

Abstract
Clostridium perfringens type C isolates cause fatal, segmental necro-hemorrhagic enteritis in animals and humans. Typically, acute intestinal lesions result from extensive mucosal necrosis and hemorrhage in the proximal jejunum. These lesions are frequently accompanied by microvascular thrombosis in affected intestinal segments. In previous studies we demonstrated that there is endothelial localization of C. perfringens type C β-toxin (CPB) in acute lesions of necrotizing enteritis. This led us to hypothesize that CPB contributes to vascular necrosis by directly damaging endothelial cells. By performing additional immunohistochemical studies using spontaneously diseased piglets, we confirmed that CPB binds to the endothelial lining of vessels showing early signs of thrombosis. To investigate whether CPB can disrupt the endothelium, we exposed primary porcine aortic endothelial cells to C. perfringens type C culture supernatants and recombinant CPB. Both treatments rapidly induced disruption of the actin cytoskeleton, cell border retraction, and cell shrinkage, leading to destruction of the endothelial monolayer in vitro. These effects were followed by cell death. Cytopathic and cytotoxic effects were inhibited by neutralization of CPB. Taken together, our results suggest that CPB-induced disruption of endothelial cells may contribute to the pathogenesis of C. perfringens type C enteritis.
The anaerobic, spore-forming bacterium Clostridium perfringens is an important Gram-positive pathogen of humans and animals (18, 42). It causes diverse gastrointestinal diseases, such as food poisoning, enterotoxemia, and enteritis, as well as wound infections and septicemias (37). The virulence of different C. perfringens strains is related to the production of a large array of exotoxins (34). C. perfringens type C isolates are defined by production of two major toxins, α-toxin (CPA) and β-toxin (CPB). In addition, type C isolates may secrete other toxins, such as β2-toxin (CPB2), perfringolysin (PFO), enterotoxin (CPE), and TpeL (2, 34, 36) C. perfringens type C strains cause severe, acute, necrotizing enteritis in livestock and humans (18, 42). Outbreaks of human type C enteritis were recorded after the Second World War in Germany (20), but this disease has been reported only sporadically in developed countries (25, 27, 39, 51). A similar disease has been diagnosed more frequently in parts of Southeast Asia (7, 17, 26), particularly in the highlands of Papua New Guinea (23), where it was a frequent cause of childhood mortality until vaccination programs were initiated (24). C. perfringens type C causes enteritis more frequently in animals, such as calves, sheep, goats, and particularly pigs (42, 43). Typically, neonatal piglets are affected from the first day of life until they are approximately 3 weeks old. The peracute to acute type of the disease affects piglets within the first few days postpartum (12, 14, 43). Macroscopic lesions at necropsy are pathognomonic, with deep, segmental mucosal necrosis and massive hemorrhage in the small intestine. In most cases the lesions are confined to the proximal jejunum; however, they can extend into the distal small intestine and the colon. This suggests that lesions are initiated in the upper small intestine and can spread rapidly throughout the intestine. In addition to these marked necro-hemorrhagic lesions, thrombosis of small vessels in the lamina propria and submucosa is a consistent finding (12, 14). A more protracted clinical course of type C enteritis is seen mainly in piglets that die when they are 1 to 3 weeks old (12, 14, 43). The pathological lesion is a segmental to diffuse fibrino-necrotizing enteritis. Histopathologically, such cases are characterized by demarcation of the deeply necrotic mucosa by marked infiltration of neutrophilic granulocytes. Similar acute and subacute forms of type C enteritis also occur in humans (6, 18, 21). In humans, however, subacute lesions are more often described as multifocal patchy necrosis of the small intestine. Again, mucosal and submucosal vascular thrombosis is a frequent finding, especially in acute lesions (20, 21).
Besides the clear epidemiological evidence for the importance of CPB in type C enteritis (41, 42), recent experimental studies using a rabbit intestinal loop model and a mouse infection model clearly demonstrated that CPB is the essential virulence factor of type C strains (38, 47, 49). In rabbit ileal loops, application of purified CPB and infection with C. perfringens type C strains caused villous tip necrosis, which indicated that there was initial intestinal epithelial damage. Vascular thrombosis in mucosal and submucosal vessels was also observed in this model. In general, the vascular damage observed in naturally occurring and experimentally induced type C enteritis is considered a secondary effect due to massive epithelial and mucosal necrosis (22, 43). However, the potential direct effects of exotoxins on vascular endothelia during type C enteritis have never been investigated.
CPB is a beta-barrel-pore-forming toxin (9) that has been shown to form oligomers in the membrane of human endothelial cells (44) and the human HL 60 cell line (31). So far, cytotoxic and cytopathic effects of CPB have been demonstrated only for HL 60 cells. HeLa, Vero, CHO, MDCK, Cos-7, P-815, and PC12 cells were not sensitive to this toxin (31, 40). These findings indicate that CPB toxicity is cell type specific and most likely occurs via binding to specific membrane receptors. Recently, we localized CPB at vascular endothelial cells in acute type C enteritis lesions in piglets and a human patient (28, 29). As a result of this, we hypothesized that direct targeting of endothelial cells and induction of local vascular damage could contribute to the rapid tissue necrosis observed in the acute form of type C enteritis. To validate our initial reports, we performed additional immunohistochemical studies with naturally diseased piglets and subsequently studied the direct cytopathic effects of CPB on cultured primary porcine endothelial cells. The objectives of this study were (i) to evaluate the susceptibility of porcine endothelial cells to CPB in vitro, (ii) to characterize early morphological changes induced by CPB in these cells, and (iii) to relate the findings obtained to pathological lesions observed in acute type C enteritis in piglets. Our results reveal for the first time that porcine aortic endothelial cell (PAEC) cultures are highly sensitive to CPB, which results in rapid disruption of the actin cytoskeleton and retraction of the cell borders progressing to marked cell shrinkage.
MATERIALS AND METHODS
Animals, histopathology, and immunohistochemistry.
Tissues from three 1-day-old piglets, which were euthanized for a routine diagnostic workup of piglet mortality in their herds, were used for this investigation. These animals were necropsied immediately after they were euthanized, and they were subsequently diagnosed with type C enteritis based on classical macroscopic (Fig. 1A) and histopathological lesions in combination with isolation of C. perfringens type C from the intestine (1, 14). Tissues from a piglet which was euthanized and diagnosed with coccidiosis based on histopathological and parasitological evaluations were used as controls. Bacteriologically, C. perfringens type A (cpa+ cpb2+), but not C. perfringens type C, was isolated from the small and large intestines. Samples of the small intestine, colon, liver, lungs, spleen, brain, and kidneys of each animal were fixed in 10% buffered formalin 15 min after the animals were euthanized. After 24 h of fixation, tissue sections were embedded in paraffin, routinely processed for histology, cut into 5-μm sections, and stained with hematoxylin and eosin (H&E). An immunohistochemistry analysis using anti-CPB and anti-CPA monoclonal antibodies as a control was performed as described previously (28).
