

Abstract
Pseudomonas aeruginosa is a pathogenic Gram-negative bacterium that causes severe opportunistic infections in immunocompromised individuals; in particular, severity of infection with P. aeruginosa positively correlates with poor prognosis in cystic fibrosis (CF) patients. Establishment of chronic infection by this pathogen is associated with downregulation of flagellar expression and of other genes that regulate P. aeruginosa motility. The current paradigm is that loss of flagellar expression enables immune evasion by the bacteria due to loss of engagement by phagocytic receptors that recognize flagellar components and loss of immune activation through flagellin-mediated Toll-like receptor (TLR) signaling. In this work, we employ bacterial and mammalian genetic approaches to demonstrate that loss of motility, not the loss of the flagellum per se, is the critical factor in the development of resistance to phagocytosis by P. aeruginosa. We demonstrate that isogenic P. aeruginosa mutants deficient in flagellar function, but retaining an intact flagellum, are highly resistant to phagocytosis by both murine and human phagocytic cells at levels comparable to those of flagellum-deficient mutants. Furthermore, we show that loss of MyD88 signaling in murine phagocytes does not recapitulate the phagocytic deficit observed for either flagellum-deficient or motility-deficient P. aeruginosa mutants. Our data demonstrate that loss of bacterial motility confers a dramatic resistance to phagocytosis that is independent of both flagellar expression and TLR signaling. These findings provide an explanation for the well-documented observation of nonmotility in clinical P. aeruginosa isolates and for how this phenotype confers upon the bacteria an advantage in the context of immune evasion.
Pseudomonas aeruginosa is an opportunistic Gram-negative bacterial pathogen that causes severe infections in immunocompromised patients and in the pulmonary compartment of patients suffering from cystic fibrosis (CF) (13, 14). In CF patients, disease severity is positively correlated with colonization by P. aeruginosa and the establishment of chronic infection. As part of the colonization process, the bacteria undergo a number of genetic changes that assist in their ability to survive in the mammalian host and to evade detection and clearance by the immune system (9, 21). One such change that has been phenotypically characterized for P. aeruginosa is loss of flagellar motility (12, 17). Furthermore, the loss of flagellar gene expression and motility function is associated with increased bacterial burdens and increased disease severity in CF patients (12, 17). While downregulation of flagellar expression has been inferred to confer a survival advantage on P. aeruginosa once it colonizes the host by evasion of both phagocytic receptors and TLR5-driven inflammatory signaling, the exact contribution of flagellum downregulation with respect to successful immune evasion is unclear (5, 17, 18).
Nonopsonic phagocytosis of P. aeruginosa by murine and human macrophages has previously been reported to require the expression of a flagellum, and the interpretation of these results concluded that the flagellum is a necessary ligand for triggering phagocytic internalization of the bacteria (18). Furthermore, flagellar expression is reported to be critical for inducing inflammation during P. aeruginosa infection, and loss of flagellar gene expression results in impaired inflammatory responses and attenuated bacterial clearance (5). Here, we provide data that challenge the current paradigm that the flagellum functions as a primary phagocytic ligand for P. aeruginosa ingestion by immune cells with the formal demonstration that motility, rather than loss of flagellar expression, confers the advantage toward P. aeruginosa evasion of phagocytosis.
In these studies, we use P. aeruginosa motility-defective mutants to assess the role of bacterial motility in regard to phagocytic recognition by innate immune cells. When present in an aqueous environment, P. aeruginosa can swim via rotation of a single polar, monotrichous flagellum (27); there is currently no evidence that P. aeruginosa bacteria produce lateral flagella or alter their cell morphology as a direct function of motility. For the purposes of this report, motility refers to flagellum-based bacterial movement in an aqueous environment unless specifically indicated. Of note, P. aeruginosa is also capable of a flagellum-independent type of motility termed twitching in which type IV pilus filaments that extend from the cell body adhere to a surface and then retract, thus propelling the bacterium forward (23). Bacterial flagellar motility occurs through a motor complex that provides energy for rotational torque of a helical filament of repeating flagellin subunits that act as a propeller. The rotor, a multimer complex composed of FliG, FliM, and FliN, acts as a molecular switch and determines clockwise or counterclockwise rotation (27). In P. aeruginosa, the stator complex, which provides a stationary housing for the rotor, is composed of at least four partially redundant integral membrane proteins, MotAB and MotCD. Deletion of all four stators allows for flagellar assembly, but the structure cannot rotate and so the mutant is nonmotile (swimming and swarming defective) (27).
Previous reports have concluded that an intact flagellum is required for phagocytic recognition of P. aeruginosa (18). However, here we demonstrate that the phagocytic resistance exhibited by swimming motility-defective bacteria is not due to loss of flagellum-mediated activation of immune cells, since bacteria expressing a nonfunctional flagellum exhibit phagocytic resistance comparable to that of flagellum-deficient bacteria and loss of MyD88 signaling in phagocytic cells does not recapitulate this defect for phagocytosis. Rather, with the use of a variety of in vitro, ex vivo, and in vivo infection models, we show that loss of P. aeruginosa motility dramatically alters immune responses to these bacteria compared to those for motile isogenic bacterial strains and that it is the loss of flagellum-mediated motility, but not flagellum expression itself, that results in dramatic bacterial resistance to phagocytosis by murine and human phagocytes. These studies provide an explanation for the clinical observation that P. aeruginosa isolates obtained from CF hosts often exhibit a nonmotile phenotype and explain how this phenotype can confer a survival advantage for bacteria that modulate or lose their motility during an active infection.
MATERIALS AND METHODS
Mice and cells.
C57BL/6 wild-type (WT) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). MyD88−/− mice were generated by Adachi et al. (1). The bone marrow-derived dendritic cell (BMDC) culture protocol used is a modification of the work of Inaba et al. (11) as previously described (4). For these studies, the Pseudomonas aeruginosa clinical isolate PA14 is the parental bacterial strain and wild-type control for all of the isogenic mutants studied. Human peripheral blood monocytes were a generous gift from the lab of Paul Guyre (Dartmouth). Monocytes were differentiated into macrophages by culturing them in the presence of 10 μg/ml granulocyte-macrophage colony-stimulating factor (GM-CSF) for 7 days.
