

Abstract
As intracellular bacteria, chlamydiae block the apoptotic pathways of their host cells. However, the infection of epithelial cells causes the loss of cell membrane integrity and can result in nonapoptotic death. Normally, cells undergoing necrosis release high-mobility group box 1 protein (HMGB1) that acts as an important proinflammatory mediator. Here, we show that in Chlamydia trachomatis-infected HeLa cells HMGB1 is not translocated from the nucleus to the cytosol and not released from injured cells in increased amounts. At 48 h after infection, degradation of HMGB1 was observed. In infected cells, poly(ADP-ribose) polymerase 1 (PARP-1), a DNA repair enzyme that also regulates HMGB1 translocation, was found to be cleaved into fragments that correspond to a necrosislike pattern of PARP-1 degradation. Cell-free cleavage assays and immunoprecipitation using purified proteolytic fractions from infected cells demonstrated that the chlamydial-protease-like activity factor (CPAF) is responsible for the cleavage of both HMGB1 and PARP-1. Proteolytic cleavage of PARP-1 was accompanied by a significant decrease in the enzymatic activity in a time-dependent manner. The loss of PARP-1 function obviously affects the viability of Chlamydia-infected cells because silencing of PARP-1 in uninfected HeLa cells with specific small interfering RNA results in increased cell membrane permeability. Our findings suggest that the Chlamydia-specific protease CPAF interferes with necrotic cell death pathways. By the degradation of HMGB1 and PARP-1, the pathogen may have evolved a strategy to reduce the inflammatory response to membrane-damaged cells in vivo.
Intracellular bacteria have evolved different strategies to establish a replication niche within eukaryotic cells. Chlamydiae invade their host cells as infectious elementary bodies, differentiate into metabolically active reticulate bodies, and replicate within an expanding vacuole, thereby forming a large intracellular inclusion. After a period of growth, reticulate bodies redifferentiate into new elementary bodies that are released from the host cell (1). Chlamydia trachomatis has a tissue tropism for ocular and urogenital epithelia. Trachoma which is due to infections by C. trachomatis serovars A to C is the most frequent cause of preventable blindness in the world (4). Urogenital infections by the serovars D to K are a major cause of infertility in women that results from salpingitis and tubal occlusions (25).
The modulation of host cell death pathways can be regarded as an important strategy of chlamydiae for maintaining the integrity of host cells during the developmental cycle (27, 36). In general, several different types of cell death are differentiated (23). Classical apoptosis defines a highly regulated process that include BH3-only proteins as triggers of apoptotic signals; Bax and Bak as effectors of mitochondrial cytochrome c release; the formation of the caspase-activating apoptosome; and the subsequent activation of effector caspases 3, 6, and 7 (10). Poly(ADP-ribose) polymerase-1 (PARP-1), an enzyme that is involved in DNA base excision repair, is cleaved and inactivated by caspase 3 (3, 5). Apoptosis is characterized by the condensation of nuclear chromatin, cleavage of DNA into oligonucleosomal fragments, loss of plasma membrane phosphatidylserine asymmetry, and in consequence the formation of apoptotic cell fragments (11). In contrast, plasma membrane lysis and release of the cellular content defines necrotic cell death that induces inflammation (11, 23). Necrosis is characterized by an overactivation of PARP-1 and its subsequent cleavage into a multifragment pattern (2, 16). High-mobility group box 1 protein (HMGB1), a nucleosome-associated protein, is released from necrotic but not from apoptotic cells and represents an important proinflammatory mediator (33).
Fan et al. (9) first described that C. trachomatis blocks effector caspase activation and apoptosis in epithelial cells after exposure to apoptotic stimuli. This activity of Chlamydia could be attributed to the blockade of mitochondrial cytochrome c release, inhibition of Bax and Bak activation, and degradation of BH3-only proteins (9, 12, 13, 14, 35). Zhong et al. (38) identified a chlamydial protease-like activity factor (CPAF) that is translocated by chlamydiae into the cytoplasm of the infected cell. CPAF is responsible for the degradation of BH3-only proteins, thereby mediating apoptosis resistance of infected cells (31). The inhibition of apoptosis may not only favor the maintenance of the intracellular environment for replication but also allow chlamydiae to escape from immune effector mechanisms directed against infected cells. However, at the end of the replication cycle cells are often lysed when new infectious elementary bodies are released (19). Although Chlamydia counter-regulates the effects of apoptosis inducers, the infection of host cells can finally result in cell death that is independent from the activation of apoptotic pathways and effector caspases (26, 28, 30, 37). Interestingly, Paschen et al. (29) showed that the transfection of epithelial cells with CPAF induces cell death that is associated with multifragmentation of PARP-1.
At present, it is not well understood how Chlamydia interferes with cellular pathways regulating nonapoptotic cell death. The aim of the present study was to investigate the role of CPAF-mediated proteolytic effects on PARP1 and HMGB1 in the modulation of C. trachomatis-induced cytopathicity.
MATERIALS AND METHODS
Cell culture and C. trachomatis infection.
HeLa cells (ATCC CCL-2) were grown in minimal essential medium (Opti-MEM; Gibco, Invitrogen, Karlsruhe, Germany) with 10% fetal calf serum (FCS; PromoCell, Heidelberg, Germany). C. trachomatis serovar D strain IC Cal 8 (obtained from the Institute of Ophthalmology, London, United Kingdom) was propagated in buffalo green monkey (BGM) cells as described previously (32). Infectivity titers of chlamydial stocks were quantified by titrating the number of inclusion-forming units (IFU) per milliliter in BGM cells. Mycoplasma contaminations in cell cultures were excluded by PCR targeting the 16S rRNA gene of Mycoplasma, Acholeplasma, and Ureaplasma species (sense primer, 5′-CCAGACTCCTACGGGAGGCA-3′; antisense primer, 5′-TGCGAGCATACTACTCAGGC-3′ [18]). For infection experiments, HeLa cells were grown in 35-mm-diameter culture wells (six-well plates) or shell-vials to ca. 70% confluence. The cells were inoculated with C. trachomatis at a multiplicity of infection (MOI) of 1 or 5. For mock-infected cultures, diluted harvests of uninfected BGM cells were added. After centrifugation at 4,000 × g at 37°C for 45 min, the inoculum was decanted, and the cells were further incubated with Opti-MEM containing 10% FCS. In some experiments, chlamydial protein synthesis was inhibited by treatment with chloramphenicol (60 μg/ml; Sigma-Aldrich, Hamburg, Germany) or doxycycline (0.16 μg/ml; twice the MIC; Sigma-Aldrich).
Cell death assays.