FIG. 1.

Pathology and immunohistochemistry. (A) Distribution of macroscopic lesions of acute C. perfringens type C enteritis in a 2-day-old piglet (stomach on left side; CO, colon). The arrows indicate the extent of typical necro-hemorrhagic lesions in the proximal jejunum. The asterisk indicates a histopathological sampling site in a macroscopically unaffected segment of the caudal jejunum. (B) H&E-stained histological sections of a macroscopically unaffected segment, showing early signs of epithelial degeneration at villous tips and thrombus formation (arrow) in an underlying vessel. (C) Immunohistochemical labeling with MAb-CPB of a serial section of the specimen shown in panel B, showing positive staining on the endothelial lining of a vessel (arrowhead). Note that no immunoreactivity was observed with epithelial cells. (D) H&E-stained control tissue from a piglet with superficial necrotizing enteritis due to I. suis infection. (E) Immunohistochemistry of sections revealed no CPB specific staining. (B and C) Magnification, ×1,000. (D and E) Magnification, ×400.
Cell cultures.
Thoracic segments of the aortas of a slaughtered 3-month-old pig and a 5-day-old piglet, which were euthanized for diagnostic purposes, were obtained under sterile conditions, placed in culture medium (Dulbecco modified Eagle medium [DMEM] containing 10% fetal calf serum [FCS], 1× antibiotic-antimycotic [Gibco], and 20 mM l-glutamine), transferred to a cell culture workbench, opened longitudinally, and washed. Thirty minutes after the animals died, endothelial cells were scraped off and transferred to fibronectin-coated tissue culture plates to establish separate primary cell cultures from the two donors. Primary porcine aortic endothelial cell (PAEC) cultures were grown to confluence at 37°C in the presence of 5% CO2 and passaged. The endothelial origin and purity of cells were verified by immunofluorescence using anti-rat PECAM-1 antibody (Millipore) (46). Cells in passage 3 were frozen in liquid nitrogen and used as stock cultures. Porcine primary fibroblasts were isolated from skin excised from a 5-day-old piglet that was euthanized for diagnostic purposes. The tissue was placed in DMEM containing 10× antibiotic-antimycotic and incubated for 6 h at 4°C. The epidermal layer was manually separated from the dermis after overnight incubation in 10 mg/ml dispase (Roche). Dermal tissue was dissociated by incubation in trypsin-EDTA (Bioconcept) with gentle agitation, and cells were seeded into tissue culture flasks in DMEM containing 10% FCS, 20 mM l-glutamine, 1 ng/ml acidic fibroblast growth factor (aFGF), and 1 ng/ml basic fibroblast growth factor (bFGF) (Gibco). Fibroblasts in passage 3 were frozen as stock cultures in liquid nitrogen. For all experiments, PAEC and fibroblasts were thawed, propagated, seeded at a density of 1.33 × 104 per cm2, and grown to confluence for 5 (PAEC) or 7 (fibroblasts) days. The cells used for experiments were exclusively passage 4 to 9 cells. To eliminate the possibility of donor-specific differences in endothelial cell responses, each experiment was performed separately with PAEC from both donors. All experiments were performed in duplicate.
Production of C. perfringens culture supernatants.
Two clinical porcine C. perfringens type C isolates from piglets with necrotizing enteritis and two C. perfringens type A isolates from healthy piglets (Table 1) were grown on blood agar plates. DNA extraction and PCR amplification of cpa, cpb, cpb2, etx, cpe, pfoA, netB, and tpeL were performed as described previously (1, 2, 8, 19). Late-log-phase culture supernatants were produced using liquid TGY anaerobic broth (3% tryptic soy broth, 2% glucose, 1% yeast extract, 0.1% l-cysteine) as described previously (8). After harvest, culture supernatants were chilled on ice, centrifuged (7,500 × g, 20 min), sterile filtered, aliquoted, and stored at −20°C.
TABLE 1.
Origins and genotypes of C. perfringens strains
| Strain | Type | Origin | Genotype | CPB concn (μg/ml) |
|---|---|---|---|---|
| JF 3719 | C | Piglet with necrotizing enteritis | cpa+cpb+cpb2+pfo+tpeL+ | 2.5 |
| JF 3721 | C | Piglet with necrotizing enteritis | cpa+cpb+cpb2+pfo+tpeL+ | 2.5 |
| JF 3693 | A | Healthy piglet | cpa+cpb2+pfo+ | |
| JF 3686 | A | Healthy piglet | cpa+ |
Expression and purification of rCPB.
The cpb gene was amplified from strain JF 3721 by performing PCR with primers adapted from primers used by Gibert et al. (10) that introduced a SmaI site at the 5′ end (TTCCCGGGGCAGCAATGATATAGGTAAAACTACTAC) and a NotI site at the 3′ end (TTGCGGCCGCCTAAATAGCTGTTACTTTGTGAG). Sequencing confirmed that there was 100% identity with the previously published gene sequence. The cpb gene was cloned into pGEM-T (Promega) and subcloned into pET 43.1a (Novagen), and recombinant CPB (rCPB) was expressed in Escherichia coli BL21 (Novagen) in a fusion with an N-terminal 495-amino-acid Nus-Tag protein. After affinity purification on Ni columns, the Nus-Tag protein was cleaved using a thrombin cleavage capture kit (Novagen). To obtain a sufficient yield of rCPB, the manufacturer's protocol was changed so that the preparation was incubated with 5 U/ml of biotinylated thrombin for 4 h at 4°C. Even under these conditions, a considerable amount of fusion protein remained uncleaved, and degradation of the fusion protein, indicated by additional bands (see Fig. 3, lanes 2 and 3), clearly occurred. Thrombin was subsequently removed using streptavidin-coated beads. rCPB was harvested at a concentration of 60 μg/ml (for quantification, see below). To eliminate toxic effects of contaminating proteins or trace amounts of thrombin, noncleaved rCPB-Nus-Tag fusion protein and mock purification from E. coli BL21 harboring the empty vector, which was incubated with thrombin as described above for rCPB, were used as controls in all experiments.
FIG. 3.