FACS-based bacterial association assay for GFP+ bacteria.
Bacterial strains expressing green fluorescent protein (GFP) were generated by transformation of the indicated strains with a multicopy plasmid (pSMC21 Ampr Kanr Carbr GFP+) that constitutively expresses GFP under the control of a derivative of the Ptac promoter (6, 15). C57BL/6 BMDCs were incubated with the indicated GFP+ bacterial strains for 45 min at 37°C or 4°C, as indicated in the text. For bacterial binding assays, BMDCs were preincubated in 10 μM cytochalasin D (Sigma) in serum-free Hanks balanced salt solution (HBSS) for 60 min at 37°C. Coincubation between BMDCs and the indicated GFP+ bacterial strains took place in the presence of 10 μM cytochalasin D for 45 min at 37°C. Cells were washed thoroughly in serum-free HBSS and then analyzed by fluorescence-activated cell sorting (FACS) for the acquisition of fluorescence as an indication of BMDC association with the bacteria. The mean fluorescence intensities of the BMDC populations were assessed and graphed to obtain relative efficiency of cellular association with the different bacterial strains.
Microscopy.
BMDCs from B6.Cg-Tg(CAG-mRFP1) mice (C57BL/6 background; purchased from Jackson Labs), which express red fluorescent protein (RFP) from the actin promoter, were cultured as previously described (5). Cells were coincubated with GFP-expressing P. aeruginosa strains as indicated for 45 min at 37°C at a multiplicity of infection (MOI) of ∼10. Cells were washed twice in 400 μl of serum-free HBSS prior to a 10-min cytospin onto glass slides at 1,000 rpm. Cells were visualized via fluorescence and differential interference contrast (DIC) microscopy. Microscopy was performed on a Zeiss LSM510 Meta microscope using a 63× lens, followed by image analysis on the LSM5 Image Browser software.
Bacterial motility swimming assay.
Bacterial motility was examined as described elsewhere (7, 27). Briefly, 0.3% agar plates were poured and the indicated bacterial strains were inoculated from liquid cultures onto the plates by puncturing inoculates halfway through the depth of the agar. Plates were incubated at room temperature for 48 h and monitored for the generation of bacterial halos as an indication of swimming motility. Plates were subsequently photographed with a digital camera to allow comparison of the relative swimming motilities of the different bacterial strains.
Western Blot analyses and Coomassie blue staining.
Analysis of total flagellin expression by the indicated bacterial strains was performed by lysis of the whole bacteria in SDS sample buffer containing 5% β-mercaptoethanol. Analysis of the presence of extracellular flagella on the ΔmotAB ΔmotCD mutant was performed as a modification of published methodology (19, 25). Briefly, a 100-ml culture of bacteria was pelleted by centrifugation, resuspended in phosphate-buffered saline (PBS), and sheared for 3 min in a blender. The sheared suspension was centrifuged at 8,000 × g for 10 min, and the protein within the supernatant was subsequently precipitated with the addition of trichloroacetic acid (TCA) to 20%. The precipitated pellet was washed with acetone and resuspended in SDS sample buffer containing 5% β-mercaptoethanol. For both the bacterial lysates and the isolated flagellin, the samples were boiled and separated on 12% SDS-PAGE gels. Parallel lanes were used either for Coomassie blue staining or for transfer onto Immobilon-P membranes (Millipore Corp.) for Western analysis. Following blotting with polyclonal anti-FliC antibody (22), Western blots were developed using enhanced chemiluminescence (ECL) (Amersham Biosciences) of horseradish peroxidase (HRP)-conjugated secondary antibodies (Jackson Immunoresearch).
In vitro gentamicin protection assays.
Phagocytosis of live P. aeruginosa bacteria was performed as a modified version of published protocols (4, 8) and as previously described (2). Briefly, overnight cultures of P. aeruginosa were washed and resuspended in serum-free medium and bacterial concentrations were determined. BMDCs of the indicated genotype (2 × 105) were incubated with bacteria at an MOI of ∼10 for 45 min at 37°C, followed by incubation in 100 μg/ml gentamicin for 20 min at 37°C. For intracellular killing assays, a modification of the previous protocol (3) was used: following a 1-h coincubation of PA14 bacteria and BMDCs, gentamicin was added and, for the following 60 min, aliquots were assayed as described above for remaining CFU. In all cases, cells were washed and subsequently lysed in 500 μl 0.1% Triton X-100 in PBS. Lysates were plated onto Pseudomonas isolation agar and incubated overnight at 37°C. The next day, colonies were counted and relative phagocytosis was determined by CFU counts.
Ex vivo peritoneal phagocyte gentamicin protection assay and FACS analyses.
Phagocytosis of live P. aeruginosa bacteria was performed as a modified version of previously described protocols (4, 8). Overnight cultures of P. aeruginosa were washed twice in 1 ml serum-free HBSS and resuspended in 10 ml HBSS. Bacterial concentrations were determined based on estimated ∼1 × 109 CFU/ml saturation levels. Naïve C57BL/6 mice were injected intraperitoneally (i.p.) with 1 ml of 4% thioglycolate and subsequently sacrificed 4 days later. The peritoneal cavity was lavaged with 6 ml of serum-free HBSS. The lavage fluid was centrifuged, and pelleted cells were washed twice in serum-free HBSS before being resuspended in 2 ml serum-free HBSS. For the gentamicin protection assay, peritoneal phagocytes and bacteria were subsequently treated as described above for in vitro gentamicin protection assays. Alternatively, peritoneal phagocytes were coincubated with GFP-expressing bacteria for 45 min at 37°C. Cells were washed twice in serum-free HBSS and resuspended in 250 μl HBSS. Bacterial association with phagocytes was determined via FACS analysis as described previously.
Ex vivo lung phagocyte gentamicin protection assay and FACS analyses.