Mock-infected and infected HeLa cells were washed with phosphate-buffered saline (PBS) at 48 h after infection and incubated with 10 μM Hoechst 34580 (Invitrogen) to stain DNA and propidium iodide (PI; Biotium, purchased from Biotrend, Cologne, Germany) to stain membrane-damaged cells. Images were taken with a confocal laser scanning microscope (Exciter 5; Zeiss, Jena, Germany). For the identification of cell membrane permeability changes by flow cytometry, an Annexin V-FLUOS staining kit (Roche Applied Science, Mannheim, Germany) was used. Culture supernatants were collected and mixed with trypsinized cells. After centrifugation at 3,000 × g for 4 min, the samples were washed in PBS and resuspended in the supplied buffer containing Annexin V-FLUOS labeling reagent and PI. After incubation at room temperature for 20 min, labeled cells were analyzed with a FACSCalibur flow cytometer and Cell QuestPro software (BD Biosciences, Heidelberg, Germany). Ten thousand cells were scored for each sample. To induce apoptosis, mock-infected and infected cells with or without chlamydial infection were treated with 1 μM staurosporine (Roche Applied Science) for 5 h. For nuclear morphology assays, the cells were washed twice with PBS, fixed with methanol for 5 min at room temperature, and stained with 10 μM Hoechst 34580 at 37°C for 30 min. After being washed three times with PBS, nuclear fragmentation was examined under a fluorescence microscope (Axioskop; Zeiss). Nuclei in 10 randomly selected visual fields were counted, and the percentage of apoptotic cells was calculated. The release of lactate dehydrogenase (LDH) from plasma membrane-damaged cells was measured with an LDH cytotoxicity detection kit (Roche Applied Science).
HMGB1 ELISA.
The levels of HMGB1 in culture supernatants were measured by enzyme-linked immunosorbent assay (ELISA; Shino-test, Kanagawa, Japan; purchased from IBL International, Hamburg, Germany) according to the manufacturer's protocol.
Immunoblot analysis of HMGB1 and PARP-1.
Cells were lysed in radioimmunoassay buffer (0.15 M NaCl, 50 mM Tris-HCl, 1% deoxycholic acid, 1% Triton X-100, 0.1% sodium dodecyl sulfate [SDS]) with phenylmethylsulfonyl fluoride (PMSF; 100 μg/ml; Serva, Heidelberg, Germany), leupeptin (2 μg/ml; Serva), and aprotinin (50 μg/ml; Sigma-Aldrich) for 30 min on ice. SDS-PAGE of cell lysates and immunoblotting were performed according to the protocol of a previous study (32). Blots were incubated with 1:2,000 dilutions of rabbit polyclonal antibodies to HMGB-1 (ab18256; Abcam, Cambridge, United Kingdom) or PARP-1 (VIC 5; Roche Applied Science) at 4°C overnight. Alkaline phosphatase-conjugated goat anti-rabbit IgG (Dianova, Hamburg, Germany) was used as secondary antibody at a dilution of 1:2,000. Blots were incubated with the secondary antibody for 2 h at room temperature. Washes between antibody additions were performed with TBS-Tween three times for 5 min each time. The bands were visualized with 5-bromo-4-chloro-3-indolylphosphate toluidine salt/p-nitroblue tetrazolium chloride (BCIP/NBT; Sigma Fast; Sigma-Aldrich). Molecular weights of proteins and cleaved fragments were calculated from a protein standard curve of log molecular weight versus distance migrated (SDS-PAGE standards broad range; Bio-Rad, Munich, Germany).
Cell-free degradation assay.
Cells grown and infected on six-well plates were trypsinized and harvested by centrifugation at 1,800 × g for 10 min at 4°C. After being washed with ice-cold PBS, the cell pellet was resuspended in 5 volumes of ice-cold buffer A (20 mM HEPES-KOH [pH 7.5], 10 mM KCl, 1.5 mM MgCl2, 1 mM sodium EDTA, 1 mM sodium EGTA, 1 mM dithiothreitol, 0.1 mM PMSF) supplemented with 50 μl of protease inhibitor cocktail (5 μg of pepstatin A/ml, 10 μg of leupeptin/ml, 2 μg of aprotinin/ml, 25 μg of N-acetyl-leucyl-norleucinal [ALLN]/ml). After incubation on ice for 15 min, the cells were disrupted by dounce homogenization. Nuclear pellets were collected by centrifugation at 1,000 × g for 10 min at 4°C and further extracted in buffer B (0.5 mM NaCl and 1% Triton X-100 in 20 mM Tris [pH 8.0]). The supernatants were centrifuged at 20,000 × g for 15 min to prepare cytosolic extracts. Cytosolic extracts collected from mock-infected and Chlamydia-infected cells were used as the source of enzyme and incubated with nuclear extracts from mock-infected HeLa cells as substrate containing PARP-1 and HMGB-1. After incubation at 37°C for 2 h, the mixtures were analyzed by SDS-PAGE and immunoblotting.
Column chromatography.
To separate proteolytic proteins that were induced in infected HeLa cells, cytosolic extracts were applied to a MonoQ ion-exchange column (GE Healthcare Life Sciences, Freiburg, Germany). An AKTA purifier 10 instrument (GE Healthcare Life Sciences) was used to run the column. Anion exchange was performed with buffer B (0.01 M Tris, 1 M NaCl [pH 7.2]), and the column was eluted with buffer A (0.01 M Tris [pH 7.2]). The proteolytic activities of the fractions against PARP-1 and HMGB-1 were measured using nuclear extracts from mock-infected cells as a substrate. Cell-free degradation assays were performed as described above. SDS-polyacrylamide gels of proteolytic fractions were analyzed by Coomassie brilliant blue staining.
Immunoblot analysis of CPAF.
For the production of CPAF-specific antibodies, the coding sequence of the CPAF gene of C. trachomatis was cloned and expressed as full-length protein in E. coli (15). The recombinant protein was purified by standard chromatographic methods. Protein purity was confirmed by SDS-PAGE, and the concentration was determined with Bradford reagent (Bio-Rad). About 30 μg of the recombinant protein was blotted onto a nitrocellulose membrane and air dried. The protein spot was cut out (0.5 cm2) and solubilized in 300 μl of dimethyl sulfoxide (DMSO). The solution was used for subcutaneous immunization of two mice. Each animal received three immunizations at 14-day intervals. Sera were collected by bleeding before immunization and 2 weeks after the third immunization. All immunization experiments were performed with 6- to 10-week-old BALB/c mice (Charles River Laboratories, Kisslegg, Germany) and approved by the government agency for animal protection. Immunoblots of chromatographic fractions from cytosolic lysates of infected HeLa were incubated with a 1:500 dilution of the anti-CPAF serum at 4°C overnight. Alkaline phosphatase-conjugated goat anti-rabbit IgG (Dianova) was used as secondary antibody at a dilution of 1:2,000. The bands were visualized with BCIP/NBT.
Immunoprecipitation.
An immunoprecipitation assay without radiolabeling was used to deplete CPAF antigen from a proteolytic chromatography fraction. The sample was incubated with 5 μg of mouse CPAF antibody per ml at 4°C overnight and then mixed with 20 μl of protein A-agarose. Mouse IgG (Abcam) was used as negative control. After incubation at 4°C overnight, the agarose pellets were spun down by centrifugation. The original chromatography fraction and the supernatants after immunoprecipitation were examined in cell-free degradation assays using human recombinant PARP-1 (Sigma-Aldrich) and HMGB1 (Abcam) as substrates. The precipitation of CPAF was confirmed by immunoblot assays of supernatants and pellets.