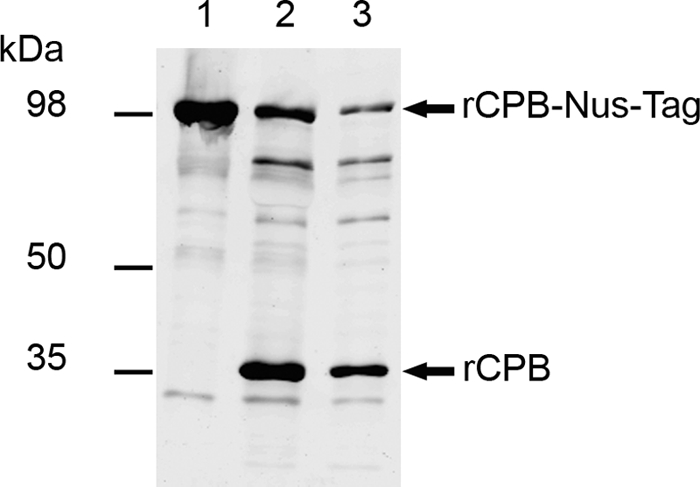
Expression of rCPB: Western blot analysis of 10-μl aliquots of purified rCPB-Nus-Tag (expected molecular mass, 98 kDa) (lane 1), rCPB (expected molecular mass, 35 kDa) after thrombin cleavage (lane 2), and rCPB after thrombin elimination (lane 3). Western blots were developed with MAb-CPB. Additional bands in lanes 2 and 3 represent degradation products of the fusion protein, likely due to excessive thrombin activity.
Quantification of rCPB.
Recombinant CPB was subjected to 10% SDS-PAGE, and protein bands were stained using Sypro Ruby protein gel staining (Sigma). The Precision Plus unstained standard (Bio-Rad) was used as a reference, and protein bands were photographed using the U:Genius gel documentation system (Syngene). Concentrations of rCPB were calculated using AIDA software (Image Analyzer for Windows). Western blotting was performed as described below.
Western blotting and quantification of CPB in culture supernatants.
C. perfringens culture supernatants were boiled in Laemmli buffer, and 20-μl samples were subjected to 10% SDS-PAGE and then transferred to nitrocellulose membranes and developed with anti-CPB monoclonal antibody 10A2 (MAb-CPB) (1:1,000 dilution; Centre for Veterinary Biologics, Ames, IA) and goat anti-mouse IRDye 800 secondary antibody (1:5,000 dilution; Li-Cor Biosciences). Serial dilutions of rCPB were used to create a standard curve for quantification of CPB in culture supernatants by Western blotting. CPB signals were visualized and scanned using an Odyssee infrared imager (Li-Cor). CPB signal intensity was measured using Odyssee 2.1 software, and concentrations were calculated and subsequently equalized to obtain a concentration of 2.5 μg CPB/ml of TGY broth (Table 1).
Cytotoxicity assays.
Confluent cells were exposed to 1:10 to 1:1,280 (vol/vol) dilutions of C. perfringens culture supernatants in serum-free endothelial cell culture medium (SFM) (Gibco). rCPB was added to SFM to obtain CPB concentrations similar to those estimated for diluted C. perfringens type C culture supernatants. Cells grown in 96-well plates were incubated with 150 μl of SFM containing different dilutions of culture supernatants or recombinant toxin preparations at 37°C in the presence of 5% CO2. At different time points, 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) cell viability tests were performed as described previously (30). SFM containing 10% TGY broth was used to determine reference values. The MTT test values for each concentration were determined in duplicate, and each experiment was performed twice for each cell culture, which resulted in four data points per group and dilution. All data were expressed as percentages of the baseline value (value obtained with serum-free medium). The effects of group and dilution on the values were assessed using two-way analysis of variance (ANOVA) (full model, including interaction). In addition, a one-way ANOVA with the post hoc Tukey-Kramer multiple-comparison routine was used to identify significant group differences for each dilution. The overall level of statistical significance (P value) used was <0.05. All statistical analyses were performed with the NCSS 2007 statistical software package (NCSS, Kaysville, UT).
Cytopathic effects and light microscopy.
Confluent PAEC and porcine fibroblasts grown in LabTek slides (Nunc) were incubated with 200 μl SFM containing the concentrations of C. perfringens culture supernatants, rCPB, and control preparations used for MTT assays. After 1 and 3 h of incubation, the medium was removed, and the cells were fixed and stained using a Diff-Quik staining kit (BD Biosciences). Cells were viewed and photographed using an Olympus BHX-51 microscope with a ProgRres C5 digital camera and ProgRres Capture Pro 2.7 software (Jenoptik).
Imaging of live cells.
Confluent PAEC cultures in collagen-coated glass bottom six-well plates (MatTek) and confluent porcine fibroblasts were incubated with SFM containing 5 μg/ml propidium iodide (33) and 32 ng/ml rCPB or JF 3721 culture supernatant diluted so that it contained the same concentration of CPB. Cells were placed on a TE2000E-PFS microscope (Nikon) equipped with a CFI Plan Fluor ELWD ×40 objective (Nikon), an Orca-ER charge-coupled device (CCD) camera (Hamamatsu), and an incubation chamber (Life Imaging Services), and images were recorded at 37°C in the presence of 5% CO2 for 20 h at 5-min intervals using the NIS Elements software (Nikon).
Immunofluorescence.
Confluent PAEC and porcine fibroblasts grown in LabTek slides (Nunc) were incubated with 200 μl SFM containing concentrations of C. perfringens culture supernatants, rCPB, and purified controls similar to the concentrations used for MTT assays. After 1 and 3 h of incubation, cells were washed with phosphate-buffered saline (PBS), fixed (1% paraformaldehyde, 20 min, room temperature), and permeabilzed (0.1% Triton X-100, 5 min). F- and G-actin were stained using Phalloidin 546 (Alexa Fluor), and nuclei were stained with Hoechst 33258 (Molecular Probes). All washing steps were performed with PBS (pH 7.5; three times for 5 min). Coverslips were mounted with fluorescence mounting medium (Dako Cytomation). Slides were viewed with a Nikon Eclipse 800 fluorescence microscope. Digital photographs were processed using the OpenLAB 5.0 software package (Improvision).
Antibody neutralization experiments.
C. perfringens culture supernatants and recombinant toxin preparations were preincubated with neutralizing mouse anti-CPB monoclonal antibody (MAb-CPB) or anti-CPA monoclonal antibody (MAb-CPA) (Centre for Veterinary Biologics) for 30 min at room temperature. For complete inhibition with excess amounts of antibody, 10 μl antibody stock solution per 100 μl SFM containing toxin was used. Serial 10-fold dilutions of the antibody were used to examine dose-dependent inhibition of CPB.
RESULTS
Histopathology and immunohistochemistry.