Phagocytosis of live P. aeruginosa bacteria was performed as a modified version of previously described protocols (4, 8). Overnight cultures of P. aeruginosa were washed twice in 1 ml serum-free HBSS and resuspended in 10 ml serum-free HBSS. Bacterial concentrations were determined based on estimated ∼1 × 109 CFU/ml saturation levels. Naïve C57BL/6 mice were treated with 150 μg lipopolysaccharide (Sigma) in PBS via tracheobronchial aspiration and were subsequently sacrificed 3 days later. The lungs were lavaged with 800 μl of PBS with 5 mM EDTA. The bronchoalveolar lavage (BAL) fluid was centrifuged, and pelleted lung cells were washed twice in 400 μl serum-free HBSS before being incubated with the indicated bacteria at an MOI of ∼10 for 45 min at 37°C. For the gentamicin protection assay, alveolar phagocytes and bacteria were subsequently treated as described above for in vitro gentamicin protection assays. Alternatively, alveolar phagocytes were coincubated with GFP-expressing bacteria for 45 min at 37°C. Cells were washed twice in serum-free HBSS and resuspended in 250 μl HBSS. Bacterial association with lung phagocytes was determined via FACS analysis as described previously.
In vivo bacterial uptake assay.
Live P. aeruginosa bacteria (5 × 106 or bacterial numbers as indicated) either were injected intraperitoneally or were oropharyngeally aspirated into C57BL/6 mice. Mice were sacrificed 1 h postinjection, and peritoneal or BAL fluid lavages, respectively, were collected. Cells were pelleted, washed twice in serum-free HBSS, and resuspended in 750 μl of HBSS. Five hundred microliters of each suspension was incubated in the presence of gentamicin for 20 min at 37°C to kill noninternalized bacteria. Cells were washed twice in serum-free HBSS and resuspended in 500 μl 0.1% Triton X-100 in PBS. Fifteen microliters of resuspended cells/bacteria was plated on Pseudomonas isolation agar plates and incubated overnight at 37°C. Colonies were counted the following morning, and CFU were calculated based on fraction of total sample plated. For peritoneal in vivo phagocytosis studies, analyses of total cell numbers and phenotypes were done in parallel. Total cell numbers harvested from peritoneal lavage samples were quantified using trypan blue staining and manual cell counts of viable cells using a hemacytometer.
Data and statistical analyses.
Sample sizes for each experiment varied and are noted in the text. For all graphs, means and standard deviations are shown. As indicated, one-way analysis of variance (ANOVA) with Tukey's multiple comparison posttest or unpaired Student's t test analysis was performed to assess statistical significance of the data. In the figures, statistical significance is represented by an asterisk and indicates P ≤ 0.05.
RESULTS
P. aeruginosa strains lacking a flagellum or flagellar stator proteins are impaired in swimming motility.
In these studies, a variety of P. aeruginosa mutants were used to assess and quantitatively compare the effect of either loss of the flagellum or flagellar function on the phagocytic recognition of these bacteria by immune cells (Table 1). PA14 is a nonmucoid wild-type P. aeruginosa clinical isolate and is the parental strain for all of the isogenic mutants described in these studies (16, 20). Two mutants (pilB and pilG) defective for type IV pilus expression and function were used as controls as strains that are impaired in twitching motility but are fully competent at swimming motility. Two different mutations (flgK, which codes for the hook-filament junction protein, and fliN, which codes for a subunit of the flagellar rotor complex) that are phenotypically impaired in proper flagellum expression were used, as well as a quadruple mutant for four flagellar stator proteins (ΔmotAB ΔmotCD) which results in intact flagellum expression but nonfunctional swimming motility (26, 27). To confirm the swimming motility phenotype of the mutant strains analyzed, we performed a standard bacterial swimming assay whereby liquid bacterial cultures are inoculated by stabbing puncture into 0.3% agar plates (see Fig. S1 in the supplemental material). Zones surrounding the point of inoculation were evaluated as an indication of swimming motility (27). In accordance with previous results, we observed complete lack of swimming motility in the flagellum-deficient mutants and the stator-deficient mutant while we observed a swimming phenotype in the parental PA14 strain and the two pilus-deficient mutant strains (Fig. S1) (26, 27).
TABLE 1.
P. aeruginosa genetic mutants used in these studies
| PA14 mutanta | WT function of gene product | Mutant phenotype for bacterial motility |
|---|---|---|
| pilB mutant | ATPase for pilus assembly | Lacks pili, twitch defective |
| pilG mutant | Pilus transcription factor | Lacks pili, twitch defective |
| flgK mutant | Flagellar hook protein | No flagellum, swimming defective |
| fliN mutant | Flagellar export/assembly protein | No flagellum, swimming defective |
| ΔmotAB ΔmotCD mutant | Flagellar stator proteins | Flagellum intact, swimming defective |
PA14 is a nonmucoid WT clinical isolate.
Swimming-deficient P. aeruginosa strains are resistant to phagocytosis by BMDCs in vitro.
To evaluate the effect of loss of bacterial motility on phagocyte recognition, we transformed parental PA14 as well as flagellum-deficient flgK and fliN mutants, the ΔmotAB ΔmotCD flagellar stator protein knockout, and the pilus-deficient pilG mutant with a GFP expression construct to allow FACS-based analysis of phagocyte-bacterium interactions. The intensity of GFP fluorescence emitted by our transformed strains did not vary significantly among the strains (see Fig. S2 in the supplemental material), allowing us to compare binding and uptake of these different strains directly in relation to each other. Using fluorescence as a quantitative indicator of bacterial association with BMDCs, we observed a marked and significant reduction in the ability of BMDCs to associate with flagellum-deficient strains (flgK and fliN) compared to the parental PA14 bacteria (Fig. 1 A and D), though the pilus-deficient mutant (pilG) associated with the BMDCs at levels comparable to those for the WT (Fig. 1A). To more rigorously assess the differential phenotypes observed with the GFP-expressing bacteria, we utilized a gentamicin protection assay to specifically and quantitatively assess in vitro phagocytosis of the respective bacterial strains. Both flagellum-deficient strains exhibited a dramatic ∼100-fold reduction in their phagocytic recognition by BMDCs in vitro compared to the parental PA14 strain (Fig. 1B). To verify that this 100-fold variance was not due to intracellular killing of P. aeruginosa, we measured the intracellular killing rate of phagocytosed PA14. Approximately 50% of ingested PA14 bacteria were killed after 2 h of incubation with BMDCs, indicating that the 100-fold differential in phagocytosis observed in a shorter duration (45 min) is not within the margin of the killing rate (Fig. 2C).