Lactacystin treatment.
Lactacystin (Enzo Life Sciences, Lörrach, Germany) was dissolved in DMSO at a concentration of 1.1 mg/ml. To inhibit CPAF activity, HeLa cells grown on six-well plates were treated with 11 μg of lactacystin after infection (final concentration, 10 μM). Control wells of mock-infected and infected cells included DMSO at an equivalent dilution. At 48 h after infection, cell lysates were prepared for SDS-PAGE. In comparison, infected cells cultured in medium alone were collected in radioimmunoassay buffer and then incubated with 11 μg of lactacystin for 45 min on ice.
Chlamydia replication assay.
Chlamydial inclusions in HeLa cells treated with or without lactacystin were quantified after immunofluorescence staining with fluorescein isothiocyanate (FITC)-conjugated antibody to the C. trachomatis major outer membrane protein (MOMP; Trinity Biotech, Lemgo, Germany). The number of inclusions per shell vial was calculated from determination of inclusions in 20 randomly selected ×400 microscopic fields. To assess the production of infectious elementary bodies during the intracellular cycle, infected cells were scraped into 500 μl of saccharose phosphate buffer (PBS with 0.2 M saccharose and 2% FCS) at 48 h after infection and lysed by sonication (ten pulses at 110 W; Branson Sonifier W-250). The inclusion-forming units (IFU) of the chlamydial suspensions were titrated in BGM cells. The chlamydial yield was calculated from the ratio of IFU to the number of inclusions.
PARP activity assay and RNA interference.
PARP activity in cell lysates was assayed by using a universal colorimetric PARP assay kit (R&D Systems, Wiesbaden-Nordenstadt, Germany) according to the manufacturer's instructions. The test principle is based on the incorporation of biotinylated poly (ADP-ribose) onto histone proteins. Poly(ADP-ribosylation) of histone proteins was detected by using streptavidin-horseradish peroxidase. HP validated small interfering RNA (siRNA) targeting PARP-1 (SI02662989) and nonsilencing All Stars Negative Control siRNA were purchased from Qiagen (Hilden, Germany). HeLa cells were transfected with 10 nM siRNA using HiPerfect transfection reagent (Qiagen) according to the manufacturer's protocol. After an initial 16 h of incubation, the transfection medium was replaced with fresh culture medium, and transfected cells were further incubated for 32 h before infection with C. trachomatis. The efficiency of the transfection method was confirmed by using Alexa Fluor 488-labeled All Stars Negative Control siRNA. Cell death induction and PARP-1 were assayed as described above.
Immunofluorescence staining of HMGB1.
Mock-infected and infected cells cultured on glass coverslips were washed with PBS, fixed with 4% paraformaldehyde for 15 min, and saturated with blocking buffer for 2 h. For immunostaining, the cells were sequentially incubated with rabbit polyclonal antibody to HMGB1 (Abcam) and Cy3-conjugated goat anti-rabbit IgG (Dianova). Cell nuclei were stained by Hoechst 34580, and Chlamydia inclusions were labeled with an FITC-conjugated MOMP antibody (Trinity Biotech). Images were taken with a confocal laser scanning microscope (Exciter 5; Zeiss). In some experiments, mock-infected and infected cells were treated with 500 μM H2O2 for 3 h to induce cell death. Mock-infected cells were treated with 300 μM DHIQ for 30 min prior to the addition of H2O2 to inhibit PARP-1 activity. Infected cells were treated with 10 μM lactacystin to inhibit CPAF activity.
Statistical analysis.
Statistical comparisons were made by using a Student t test. P values of ≤0.05 were considered to be statistically significant.
RESULTS
C. trachomatis infection of HeLa cells is accompanied by changes of host cell membrane permeability.
HeLa cells were infected with C. trachomatis serovar D at an MOI of 1 and double stained with Hoechst and PI. The loss of membrane integrity allows the uptake of PI, an intercalating DNA stain, while intact membranes exclude it. As shown in Fig. 1 A, cells containing a chlamydial inclusion were found to be PI positive, indicating that plasma membrane permeability is enhanced in infected cells. For flow cytometry, double labeling of the cells with annexin V and PI was used. Annexin V binds phosphatidylserine, which is typically exposed on the outer leaflet of apoptotic cell membranes. Phosphatidylserine can also be detected within cells during necrotic cell death when annexin V enters the cell after membrane damage. A significant increase in PI and double-positive cells was observed at 24 and 48 h after infection, demonstrating that cell membrane integrity is lost in C. trachomatis-infected cells (Fig. 1B and C). Membrane disintegration may result in necrotic cell death. To demonstrate that the cytopathic effects of C. trachomatis are independent from the ability to mediate resistance to apoptotic stimuli, HeLa cells were treated with 1 μM staurosporine for 5 h, and the number of apoptotic cells was quantified after Hoechst staining. When the cells were treated with staurosporine at 43 h after infection, the percentage of cells with apoptotic nuclei was reduced to 2%, compared to 23% positive cells in mock-infected cultures (Fig. 1D).
FIG. 1.

Increased cell membrane permeability of HeLa cells after C. trachomatis infection. (A) HeLa cells were infected at an MOI of 1 and stained with Hoechst 34580 (blue) and PI (pink) at 48 h after infection. Arrowhead, chlamydial inclusion. Bars, 15 μm. (B) Dot blots of mock-infected and infected cells analyzed by flow cytometry with annexin V/PI double labeling. (C) Time-dependent increase in cell membrane permeability of Chlamydia-infected cells. The results are given as the percentage of PI or annexin V-positive cells (with standard deviations) as measured by flow cytometry. *, P ≤ 0.03; **, P ≤ 0.001; and ***, P ≤ 0.05 (compared to values for infected cells at 10 h; n = 4). (D) Inhibition of staurosporine-induced apoptosis in C. trachomatis-infected HeLa cells. Cells infected at an MOI of 5 were incubated in medium and treated with staurosporine for 5 h at the indicated time points after infection. The percentage of cells with apoptotic nuclei was calculated from the determination of nuclear apoptosis after Hoechst staining. ****, P ≤ 0.01 (compared to values for mock-infected cells and infected cells at 10 h; n = 4).
C. trachomatis-induced cytopathic effects are characterized by the cleavage of HMGB1 and PARP-1.
The cytopathicity of C. trachomatis was further characterized by analysis of HMBG1 and PARP-1, which serve as markers to differentiate between apoptosis and necrosis (2). HMGB1 is a nuclear protein that is leaked out when cell membrane permeability is enhanced and the cells undergo necrosis (33). Surprisingly, at 48 h after infection, lower levels of HMGB1 were measured in supernatants of C. trachomatis-infected cells compared to mock-infected cultures (Fig. 2A). Immunoblot analysis revealed that the HMGB1 full-length protein of ∼25 kDa disappeared at 48 h after infection, whereas a smaller band of ∼17 kDa was detected (Fig. 2B).
FIG. 2.

Degradation of HMGB1 and PARP-1 in HeLa cells after infection with C. trachomatis. (A) Time course of HMGB-1 release from mock-infected and infected cells. HMGB1 levels in culture supernatants were determined by ELISA. *, P ≤ 0.05 (compared to values for mock-infected cells at 48 h; n = 3). (B) HMGB1 levels in mock-infected and infected cells. Whole-cell extracts were prepared at different time points after infection and analyzed by immunoblotting. (C) Time course of PARP-1 cleavage after chlamydial infection. The apoptotic cleavage in mock-infected cells induced by staurosporine treatment was used as control. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was stained as a reference band. (D) Presence of the proteolytic activity against HMGB1 and PARP-1 in cytosolic and nuclear fractions of Chlamydia-infected cells but not mock-infected cells. Cytosolic and nuclear fractions were prepared from cell samples collected at 48 h after infection. Cytosolic fractions from mock-infected or infected cells were tested in a cell-free cleavage assay using nuclear fractions from mock-infected cells as substrate (lanes 5 and 6). HMGB1 and PARP-1 were analyzed by immunoblotting. GAPDH was stained as a loading control of cytosolic extracts. NE, nuclear extract; CE, cytosolic extract; M, mock-infected cells; C, Chlamydia-infected cells. (E) Effects of chloramphenicol (Chl) and doxycycline (Dox) treatment on HMGB1 and PARP-1 cleavage in infected cells (MOI of 5). Antibiotics were added at 1 h after inoculation of chlamydiae. Cell lysates were prepared at 48 h after infection.
Upon C. trachomatis infection, the full-length PARP-1 protein of 113 kDa was found to be cleaved into a ladder with predominant bands at 71, 55, and 42 kDa (Fig. 2C). Compared to staurosporine-treated cells, the cleavage bands in Chlamydia-infected cells were completely different from those generated during apoptosis (Fig. 2C). The degradation of PARP-1 started at approximately 16 h after infection, a time point at which chlamydiae are in the replicative phase.
Cell-free degradation assays were used to confirm the presence of a specific HMGB1 and PARP-1-degrading protease in Chlamydia-infected cells. Both cytosolic and nuclear extracts from infected cells contained lower amounts of HMGB1 than extracts from mock-infected cells (Fig. 2D). Furthermore, the incubation of nuclear extracts from mock-infected cells with cytosolic extracts from infected cells resulted in a complete degradation of the HMGB1 full-length protein (Fig. 2D). Similar results were obtained for PARP-1 (Fig. 2D). Although PARP-1 fragments of 55 and 42 kDa were found in most of the experiments, the degradation patterns showed some variations in band sizes, indicating that the responsible protease may have multiple binding sites on PARP-1. Treatment of infected cells with chloramphenicol or doxycycline indicated that HMGB1 and PARP-1 degradation was dependent on bacterial protein synthesis (Fig. 2E).
HMGB1 and PARP-1 cleavage in C. trachomatis-infected HeLa cells is caused by CPAF.
Because Paschen et al. (29) observed that PARP-1 is degraded in cells transfected with the chlamydial protease CPAF, it could be hypothesized that CPAF represents the protease cleaving both HMGB1 and PARP-1 in Chlamydia-infected HeLa cells. For the identification of a Chlamydia-specific proteolytic factor, a MonoQ column chromatography approach was used to purify fractions from cytosolic extracts of infected HeLa cells. A total of 50 fractions eluted from MonoQ columns were tested for proteolytic activities in cell-free degradation assays. Fractions 23 to 26 showed a strong proteolytic activity against both HMGB1 and PARP-1 (Fig. 3A). In SDS-PAGE gels, a protein double band of about 29 and 34 kDa was detected in fractions 24 to 26 (Fig. 3B). Immunoblots of proteolytic fractions that were stained with a mouse polyclonal CPAF antibody confirmed that the bands represented proteins of CPAF dimers (Fig. 3C). In fraction 23 this double band could not be detected, but this may be due to limited sensitivities of Coomassie and antibody staining.
FIG. 3.

CPAF is responsible for HMGB1 and PARP-1 degradation in C. trachomatis-infected HeLa cells. (A) Cytosolic extracts of cells infected at an MOI of 5 were fractionated by MonoQ column chromatography. Fractions were examined for HMGB1 and PARP-1 cleavage in a cell-free assay using nuclear fractions of mock-infected cells as a substrate. Cleavage was detected by immunoblotting. (B) Protein profiles of proteolytic fractions were monitored by Coomassie staining of SDS gels. Note that proteolytic fractions showed two bands of 29 and 34 kDa that were absent in fractions without proteolytic activity. (C) Immunoblots of MonoQ column fractions were stained with a mouse polyclonal antibody to CPAF. (D) Immunoprecipitation (IP) of fraction 25 with a mouse polyclonal CPAF antibody (Ab). The proteolytic activity of the supernatant was determined by a cell-free cleavage assay using human recombinant HMGB1 and PARP-1 as substrates. Mouse IgG was used as an IP control. The supernatant and pellet of the precipitates were analyzed for the presence of CPAF by immunoblotting.
To verify that CPAF was necessary and sufficient to degrade HMGB1 and PARP-1, immunoprecipitation was used to deplete CPAF from chromatography fraction 25. Residual supernatants after precipitation with the CPAF antibody lost the ability to cleave human recombinant HMGB1 and PARP-1, whereas immunoprecipitation with mouse IgG that was used as a control had no effect on the proteolytic activity of the fraction (Fig. 3D).
Lactacystin, an inhibitor of the 26S proteasome, has also been identified to inhibit the activity of CPAF (38). When lactacystin was added to infected cultures at a concentration of 10 μM, HMGB1 and PARP-1 could be protected from cleavage (Fig. 4A). To exclude that PARP-1 and HMGB1 degradation is artificially caused by CPAF during the preparation of cell lysates for immunoblotting, infected cells cultured in medium alone were collected and treated with lactacystin during incubation in lysis buffer. In this case, lactacystin did not prevent PARP-1 and HMGB1 cleavage (Fig. 4A).
FIG. 4.
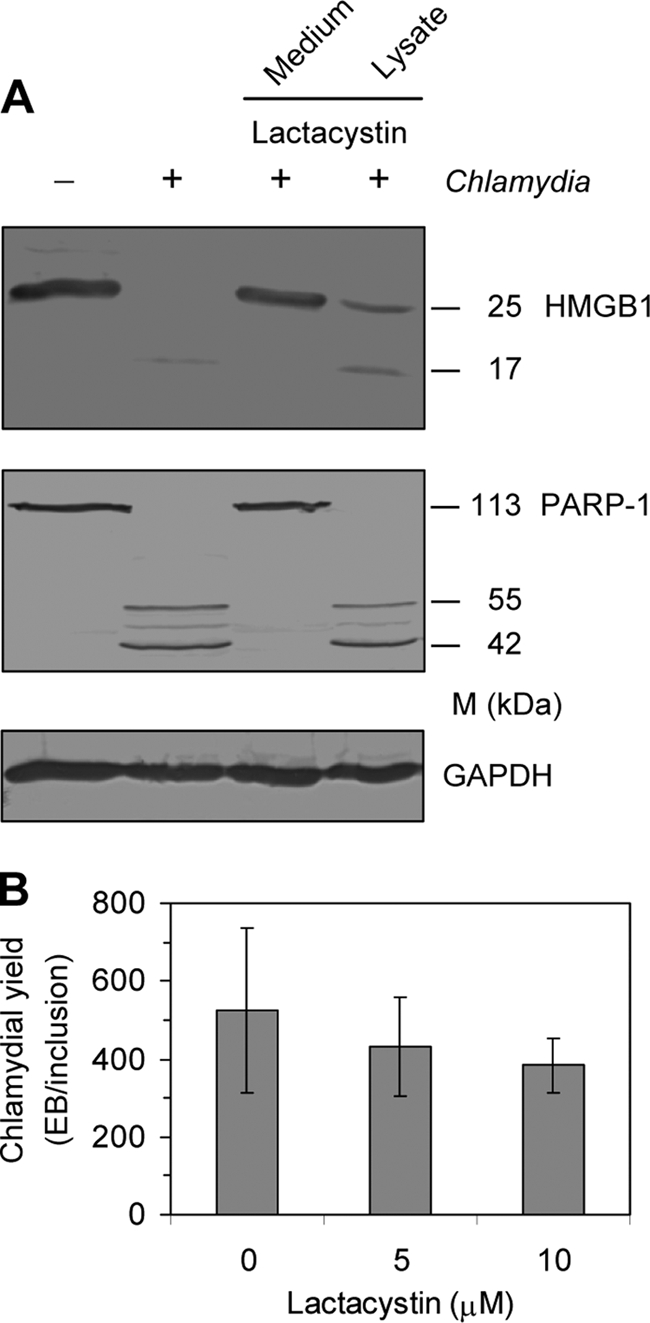
Effects of the CPAF inhibitor lactacystin on C. trachomatis-infected HeLa cells. (A) Cells were infected at an MOI of 5. A total of 11 μg of lactacystin per 35-mm-diameter well was added to the culture medium after infection (final concentration, 10 μM; lane 3) or to cell lysates during preparation for SDS-PAGE (lane 4). Cells were collected at 48 h after infection and analyzed by immunoblotting. GAPDH was stained as a reference band. (B) Replication of C. trachomatis in HeLa cells treated with lactacystin. Chlamydial yield was calculated from the determination of inclusions and titration of newly produced infectious elementary bodies.
Because large amounts of active PARP-1 can cause a critical decline in the ATP level its degradation might promote chlamydial replication. However, inhibition of CPAF activity by lactacystin had no significant effect on the number of chlamydial elementary bodies produced during the replication cycle (Fig. 4B).
PARP-1 cleavage in C. trachomatis-infected HeLa cells is associated with the loss of polymerase activity, and PARP-1 silencing causes cytopathic effects.
It could be supposed that the degradation of full-length PARP-1 into multiple fragments results in a loss of (ADP-ribose)polymerase activity. Extracts from infected HeLa cells were collected at different time points after infection, and the activity of PARP-1 was calculated by measuring the incorporation of biotinylated poly(ADP-ribose) onto histone proteins. C. trachomatis infection caused a significant decrease in PARP-1 enzymatic activity at 24 and 48 h, corresponding to the degree of PARP-1 cleavage observed in immunoblots (Fig. 5A).
FIG. 5.
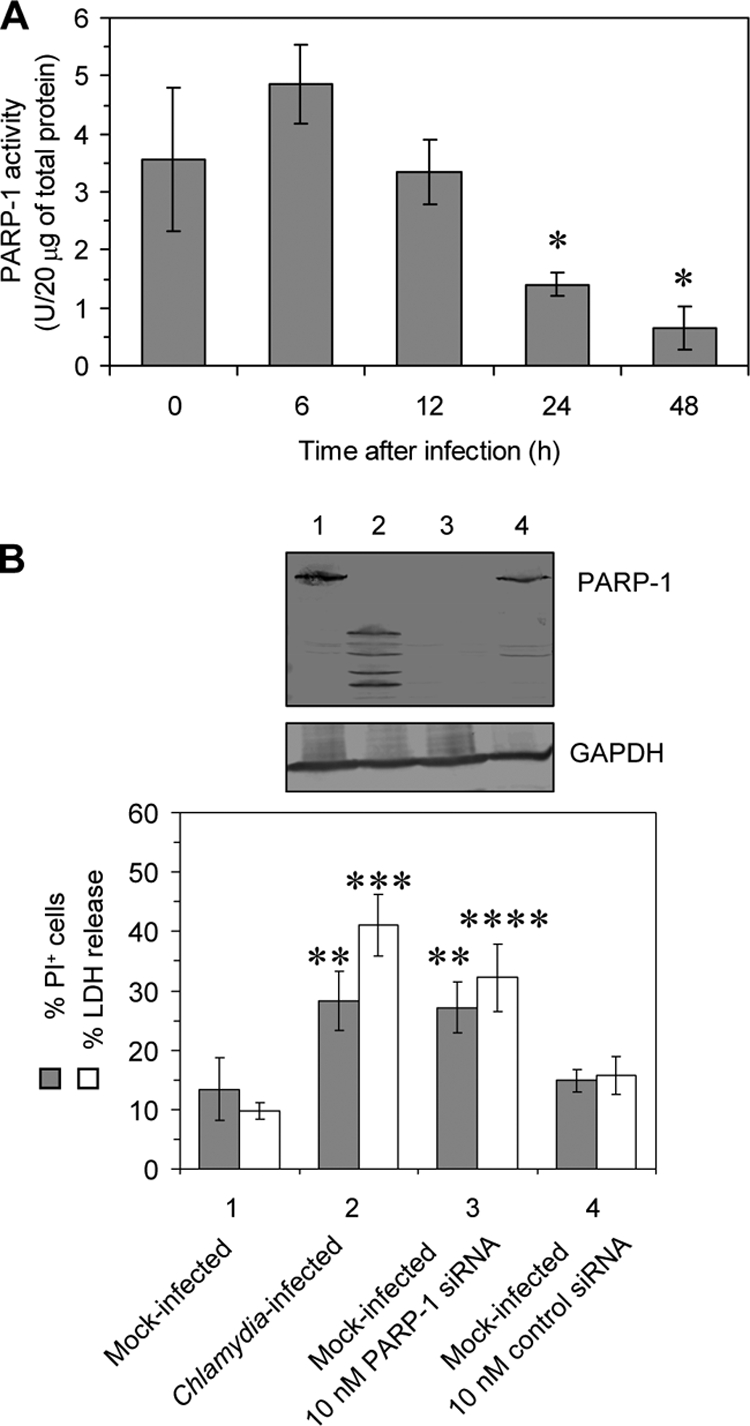
Silencing of PARP-1 in HeLa cells. (A) Decrease in PARP-1 activity in HeLa cells upon infection with C. trachomatis. Total cell lysates were analyzed by a colorimetric enzyme activity assay. *, P ≤ 0.01 (compared to values for infected cells at 0 and 6 h; n = 3). (B) Downregulation of PARP-1 protein in HeLa cells by siRNA transfection. Cells were transfected with PARP-1-specific and nonsilencing siRNA and incubated for 48 h. Chlamydia-infected cells served as a control. PARP-1 was analyzed by immunoblotting. GAPDH was stained as a reference band. Cell death was measured by flow cytometry following PI labeling and quantification of LDH release. LDH activities are expressed as the percentage of the maximum releasable LDH activity in the cells that was measured after Triton lysis. **, P ≤ 0.03; ***, P ≤ 0.01; ****, P ≤ 0.04 (compared to values for mock-infected cells incubated in medium alone or transfected with control siRNA; n = 4).
PARP-1 activation and degradation are involved in the regulation of cell death; however, it is unknown whether the loss of functional PARP-1 results in increased cell membrane permeability. HeLa cells were transfected with specific siRNA to silence PARP-1 expression. Nonsilencing siRNA served as a negative control. To validate the transfection procedure, we used Alexa Fluor 488-labeled control siRNA and assessed the number of cells containing siRNA by using confocal microscopy. At 24 h after the addition 93% of the cells were found to be positive. Transfection with 10 nM the specific siRNA was sufficient to inhibit PARP-1 protein expression in HeLa cells, whereas PARP-1 was present after control siRNA transfection (Fig. 5B). As shown by flow cytometry, PARP-1 inhibition in mock-infected HeLa cells induced ca. 30% PI-positive cells. When the cultures were infected at an MOI of 5, which resulted in 82% inclusion-containing cells, a similar percentage of PI-positive cells was found (Fig. 5B). Moreover, the release of LDH from the cells was significantly increased after infection or PARP-1 siRNA transfection (Fig. 5B).
HMGB1 is not translocated from the nucleus to the cytosol in Chlamydia-infected cells.
During necrosis, PARP-1 activation induces the nuclear-to-cytosolic translocation of HMGB1 that serves the ability of membrane-damaged cells to release this inflammatory mediator (6). Immunofluorescence staining was used to analyze the distribution of HMGB1 in mock-infected and infected HeLa cells. The results are summarized in Fig. 6A. When necrosis was induced in mock-infected cells by treatment with H2O2, the translocation of HMGB1 into the cytosol was observed. The addition of DHIQ, an inhibitor of PARP-1, prevented nuclear release of HMGB1. In Chlamydia-infected cells, HMGB1 was retained in the nucleus. At 24 h after infection the level of nuclear HMBG1 remained unchanged compared to the cells in mock-infected cultures. When H2O2 was added to infected cultures HMGB1 translocation into the cytosol could be found in Chlamydia-negative but not in inclusion-positive cells. This finding corresponds to the observation that PARP-1 is already degraded at 24 h after infection. The inhibition of CPAF activity by lactacystin in infected cells restored the ability to translocate HMGB1 into the cytosol after exposure to H2O2. At 48 h after infection, the fluorescence intensity of nuclear HMGB1 was clearly decreased in inclusion-positive cells but not in neighboring uninfected cells, confirming that HMGB1 is degraded at a late stage of chlamydial infection.
FIG. 6.

Distribution of HMGB1 in Chlamydia-infected HeLa cells compared to mock-infected cells after H2O2 treatment. Cells were infected with C. trachomatis at an MOI of 1. H2O2 was added to mock-infected or infected cells at a concentration of 500 μM for 3 h. The PARP-1 inhibitor DHIQ was added to mock-infected cells at a concentration of 300 μM for 30 min prior to H2O2 treatment. Lactacystin was added at a concentration of 10 μM after infection. (A) Cell monolayers were costained with Hoechst for DNA (blue), HMGB1 antibody (probed with a Cy3-conjugated secondary antibody; red), and FITC-conjugated C. trachomatis MOMP antibody (green). Bar, 15 μm. (B) Intracellular HMGB1 levels were determined by immunoblotting. Cell lysates were prepared at 24 h after infection. (C) Extracellular HMGB1 was quantified in culture supernatants by ELISA. *, P ≤ 0.01 (compared to values for mock-infected cells cultured in medium alone); **, P ≤ 0.02 (compared to values for mock-infected cells treated with H2O2); ***, P ≤ 0.01 (compared to values for infected cells treated with H2O2 alone) (n = 4).
Microscopic observations were verified by determination of intracellular and extracellular levels of HMGB1. Immunoblot analysis of whole-cell lysates revealed that H2O2 treatment caused a decrease in the HMGB1 levels in mock-infected cells but not in infected cells at 24 h after inoculation of Chlamydia (Fig. 6B). When lactacystin was added, only low amounts of intracellular HMGB1 could be found in infected cells after H2O2 exposure (Fig. 6B). The levels of HMGB1 in cell lysates were inversely correlated to its concentrations in culture supernatants (Fig. 6C). HMGB1 release after H2O2 addition was upregulated in mock-infected cultures but not in infected cultures. However, H2O2 induced the release of HMGB1 from infected cells in the presence of lactacystin.
DISCUSSION
Chlamydiae are obligate intracellular bacteria that modulate host cell death pathways during the replication cycle. We show here that C. trachomatis infection results in cytopathic effects that are characterized by increased cell membrane permeability and cleavage of PARP-1 and HMGB1 by the chlamydial protease CPAF. PARP-1 is a highly conserved multifunctional enzyme. After activation by DNA strand breaks, it catalyzes the synthesis of poly(ADP-ribose) at the expense of NAD+ and transfers these polymers onto acceptor proteins (5). The molecular structure of this protein comprises three main distinct regions: an N-terminal DNA-binding domain, a central automodification domain, and a C-terminal catalytic domain (5). Under homeostatic conditions, PARP-1 participates in genome repair, DNA replication, and the regulation of transcription (24). During apoptosis, activated caspase 3 cleaves the 113-kDa form of PARP-1 at the DEVD site to generate two fragments of 85 and 24 kDa (3, 21). This cleavage inactivates the enzyme by separation of the two zinc-finger DNA-binding motifs from the automodification and catalytic domains and destroys its ability to respond to DNA strand breaks. In contrast, PARP-1 overactivation results in extensive poly(ADP-ribosyl)ation of nuclear proteins, which in turn causes the depletion of NAD+ and ATP energy stores inside the cells, thereby inducing necrosis (2). In necrotic cells, PARP-1 is subsequently degraded into multiple fragments by cathepsins (16). Interestingly, the Chlamydia-specific protease CPAF causes a similar multifragmentation of PARP-1 in infected cells. CPAF is synthesized as one polypeptide of 70 kDa and rapidly processed into a 35-kDa C-terminal and a 29-kDa N-terminal subunit, which assemble to intramolecular dimers (7, 8, 38). The protease is secreted from the chlamydial inclusion into the host cell cytosol and degrades several host cell proteins (22, 31, 38). At present it is unclear by which mechanism CPAF is translocated into the nucleus to degrade nuclear proteins. C. trachomatis-infected epithelial cells undergo mitosis without subsequent cell division (17). It can be supposed that CPAF also contacts nucleus-associated proteins during mitotic processes.
By the cleavage of proapoptotic BH3-only proteins, CPAF plays an important role in Chlamydia-mediated apoptosis inhibition. On the other hand, Paschen et al. (29) reported that the transfection of epithelial cells with CPAF induces cell death and the degradation of several host cell proteins, including PARP-1. We show here that the CPAF-caused cleavage of PARP-1 is accompanied by a continuing decline of its enzyme activity during the chlamydial replication cycle. As shown by the effects of PARP-1 silencing on the viability of mock-infected HeLa cells, the loss of PARP-1 function promotes plasma membrane disintegration and LDH release and therefore may contribute to Chlamydia-induced cytopathicity. Because high activities of PARP-1 can result in NAD+ and ATP depletion one may speculate that its degradation may improve the availability of energy metabolites for chlamydiae during the replication cycle. However, treatment of infected HeLa cells with the CPAF inhibitor lactacystin had no significant effect on the progeny of chlamydial elementary bodies, indicating that PARP-1 degradation may be linked to another strategy of chlamydiae to modulate host cell functions.
Jungas et al. (20) have suggested that cell death after Chlamydia infection is characterized by the release of the nuclear protein HMGB1 because the amount of cell-associated HMGB1 was found to decrease upon infection. The present study demonstrates that the disappearance of HMGB1 in C. trachomatis-infected HeLa cells does not result from its release into the supernatant but from CPAF-mediated degradation. In contrast to PARP-1, degradation of HMGB1 is restricted to the late stage of infection. Because this protein is histone associated, it is possible that CPAF can only attack HMGB1 when it is displaced from this complex. HMGB1 evolves several biological functions: the intracellular form facilitates DNA binding, stabilizes nucleosome formation, and acts as a transcriptional regulator, whereas extracellular HMGB-1 that is released from membrane-damaged cells has been recognized as an important mediator inducing the inflammatory response to dying cells (34). HMGB1 must be translocated from the nucleus into the cytosol before it can be released from cells that lost their plasma membrane integrity. PARP-1 activation has been shown to regulate this translocation (6). In Chlamydia-infected cells the loss of plasma membrane integrity can be observed at 24 h after infection. Although HMGB1 is not degraded at this time point, it cannot be found in increased quantities outside the cells. Because PARP-1 is already degraded at 24 h after infection, the pathway mediating HMGB1 translocation is interrupted in Chlamydia-infected cells. This hypothesis could be confirmed by the observation that H2O2, an inducer of necrosis, causes the translocation of HMGB1 in mock-infected cells but not in cells containing a chlamydial inclusion. The findings of the present study suggest that C. trachomatis prevents the release of the inflammatory mediator HMGB1 from injured host cells by two mechanisms: inhibition of HMGB1 translocation from the nucleus to the cytosol and degradation of HMGB1 at a late time point of the infection cycle. Both effects are mediated by the proteolytic activity of CPAF.
In summary, we show here that although host cell infection results in the loss of cell membrane integrity, C. trachomatis interferes with cellular pathways involved in necrotic cell death by CPAF-mediated degradation of HMGB1 and PARP-1. The suppression of the release of HMGB1 from infected cells, and its subsequent cleavage, may contribute to immune evasion strategies of Chlamydia that limit the inflammatory response following epithelial cell damage.
Acknowledgments
This study was supported by a grant from the International Leibniz Research School of Microbial and Biomolecular Interactions, Jena, Germany, to H.Y. and by grant 01KI0726 from the Bundesministerium für Bildung und Forschung (Germany) to E.S. and J.R.
We are grateful to Karl-Hermann Schmidt (Institute of Medical Microbiology, University of Jena) for his kind help with the column chromatography.
Editor: R. P. Morrison
Footnotes
Published ahead of print on 26 April 2010.
REFERENCES
- 1.Abdelrahman, Y. M., and R. J. Belland. 2005. The chlamydial developmental cycle. FEMS Microbiol. Rev. 29:949-959. [DOI] [PubMed] [Google Scholar]
- 2.Bouchard, V. J., M. Rouleau, and G. G. Poirier. 2003. PARP-1, a determinant of cell survival in response to DNA damage. Exp. Haematol. 31:446-454. [DOI] [PubMed] [Google Scholar]
- 3.Boulares, A. H., G. Y. Yakovlevl, V. Ivanova, B. A. Stoica, G. Wang, S. Iyer, and M. Smulson. 1999. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis: caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J. Biol. Chem. 274:22932-22940. [DOI] [PubMed] [Google Scholar]
- 4.Burton, M. J. 2007. Trachoma: an overview. Br. Med. Bull. 84:99-116. [DOI] [PubMed] [Google Scholar]
- 5.D'Amours, D., S. Desnoyers, I. D' Silva, and G. G. Poirier. 1999. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 342:249-268. [PMC free article] [PubMed] [Google Scholar]
- 6.Ditsworth, D., W. X. Zong, and C. B. Thompson. 2007. Activation of poly(ADP)-ribose polymerase (PARP-1) induces release of the pro-inflammatory mediator HMGB1 from the nucleus. J. Biol. Chem. 282:17845-17854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dong, F., J. Sharma, Y. Xiao, Y. Zhong, and G. Zhong. 2004. Intramolecular dimerization is required for the Chlamydia-secreted protease CPAF to degrade host transcriptional factors. Infect. Immun. 72:3869-3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong, F., M. Pirbhai, Y. Zhong, and G. Zhong. 2004. Cleavage-dependent activation of a Chlamydia-secreted protease. Mol. Microbiol. 52:1487-1494. [DOI] [PubMed] [Google Scholar]
- 9.Fan, T., H. Lu, H. Hu, L. Shi, G. A. McClarty, D. M. Nance, A. H. Greenberg, and G. Zhong. 1998. Inhibition of apoptosis in Chlamydia-infected cells: blockade of mitochondrial cytochrome c release and caspase activation. J. Exp. Med. 187:487-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fiers, W., R. Beyaert, W. Declercq, and P. Vandenabeele. 1999. More than one way to die: apoptosis, necrosis and reactive oxygen damage. Oncogene 18:7719-7730. [DOI] [PubMed] [Google Scholar]
- 11.Fink, S. L., and B. T. Cookson. 2005. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 73:1907-1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fischer, S. F., C. Schwarz, J. Vier, and G. Häcker. 2001. Characterization of antiapoptotic activities of Chlamydia pneumoniae in human cells. Infect. Immun. 69:7121-7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fischer, S. F., J. Vier, S. Kirschnek, A. Klos, S. Hess, S. Ying, and G. Häcker. 2004. Chlamydia inhibit host cell apoptosis by degradation of proapoptotic BH3-only proteins. J. Exp. Med. 200:905-916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fischer, S. F., T. Harlander, J. Vier, and G. Häcker. 2004. Protection against CD95-induced apoptosis by chlamydial infection at a mitochondrial step. Infect. Immun. 72:1107-1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forsbach-Birk, V., U. Simnacher, K.-I. Pfrepper, E. Soutschek, A. O. Kiselev, M. F. Lampe, T. Meyer, E. Straube, and A. Essig. Identification and evaluation of a combination of chlamydial antigens to support the diagnosis of severe and invasive Chlamydia trachomatis infections. Clin. Microbiol. Infect. doi: 10.1111/j.1469-0691.2009.03041.x. [DOI] [PubMed]
- 16.Gobeil, S., C. C. Boucher, D. Nadeau, and G. G. Poirier. 2001. Characterization of the necrotic cleavage of poly(ADP-ribose) polymerase (PARP-1): implication of lysosomal protease. Cell Death Differ. 8:588-594. [DOI] [PubMed] [Google Scholar]
- 17.Greene, W., and G. Zhong. 2003. Inhibition of host cell cytokinesis by Chlamydia trachomatis infection. J. Infect. 47:45-51. [DOI] [PubMed] [Google Scholar]
- 18.Hotzel, H., and K. Sachse. 1998. Verbesserung und Beschleunigung der Rinderlungenseuche-Diagnostik durch Direktnachweis des Erregers mittels Polymerase-Kettenreaktion (PCR). Berl. Munch. Tierarztl. Wochenschr. 111:268-272. [PubMed] [Google Scholar]
- 19.Hybiske, K., and R. S. Stephens. 2007. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc. Natl. Acad. Sci. U. S. A. 104:11430-11435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jungas, T., P. Verbeke, T. Darville, and D. M. Ojcius. 2004. Cell death, Bax activation, and HMGB1 release during infection with Chlamydia. Microb. Infect. 6:1145-1155. [DOI] [PubMed] [Google Scholar]
- 21.Kaufmann, S. H., S. Desnoyers, Y. Ottaviano, N. E. Davidson, and G. G. Poirier. 1993. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res. 53:3976-3985. [PubMed] [Google Scholar]
- 22.Kawana, K., A. J. Quayle, M. Ficarra, J. A. Ibana, L. Shen, Y. Kawana, H. Yang, L. Marrero, S. Yavagal, S. J. Greene, Y.-X. Zhang, R. B. Pyles, R. S. Blumberg, and D. J. Schust. 2007. CD1d degradation in Chlamydia trachomatis-infected epithelial cells is the result of both cellular and chlamydial proteasomal activity. J. Biol. Chem. 282:7368-7375. [DOI] [PubMed] [Google Scholar]
- 23.Kroemer, G., L. Galluzzi, P. Vandenabeele, J. Abrams, E. S. Alnemri, E. H. Baehrecke, M. V. Blagosklonny, W. S. El-Deiry, P. Golstein, D. R. Green, M. Hengartner, R. A. Knight, S. Kumar, S. A. Lipton, W. Malorni, G. Nuñez, M. E. Peter, J. Tschopp, J. Yuan, M. Piacentini, B. Zhivotovsky, and G. Melino. 2009. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 16:3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindahl, T., M. S. Satoh, G. G. Poirier, and A. Klungland. 1995. Post-translational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends Biochem. Sci. 20:405-411. [DOI] [PubMed] [Google Scholar]
- 25.Mårdh, P. A. 2004. Tubal factor infertility, with special regard to chlamydial salpingitis. Curr. Opin. Infect. Dis. 17:49-52. [DOI] [PubMed] [Google Scholar]
- 26.Marino, J., I. Stoeckli, M. Walch, S. Latinovic-Golic, H. Sundstroem, P. Groscurth, U. Ziegler, and C. Dumrese. 2008. Chlamydophila pneumoniae derived from inclusions late in the infectious cycle induce aponecrosis in human aortic endothelial cells. BMC Microbiol. 8:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miyairi, I., and G. I. Byrne. 2006. Chlamydia and programmed cell death. Curr. Opin. Microbiol. 9:102-108. [DOI] [PubMed] [Google Scholar]
- 28.Ojcius, D., P. Souque, J. L. Perfettini, and A. Dautry-Varsat. 1998. Apoptosis of epithelial cells and macrophages due to infection with the obligate intracellular pathogen Chlamydia psittaci. J. Immunol. 161:4220-4226. [PubMed] [Google Scholar]
- 29.Paschen, S. A., J. G. Christian, J. Vier, F. Schmidt, A. Walch, D. M. Ojcius, and G. Häcker. 2008. Cytopathicity of Chlamydia is largely reproduced by expression of a single chlamydial protease. J. Cell Biol. 182:117-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perfettini, J. L., J. C. Reed, N. Israël, J. C. Martinou, A. Dautry-Varsat, and D. M. Ojcius. 2002. Role of BCL-2 family members in caspase-independent apoptosis due to Chlamydia infection. Infect. Immun. 70:55-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pirbhai, M., F. Dong, Y. Zhong, K. Z. Pan, and G. Zhong. 2006. The secreted protease factor CPAF is responsible for degrading proapoptotic BH3-only proteins in Chlamydia trachomatis-infected cells. J. Biol. Chem. 281:31495-31501. [DOI] [PubMed] [Google Scholar]
- 32.Rödel, J., M. Woytas, A. Groh, K.-H. Schmidt, M. Hartmann, M. Lehmann, and E. Straube. 2000. Production of basic fibroblast growth factor and interleukin 6 by human smooth muscle cells following infection with Chlamydia pneumoniae. Infect. Immun. 68:3635-3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scaffidi, P., T. Misteli, and M. E. Bianchi. 2002. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418:191-195. [DOI] [PubMed] [Google Scholar]
- 34.Ulloa, L., and D. Messmer. 2006. High-mobility group box 1 (HMGB1) protein: friend and foe. Cytokine Growth Factor Rev. 17:189-201. [DOI] [PubMed] [Google Scholar]
- 35.Xiao, Y., Y. Zhong, W. Greene, F. Dong, and G. Zhong. 2004. Chlamydia trachomatis infection inhibits both Bax and Bak activation induced by staurosporine. Infect. Immun. 72:5470-5474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ying, S., M. Pettengill, E. R. Latham, A. Walch, D. M. Ojcius, and G. Häcker. 2008. Premature apoptosis of Chlamydia-infected cells disrupts chlamydial development. J. Infect. Dis. 198:1536-1544. [DOI] [PubMed] [Google Scholar]
- 37.Ying, S., S. F. Fischer, M. Pettengill, D. Conte, S. A. Paschen, D. M. Ojcius, and G. Häcker. 2006. Characterization of host cell death induced by Chlamydia trachomatis. Infect. Immun. 74:6057-6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhong, G., P. Fan, H. Ji, F. Dong, and Y. Huang. 2001. Identification of a chlamydial protease-like activity factor responsible for the degradation of host transcription factors. J. Exp. Med. 193:935-942. [DOI] [PMC free article] [PubMed] [Google Scholar]