Our previous studies demonstrated that CPB bound to vascular endothelial cells in piglets with acute C. perfringens type C enteritis terminal lesions. To eliminate the possibility of postmortem binding of CPB to necrotic vessels, we investigated three piglets which were euthanized and subsequently diagnosed with typical acute C. perfringens type C enteritis (Fig. 1A). We specifically examined macroscopically unaffected distal segments of the jejunum, because these areas are the areas most likely to exhibit lesions indicative of progression of necrosis along the small intestine. Histopathology and immunohistochemistry analyses of jejunal segments with fully developed necro-hemorrhagic lesions revealed widespread vascular thrombosis and endothelial localization of CPB, as described previously (28) (data not shown). In caudal, macroscopically unaffected jejunal segments there was only multifocal loss of epithelial cells and occasional villous tip necrosis. These segments also exhibited early signs of fibrin thrombus formation in vessels in the lamina propria (Fig. 1B). Strong CPB staining was seen on the endothelial lining of these vessels but not on epithelial cells (Fig. 1C). No lesions or evidence of CPB was seen in colon, liver, lung, kidney, and brain sections (data not shown). Control sections from a piglet with superficial necrotizing enteritis due to Isospora suis infection did not show a CPB-positive reaction (Fig. 1D and E). These results suggested that CPB-mediated damage to vascular endothelial cells of mucosal vessels could play a role during the rapid spread of intestinal necrosis in C. perfringens type C enteritis.
Cytotoxicity of C. perfringens type C culture supernatants.
We compared the susceptibilities of PAEC and porcine fibroblasts to serial dilutions (1:10 to 1:1,280) of supernatants from C. perfringens type C and type A isolates described in Table 1. Both type C strain supernatants (JF 3719 and JF 3721) resulted in a marked, dose-dependent reduction in cell viability in PAEC cultures (Fig. 2A). Marked cytotoxic effects were demonstrated by the MTT test after 5 h. Markedly reduced cell viability was still evident when culture supernatants were diluted so that they contained 16 ng/ml of CPB (160-fold dilution). Incubation with diluted type A strain supernatants (JF 3693 and JF 3686) had no effect on the viability of PAEC at 5 h (Fig. 2A) or 20 h (data not shown). There was no difference between PAEC from the two different donors. The viability of porcine fibroblasts was not affected under the same experimental conditions (data not shown). These results indicated that type C culture supernatants were highly toxic to PAEC.
FIG. 2.

Cytotoxicity of C. perfringens type C culture supernatants for PAEC. (A) MTT cell viability tests using serial dilutions of culture supernatants revealed that JF 3719 and JF 3721 (type C), but not JF 3693 or JF 3686 (type A), induced dose-dependent cytotoxicity in PAEC. (B) Preincubation of JF 3719 and JF 3721 supernatants (1:20 [vol/vol] dilutions; 125 ng/ml CPB) with different amounts of MAb-CPB inhibited cytotoxicity for PAEC in a dose-dependent manner. Preincubation with MAb-CPA did not have an inhibitory effect. The exposure time was 5 h. MTT values were obtained in duplicate for each time point. The symbols indicate the means for two separate experiments performed with PAEC from one donor. The error bars indicate the standard errors of the means. Asterisks indicate significant differences between type C supernatants and control type A supernatants (A) or between preincubation with MAb-CPB and preincubation with MAb-CPA (B) (P < 0.05).
CPB neutralization inhibits cytotoxicity of C. perfringens type C culture supernatants.
We further elucidated the essential contribution of CPB to cytotoxic effects on PAEC by neutralization with an anti-CPB monoclonal antibody. In both type C culture supernatants, preincubation with the antibody inhibited cytotoxic effects on endothelial cells in a dose-dependent manner (Fig. 2B). Preincubating the same supernatants with a CPA-neutralizing monoclonal antibody had no effect on cytotoxicity (Fig. 2B). This indicated that CPB was an essential factor for cytotoxicity in type C culture supernatants.
Cytotoxicity of recombinant CPB.
To prove that CPB was not only essential but also sufficient to induce cytotoxic effects in PAEC, we used recombinant CPB expressed in E. coli (Fig. 3). Incubation of PAEC from both donors with rCPB resulted in a dose-dependent reduction in cell viability (Fig. 4A). In contrast to the results for type C culture supernatants, cytotoxicity was detected by the MTT test after 12 h of incubation (Fig. 4A), but not at earlier time points (5 h and 8 h) (data not shown). Cytotoxic effects were also evident when 13 ng/ml rCPB was used (Fig. 4A). Control incubations using uncleaved rCPB-Nus-Tag fusion protein or Nus-Tag mock purification did not affect the viability of PAEC. Furthermore, neutralization of rCPB inhibited cytotoxicity for PAEC (Fig. 4B), whereas preincubation of rCPB with anti-CPA monoclonal antibodies did not inhibit cytotoxicity (data not shown). Porcine fibroblasts incubated with rCPB did not exhibit reduced viability (data not shown).
FIG. 4.

Cytotoxicity of rCPB for PAEC. (A) Exposure of PAEC to rCPB, but not exposure of PAEC to rCPB-Nus-Tag or mock-purified Nus-Tag, resulted in a dose-dependent reduction in cell viability. (B) Preincubation of rCPB with MAb-CPB inhibited cytotoxicity for PAEC in a dose-dependent manner. The exposure time was 12 h. MTT values were obtained from two separate experiments performed in duplicate for each time point. The symbols indicate the means for two separate experiments with PAEC from one donor. The error bars indicate the standard errors of the means. The asterisks in panel A indicate statistical significance for comparisons of rCPB with both control preparations (P < 0.05). The asterisks in panel B indicate statistical significance for comparisons of rCPB with 1,000- and 10,000-fold dilutions of MAb-CPB and control preparations (P < 0.05).
CPB has early morphological effects in endothelial cells.
