
The general belief that M. tuberculosis, and H37Rv in particular, has a comparatively stable genome is due in part to the low diversity observed among isolates worldwide (typically differing by <0.01% at the genetic level). However, this should not be misinterpreted as evidence for a low mutation rate, as the frequency of selection of mutants resistant to various drugs falls in the range of 10−5 to 10−8, which is typical of other prokaryotes (22). Instead, the low diversity has been attributed to a population bottleneck estimated at occurring 15,000 to 20,000 years ago (41). M. tuberculosis continues to evolve, as evidenced by independent outbreaks of drug resistance (47); loss of deletion regions (44); and relocation of transposons, changes in copy number of tandem repeats, and other genomic characteristics often exploited for genotyping (30). Ongoing evolution has also been observed in vitro via accumulation of genetic differences during serial passaging of cultures (32). Genetic variation as a result of culturing has been observed in many other bacterial species as well (15), including Escherichia coli (1, 19), Streptococcus (28), Campylobacter jejuni (16), and Helicobacter pylori (46), often leading to phenotypic changes in virulence. Some evolution is driven by selection largely for mutations that accelerate growth in vitro. For example, loss of PDIM synthesis, which is often associated with reduced virulence, results in more rapid growth in laboratory media (11). Moreover, changes in Mycobacterium bovis passaged over multiple generations resulted in the strain, M. bovis BCG, that is no longer able to cause disease (2) and therefore was chosen as a vaccine and, prior to the availability of methods for preserving strains, was further propagated. However, while some characteristics are selected, many genetic changes likely occur due to simple genetic drift. For example, while M. bovis BCG has some changes that result in decreased virulence, the majority of alterations have no known consequence (4).
Our whole-genome sequencing results show that differences among H37Rv strains have also arisen over time. The differences are few (on the order of 5 to 10 polymorphisms per strain, after errors in the original reference sequence are ruled out) but potentially functionally relevant, such as nonsynonymous mutations in PDIM biosynthesis genes and possibly other hypothetical proteins whose functions are currently unknown. The variation we see among isolates passaged for a limited amount of time in separate laboratories could represent a mixture of selected and random events that become fixed with the genetic bottleneck associated with transfer of clones among investigators. Thus, the notion of H37Rv as a standard reference strain should be used with some caution, as experimental results derived with “H37Rv” may depend on the laboratory in which it is maintained and the associated unique genetic characteristics. Knowledge of the genome sequences of individual strains used in each lab might be helpful for explaining conflicting experimental data generated in different labs (e.g., differences in MICs or growth rates), as well as for distinguishing relevant SNPs associated with phenotypes in isogenic mutants (e.g., for identifying drug targets).
Acknowledgments
Funding was provided in part by the Robert A. Welch Foundation (to J.C.S.). This work was also funded in part by a grant from the Bill & Milinda Gates Foundation.
We thank David Roberts, Bavesh Kana, and Bhavna Gordhan for assistance with genomic DNA preparation.
Footnotes
Published ahead of print on 14 May 2010.
REFERENCES
- 1.Barrick, J. E., D. S. Yu, S. H. Yoon, H. Jeong, T. K. Oh, D. Schneider, R. E. Lenski, and J. F. Kim. 2009. Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature 461:1243-1247. [DOI] [PubMed] [Google Scholar]
- 2.Behr, M. A., M. A. Wilson, W. P. Gill, H. Salamon, G. K. Schoolnik, S. Rane, and P. M. Small. 1999. Comparative genomics of BCG vaccines by whole-genome DNA microarray. Science 284:1520-1523. [DOI] [PubMed] [Google Scholar]
- 3.Bifani, P., S. Moghazeh, B. Shopsin, J. Driscoll, A. Ravikovitch, and B. N. Kreiswirth. 2000. Molecular characterization of Mycobacterium tuberculosis H37Rv/Ra variants: distinguishing the mycobacterial laboratory strain. J. Clin. Microbiol. 38:3200-3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brosch, R., S. V. Gordon, T. Garnier, K. Eiglmeier, W. Frigui, P. Valenti, S. Dos Santos, S. Duthoy, C. Lacroix, C. Garcia-Pelayo, J. K. Inwald, P. Golby, J. N. Garcia, R. G. Hewinson, M. A. Behr, M. A. Quail, C. Churcher, B. G. Barrell, J. Parkhill, and S. T. Cole. 2007. Genome plasticity of BCG and impact on vaccine efficacy. Proc. Natl. Acad. Sci. U. S. A. 104:5596-5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brudey, K., J. R. Driscoll, L. Rigouts, W. M. Prodinger, A. Gori, et al. 2006. Mycobacterium tuberculosis complex genetic diversity: mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology. BMC Microbiol. 6:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Camus, J. C., M. J. Pryor, C. Médigue, and S. T. Cole. 2002. Re-annotation of the genome sequence of Mycobacterium tuberculosis H37Rv. Microbiology 148:2967-2973. [DOI] [PubMed] [Google Scholar]
- 7.Chesne-Seck, M. L., N. Barilone, F. Boudou, J. Gonzalo Asensio, P. E. Kolattukudy, C. Martín, S. T. Cole, B. Gicquel, D. N. Gopaul, and M. Jackson. 2008. A point mutation in the two-component regulator PhoP-PhoR accounts for the absence of polyketide-derived acyltrehaloses but not that of phthiocerol dimycocerosates in Mycobacterium tuberculosis H37Ra. J. Bacteriol. 190:1329-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cole, S. T., R. Brosch, J. Parkhill, T. Garnier, C. Churcher, et al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537-544. [DOI] [PubMed] [Google Scholar]
- 9.Constant, P., E. Perez, W. Malaga, M. A. Lanéelle, O. Saurel, M. Daffé, and C. Guilhot, C. 2002. Role of the pks15/1 gene in the biosynthesis of phenolglycolipids in the Mycobacterium tuberculosis complex. Evidence that all strains synthesize glycosylated p-hydroxybenzoic methyl esters and that strains devoid of phenolglycolipids harbor a frameshift mutation in the pks15/1 gene. J. Biol. Chem. 277:38148-38158. [DOI] [PubMed] [Google Scholar]
- 10.Cox, J. S., B. Chen, M. McNeil, and W. R. Jacobs, Jr. 1999. Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature 402:79-83. [DOI] [PubMed] [Google Scholar]
- 11.Domenech, P., and M. B. Reed. 2009. Rapid and spontaneous loss of phthiocerol dimycocerosate (PDIM) from Mycobacterium tuberculosis grown in vitro: implications for virulence studies. Microbiology 155:3532-3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Filliol, I., A. S. Motiwala, M. Cavatore, W. Qi, M. H. Hazbón, et al. 2006. Global phylogeny of Mycobacterium tuberculosis based on single nucleotide polymorphism (SNP) analysis: insights into tuberculosis evolution, phylogenetic accuracy of other DNA fingerprinting systems, and recommendations for a minimal standard SNP set. J. Bacteriol. 188:759-772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fortune, S. M., A. Jaeger, D. A. Sarracino, M. R. Chase, C. M. Sassetti, D. R. Sherman, B. R. Bloom, and E. J. Rubin. 2005. Mutually dependent secretion of proteins required for mycobacterial virulence. Proc. Natl. Acad. Sci. U. S. A. 102:10676-10681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Froman, S., D. W. Will, A. Blisse, L. J. Conde, I. Krasnow, and E. Bogen. 1955. Bacteriophage susceptibility and cultural characteristics of BCG and other tubercle bacilli. Dis. Chest 28:377-390. [DOI] [PubMed] [Google Scholar]
- 15.Fux, C. A., M. Shirtliff, P. Stoodley, and J. W. Costerton. 2005. Can laboratory reference strains mirror ‘real-world’ pathogenesis? Trends Microbiol. 13:58-63. [DOI] [PubMed] [Google Scholar]
- 16.Gaynor, E. C., S. Cawthraw, G. Manning, J. K. MacKichan, S. Falkow, and D. G. Newell. 2004. The genome-sequenced variant of Campylobacter jejuni NCTC 11168 and the original clonal clinical isolate differ markedly in colonization, gene expression, and virulence-associated phenotypes. J. Bacteriol. 186:503-517. (Erratum, 186:8159.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonzalo-Asensio, J., C. Y. Soto, A. Arbués, J. Sancho, M. del Carmen Menéndez, M. J. García, B. Gicquel, and C. Martín. 2008. The Mycobacterium tuberculosis phoPR operon is positively autoregulated in the virulent strain H37Rv. J. Bacteriol. 190:7068-7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gordon, S. V., B. Heym, J. B. Parkhill, and S. T. Cole. 1999. New insertion sequences and a novel repeated sequence in the genome of Mycobacterium tuberculosis H37Rv. Microbiology 145:881-892. [DOI] [PubMed] [Google Scholar]
- 19.Herring, C. D., A. Raghunathan, C. Honisch, T. Patel, M. K. Applebee, A. R. Joyce, T. J. Albert, F. R. Blattner, D. van den Boom, C. R. Cantor, and B. Ø. Palsson. 2006. Comparative genome sequencing of Escherichia coli allows observation of bacterial evolution on a laboratory timescale. Nat. Genet. 38:1406-1412. [DOI] [PubMed] [Google Scholar]
- 20.Kamerbeek, J., L. Schouls, A. Kolk, M. van Agterveld, D. van Soolingen, S. Kuijper, A. Bunschoten, H. Molhuizen, R. Shaw, M. Goyal, and J. van Embden. 1997. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J. Clin. Microbiol. 35:907-914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kana, B. D., B. G. Gordhan, K. J. Downing, N. Sung, G. Vostroktunova, E. E. Machowski, L. Tsenova, M. Young, A. Kaprelyants, G. Kaplan, and V. Mizrahi. 2008. The resuscitation-promoting factors of Mycobacterium tuberculosis are required for virulence and resuscitation from dormancy but are collectively dispensable for growth in vitro. Mol. Microbiol. 67:672-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kapur, V., T. S. Whittam, and J. M. Musser. 1994. Is Mycobacterium tuberculosis 15,000 years old? J. Infect. Dis. 170:1348-1349. [DOI] [PubMed] [Google Scholar]
- 23.Kurepina, N. E., S. Sreevatsan, B. B. Plikaytis, P. J. Bifani, N. D. Connell, R. J. Donnelly, D. van Sooligen, J. M. Musser, and B. N. Kreiswirth. 1998. Characterization of the phylogenetic distribution and chromosomal insertion sites of five IS6110 elements in Mycobacterium tuberculosis: non-random integration in the dnaA-dnaN region. Tuber. Lung Dis. 79:31-42. [DOI] [PubMed] [Google Scholar]
- 24.Kurtz, S., A. Phillippy, A. L. Delcher, M. Smoot, M. Shumway, C. Antonescu, and S. L. Salzberg. 2004. Versatile and open software for comparing large genomes. Genome Biol. 5:R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lari, N., L. Rindi, and C. Garzelli. 2001. Identification of one insertion site of IS6110 in Mycobacterium tuberculosis H37Ra and analysis of the RvD2 deletion in M. tuberculosis clinical isolates. J. Med. Microbiol. 50:805-811. [DOI] [PubMed] [Google Scholar]
- 26.Larsen, M. H., K. Biermann, S. Tandberg, T. Hsu, and W. R. Jacobs, Jr. 2007. Genetic Manipulation of Mycobacterium tuberculosis. Curr. Protoc. Microbiol. 10:A2.1-A2.21. [DOI] [PubMed] [Google Scholar]
- 27.Larson, C. L., and W. C. Wicht. 1964. Infection of mice with Mycobacterium tuberculosis, strain H37Ra. Am. Rev. Respir. Dis. 90:742-748. [DOI] [PubMed] [Google Scholar]
- 28.Leonard, B. A., M. Woischnik, and A. Podbielski. 1998. Production of stabilized virulence factor-negative variants by group A streptococci during stationary phase. Infect. Immun. 66:3841-3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marshak, A. 1951. Differences in response of a virulent strain of the tubercle bacillus and its avirulent variant to metabolites and their genetic significance. J. Bacteriol. 61:1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mathema, B., N. E. Kurepina, P. J. Bifani, and B. N. Kreiswirth. 2006. Molecular epidemiology of tuberculosis: current insights. Clin. Microbiol. Rev. 19:658-685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McEvoy, C. R., A. A. Falmer, N. C. Gey van Pittius, T. C. Victor, P. D. van Helden, and R. M. Warren. 2007. The role of IS6110 in the evolution of Mycobacterium tuberculosis. Tuberculosis (Edinburgh) 87:393-404. [DOI] [PubMed] [Google Scholar]
- 32.Molina-Torres, C. A., J. Castro-Garza, J. Ocampo-Candiani, M. Monot, S. T. Cole, and L. Vera-Cabrera. 2010. Effect of serial subculturing on the genetic composition and cytotoxic activity of Mycobacterium tuberculosis. J. Med. Microbiol. 59:384-391. [DOI] [PubMed] [Google Scholar]