FIG. 1.

P. aeruginosa mutants deficient for flagellum or flagellar stator proteins are resistant to phagocytosis by BMDCs in vitro. (A) C57BL/6 BMDCs were analyzed by FACS for relative association with GFP-transformed PA14, the pilus-deficient pilG mutant, or the flagellum-deficient flgK and fliN mutants. (B) C57BL/6 BMDCs were assayed by gentamicin protection assay for relative phagocytic uptake of PA14 or flagellum-deficient flgK and fliN mutants. The graph is plotted on a logarithmic scale. (C) FACS analysis (left) and gentamicin protection assay (right) of C57BL/6 BMDCs coincubated with either WT PA14 or the ΔmotAB ΔmotCD flagellar stator mutant. (D) FACS histogram of total BMDC cellular association with GFP-transformed P. aeruginosa strains after 45 min of coincubation. (E) Fluorescence microscopy of RFP-expressing BMDCs coincubated with GFP-transformed PA14 or the ΔmotAB ΔmotCD mutant viewed with DIC overlay at ×65 magnification. For all graphs, phagocytic uptake levels were normalized as percentages of the mean WT phagocytosis. For all genotypes, n is ≥9; means, standard deviations, and statistical significance (asterisks) are shown.
FIG. 2.

Phagocytic resistance of nonswimming P. aeruginosa is not due to bactericidal activity or differential flagellar expression. (A) WT PA14 (PA14) and the ΔmotAB ΔmotCD (mot) strain express similar total levels of flagellin as assessed by Western analysis for the FliC protein (right). Coomassie blue staining of parallel lanes is shown as a control for protein load (left). (B) To confirm that the ΔmotAB ΔmotCD mutant strain has intact extracellular flagella, the flagella were mechanically sheared from the bacteria and assessed by Coomassie blue staining (left) and Western analysis (right). (C) The BMDC killing rate of P. aeruginosa was assayed by gentamicin protection assay. Following a 1-h coincubation of PA14 with BMDCs, gentamicin was added, and at the indicated time points following gentamicin addition, aliquots were harvested and lysed to assess the death of internalized bacteria over the course of the assay. CFU were plotted relative to initial recovery. For each time point, n is ≥6; standard deviations are shown.
With the use of both assays, we next tested if phagocyte recognition and engulfment of P. aeruginosa were dependent on a fully assembled flagellum by coincubating BMDCs with the ΔmotAB ΔmotCD flagellar stator mutant and assaying via flow cytometry and gentamicin protection. The ΔmotAB ΔmotCD mutant was substantially and significantly reduced in both its overall association (FACS assay [Fig. 1C, left]) and subsequent engulfment (gentamicin protection assay [Fig. 1C, right]) by BMDCs, similarly to the flgK and fliN mutants (compare Fig. 1A and B with C). These results were then confirmed by fluorescence microscopy, which similarly demonstrated the dramatic deficit in the association of swimming-defective but flagellum-expressing P. aeruginosa with BMDCs in vitro (Fig. 1E). This deficit was not due to observable differences in total flagellar expression between the ΔmotAB ΔmotCD mutant and the parental wild type as detected by Coomassie blue staining and Western blot analysis (Fig. 2A), and in biochemical confirmation of a previous report utilizing microscopy (27), we demonstrate that the ΔmotAB ΔmotCD mutant has an intact extracellular flagellum (Fig. 2B). The observation that the ΔmotAB ΔmotCD mutant, but not the pilG mutant, recapitulates the dramatic phagocytic deficit seen for the flagellum-deficient strains demonstrates that the resistance to phagocytosis by these strains is not due to loss of an intact flagellum or pili but the loss of functional flagellum-based motility. Additionally, this deficit is specific to flagellum-based swimming motility, since loss of pilus-based twitching motility did not confer measurable phagocytic evasion.
P. aeruginosa mutants deficient for flagellum or flagellar stator proteins are resistant to phagocytosis by human macrophages in vitro.
To evaluate whether swimming-defective P. aeruginosa strains were also resistant to phagocytosis by human cells, we generated in vitro peripheral blood mononuclear cell-derived human macrophages to assay them by gentamicin protection assay for the relative phagocytic uptake of PA14 and its isogenic pilB, pilG, flgK, fliN, and ΔmotAB ΔmotCD mutants (Fig. 3). The results with human macrophages recapitulated our data with murine BMDCs, whereby we observed a 30- to 100-fold impairment for the phagocytic recognition of swimming-defective P. aeruginosa strains by in vitro-cultured human macrophages compared to recognition of the wild-type parental bacterial strain (Fig. 3). The recapitulation of the murine data with human macrophages indicates that the loss of motility by P. aeruginosa bacteria is likely a universal evasion strategy from phagocytosis by immune cells that spans multiple cell types and species.
FIG. 3.

P. aeruginosa mutants deficient for flagellum or flagellar motor proteins are resistant to phagocytosis by human macrophages in vitro. Peripheral blood mononuclear cell (PBMC)-derived human macrophages were cultured and assayed by gentamicin protection assay for relative in vitro uptake of PA14, pilus-deficient pilB and pilG mutants, flagellum-deficient flgK and fliN mutants, and the flagellar stator protein-deficient ΔmotAB ΔmotCD mutant. Phagocytic uptake levels were normalized as percentages of the mean WT phagocytosis. Statistical significance (P < 0.05) of differences from wild-type levels is indicated (asterisks). Graphs are plotted on a log scale. For all genotypes, n is ≥9; means and standard deviations are shown.
Loss of MyD88 signaling fails to recapitulate the phagocytic deficit for a flagellum-deficient P. aeruginosa strain.
Our data with the ΔmotAB ΔmotCD P. aeruginosa strain indicated that the deficit for flagellum-deficient strains is not an intrinsic function of lack of flagellum expression itself or the loss of flagellum-mediated activation of immune cells. To test this hypothesis more stringently, we generated BMDCs from MyD88-deficient (MyD88−/−) mice to determine if loss of MyD88 signaling recapitulates the phagocytic deficit observed for flagellum-deficient bacteria. We analyzed the relative association of the GFP-expressing wild-type PA14, flgK, and ΔmotAB ΔmotCD strains with WT or MyD88−/− BMDCs in parallel. Loss of MyD88 signaling did not change the relative level of bacterial association with BMDCs (Fig. 4A). Furthermore, WT and MyD88−/− BMDCs quantitatively assayed in parallel for in vitro phagocytosis of wild-type or swimming-defective P. aeruginosa supported the FACS analysis in that loss of MyD88 signaling did not recapitulate the phagocytic deficit seen with flgK and ΔmotAB ΔmotCD bacteria (Fig. 4B). These results strongly support the idea that the phagocytic deficit is independent of Toll-like receptor (TLR)/MyD88 signaling during immune recognition of flagellum-deficient bacteria.
FIG. 4.

The phagocytic deficiency for nonmotile P. aeruginosa strains is independent of MyD88 signaling. (A) WT or MyD88−/− BMDCs were assayed for relative in vitro cellular association of GFP-PA14, GFP-flgK, or GFP-ΔmotAB ΔmotCD P. aeruginosa strains by FACS analysis. Data are represented as the mean fluorescence intensity (MFI) of each experimental group. (B) WT or MyD88−/− BMDCs were assayed for in vitro phagocytosis of PA14 or flgK or ΔmotAB ΔmotCD P. aeruginosa strains by gentamicin protection assay. Phagocytic uptake levels were normalized as percentages of mean WT PA14 phagocytosis. The graph is plotted on a logarithmic scale. For all genotypes, n is ≥6 and means, standard deviations, and statistical significance (asterisks) are shown.
Loss of motility is sufficient to confer P. aeruginosa phagocytic resistance to primary peritoneal and alveolar murine cells ex vivo.
Having identified a deficit for the phagocytosis of stator protein-deficient P. aeruginosa strains by murine BMDCs and human cultured macrophages in vitro, we next tested whether or not this observation could be recapitulated by primary cells ex vivo. Peritoneal phagocytes were harvested and incubated with the various GFP-transformed bacterial strains, and cellular association with the bacteria was analyzed by FACS (Fig. 5A). Peritoneal phagocytes were also assayed for their ability to phagocytose bacteria, assessed by gentamicin protection (Fig. 5B). Consistent with our data derived from in vitro-cultured cells, nonmotile P. aeruginosa strains were dramatically resistant to phagocytosis by ex vivo primary peritoneal macrophages. As a control to assess whether this massive phagocytic differential was merely reflective of a cell surface binding deficit, we measured cell surface binding of bacteria to murine peritoneal macrophages pretreated with cytochalasin D to inhibit phagocytic uptake. Cytochalasin D treatment of P. aeruginosa did not alter its motility (data not shown). No statistical difference in P. aeruginosa cell surface binding was observed among the bacterial genotypes (see Fig. S3 in the supplemental material); this is in direct contrast to parallel association experiments using untreated, phagocytically competent macrophages (Fig. 5A). These data support the idea that it is the evasion of phagocytic engulfment and not initial bacterium-phagocyte contact that provides for the phagocytic resistance phenotype of nonmotile P. aeruginosa.
FIG. 5.

Motor protein-deficient P. aeruginosa strains are resistant to phagocytosis by ex vivo primary phagocytic cells from the peritoneum and the lung. (A) C57BL/6 peritoneal exudate cells were harvested by lavage and assayed by FACS ex vivo for relative cellular association with the GFP-expressing bacterial strains indicated. MFI, mean fluorescence intensity. (B) Peritoneal phagocytes were assayed for relative phagocytosis of WT PA14, flgK, and ΔmotAB ΔmotCD bacteria by gentamicin protection assay. (C) C57BL/6 lung phagocytes were harvested via bronchoalveolar lavage and subsequently assayed by FACS ex vivo for relative cellular association with GFP-transformed bacteria as indicated. (D) Lung phagocytes were assayed by gentamicin protection assay ex vivo for relative uptake of WT, flagellum-deficient mutant flgK, and flagellar motor protein-deficient mutant ΔmotAB ΔmotCD bacteria. For all graphs, n is ≥4 and means, standard deviations, and statistical significance (asterisks) are shown.
The lungs are a major site of clinical infection by P. aeruginosa (14, 21), and the prevalence of pulmonary P. auruginosa isolates lacking motility correlates with poorer clinical condition in CF patients (12). Therefore, to address whether our previous results extended to pulmonary cells, we assayed bacterial association and phagocytosis by primary lung phagocytes ex vivo. We harvested murine lung cells by bronchial lavage and assessed phagocyte association with GFP-expressing bacteria by FACS analysis (Fig. 5C) and assayed for bacterial uptake by gentamicin protection assay (Fig. 5D). In these assays, flagellum-deficient and stator protein-deficient P. aeruginosa strains were highly resistant to phagocytosis and overall association with ex vivo alveolar cells. These findings demonstrate that the loss of bacterial motility by P. aeruginosa confers resistance to phagocytosis by multiple types of primary murine immune cells.
Stator protein-deficient P. aeruginosa strains are resistant to phagocytosis in vivo.
Having determined that loss of motility protects P. aeruginosa from phagocytosis in vitro and ex vivo, we next tested the in vivo relevance of our findings. To address whether loss of motility in P. aeruginosa enables immune evasion during infection of a host organism, we determined the relative clearance of the PA14, flgK, and ΔmotAB ΔmotCD bacterial strains by both peritoneal and pulmonary phagocytes in vivo. With the use of a previously described in vivo gentamicin protection assay (2), we observed a dramatic 10-fold reduction in the ability of peritoneal phagocytes to ingest flgK and ΔmotAB ΔmotCD mutants in vivo compared to uptake of the parental PA14 strain (Fig. 6A). The differences in the phagocytosis of the various bacterial strains were not due to differential recruitment of cells to the peritoneal cavity during the assay, since quantification of total peritoneal cells from treated mice showed no discrepancy in total peritoneal cell numbers between mice injected with the parental P. aeruginosa strain and mice injected with the mutant P. aeruginosa strains (Fig. 6B, bottom). Extending these findings, and with direct clinical relevance to P. aeruginosa colonization of the lung, we assessed the relative clearance of the PA14 and ΔmotAB ΔmotCD bacterial strains by pulmonary phagocytes in vivo. Consistent with our findings with peritoneal phagocytes, lung phagocytes ingested motile PA14 10-fold better than did nonmotile PA14 (Fig. 6B, top). From these results, we conclude that downregulation of motility, independent of structural loss of flagella, by P. aeruginosa confers a dramatic resistance to recognition by the host immune system in vivo and that this provides a mechanism for immune evasion.
FIG. 6.
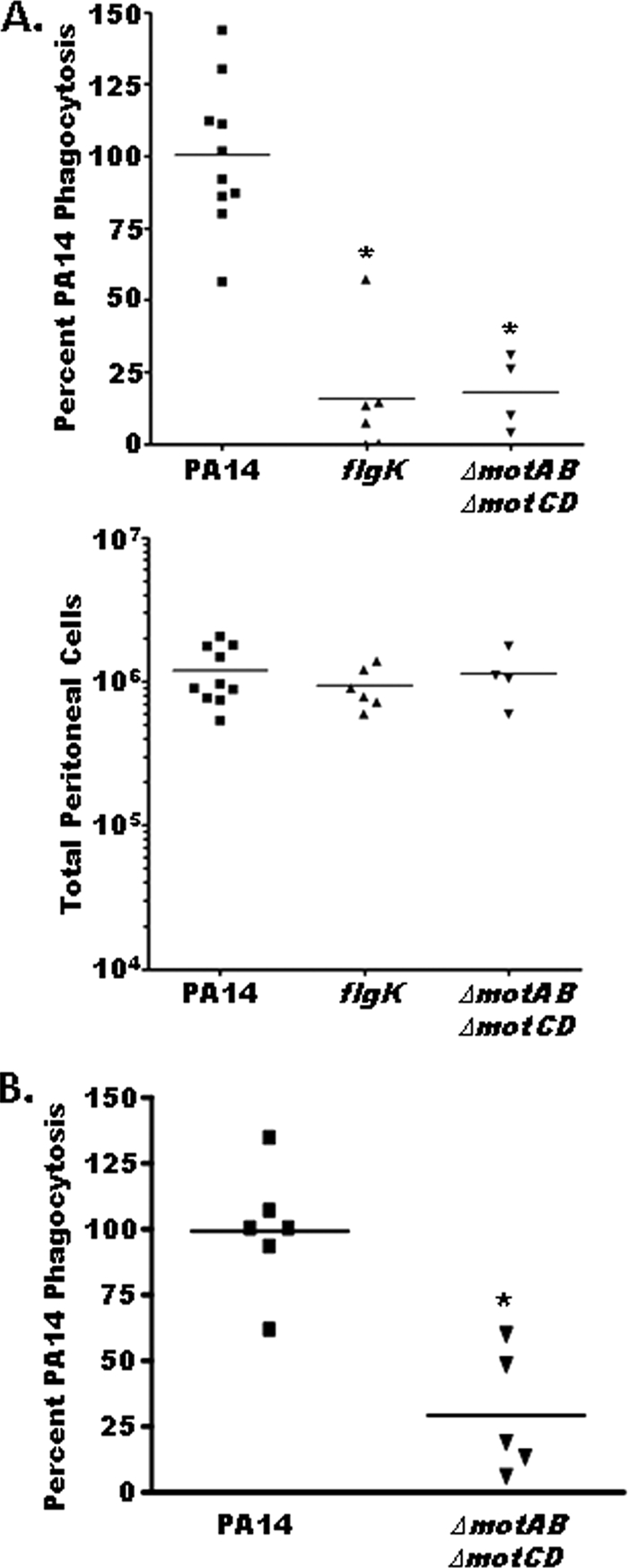
Flagellum-deficient and stator protein-deficient P. aeruginosa strains are resistant to phagocytosis in vivo. (A) (Top) An in vivo gentamicin protection assay was used to determine relative uptake of PA14, flgK, and ΔmotAB ΔmotCD bacterial strains by peritoneal phagocytes. Phagocytic uptake levels were normalized as percentages of the mean WT phagocytosis levels. (Bottom) Total peritoneal cells were quantified from lavage samples of mice treated with PA14, flgK, or ΔmotAB ΔmotCD bacteria, to control for differential immune cell recruitment by the different bacterial strains. (B) Gentamicin protection assay results following in vivo oropharyngeal aspiration of WT or ΔmotAB ΔmotCD PA14 are consistent with the data shown in panel A, with nonmotile PA14 being ∼10-fold more resistant to phagocytosis than motile PA14. Individual data points (mice), means, and statistical significance (asterisks) are shown.
DISCUSSION
Pseudomonas aeruginosa is an opportunistic Gram-negative pathogenic bacterium that poses a significant clinical concern for immunocompromised individuals (9, 13, 14, 21). Two prominent clinical correlations that have been identified in CF patients are a strong positive correlation between the extent of the P. aeruginosa infection and the severity of the prognosis for the patient (12, 17) and a marked phenotypic shift toward the loss of bacterial motility and downregulation of flagellum expression that correlates with the severity of CF disease (12, 17). Therefore, there is considerable interest in understanding the basis for the latter phenotypic observation, especially in relation to the advantage that it may confer on the bacteria and, consequently, how we may more effectively treat the infection.
Previous reports have shown that loss of flagellar expression in clinical bacterial isolates results in dramatic decreases in nonopsonic phagocytic recognition by murine and human macrophages in vitro (17, 18). Other reports have indicated that the flagellum itself, and not the loss of bacterial motility, is the primary determinant in controlling the host immune response to P. aeruginosa (5). In these studies we have utilized a bacterial and mammalian genetic approach to show that P. aeruginosa strains that are defective for swimming motility are resistant to phagocytosis in vitro and in vivo. Furthermore, we demonstrate that this resistance to phagocytosis is independent of flagellum expression and not dependent on MyD88 signaling, in response to these bacteria. Our results indicate that it is the functional loss of flagellum-mediated swimming motility in P. aeruginosa that confers phagocytic resistance on these bacteria. These data are the first demonstration that the loss of flagellar function, not the loss of the flagellum apparatus as an activation or association ligand, is responsible for conferring resistance to phagocytosis and that this phenotype has an important role in immune evasion by P. aeruginosa of both mouse and human immune cells.
Based on the observations that P. aeruginosa tends to downregulate flagellar gene expression and functional motility in CF patients, it has been proposed that P. aeruginosa undergoes these phenotypic changes as part of an immune evasion strategy to avoid detection by phagocytic and cell-activating receptors during chronic infections (12, 17). Furthermore, nonopsonic phagocytosis of P. aeruginosa has been reported to require the expression of an intact flagellum, as flagellum-deficient bacteria are reported to be resistant to phagocytosis by murine and human macrophages in vitro (18). The ΔmotAB ΔmotCD strain used in these studies is deficient for four stator genes which comprise the stationary components of the P. aeruginosa flagellar motor (27). In this strain, flagellar structures are intact on the surface of the bacteria but are not functional and thus these bacteria are impaired in swimming motility (27). Based on previous electron micrograph studies and our confirmatory biochemical studies here, the flagellar structure in the ΔmotAB ΔmotCD mutant is intact and does not appear to be different from its wild-type counterpart (27). By demonstrating that the ΔmotAB ΔmotCD strain is equally as resistant to phagocytosis as the flagellum-deficient strains, we conclude that the flagellum itself is not a critical ligand for nonopsonic phagocytosis of P. aeruginosa in our experimental system. Intriguingly, a previous report found that loss of the stator genes motAB in a different human clinical P. aeruginosa isolate resulted in resistance to phagocytosis by macrophages in vitro (24). However, this did not affect measurable motility and the authors noted that the parental strain used for their studies exhibited an abnormal phenotype which possibly could confound the interpretation of the data (24). In the PA14 P. aeruginosa strain used in our studies, only loss of all four bacterial stator proteins (ΔmotAB ΔmotCD) results in swimming motility defects under standard experimental conditions (27) and the isogenic ΔmotAB PA14 mutant did not recapitulate the phagocytic defect observed in the ΔmotAB ΔmotCD mutant (data not shown).
TLR5 has been recognized as a major pattern recognition receptor (PRR) in innate immune cells, binding bacterial flagellin and activating the MyD88 cell signaling pathway (10, 25). We observed only a minor deficit for P. aeruginosa phagocytosis in MyD88−/− BMDCs that did not recapitulate the dramatic deficit seen for phagocytic recognition of flagellum-deficient bacteria. Similarly, bacterial association with MyD88−/− BMDCs was not any lower than that with WT BMDCs. These findings are particularly relevant since previously published hypotheses have argued that flagellum-deficient bacteria escape immune detection primarily through the loss of flagellum-mediated immune activation of TLR5 (5, 17, 18). Because activation via TLR5 is MyD88 dependent, we believe that it is not a major contributor in the phagocytic resistance phenomenon observed in the nonmotile PA14 mutants. This is supported by similar degrees of phagocytic resistance in both flagellated and nonflagellated nonmotile strains. However, it is reasonable to suggest that TLR5 and other MyD88-dependent PRRs likely do play a role in other aspects of the overall immune response to PA14 infection. Indeed, it has been demonstrated with an alternative P. aeruginosa strain that the TLR5-flagellin interaction is sufficient to activate alveolar macrophage upregulation of cytokine production (5). Additionally, we have previously reported that cytokine responses to bacterial challenge are not necessarily directly coupled to the phagocytosis of those bacteria (2). Overall, it is now clear that effective bacterial clearance of nonopsonized PA14 via phagocytosis hinges on motility-based factors outside of simple recognition of flagellin as a stimulating ligand.
In summary, our data demonstrate that loss of swimming motility in P. aeruginosa bacteria results in dramatic resistance to phagocytic recognition by innate immune cells in vitro and in vivo. This phagocytic resistance was observed for both mouse and human cells and was not due to the specific loss of flagellar expression, since bacteria that expressed nonfunctional flagella were able to recapitulate the same phagocytic resistance exhibited by flagellum-deficient bacteria. Furthermore, genetic deletion of MyD88 in phagocytic cells was not able to recapitulate the defect for phagocytosis of flagellum-deficient bacteria, demonstrating that loss of flagellum-mediated TLR5 signaling is not responsible for the impaired phagocytosis of flagellum-deficient P. aeruginosa bacteria. We found that motility-deficient P. aeruginosa strains are resistant to phagocytosis by both human and mouse phagocytes in vitro, as well as primary mouse cells ex vivo and in vivo. These data provide new insights into important clinical observations about P. aeruginosa and the loss of bacterial motility and are the first demonstration that P. aeruginosa strains defective for swimming motility are resistant to phagocytosis in vivo. These findings provide an important new understanding of the immunological advantage in vivo for the loss of motility by P. aeruginosa during chronic infections.
Supplementary Material
Acknowledgments
We thank Shizuo Akira (Osaka University) and Paul Guyre, Harry Higgs, and J. Ma Collins (Dartmouth) for reagents; Julie Acker and Ryan Collins for technical assistance; the Dartmouth Medical School FACS and microscopy core facilities; and Matt Wargo, Laurie Whittaker, and Jenna Allard (UVM) for technical advice.
This research was supported by RO1 AI067405 and a CF Foundation RDP training grant (B.B.) and NIH training grants T32 AI07363 (E.A.) and NIGMS GM008704 (R.R.L.).
Editor: A. Camilli
Footnotes
Published ahead of print on 10 May 2010.
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1.Adachi, O., T. Kawai, K. Takeda, M. Matsumoto, H. Tsutsui, M. Sakagami, K. Nakanishi, and S. Akira. 1998. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 9:143-150. [DOI] [PubMed] [Google Scholar]
- 2.Amiel, E., J. L. Acker, R. M. Collins, and B. Berwin. 2009. Uncoupling scavenger receptor A-mediated phagocytosis of bacteria from endotoxic shock resistance. Infect. Immun. 77:4567-4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amiel, E., A. Alonso, S. Uematsu, S. Akira, M. E. Poynter, and B. Berwin. 2009. Pivotal advance: Toll-like receptor regulation of scavenger receptor-A-mediated phagocytosis. J. Leukoc. Biol. 85:595-605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amiel, E., S. Nicholson-Dykstra, J. J. Walters, H. Higgs, and B. Berwin. 2007. Scavenger receptor-A functions in phagocytosis of E. coli by bone marrow dendritic cells. Exp. Cell Res. 313:1438-1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balloy, V., A. Verma, S. Kuravi, M. Si-Tahar, M. Chignard, and R. Ramphal. 2007. The role of flagellin versus motility in acute lung disease caused by Pseudomonas aeruginosa. J. Infect. Dis. 196:289-296. [DOI] [PubMed] [Google Scholar]
- 6.Bloemberg, G. V., G. A. O'Toole, B. J. Lugtenberg, and R. Kolter. 1997. Green fluorescent protein as a marker for Pseudomonas spp. Appl. Environ. Microbiol. 63:4543-4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braun, T. F., S. Poulson, J. B. Gully, J. C. Empey, S. Van Way, A. Putnam, and D. F. Blair. 1999. Function of proline residues of MotA in torque generation by the flagellar motor of Escherichia coli. J. Bacteriol. 181:3542-3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duncan, M. J., G. Li, J. S. Shin, J. L. Carson, and S. N. Abraham. 2004. Bacterial penetration of bladder epithelium through lipid rafts. J. Biol. Chem. 279:18944-18951. [DOI] [PubMed] [Google Scholar]
- 9.Gibson, R. L., J. L. Burns, and B. W. Ramsey. 2003. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am. J. Respir. Crit. Care Med. 168:918-951. [DOI] [PubMed] [Google Scholar]
- 10.Hayashi, F., K. D. Smith, A. Ozinsky, T. R. Hawn, E. C. Yi, D. R. Goodlett, J. K. Eng., S. Akira, D. M. Underhill, and A. Aderem. 2001. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410:1099-1103. [DOI] [PubMed] [Google Scholar]
- 11.Inaba, K., M. Inaba, M. Deguchi, K. Hagi, R. Yasumizu, S. Ikehara, S. Muramatsu, and R. M. Steinman. 1993. Granulocytes, macrophages, and dendritic cells arise from a common major histocompatibility complex class II-negative progenitor in mouse bone marrow. Proc. Natl. Acad. Sci. U. S. A. 90:3038-3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luzar, M. A., M. J. Thomassen, and T. C. Montie. 1985. Flagella and motility alterations in Pseudomonas aeruginosa strains from patients with cystic fibrosis: relationship to patient clinical condition. Infect. Immun. 50:577-582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lyczak, J. B., C. L. Cannon, and G. B. Pier. 2000. Establishment of Pseudomonas aeruginosa infection: lessons from a versatile opportunist. Microbes Infect. 2:1051-1060. [DOI] [PubMed] [Google Scholar]
- 14.Lyczak, J. B., C. L. Cannon, and G. B. Pier. 2002. Lung infections associated with cystic fibrosis. Clin. Microbiol. Rev. 15:194-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.MacFerrin, K. D., M. P. Terranova, S. L. Schreiber, and G. L. Verdine. 1990. Overproduction and dissection of proteins by the expression-cassette polymerase chain reaction. Proc. Natl. Acad. Sci. U. S. A. 87:1937-1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahajan-Miklos, S., M. W. Tan, L. G. Rahme, and F. M. Ausubel. 1999. Molecular mechanisms of bacterial virulence elucidated using a Pseudomonas aeruginosa-Caenorhabditis elegans pathogenesis model. Cell 96:47-56. [DOI] [PubMed] [Google Scholar]
- 17.Mahenthiralingam, E., M. E. Campbell, and D. P. Speert. 1994. Nonmotility and phagocytic resistance of Pseudomonas aeruginosa isolates from chronically colonized patients with cystic fibrosis. Infect. Immun. 62:596-605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahenthiralingam, E., and D. P. Speert. 1995. Nonopsonic phagocytosis of Pseudomonas aeruginosa by macrophages and polymorphonuclear leukocytes requires the presence of the bacterial flagellum. Infect. Immun. 63:4519-4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Montie, T. C., R. C. Craven, and I. A. Holder. 1982. Flagellar preparations from Pseudomonas aeruginosa: isolation and characterization. Infect. Immun. 35:281-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rahme, L. G., E. J. Stevens, S. F. Wolfort, J. Shao, R. G. Tompkins, and F. M. Ausubel. 1995. Common virulence factors for bacterial pathogenicity in plants and animals. Science 268:1899-1902. [DOI] [PubMed] [Google Scholar]
- 21.Sadikot, R. T., T. S. Blackwell, J. W. Christman, and A. S. Prince. 2005. Pathogen-host interactions in Pseudomonas aeruginosa pneumonia. Am. J. Respir. Crit. Care Med. 171:1209-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sauer, K., and A. K. Camper. 2001. Characterization of phenotypic changes in Pseudomonas putida in response to surface-associated growth. J. Bacteriol. 183:6579-6589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Semmler, A. B., C. B. Whitchurch, and J. S. Mattick. 1999. A re-examination of twitching motility in Pseudomonas aeruginosa. Microbiology 145:2863-2873. [DOI] [PubMed] [Google Scholar]
- 24.Simpson, D. A., and D. P. Speert. 2000. RpmA is required for nonopsonic phagocytosis of Pseudomonas aeruginosa. Infect. Immun. 68:2493-2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith, K. D., and A. Ozinsky. 2002. Toll-like receptor-5 and the innate immune response to bacterial flagellin. Curr. Top. Microbiol. Immunol. 270:93-108. [DOI] [PubMed] [Google Scholar]
- 26.Toutain, C. M., N. C. Caizza, M. E. Zegans, and G. A. O'Toole. 2007. Roles for flagellar stators in biofilm formation by Pseudomonas aeruginosa. Res. Microbiol. 158:471-477. [DOI] [PubMed] [Google Scholar]
- 27.Toutain, C. M., M. E. Zegans, and G. A. O'Toole. 2005. Evidence for two flagellar stators and their role in the motility of Pseudomonas aeruginosa. J. Bacteriol. 187:771-777. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.