Even before reduced cell viability was detected by the MTT test, PAEC showed striking morphological changes when they were exposed to CPB. After 3 h of exposure to type C culture supernatants (JF 3719 and JF 3721) (Fig. 5) diluted so that the medium contained 32 ng CPB/ml, methanol-fixed cells exhibited marked cell shrinkage and karyopyknosis. Incubation of PAEC with 32 ng rCPB/ml medium induced similar, but less severe, morphological changes (Fig. 5). Live cell imaging revealed that after 15 min of exposure to type C supernatants the intercellular spaces between PAEC started to widen (see Fig. S1 in the supplemental material). These spaces expanded with time, as PAEC exhibited rapid and progressive cell border retraction, cell shrinkage, cytoplasmic blebbing, and cell rounding. Similar concentrations of rCPB induced the same morphological changes, albeit less rapidly (see Fig. S2 in the supplemental material). Cytoplasmic blebbing was observed only at higher concentrations (1 μg/ml rCPB) (data not shown). Corresponding to the results of the cell viability test, propidium iodide influx into the nuclei of the majority of cells was observed between 2 and 4 h after the initial exposure to type C supernatants and 10 to 12 h after the initial exposure to 32 ng rCPB/ml medium (see Fig. S1 and S2 in the supplemental material). Cytopathic effects were eliminated by preincubation with anti-CPB monoclonal antibody (Fig. 5) but not by preincubation with anti-CPA antibodies (data not shown). Control type A supernatants (Fig. 5), rCPB-Nus-Tag, and Nus-Tag mock purification (data not shown) did not induce cytopathic effects, even after prolonged incubation (up to 20 h) (data not shown). Porcine fibroblasts did not show comparable morphological alterations under the same experimental conditions (see Fig. S3 and S4 in the supplemental material). These results demonstrated that CPB not only was highly active with PAEC but was the essential toxin that induced rapid morphological changes in these cells.
FIG. 5.
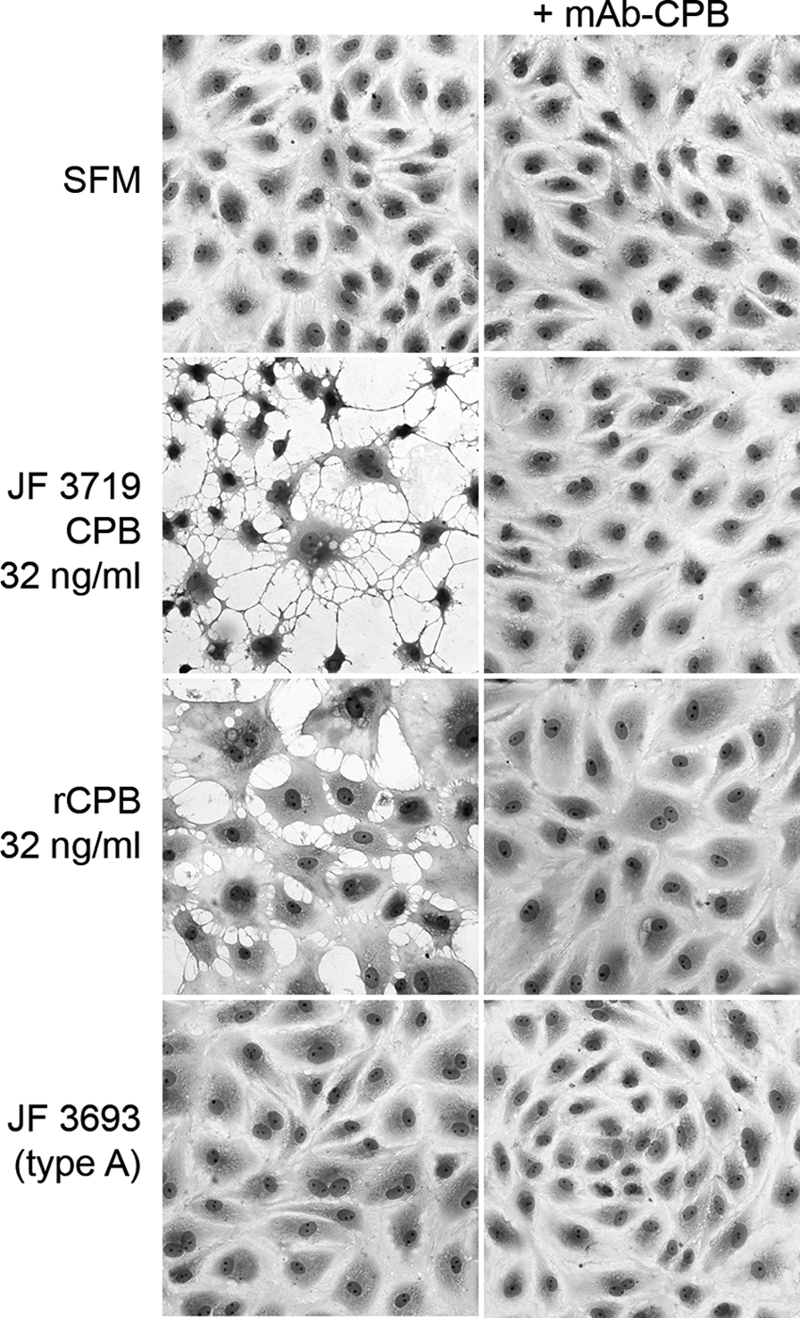
CPB-induced cytopathic effects on PAEC: photomicrographs of PAEC after 3 h of exposure to serum-free medium (SFM), a JF 3719 culture supernatant dilution containing 32 ng/ml CPB, 32 ng/ml rCPB, and a 1:10 (vol/vol) dilution of JF 3693 culture supernatant. Only CPB-containing media induced cell shrinkage and widening of intercellular gaps. Preincubation with neutralizing anti-CPB monoclonal antibodies (mAb-CPB) inhibited the cytopathic effects. Cells were fixed in methanol and stained with Diff-Quik. Magnification, ×400.
CPB induces rapid cytoskeletal disruption in endothelial cells.
One hour after PAEC were exposed to rCPB (at concentrations ranging from 32 ng/ml to 1 μg/ml), marked disruption of actin filaments was observed (Fig. 6). This effect progressed over time (Fig. 6), and preincubation of rCPB with MAb-CPB inhibited this effect. Uncleaved rCPB-Nus-Tag fusion protein did not induce cytoskeletal disruption, even at concentrations up to 3 μg/ml (Fig. 6). Type C culture supernatants, but not type A culture supernatants, induced disruption of the actin cytoskeleton in PAEC (data not shown). Again, preincubation with neutralizing anti-CPB antibody inhibited the cytoskeletal changes (data not shown). Porcine fibroblasts were not affected under the same experimental conditions (Fig. 6). These results indicated that CPB induced rapid disruption of the actin cytoskeleton in PAEC.
FIG. 6.

rCPB-induced disruption of the actin cytoskeleton in PAEC: fluorescent staining of F-actin in PAEC after 1 and 3 h and in porcine fibroblasts after 3 h of incubation with serum-free medium (SFM), 32 ng/ml rCPB, 32 ng/ml rCPB preincubated with MAb-CPB, and 3 μg/ml rCPB-Nus-Tag fusion protein. Disruption of the actin cytoskeleton was visible 1 h after exposure to rCPB. The effect increased over time (3 h) and was inhibited by neutralization of rCPB. The actin cytoskeleton of porcine fibroblasts was not affected.
DISCUSSION
Acute C. perfringens type C enteritis in animals and humans is characterized by segmental mucosal necrosis and hemorrhage of the small intestine and, in fewer cases, the large intestine (6, 14, 20, 23). Vascular thrombosis of small and medium-sized vessels occurs in the mucosa of affected small-intestine segments in the acute form of type C enteritis in piglets and humans (12, 14, 20, 21). This observation has received little attention because vascular damage has been considered a direct consequence of widespread epithelial necrosis (21, 43). Our recent observation that CPB was consistently localized at endothelial cells in acute lesions of spontaneous type C enteritis in piglets (28) led us to hypothesize that disruption of endothelial cells by CPB could contribute to the pathogenesis of type C enteritis. Therefore, in the present study, we tested whether CPB has cytopathic and cytotoxic effects on cultured porcine endothelial cells in vitro.
Our results show that PAEC were highly sensitive to CPB-containing type C culture supernatants. The immediate cytopathic effects in PAEC were characterized by rapid disruption of the actin cytoskeleton and retraction of the cell borders, which progressed to marked cell shrinkage. These effects were dose dependent and induced by type C culture supernatants but not by type A culture supernatants. To prove that CPB was the essential factor causing the cytopathic effects in PAEC in vitro, we used two independent approaches: (i) neutralization of CPB by monoclonal antibodies (38, 47, 49) and (ii) use of recombinant CPB. Neutralization of CPB inhibited cytopathic effects on PAEC in a dose-dependent manner. Additionally, rCPB induced cytopathic and cytotoxic effects in endothelial cells similar to those induced by type C culture supernatants. Again, these effects were inhibited by neutralization of CPB. Importantly, cytoskeletal disruption and cell border retraction occurred as early as 1 h after exposure of PAEC to very low concentrations (32 ng/ml) of rCPB. Loss of cell viability and propidium iodide influx, indicating cell death, became evident later, after 8 to 12 h of exposure. This was in contrast to the more rapid cell death that occurred between 2 and 5 h after exposure to type C culture supernatants. The difference most likely was due to synergistic effects of other toxins or enzymes secreted by type C strains into culture supernatants.
Endothelial cells form a vital barrier that controls the exchange of cells, macromolecules, and fluids between the vascular lumen and the surrounding tissue. They also maintain the normal blood flow due to their antiplatelet, anticoagulant, and fibrinolytic properties (5, 48). Disruption of the endothelial barrier leads to increased vascular permeability along with tissue edema and hemorrhage. Furthermore, it augments local coagulation and vascular thrombosis (5, 48). Cytopathic effects on endothelial cells, such as those observed in our study, could therefore contribute to vascular thrombosis. Johannsen et al. (15, 16) demonstrated that in experimental type C infections in piglets endothelial damage and microvascular thrombosis in small-intestine villi occurred shortly after ultrastructural alterations in enterocytes but before marked epithelial necrosis became evident. Our in vitro results indicate for the first time that direct interaction of CPB with endothelial cells could be involved in this process.
In our study, we used readily obtainable aortic endothelial cells as a model for porcine vascular endothelial cells. By performing all experiments with PAEC from two different pigs we eliminated donor-specific differences in cellular reactions (4). However, cultured macrovascular endothelial cells can differ from microvascular endothelial cells in phenotype and in the response to toxins (35). Therefore, we could not eliminate differences in the susceptibility of small-intestine microvascular endothelial cell cultures to CPB. Nevertheless, as reported previously for E. coli Shiga toxin, target organ-derived microvascular endothelial cell cultures could be expected to be more sensitive to toxins than aortic endothelial cells (13, 32). More importantly, our in situ investigations demonstrated that there was binding of CPB to endothelial cells in small-intestine mucosal vessels. Thus, it is conceivable that our in vitro results can be extrapolated to intestinal microvascular endothelial cells. Consistent with our previous study (28), CPB was detected by immunohistochemistry only in or adjacent to small-intestine lesions. This suggests that CPB acts mainly on endothelial cells close to damaged mucosa. A possible explanation for this is that once CPB is absorbed by the bloodstream, rapid degradation and dilution might prevent accumulation of the toxin at concentrations required for marked systemic endothelial damage and thrombosis. The proposed pathogenic mechanism for endothelial damage involves initial uptake of CPB through the intestinal epithelial barrier. The experimental trials with piglets carried out by Johannsen et al. (15, 16) and the recent studies using the rabbit ileal loop model (38, 49) indicated that epithelial cell necrosis is an initial event during the pathogenesis of type C enteritis. Additionally, C. perfringens type C damages intestinal epithelial cell-like Caco-2 cells in vitro (50). It is therefore conceivable that the initial overgrowth of C. perfringens type C and toxin production damage the intestinal epithelial barrier, leading to increased absorption of CPB and other toxins from the intestine. Interestingly, we could not demonstrate binding of CPB to epithelial cells in the intestines of naturally diseased piglets. However, the lesions in these animals represent the final stage of the acute disease, where necrosis has already occurred to a large extent. The 6-h experimental infections in rabbit intestinal loops, therefore, more closely represent the initial stages of the disease. Thus, widespread small-intestine microvascular thrombosis could be a characteristic lesion only during later phases of the disease, when mucosal necrosis extends to large areas of the intestine. Based on our results, we hypothesize that when CPB is taken up after initial damage of the jejunal epithelium, it diffuses toward and binds to endothelial cells of blood vessels within villi. Binding to unknown receptors would result in oligomerization, membrane pore formation, and cytopathic effects leading to disruption of the endothelial barrier. Immediate consequences of this would be tissue edema and extravasation of erythrocytes into the surrounding tissue. If sufficient amounts of CPB can reach the local vasculature, cytopathic effects on endothelial cells could contribute to rapid microvascular thrombosis and ischemic mucosal infarction. This in turn would favor clostridial proliferation and further toxin secretion. Direct endothelial toxicity and potentially epithelial toxicity, as well as hematogenous spread of CPB and other toxins, could then lead to the fulminant and progressive tissue necrosis observed in the acute form of C. perfringens type C enteritis. Effects of CPB on cultured porcine small-intestine epithelial cells are currently being investigated in our laboratory. Preliminary results obtained using similar experimental approaches indicate that intestinal epithelial cells are less sensitive to rCPB and type C culture supernatants than endothelial cells are (unpublished data). However, detailed studies on the effect of CPB on additional parameters, such as paracellular permeability in epithelial layers, should be performed.
Different histotoxic clostridial infections frequently cause massive hemorrhaging and necrosis in affected tissues (45). For example, C. perfringens type A-induced gas gangrene is characterized by rapid spread of myonecrosis, which for the most part involves rapid expansion of vascular thrombosis (5). C. perfringens can produce several toxins, such as CPA and PFO, which induce a procoagulative state locally (3, 5, 11), and it is possible that they also contribute to vascular thrombosis in type C enteritis. Nevertheless, direct endothelial damage mediated by CPB or similar toxins might be an important mechanism by which clostridia induce fulminant progression of ischemic tissue necrosis.
Taken together, our results suggest that CPB-mediated disruption of the endothelial barrier contributes to the pathogenesis of C. perfringens type C enteritis. Further analysis of the molecular interaction of the bacterium and its toxins with intestinal epithelial cells and intestinal microvascular endothelial cells is needed to unravel the complex pathogenesis of the fulminant intestinal necrosis observed in this disease. Because hemorrhaging and ischemic necrosis are typical features of many histotoxic clostridial infections, targeted disruption of the endothelium by exotoxins might be a common pathogenic mechanism used by these pathogens. Our approach, combining in vitro studies based on potential target cell cultures derived from natural hosts with in situ investigation of spontaneously affected animals, is an extension of current experimental animal models and provides a basis for more in-depth experimental studies, including infectious trials with pigs. Due to the similarities between spontaneous animal and human clostridial diseases, such studies should improve our understanding of the pathogenesis of these diseases in animals and humans.
Supplementary Material
Acknowledgments
This project was supported by a young investigator grant from the University of Bern (H.P.). F.P. was a recipient of a pathology training fellowship from Pfizer, Ltd., Sandwich, United Kingdom.
We thank E. Vilei, Y. Schlatter, and A. Thomann for help with bacteriological work, B. Grabscheid for immunohistochemical staining, M. Doherr for statistical analysis, and R. Kalaji for editorial changes.
Editor: S. R. Blanke
Footnotes
Published ahead of print on 19 April 2010.
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1.Albini, S., I. Brodard, A. Jaussi, N. Wollschlaeger, J. Frey, R. Miserez, and C. Abril. 2008. Real-time multiplex PCR assays for reliable detection of Clostridium perfringens toxin genes in animal isolates. Vet. Microbiol. 127:179-185. [DOI] [PubMed] [Google Scholar]
- 2.Amimoto, K., T. Noro, E. Oishi, and M. Shimizu. 2007. A novel toxin homologous to large clostridial cytotoxins found in culture supernatant of Clostridium perfringens type C. Microbiology 153:1198-1206. [DOI] [PubMed] [Google Scholar]
- 3.Awad, M. M., D. M. Ellemor, R. L. Boyd, J. J. Emmins, and J. I. Rood. 2001. Synergistic effects of alpha-toxin and perfringolysin O in Clostridium perfringens-mediated gas gangrene. Infect. Immun. 69:7904-7910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bouis, D., G. A. Hospers, C. Meijer, G. Molema, and N. H. Mulder. 2001. Endothelium in vitro: a review of human vascular endothelial cell lines for blood vessel-related research. Angiogenesis 4:91-102. [DOI] [PubMed] [Google Scholar]
- 5.Bryant, A. E. 2003. Biology and pathogenesis of thrombosis and procoagulant activity in invasive infections caused by group A streptococci and Clostridium perfringens. Clin. Microbiol. Rev. 16:451-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooke, R. 1979. The pathology of pig bel. P. N. G. Med. J. 22:35-38. [PubMed] [Google Scholar]
- 7.Eason, R. J., and R. van Rij. 1984. Pigless pigbel: enteritis necroticans in the Solomon Islands. P. N. G. Med. J. 27:42-44. [PubMed] [Google Scholar]
- 8.Fisher, D. J., M. E. Fernandez-Miyakawa, S. Sayeed, R. Poon, V. Adams, J. I. Rood, F. A. Uzal, and B. A. McClane. 2006. Dissecting the contributions of Clostridium perfringens type C toxins to lethality in the mouse intravenous injection model. Infect. Immun. 74:5200-5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geny, B., and M. R. Popoff. 2006. Bacterial protein toxins and lipids: pore formation or toxin entry into cells. Biol. Cell 98:667-678. [DOI] [PubMed] [Google Scholar]
- 10.Gibert, M., C. Jolivet-Reynaud, and M. R. Popoff. 1997. Beta2 toxin, a novel toxin produced by Clostridium perfringens. Gene 203:65-73. [DOI] [PubMed] [Google Scholar]
- 11.Hickey, M. J., R. Y. Kwan, M. M. Awad, C. L. Kennedy, L. F. Young, P. Hall, L. M. Cordner, D. Lyras, J. J. Emmins, and J. I. Rood. 2008. Molecular and cellular basis of microvascular perfusion deficits induced by Clostridium perfringens and Clostridium septicum. PLoS Pathog. 4:e1000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hogh, P. 1969. Necrotizing infectious enteritis in piglets, caused by Clostridium perfringens type C. 3. Pathological changes. Acta Vet. Scand. 10:57-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacewicz, M. S., D. W. Acheson, D. G. Binion, G. A. West, L. L. Lincicome, C. Fiocchi, and G. T. Keusch. 1999. Responses of human intestinal microvascular endothelial cells to Shiga toxins 1 and 2 and pathogenesis of hemorrhagic colitis. Infect. Immun. 67:1439-1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaggi, M., N. Wollschlager, C. Abril, S. Albini, C. Brachelente, M. Wyder, and H. Posthaus. 2009. Retrospective study on necrotizing enteritis in piglets in Switzerland. Schweiz. Arch. Tierheilkd. 151:369-375. [DOI] [PubMed] [Google Scholar]
- 15.Johannsen, U., S. Menger, W. Erwerth, and B. Kohler. 1986. Clostridium perfringens type C enterotoxemia (necrotizing enteritis) in suckling pigs. 2. Light and electron microscopic studies of the pathology and pathogenesis of experimental Clostridium perfringens type C toxin poisoning. Arch. Exp. Veterinaermed. 40:881-894. [PubMed] [Google Scholar]
- 16.Johannsen, U., S. Menger, W. Erwerth, and B. Kohler. 1986. Clostridium perfringens type C enterotoxemia (necrotizing enteritis) of suckling pigs. 3. Light and electron microscopic studies of the pathology and pathogenesis of experimental Clostridium perfringens type C infection. Arch. Exp. Veterinaermed. 40:895-909. [PubMed] [Google Scholar]
- 17.Johnson, S., P. Echeverria, D. N. Taylor, S. R. Paul, R. Coninx, J. Sakurai, B. Eampokalap, P. Jimakorn, R. A. Cooke, G. W. Lawrence, et al. 1987. Enteritis necroticans among Khmer children at an evacuation site in Thailand. Lancet ii:496-500. [DOI] [PubMed] [Google Scholar]
- 18.Johnson, S., and D. N. Gerding. 1997. Enterotoxemic infections, p. 117-140. In J. I. Rood, B. A. McClaine, J. G. Songer, and R. W. Titball (ed.), The clostridia: molecular biology and pathogenesis. Academic Press, San Diego, CA.
- 19.Keyburn, A. L., J. D. Boyce, P. Vaz, T. L. Bannam, M. E. Ford, D. Parker, A. Di Rubbo, J. I. Rood, and R. J. Moore. 2008. NetB, a new toxin that is associated with avian necrotic enteritis caused by Clostridium perfringens. PLoS Pathog. 4:e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kreft, B., K. Dalhoff, and K. Sack. 2000. Necrotizing enterocolitis: a historical and current review. Med. Klin. 95:435-441. [DOI] [PubMed] [Google Scholar]
- 21.Lawrence, G. 1979. The pathogenesis of pig-bel in Papua New Guinea. P. N. G. Med. J. 22:39-49. [PubMed] [Google Scholar]
- 22.Lawrence, G. 2005. The pathogenesis of pig-bel in Papua New Guinea. 1979. P. N. G. Med. J. 48:39-49. [PubMed] [Google Scholar]
- 23.Lawrence, G., and P. D. Walker. 1976. Pathogenesis of enteritis necroticans in Papua New Guinea. Lancet i:125-126. [DOI] [PubMed] [Google Scholar]
- 24.Lawrence, G. W., D. Lehmann, G. Anian, C. A. Coakley, G. Saleu, M. J. Barker, and M. W. Davis. 1990. Impact of active immunisation against enteritis necroticans in Papua New Guinea. Lancet 336:1165-1167. [DOI] [PubMed] [Google Scholar]
- 25.Li, D. Y., A. O. Scheimann, J. G. Songer, R. E. Person, M. Horwitz, L. Resar, and K. B. Schwarz. 2004. Enteritis necroticans with recurrent enterocutaneous fistulae caused by Clostridium perfringens in a child with cyclic neutropenia. J. Pediatr. Gastroenterol. Nutr. 38:213-215. [DOI] [PubMed] [Google Scholar]
- 26.Mandrella, B. 2007. A recent outbreak of necrotizing enteritis in eastern Sri Lanka. Trop. Dr. 37:52-54. [DOI] [PubMed] [Google Scholar]
- 27.Matsuda, T., Y. Okada, E. Inagi, Y. Tanabe, Y. Shimizu, K. Nagashima, J. Sakurai, M. Nagahama, and S. Tanaka. 2007. Enteritis necroticans ‘pigbel’ in a Japanese diabetic adult. Pathol. Int. 57:622-626. [DOI] [PubMed] [Google Scholar]
- 28.Miclard, J., M. Jaggi, E. Sutter, M. Wyder, B. Grabscheid, and H. Posthaus. 2009. Clostridium perfringens beta-toxin targets endothelial cells in necrotizing enteritis in piglets. Vet. Microbiol. 137:320-325. [DOI] [PubMed] [Google Scholar]
- 29.Miclard, J., J. van Baarlen, M. Wyder, B. Grabscheid, and H. Posthaus. 2009. Clostridium perfringens beta-toxin binding to vascular endothelial cells in a human case of enteritis necroticans. J. Med. Microbiol. 58:826-828. [DOI] [PubMed] [Google Scholar]
- 30.Mosmann, T. 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65:55-63. [DOI] [PubMed] [Google Scholar]
- 31.Nagahama, M., S. Hayashi, S. Morimitsu, and J. Sakurai. 2003. Biological activities and pore formation of Clostridium perfringens beta toxin in HL 60 cells. J. Biol. Chem. 278:36934-36941. [DOI] [PubMed] [Google Scholar]
- 32.Obrig, T. G., C. B. Louise, C. A. Lingwood, B. Boyd, L. Barley-Maloney, and T. O. Daniel. 1993. Endothelial heterogeneity in Shiga toxin receptors and responses. J. Biol. Chem. 268:15484-15488. [PubMed] [Google Scholar]
- 33.Petit, L., M. Gibert, A. Gourch, M. Bens, A. Vandewalle, and M. R. Popoff. 2003. Clostridium perfringens epsilon toxin rapidly decreases membrane barrier permeability of polarized MDCK cells. Cell. Microbiol. 5:155-164. [DOI] [PubMed] [Google Scholar]
- 34.Petit, L., M. Gibert, and M. R. Popoff. 1999. Clostridium perfringens: toxinotype and genotype. Trends Microbiol. 7:104-110. [DOI] [PubMed] [Google Scholar]
- 35.Ribatti, D., B. Nico, A. Vacca, L. Roncali, and F. Dammacco. 2002. Endothelial cell heterogeneity and organ specificity. J. Hematother. Stem Cell Res. 11:81-90. [DOI] [PubMed] [Google Scholar]
- 36.Rood, J. I. 1998. Virulence genes of Clostridium perfringens. Annu. Rev. Microbiol. 52:333-360. [DOI] [PubMed] [Google Scholar]
- 37.Rood, J. I., B. A. McClaine, J. G. Songer, and R. W. Titball (ed.). 1997. The clostridia: molecular biology and pathogenesis. Academic Press, San Diego, CA.
- 38.Sayeed, S., F. A. Uzal, D. J. Fisher, J. Saputo, J. E. Vidal, Y. Chen, P. Gupta, J. I. Rood, and B. A. McClane. 2008. Beta toxin is essential for the intestinal virulence of Clostridium perfringens type C disease isolate CN3685 in a rabbit ileal loop model. Mol. Microbiol. 67:15-30. [DOI] [PubMed] [Google Scholar]
- 39.Severin, W. P., A. A. de la Fuente, and M. F. Stringer. 1984. Clostridium perfringens type C causing necrotising enteritis. J. Clin. Pathol. 37:942-944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shatursky, O., R. Bayles, M. Rogers, B. H. Jost, J. G. Songer, and R. K. Tweten. 2000. Clostridium perfringens beta-toxin forms potential-dependent, cation-selective channels in lipid bilayers. Infect. Immun. 68:5546-5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Songer, J. G. 2010. Clostridia as agents of zoonotic disease. Vet. Microbiol. 140:399-404. [DOI] [PubMed] [Google Scholar]
- 42.Songer, J. G. 1996. Clostridial enteric diseases of domestic animals. Clin. Microbiol. Rev. 9:216-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Songer, J. G., and F. A. Uzal. 2005. Clostridial enteric infections in pigs. J. Vet. Diagn. Invest. 17:528-536. [DOI] [PubMed] [Google Scholar]
- 44.Steinthorsdottir, V., H. Halldorsson, and O. S. