

Abstract
The mechanisms of the primary adaptive immune response to Coxiella burnetii are not well known. Following inoculation of the lungs with C. burnetii Nine Mile phase I (NMI), SCID mice developed pneumonia and splenomegaly and succumbed to infection, whereas wild-type mice cleared the infection by 24 days. SCID mice reconstituted with either CD4+ T cells or CD8+ T cells alone were able to control the infection, indicating that the presence of either type of T cells was sufficient to control infection, and B cells were not necessary for primary immunity. Similarly, wild-type mice depleted of either CD4+ T cells or CD8+ T cells controlled infections in their lungs, but these mice were highly susceptible if they were depleted of both types of T cells. However, compared to CD4+ T-cell-dependent protection, CD8+ T-cell-dependent protection resulted in less inflammation in the lungs and less growth of bacteria in the spleens.
Coxiella burnetii, the etiologic agent of Q fever, is thought to be a widely underdiagnosed cause of pneumonia. Acute infections with this organism commonly result in a self-limiting, febrile illness with pulmonary involvement, reflecting the typical acquisition of the infection by the aerosol route. Complications associated with such infections include development of a chronic phase in certain susceptible individuals which presents as endocarditis and has a high fatality rate in the absence of appropriate treatment. Interest in this organism has recently been piqued by its inclusion on the list of potential bioterror agents. Notwithstanding the relatively low mortality rate associated with C. burnetii infections, this organism is highly infectious and has the capacity to cause significant morbidity (16, 23).
Two phase variants C. burnetii have been found; phase I is highly virulent and is the naturally occurring variant, and phase II occurs following repeated passage through cell cultures. The two phases differ in lipopolysaccharide (LPS) structure. Phase I C. burnetii encodes a complete LPS with an O side chain, while phase II C. burnetii expresses a truncated LPS lacking the O side chain and some additional sugar residues (10). Andoh et al. (2) examined the comparative virulence of the two variants in SCID and immunocompetent mice using the intraperitoneal (i.p.) route of inoculation and demonstrated that some replication of C. burnetii Nine Mile phase II (NMII) took place in immunocompromised mice but not in immunocompetent mice. This finding suggests that an acquired immune system is required for control of infection with this organism (3).
Aerosols are thought to be the most common cause of transmission of Coxiella to humans and other mammals. However, very little is known about the effects of this route of infection at the cellular level. To date, most studies using animal models of C. burnetii infection have utilized the intraperitoneal (i.p.) route of inoculation, and while these studies have provided important data, this route of infection may not entirely reproduce the pulmonary sequelae of most human infections (3). Studies of pulmonary Coxiella infection in guinea pigs (15) and in BALB/c and SCID mice (21) demonstrated that lymphocytes accumulate early during primary lung infection. However, the subsets of lymphocytes elicited in the primary pulmonary response and the role of each subset were not clearly defined. The availability of a protective vaccine against C. burnetii has enabled studies of the immune response to postvaccine Coxiella challenge, which showed that the adaptive immune response is involved in successful resolution of postvaccination Coxiella infections. Indeed, studies using vaccination models have suggested that the predominant immune response to i.p. infection is T cell mediated (12). Immunized B-cell-deficient mice are capable of clearing i.p. delivered C. burnetii NMI, although the mice exhibit histopathological changes, suggesting that B cells may be important for controlling inflammatory damage during a secondary response, possibly through production of interleukin-10 (IL-10) (3).
MATERIALS AND METHODS
Bacterial strains.
A C. burnetii Nine Mile phase I (NMI) strain (strain RSA493) was kindly donated by Robert Heinzen (Rocky Mountain Labs, Hamilton, MT).
Animals.
All procedures performed were approved by the Institutional Animal Care and Use Committee at Montana State University, Bozeman, MT. The mice used for the NMI depletion experiment were 7-week-old male BALB/c and SCID mice obtained from Simonsen, Gilroy CA. The mice used for the NMI reconstitution experiment were 6-week-old male SCID mice obtained from NCI, Rockville MD.
Reconstitution of CD4+ and CD8+ T cells.
At 4 days and 1 day prior to reconstitution, BALB/c donor mice were depleted of either CD4+ T cells or CD8+ T cells by injection of 500 μl of either Tib-210 (ATCC, Manassas, VA) to deplete CD8+ T cells or Gk1.5 (ATCC, Manassas, VA) to deplete CD4+ T cells. Whole-spleen-cell donor mice were not depleted of either CD4+ T cells or CD8+ T cells. Donor mice were asphyxiated with CO2 gas, and their spleens were excised, homogenized by passage through a wire mesh screen, and filtered. CD4+ T-cell-depleted or CD8+ T-cell-depleted donor spleens were resuspended in sterile Hanks' buffered saline solution (HBSS) containing 10% fetal bovine serum (FBS), and the whole donor spleens were resuspended in sterile HBSS. Cells were pelleted by centrifugation at 1,000 rpm for 8 min. The supernatant was discarded, and the red cells were lysed in 5 ml sterile filtered ACK lysis buffer (0.15 M NH4Cl, 1.0 mM KHCO3, 0.1 mM Na2EDTA; pH 7.2 to 7.4). Homogenates were washed in sterile HBSS with 10% FBS, and the cells were pelleted by centrifugation at 1,000 rpm for 8 min. CD8+ T-cell donor spleen cells were resuspended in 2 ml column wash buffer, and CD4+ T-cell donor spleen cells were resuspended in 4 ml column wash buffer; then the cells were enumerated. Cells were then purified using an R&D Systems (Minneapolis, MN) column kit for the mouse T-cell CD4+/CD8+ subset according to the manufacturer's instructions. Cells were enumerated prior to reconstitution and checked for purity by fluorescence-activated cell sorting (FACS) analysis, and they were found to be >98% pure. Two groups of SCID recipient mice were then reconstituted with either 3.27 × 106 CD4+ T cells or 2.033 × 106 CD8+ T cells. A third group of SCID mice were given a combination of 3.27 × 105 CD4+ T cells and 2.033 × 105 CD8+ T cells. Cells were delivered in 200 to 300 μl intravenously (i.v.) in the recipient mouse tail vein 1 day prior to infection with C. burnetii. Following euthanasia the lungs of CD4+ T-cell-reconstituted mice contained about 88% CD4+ T cells, and the lungs of CD8+ T-cell-reconstituted mice contained about 90% CD8+ T cells.
Depletion of CD4+ T cells and/or CD8+ T cells.
Cells in wild-type BALB/c mice were depleted by intraperitoneal inoculation of 300 μg of Tib-210, 300 μg of GK1.5, or both Tib-210 and GK1.53 days prior to intratracheal (i.t.) inoculation of NMI. The depletion treatments were repeated at 3-day intervals throughout the experiment. Bronchoalveolar lavage fluid (BALF) cells were analyzed after euthanasia, and the numbers of CD4+ T cells and CD8+ T cells were determined by FACS analysis, which indicated that in replicate experiments there was 82.4 to 100% depletion of the targeted cell subset accumulating in the lungs of infected mice.
i.t. infection of mice.
Mice were infected intratracheally (i.t.) with 103 genome copies of NMI in 100 μl phosphate-buffered saline (PBS) per mouse. Body weights were recorded at approximately 3-day intervals. At 23 and 24 days postinfection (p.i.) (depleted and reconstituted groups, respectively), mice were euthanized by phenobarbital anesthesia, and this was followed by exsanguination. Spleens were removed and weighed, and then they were divided in half and weighed again. Finally, they were homogenized in Dulbecco modified Eagle medium (DMEM) with 10% FBS. One-half of each lung was homogenized in medium as described above for the spleen. Homogenates of spleens and lungs were snap-frozen in liquid nitrogen and stored for future DNA extraction and quantification of C. burnetii by quantitative real-time PCR (RT-PCR).
Bronchoalveolar lavage.
Mice were euthanized by phenobarbital anesthesia, and this was followed by exsanguination. A tube was inserted through a small hole in the trachea, and five 1-ml aliquots of Hanks' balanced salt solution with 3 mM/liter EDTA were used to lavage the lungs. Total cell counts were determined for an aliquot of the lavage fluid, and 100 μl of the lavage fluid was applied to a slide using a cytospin centrifuge and stained with Diff-Quik dye (Dade Behring, Newark, DE). The proportion of each cell type was determined microscopically (22). Lavage fluid was then centrifuged at 1,000 × g for 8 min, and the cell pellets were reserved for FACS analysis.
Quantitative PCR.
DNA was extracted from lung and spleen homogenates using a Qiagen DNeasy blood and tissue kit according to manufacturer's instructions. DNA was used as a template for quantitative real-time PCR with SYBR green PCR master mixture (Applied Biosystems, Foster City, CA), in which the number of C. burnetii genome copies was determined from the number of amplified rpoS gene copies (7). PCRs were performed with an Applied Biosystems 7500 real-time PCR system (Applied Biosystems). The number of genome copies per organ was determined as described previously (7).
Histopathology.
The left lobe of a lung and one-half of a spleen were collected in 10% buffered formalin. Fixed tissues were mounted in paraffin, sectioned, and stained with hematoxylin and eosin (H&E).
Statistical analysis.
Results are expressed below as means and standard errors of the means. PCR data were log transformed, and a statistical analysis was conducted using a one-way, nonparametric analysis of variance (ANOVA) test, followed by posttest Tukey analysis for experiments performed with more than two groups. Time course experiments were analyzed using a two-way ANOVA with Bonferroni posttests. A difference was considered significant if the P value was <0.05.
RESULTS
NMI infection in immunocompetent and immunocompromised mice.
Immunocompetent BALB/c mice and immunocompromised SCID mice were infected with 103 genome copies of NMI intratracheally, and body weights were monitored at approximately 3-day intervals from zero time to 24 days postinfection (p.i.). At day 6 p.i., both groups of mice had lost weight (Fig. 1A). SCID mice had lost 14.7% of their initial body weight, and BALB/c mice had lost 6.5% of their initial body weight. The difference was significant (P < 0.001). Both SCID and BALB/c mice gained weight between days 6 and 9 p.i. However, SCID mice then continued to lose weight until 24 days p.i., when these mice were deemed moribund and showed clinical signs of infection, including dehydration and immobility. BALB/c mice gained weight steadily until 14 days p.i., when there was a slight decrease in weight for this group. BALB/c mice then continued to gain weight until 24 days p.i., when the experiment was terminated. Mice were euthanized at 2, 9, 16, and 24 days p.i., and the spleens were examined to determine if there were any changes. Splenomegaly, determined based on the spleen weight as a percentage of the total body weight, was detected in both BALB/c and SCID mice by day 9 p.i. (Fig. 1B), but at day 16 p.i. the splenomegaly in SCID mice was significantly greater than that in BALB/c mice (P < 0.001). The sizes of the spleens of SCID mice continued to increase, and at 24 days p.i. the spleens were still significantly larger than those of BALB/c mice (P < 0.001). At 24 days the SCID mice were deemed moribund, and the spleens were accounted for an average of 6.1% of the total mouse body weight (Fig. 1B). The BALB/c mice exhibited splenomegaly at a much lower rate than the SCID mice, although changes in spleen size were detected by 9 days p.i. In contrast to the results obtained for the SCID mice, splenomegaly peaked in the BALB/c mice by 16 days p.i. and the spleen weight accounted for 1.68% of the total body weight, and by day 24 p.i. the weights of the spleens of the BALB/c mice had decreased (Fig. 1B).
FIG. 1.
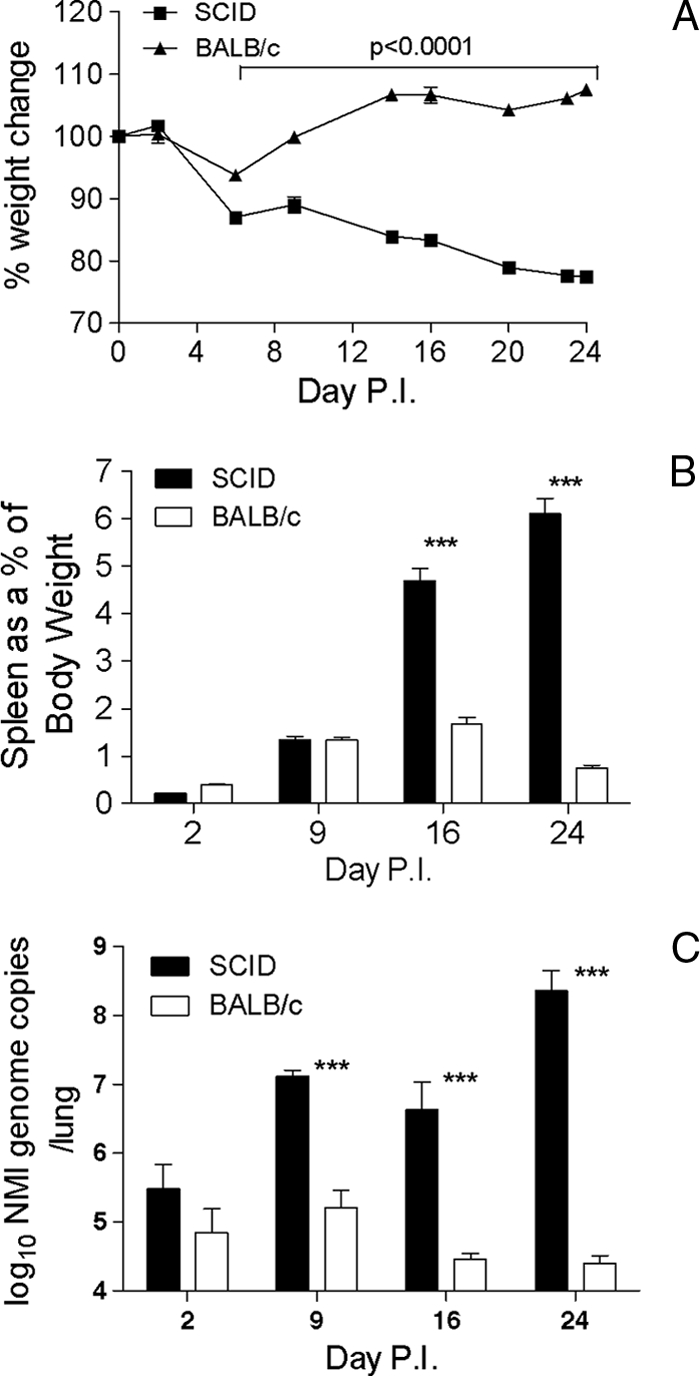
Changes in body weights, spleen weights, and NMI burdens in the lungs of BALB/c mice and SCID mice infected with 103 genome copies of NMI. (A) Body weights expressed as a percentage of the initial body weight. Significant differences were found at each time point between 6 and 24 days. (B) Splenomegaly: spleen weights expressed as a percentage of body weight (P < 0.001). (C) NMI burdens in lungs of BALB/c mice and SCID mice infected with 103 genome copies of NMI (expressed as number of genome copies per lung) determined by RT-PCR (P < 0.001). The data are representative of the results of one of at least two independent experiments performed with five or six mice per group.
Examination of the genome copy numbers in the lungs of BALB/c and SCID mice at 2, 9, 16, and 24 days postinfection indicated that by 2 days p.i. the initial NMI 103-genome copy inoculant had replicated in the lungs of both BALB/c and SCID mice (Fig. 1C) so that there were >105 genome copies. Between days 2 and 9 p.i. in immunocompetent BALB/c mice the bacterial genome copy number remained constant. However, in these mice at day 16 p.i. there was a sharp decrease in the NMI genome copy number, indicating that there was clearance of the pathogen from the lungs. Between days 16 and 24 p.i., however, the NMI genome copy number remained constant, indicating that the infection was not completely resolved, although the mice did not exhibit any overt clinical signs of disease (Fig. 1C). In SCID mice, replication of NMI occurred between days 2 and 9 p.i., but between days 9 and 16 p.i. the NMI genome copy number remained constant. However, between days 16 and 24 p.i. there was significant bacterial replication again (Fig. 1C).
Development of NMI-associated pneumonia in immunocompetent and immunocompromised mice.
Examination of the lungs of immunocompetent and SCID mice at 2, 9, 16, and 24 days p.i. indicated that there were a number of changes. At day 9 SCID mice exhibited airway epithelial hypertrophy (Fig. 2A). Inflammatory changes characterized by marked cell infiltration in the alveoli, as well as the perivascular and peribronchiolar interstitium, were also observed. By day 16 p.i., the bronchiolar epithelial hypertrophy had increased and edema and perivascular and peribronchiolar cuffing persisted. In addition, these mice exhibited signs of bronchiolization and partial lung consolidation (Fig. 2B). The lung pathology in SCID mice at 24 days p.i. resembled that seen at day 16 p.i. Epithelial hypertrophy and extensive cell infiltration and bronchiolization were clearly apparent (Fig. 2C).
FIG. 2.

Changes in lungs of SCID and BALB/c mice infected with 103 genome copies of NMI. At each time point lungs were fixed in formalin and stained with H&E. (A) Epithelial hypertrophy in a lung of a SCID mouse at 9 days p.i. (indicated by an arrow). (B) Epithelial hypertrophy (arrow a), edema (arrow b), bronchiolization (arrow c), and cell infiltration (arrow d) in a lung of a SCID mouse at 16 days p.i. (C) Bronchiolization in a lung of a SCID mouse at 24 days p.i. (indicated by an arrow). (D). Epithelial hypertrophy (arrow a) and cell infiltration (arrow b) in a lung of a BALB/c mouse at 9 days p.i. (E) Reduced epithelial hypertrophy (arrow a) and some cell infiltration (arrow b) in a lung of a BALB/c mouse at 16 days p.i. (F) Lung of a BALB/c mouse at 24 days p.i. (G). Lung of an uninfected BALB/c mouse. Original magnification, ×100.
In immunocompetent mice, the inflammatory changes peaked at day 9 p.i. Epithelial hypertrophy was also visible in these mice, and cell infiltration leading to moderate perivascular and peribronchiolar cuffing was clearly observed (Fig. 2D). By days 16 and 24 p.i. the inflammatory changes in the lung were considerably reduced (Fig. 2E and F). Airway epithelial hypertrophy was present but to a much lesser extent than in the same mice at day 9 p.i. and in SCID mice at all time points. At both 16 and 24 days p.i. there was some evidence of cell infiltration concentrated in the interstitial spaces between the bronchioles. Bronchiolization was not observed in immunocompetent mice at any time point.
Effect of depletion of CD4+ T cells and CD8+ T cells on translocation of NMI from the lung to the spleen.
BALB/c mice were depleted of CD4+ T cells, CD8+ T cells, or both T-cell subsets, and the resulting mice, in addition to SCID mice, were infected with 103 NMI genome copies. After 23 days, both the SCID mice and the BALB/c mice depleted of both T-cell subsets were deemed moribund. Previous experiments indicated that by this time point infections in immunocompetent mice are largely resolved, although bacteria are still detectable. By day 23 p.i., both SCID mice and BALB/c mice depleted of both CD4+ T cells and CD8+ T cells showed splenomegaly that was significantly greater than that in infected undepleted wild-type mice (P < 0.001 and P < 0.001, respectively) (Fig. 3A). Splenomegaly was significantly greater in the mice depleted of both types of T cells than in the SCID mice (P < 0.001). Depletion of CD8+ T cells led to splenomegaly that was significantly more severe than that in the infected wild-type mice (P < 0.05). There was no significant difference in spleen weight between mice depleted of CD4+ T cells and infected undepleted wild-type mice. The data indicate that while either CD4+ T cells or CD8+ T cells are sufficient to control splenomegaly due to NMI infection, CD8+ T cells may control splenomegaly and translocation from the lungs to the spleen more effectively.
FIG. 3.
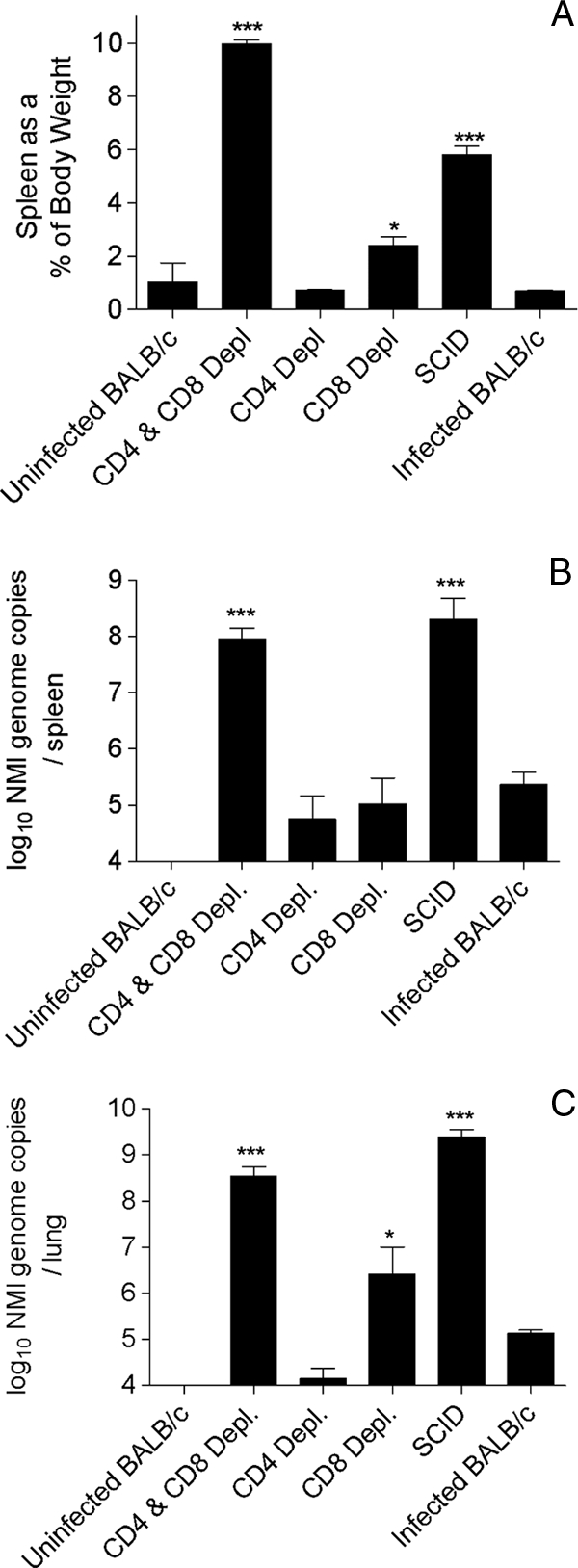
Infection of BALB/c mice, T-cell-depleted mice, and SCID mice with 103 genome copies of NMI. Mice were euthanized at 23 days p.i. Spleen weights were recorded, and bacterial burdens in the spleens and lungs were determined. (A) Spleen weights expressed as a percentage of the mouse body weight at the time of euthanasia. *, P < 0.05; ***, P < 0.001. Depl, depleted. (B) NMI bacterial burdens in the spleen determined by quantitative RT-PCR. ***, P < 0.001. (C) NMI bacterial burdens in the lungs of BALB/c and T-cell-depleted BALB/c mice as determined by quantitative RT-PCR. *, P < 0.05; ***, P < 0.001. The data are representative of the results of one of at least two independent experiments performed with five mice per group.
In the spleens of SCID mice and mice depleted of both CD4+ T cells and CD8+ T cells there was significantly more bacterial DNA than there was in the spleens of in the infected wild-type mice (P < 0.001 and P < 0.001) (Fig. 3B). One sample from the doubly depleted group was lost and could not be amplified by PCR and thus was not included in the spleen PCR data set. The bacterial loads in mice depleted of either CD4+ T cells or CD8+ T cells were not significantly different from those in the infected wild-type mice. In conclusion, either CD4+ T cells or CD8+ T cells were sufficient to control bacterial translocation to the spleen and/or to control extensive bacterial replication in the spleen.
Effect of depleting CD4+ T cells and CD8+ T cells on resolution of NMI infection in the lung.
Mice infected with 103 genome copies of NMI had detectable amounts of bacterial DNA in their lungs at day 23 p.i. (Fig. 3C). The bacterial loads were significantly greater in mice depleted of both CD4+ T cells and CD8+ T cells and in SCID mice than in infected undepleted wild-type mice (P < 0.001 and P < 0.001, respectively). Mice depleted of CD8+ T cells had more bacterial DNA in their lungs than infected undepleted wild-type mice (P < 0.05). In this experiment either CD4+ T cells or CD8+ T cells were able to control NMI loads in the lungs of infected mice.
Development of NMI-associated pneumonia in CD4+ T-cell-depleted mice and CD8+ T-cell-depleted mice.
Consistent with the PCR and splenomegaly data, mice depleted of CD4+ T cells exhibited lower levels of inflammation than mice depleted of CD8+ T cells (Fig. 4). There were also minimal cell infiltration and minimal epithelial cell hypertrophy in the CD4+ T-cell-depleted mice, indicating that CD8+ T-cell-mediated protection resulted in less pathology than CD4+ T-cell-mediated protection (Fig. 4A). Thus, in mice depleted of CD8+ T cells there was increased cell infiltration in the perivascular and peribronchiolar interstitium, and airway epithelial hypertrophy was also slightly increased compared with that in mice depleted of CD4+ T cells (Fig. 4B). Mice depleted of both T-cell subsets had extensive inflammatory changes, which were characterized by edema and cell infiltration in the perivascular and peribronchiolar interstitium (Fig. 4C). Bronchiolization was also detected in mice depleted of both CD4+ T cells and CD8+ T cells. Undepleted BALB/c mice exhibited minimal pathology associated with pulmonary infection. Some cell infiltration was apparent in the interstitial spaces between airways; however, there was no edema, and there was minimal hypertrophy of airway epithelial cells (Fig. 4D). SCID mice exhibited bronchiolization, epithelial hypertrophy, and cell infiltration in the perivascular and peribronchiolar interstitium (Fig. 4E).
FIG. 4.

Changes in lungs of BALB/c, T-cell-depleted BALB/c mice, and SCID mice infected with 103 genome copies of NMI. Mice were euthanized at 23 days p.i., and lungs were fixed in formalin and stained with H&E. (A) Lung of a CD4+ T-cell-depleted mouse showing minimal epithelial hypertrophy (arrow a) and cell infiltration (arrow b). (B) Lung of a CD8+ T-cell-depleted mouse showing modest cellular infiltration (arrow a) and marked epithelial hypertrophy (arrow b). (C) Lung of a BALB/c mouse depleted of both CD4+ T cells and CD8+ T cells showing extensive cellular infiltration (arrow a), epithelial cell hypertrophy (arrow b), and bronchiolization (arrow c). (D) Lung of an undepleted BALB/c mouse showing minimal cell infiltration and epithelial hypertrophy. (E) Lung of a SCID mouse showing extensive cell infiltration (arrow a), edema (arrow b), epithelial cell hypertrophy (arrow c), and bronchiolization (arrow d). (F) Lung of an uninfected BALB/c mouse. Original magnification 100x.
Effect of CD4+ T-cell reconstitution and CD8+ T-cell reconstitution on resolution of infection in the spleen following a pulmonary NMI infection.
SCID mice reconstituted with CD4+ T cells, CD8+ T cells, or both CD4+ and CD8+ T cells were infected with NMI i.t. and euthanized after 24 days. The spleens of the infected SCID mice accounted for 5.3% of the total body weight at 24 days p.i. (Fig. 5A) and were significantly larger than the spleens of infected BALB/c mice and SCID mice reconstituted with either CD8+ T cells or both CD8+ T cells and CD4+ T cells (P < 0.001). However, the sizes of the spleens of infected SCID mice reconstituted with CD4+ T cells were comparable to the sizes of the spleens of the infected SCID mice; the spleens accounted for 4.2% of the total body weight but were still significantly smaller than those of infected SCID mice (P < 0.05). This result is similar to that obtained for the infected BALB/c mice depleted of CD8+ T cells, again indicating that CD4+ T-cell-mediated protection results in greater splenomegaly than CD8+ T-cell-mediated protection. By day 24 p.i., bacteria had translocated to the spleen in all groups of mice; however, levels of bacteria in the spleens of the BALB/c mice or the mice reconstituted with CD4+ T cells, CD8+ T cells, or both CD4+ T cells and CD8+ T cells were significantly lower than the levels in the spleens of the infected SCID mice (P < 0.001) (Fig. 5B).
FIG. 5.
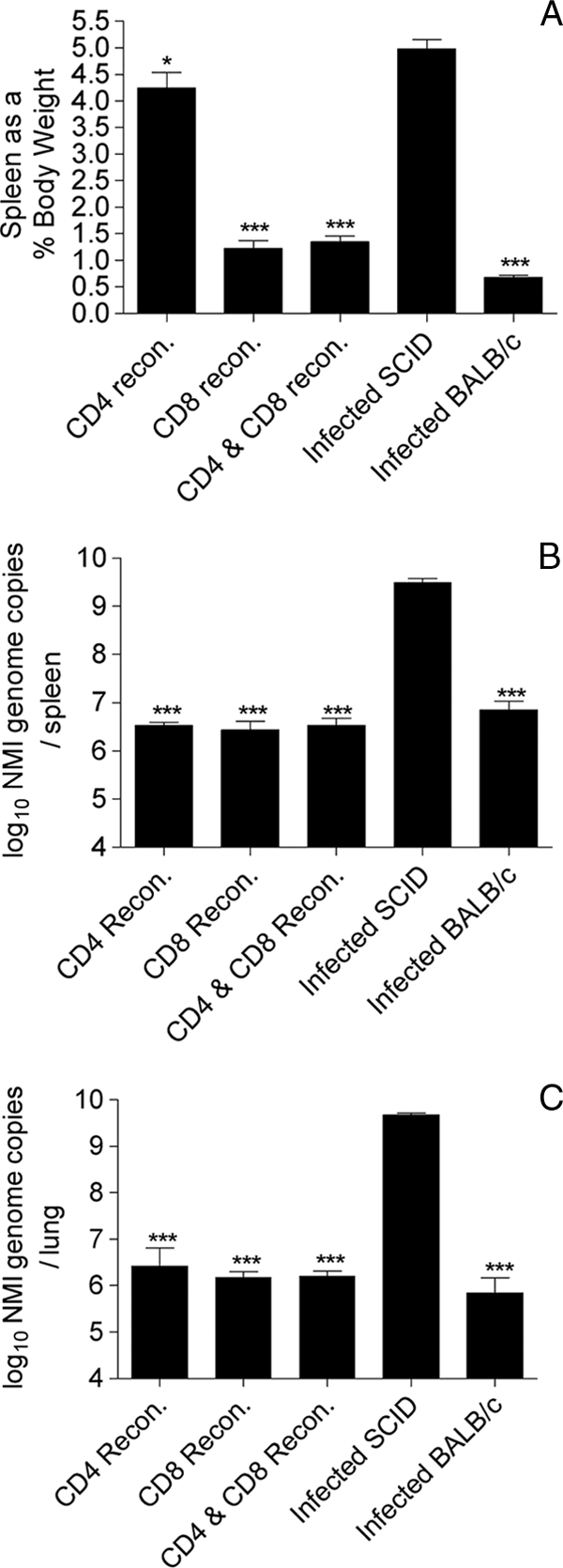
Infection of BALB/c, SCID, and T-cell-reconstituted SCID mice with 103 genome copies of NMI. Mice were euthanized at 24 days p.i., spleen weights were recorded, and the bacterial burdens in both the spleens and lungs were determined. (A) Spleen weights expressed as a percentage of the mouse body weight at the time of euthanasia. ***, P < 0.001. recon., reconstituted. (B) NMI bacterial burdens in the spleen determined by quantitative RT-PCR. ***, P < 0.001. (C) NMI bacterial burdens in the lungs of BALB/c and T-cell-depleted BALB/c mice determined by quantitative RT-PCR. ***, P < 0.001. The data are representative of the results of one of at least two independent experiments performed with six mice per group.
Mice infected with NMI and reconstituted with CD4+ T cells, CD8+ T cells, or both CD4+ T cells and CD8+ T cells were able to control the infection in their spleens, indicating that either type of cells can confer protection. However, SCID mice reconstituted with CD4+ T cells alone exhibited greater splenomegaly and had a greater bacterial burden than mice reconstituted with CD8+ T cells alone, suggesting that CD8+ T cells may play a more significant role in controlling splenomegaly and/or translocation of bacteria from the lung to the spleen.
Effect of CD4+ T-cell reconstitution and CD8+ T-cell reconstitution on resolution of infection in the lungs of mice infected with NMI.
Infection of SCID mice with NMI resulted in high numbers of bacteria in the lungs at 24 days p.i. (Fig. 5C). In contrast, reconstitution of SCID mice with CD4+ T cells, CD8+ T cells, or both CD4+ T cells and CD8+ T cells controlled the number of bacteria, and the NMI burden in the lungs was significantly lower than that in the lungs of infected unreconstituted SCID mice (P < 0.001).
Development of NMI-associated pneumonia in CD4+ T-cell-reconstituted mice and CD8+ T-cell-reconstituted mice.
NMI-infected SCID mice reconstituted with only CD4+ T cells exhibited more histopathological changes than mice reconstituted with only CD8+ T cells. Cellular hypertrophy was visible along with extensive cell infiltration in the perivascular and peribronchiolar interstitium and was accompanied by edema (Fig. 6A). There was minimal cell infiltration and epithelial cell hypertrophy in the CD8+ T-cell-reconstituted mice, indicating that CD8+ T-cell-mediated protection resulted in less pathology than CD4+ T-cell-mediated protection (Fig. 6B). Mice reconstituted with both T-cell subsets had minimal inflammatory changes, and their lungs resembled the lungs of infected wild-type mice (Fig. 6C). SCID mice exhibited clear signs of bronchiolization, marked epithelial hypertrophy, and inflammatory cell infiltration in the perivascular and peribronchiolar interstitium (Fig. 6D). Infected BALB/c mice exhibited minimal cell infiltration that was confined to the interstitial spaces between airways. However, there was no visible edema, and there was minimal hypertrophy of airway epithelial cells (Fig. 6E).
FIG. 6.

Changes in lungs of SCID mice reconstituted with CD4+ T cells, CD8+ T cells, or both CD4+ T cells and CD8+ T cells, nonreconstituted SCID mice, and BALB/c mice infected with 103 genome copies of NMI. Mice were euthanized at 24 days p.i., and lungs were fixed in formalin and stained with H&E. (A) Lung of a CD4+ T-cell-reconstituted mouse showing inflammatory cell infiltration (arrow a) and marked epithelial cell hypertrophy (arrow b). (B) Lung of a CD8+ T-cell-reconstituted mouse showing some cellular infiltration (arrow a) and minimal epithelial cell hypertrophy (arrow b). (C) Lung of a mouse reconstituted with both CD4+ T cells and CD8+ T cells exhibiting some minimal cell infiltration (arrow a) and epithelial cell hypertrophy (arrow b). (D) Lung of a SCID mouse showing extensive inflammatory changes, including marked cellular infiltration (arrow a), edema (arrow b), airway epithelial cell hypertrophy (arrow c), and bronchiolozation (arrow d). (E) Lung of an infected BALB/c mouse showing minimal cell infiltration (arrow a) and minimal changes in airway epithelial cells (arrow b). (F) Lung of an uninfected BALB/c mouse. Original magnification, ×100.
DISCUSSION
Previous studies by other groups suggested that T cells are important in resolving infection with either NMI or NMII (1-3, 6, 8, 11, 14). By either reconstituting immunodeficient SCID mice with CD4+ T cells or CD8+ T cells or depleting immunocompetent BALB/c mice of T-cell subsets, we found that either CD4+ T cells alone or CD8+ T cells alone were sufficient to control lung infections with NMI; comparable results were obtained for lung infections with NMII (data not shown).
Resolution of C. burnetii pneumonia in SCID mice reconstituted with purified CD4+ T cells suggested that certain mechanisms are used for clearing C. burnetii. First, CD4+ T cells, when activated in a major histocompatibility complex (MHC) class II-restricted manner, produce gamma interferon (IFN-γ), which among other things can lead to activation of macrophages, resulting in clearance of infectious agents. CD4+ T cells can recruit phagocytic cells to sites of infection by production of hematopoietic factors, which lead to the upregulation of production of phagocytic cells and chemoattractants that recruit phagocytes. Second, CD4+ T cells can exhibit cytolytic activity (4, 5, 9, 24). It is thought that the number of cytotoxic CD4+ T cells in uninfected individuals is relatively low, although evidence from HIV patients suggests that the levels can increase dramatically (18). It has been proposed that CD4+ T cells have cytolytic activity when they become highly differentiated and that they lose a number of surface markers and subsequently facilitate killing of target cells by a perforin-dependent method (24). Further research is required to examine the potential role of cytotoxic CD4+ T cells in C. burnetii infection. Third, CD4+ T cells play an important role in signaling B cells and subsequently triggering an antibody response against pathogens. However, we demonstrate here that B cells are not required for controlling primary C. burnetii infections as CD4+ T cells (or CD8+ T cells) alone can adequately control an infection. However, this does not mean that when B cells are present, they do not contribute to resistance. Interestingly, as observed for CD8+ T-cell-depleted BALB/c mice or CD4+ T-cell-reconstituted SCID mice, in the absence of CD8+ T cells there were more inflammatory changes in the lungs. Previous studies with other pulmonary pathogens, including Pneumocystis and Mycoplasma, have indicated that in the absence of CD8+ T cells, CD4+ T cells cause considerably more tissue damage (13, 22). This observation has a number of implications for the role that CD8+ T cells play in the immune response to pulmonary C. burnetii infection. It is reasonable to hypothesize that in immunocompetent mice infected with NMI CD8+ T cells play an immunomodulatory role, controlling a damaging CD4+ T-cell response, or, alternatively, that the CD8+ T-cell response to such an infection is less damaging.
In the absence of both CD4+ T cells and B cells, CD8+ T cells were also able to control Coxiella pneumonia. CD8+ T cells are MHC class I restricted and can be cytolytic. When activated, CD8+ T cells can release cytolytic compounds such as perforin, which disrupts the host cell membrane, or granzyme, which can induce apoptosis of infected cells. Apoptosis can also be induced through interactions between a CD8+ T cell and Fas on the surface of a target infected cell. CD8+ T cells can produce type 1 cytokines, including IFN-γ, which can lead to macrophage activation and have other effects, and tumor necrosis factor alpha (TNF-α), which contributes to the pulmonary inflammatory response. CD8+ T cells also played an important role in the development of disease in the spleen. Upon depletion of CD8+ T cells, the infection was controlled. However, the mice had greater splenomegaly and an increased bacterial burden in the spleen. It is not possible to discern from the data presented here whether the CD8+ T cells directly impacted the translocation of bacteria from the lung or whether they enhanced bacterial clearance in the spleen.
These findings also suggested that other immune cells, such as gamma delta T cells, are not likely to be required for controlling primary pulmonary NMI infections. In this regard, BALB/c mice depleted of both CD4+ T cells and CD8+ T cells still contained gamma delta T cells; however, these mice were just as susceptible to infection as SCID mice. The role of B cells in controlling primary infections caused by C. burnetii has not been well studied. Studies of NMI i.p. infections have suggested that B cells may play an important role in controlling tissue damage (3). Antibodies clearly provide protection against NMI i.p. infections since mice immunized with formalin-inactivated NMI exhibited a protective antibody response against further i.p. infection with NMI. Adoptive transfer of sera from immunized mice also conferred protection against a subsequent i.p. NMI infection (25). Collectively, the data presented here indicate that B cells are not required for resolving primary NMI lung infections as SCID mice reconstituted with only T cells, in the absence of B cells, were able to control infections as well as mice reconstituted with both T cells and B cells. It is possible that antibodies do not play a role in resistance to primary infection because the antibody response is not initiated until after the Coxiella cells have infected macrophages, and thus the antibodies would not have access to the organisms. However, antibodies induced by vaccination or primary infection could be effective in clearing a subsequent Coxiella challenge before the bacteria enter macrophages.
Mice are clearly not as susceptible as guinea pigs to pulmonary NMI (20), and this is one of the strengths of the mouse model for studying the immune response, since mechanisms of a highly efficacious immune response to Coxiella can be investigated. Coxiella infections in humans are highly variable but commonly present as a self-limiting, flu-like illness. Frequently exposed individuals may even remain asymptomatic. Thus, serological surveys of blood donors in Netherlands suggested that up to 73% of male donors were seropositive (19), suggesting that exposure to Q fever is widespread. However, in general, there are few reports of the disease and few patients are admitted to hospitals with acute Q fever, with the exception of some outbreaks. Also, for patients diagnosed with acute Q fever the rate of admission to the hospital is only around 5% (17). This suggests that many humans are able to mount a strong primary immune response to Coxiella after infection and that severe underlying pathology and disease do not occur. Thus, the ability of mice to mount a successful primary immune response to pulmonary Coxiella infection provides an opportunity to closely examine this response to pulmonary NMI and determine why the response is so successful. Certainly, as we show in this paper, mice are extremely susceptible to NMI when they are deficient in T cells, which indicates the efficacy of their acquired immune response against Coxiella.
In summary, in a primary adaptive immune response to NMI in mice, either CD4+ T cells or CD8+ T cells are sufficient to resolve an NMI infection, whereas B cells are not required. However, CD8+ T-cell-dependent immunity results in less lung inflammation and less growth of Coxiella in the spleens of intratracheally inoculated mice than CD4+ T-cell-dependent immunity. Previous studies by Zhang et al. (25) indicated that antibodies are important in secondary immunity and that efficacious Coxiella vaccines should target antibody production. Our results suggest that CD8+ T-cell immunity may also be a target of effective vaccination against Coxiella.
Acknowledgments
This work was supported by grants US4AI065357, P20RR020185, and P20RR16455 from the Rocky Mountain Regional Centre of Excellence, Montana State University COBRE, and Montana INBRE, respectively.
We thank Tamara Marcotte, Trent Bushmaker, Matt Calverley, and Ann Harmsen for technical assistance and Laura Richert for critical review of the manuscript.
Editor: R. P. Morrison
Footnotes
Published ahead of print on 29 March 2010.
REFERENCES
- 1.Andoh, M., T. Naganawa, A. Hotta, T. Yamaguchi, H. Fukushi, T. Masegi, and K. Hirai. 2003. SCID mouse model for lethal Q fever. Infect. Immun. 71:4717-4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andoh, M., K. E. Russell-Lodrigue, G. Zhang, and J. E. Samuel. 2005. Comparative virulence of phase I and II Coxiella burnetii in immunodeficient mice. Ann. N. Y. Acad. Sci. 1063:167-170. [DOI] [PubMed] [Google Scholar]
- 3.Andoh, M., G. Zhang, K. E. Russell-Lodrigue, H. R. Shive, B. R. Weeks, and J. E. Samuel. 2007. T cells are essential for bacterial clearance, and gamma interferon, tumor necrosis factor alpha, and B cells are crucial for disease development in Coxiella burnetii infection in mice. Infect. Immun. 75:3245-3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Appay, V. 2004. The physiological role of cytotoxic CD4(+) T-cells: the holy grail? Clin. Exp. Immunol. 138:10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Appay, V., J. J. Zaunders, L. Papagno, J. Sutton, A. Jaramillo, A. Waters, P. Easterbrook, P. Grey, D. Smith, A. J. McMichael, D. A. Cooper, S. L. Rowland-Jones, and A. D. Kelleher. 2002. Characterization of CD4(+) CTLs ex vivo. J. Immunol. 168:5954-5958. [DOI] [PubMed] [Google Scholar]
- 6.Atzpodien, E., W. Baumgartner, A. Artelt, and D. Thiele. 1994. Valvular endocarditis occurs as a part of a disseminated Coxiella burnetii infection in immunocompromised BALB/cJ (H-2d) mice infected with the nine mile isolate of C. burnetii. J. Infect. Dis. 170:223-226. [DOI] [PubMed] [Google Scholar]
- 7.Coleman, S. A., E. R. Fischer, D. Howe, D. J. Mead, and R. A. Heinzen. 2004. Temporal analysis of Coxiella burnetii morphological differentiation. J. Bacteriol. 186:7344-7352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Criley, J. M., A. J. Carty, C. L. Besch-Williford, and C. L. Franklin. 2001. Coxiella burnetii infection in C.B-17 scid-bg mice xenotransplanted with fetal bovine tissue. Comp. Med. 51:357-360. [PubMed] [Google Scholar]
- 9.Fleischer, B. 1984. Acquisition of specific cytotoxic activity by human T4+ T lymphocytes in culture. Nature 308:365-367. [DOI] [PubMed] [Google Scholar]
- 10.Hackstadt, T., M. G. Peacock, P. J. Hitchcock, and R. L. Cole. 1985. Lipopolysaccharide variation in Coxiella burnetti: intrastrain heterogeneity in structure and antigenicity. Infect. Immun. 48:359-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hall, W. C., J. D. White, R. A. Kishimoto, and R. E. Whitmire. 1981. Aerosol Q fever infection of the nude mouse. Vet. Pathol. 18:672-683. [DOI] [PubMed] [Google Scholar]
- 12.Izzo, A. A., B. P. Marmion, and T. Hackstadt. 1991. Analysis of the cells involved in the lymphoproliferative response to Coxiella burnetii antigens. Clin. Exp. Immunol. 85:98-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones, H. P., L. Tabor, X. Sun, M. D. Woolard, and J. W. Simecka. 2002. Depletion of CD8+ T cells exacerbates CD4+ Th cell-associated inflammatory lesions during murine mycoplasma respiratory disease. J. Immunol. 168:3493-3501. [DOI] [PubMed] [Google Scholar]
- 14.Kishimoto, R. A., H. Rozmiarek, and E. W. Larson. 1978. Experimental Q fever infection in congenitally athymic nude mice. Infect. Immun. 22:69-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kishimoto, R. A., B. J. Veltri, F. G. Shirey, P. G. Canonico, and J. S. Walker. 1977. Fate of Coxiella burnetti in macrophages from immune guinea pigs. Infect. Immun. 15:601-607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Madariaga, M. G., K. Rezai, G. M. Trenholme, and R. A. Weinstein. 2003. Q fever: a biological weapon in your backyard. Lancet Infect. Dis. 3:709-721. [DOI] [PubMed] [Google Scholar]
- 17.Maurin, M., and D. Raoult. 1999. Q fever. Clin. Microbiol. Rev. 12:518-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Norris, P. J., M. Sumaroka, C. Brander, H. F. Moffett, S. L. Boswell, T. Nguyen, Y. Sykulev, B. D. Walker, and E. S. Rosenberg. 2001. Multiple effector functions mediated by human immunodeficiency virus-specific CD4+ T-cell clones. J. Virol. 75:9771-9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richardus, J. H., A. Donkers, A. M. Dumas, G. J. Schaap, J. P. Akkermans, J. Huisman, and H. A. Valkenburg. 1987. Q fever in the Netherlands: a sero-epidemiological survey among human population groups from 1968 to 1983. Epidemiol. Infect. 98:211-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Russell-Lodrigue, K. E., G. Q. Zhang, D. N. McMurray, and J. E. Samuel. 2006. Clinical and pathologic changes in a guinea pig aerosol challenge model of acute Q fever. Infect. Immun. 74:6085-6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stein, A., C. Louveau, H. Lepidi, F. Ricci, P. Baylac, B. Davoust, and D. Raoult. 2005. Q fever pneumonia: virulence of Coxiella burnetii pathovars in a murine model of aerosol infection. Infect. Immun. 73:2469-2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swain, S. D., N. N. Meissner, and A. G. Harmsen. 2006. CD8 T cells modulate CD4 T-cell and eosinophil-mediated pulmonary pathology in pneumocystis pneumonia in B-cell-deficient mice. Am. J. Pathol. 168:466-475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tigertt, W. D., A. S. Benenson, and W. S. Gochenour. 1961. Airborne Q fever. Bacteriol. Rev. 25:285-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams, N. S., and V. H. Engelhard. 1996. Identification of a population of CD4+ CTL that utilizes a perforin- rather than a Fas ligand-dependent cytotoxic mechanism. J. Immunol. 156:153-159. [PubMed] [Google Scholar]
- 25.Zhang, G., K. E. Russell-Lodrigue, M. Andoh, Y. Zhang, L. R. Hendrix, and J. E. Samuel. 2007. Mechanisms of vaccine-induced protective immunity against Coxiella burnetii infection in BALB/c mice. J. Immunol. 179:8372-8380. [DOI] [PubMed] [Google Scholar]