- 33.Parish, T., D. A. Smith, S. Kendall, N. Casali, G. J. Bancroft, and N. G. Stoker. 2003. Deletion of two-component regulatory systems increases the virulence of Mycobacterium tuberculosis. Infect. Immun. 71:1134-1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Plikaytis, B. B., J. L. Marden, J. T. Crawford, C. L. Woodley, W. R. Butler, and T. M. Shinnick. 1994. Multiplex PCR assay specific for the multidrug-resistant strain W of Mycobacterium tuberculosis. J. Clin. Microbiol. 32:1542-1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raman, S., X. Puyang, T. Y. Cheng, D. C. Young, D. B. Moody, and R. N. Husson. 2006. Mycobacterium tuberculosis SigM positively regulates Esx secreted protein and nonribosomal peptide synthetase genes and down regulates virulence-associated surface lipid synthesis. J. Bacteriol. 188:8460-8468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosenberg, N. A., A. G. Tsolaki, and M. M. Tanaka. 2003. Estimating change rates of genetic markers using serial samples: applications to the transposon IS6110 in Mycobacterium tuberculosis. Theor. Popul. Biol. 63:347-363. [DOI] [PubMed] [Google Scholar]
- 37.Sampson, S., R. Warren, M. Richardson, G. van der Spuy, and P. van Helden. 2001. IS6110 insertions in Mycobacterium tuberculosis: predominantly into coding regions. J. Clin. Microbiol. 39:3423-3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singh, K. K., N. Sharma, D. Vargas, Z. Liu, J. T. Belisle, V. Potharaju, A. Wanchu, D. Behera, and S. Laal. 2009. Peptides of a novel Mycobacterium tuberculosis-specific cell wall protein for immunodiagnosis of tuberculosis. J. Infect. Dis. 200:571-581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh, P. P., M. Parra, N. Cadieux, and M. J. Brennan. 2008. A comparative study of host response to three Mycobacterium tuberculosis PE_PGRS proteins. Microbiology 154:3469-3479. [DOI] [PubMed] [Google Scholar]
- 40.Speirs, R. J., J. T. Welch, and M. H. Cynamon. 1995. Activity of n-propyl pyrazinoate against pyrazinamide-resistant Mycobacterium tuberculosis: investigations into mechanism of action of and mechanism of resistance to pyrazinamide. Antimicrob. Agents Chemother. 39:1269-1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sreevatsan, S., X. Pan, K. E. Stockbauer, N. D. Connell, B. N. Kreiswirth, T. S. Whittam, and J. M. Musser. 1997. Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global dissemination. Proc. Natl. Acad. Sci. U. S. A. 94:9869-9874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steenken, W. 1935. Lysis of tubercle bacilli in vitro. Proc. Soc. Expl. Biol. Med. 33:253-255. [Google Scholar]
- 43.Steenken, W., W. H. Oatway, and S. A. Petroff. 1934. Biological studies of the tubercle bacillus. III. Dissociation and pathogenicity of the R and S variants of the human tubercle bacillus (H37). J. Exp. Med. 60:515-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsolaki, A. G., A. E. Hirsh, K. DeRiemer, J. A. Enciso, M. Z. Wong, M. Hannan, Y. O. Goguet de la Salmoniere, K. Aman, M. Kato-Maeda, and P. M. Small. 2004. Functional and evolutionary genomics of Mycobacterium tuberculosis: insights from genomic deletions in 100 strains. Proc. Natl. Acad. Sci. U. S. A. 101:4865-4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vera-Cabrera, L., M. A. Hernández-Vera, O. Welsh, W. M. Johnson, and J. Castro-Garza. 2001. Phospholipase region of Mycobacterium tuberculosis is a preferential locus for IS6110 transposition. J. Clin. Microbiol. 39:3499-3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang, G., M. Z. Humayun, and D. E. Taylor. 1999. Mutation as an origin of genetic variability in Helicobacter pylori. Trends Microbiol. 7:488-493. [DOI] [PubMed] [Google Scholar]
- 47.World Health Organization. 2008. Anti-tuberculosis drug resistance in the world, report no. 4. http://www.who.int/tb/publications/2008/drs_report4_26feb08.pdf.
- 48.Zheng, H., L. Lu, B. Wang, S. Pu, X. Zhang, G. Zhu, W. Shi, L. Zhang, H. Wang, S. Wang, G. Zhao, and Y. Zhang. 2008. Genetic basis of virulence attenuation revealed by comparative genomic analysis of Mycobacterium tuberculosis strain H37Ra versus H37Rv. PLoS One 3:e2375. [DOI] [PMC free article] [PubMed] [Google Scholar]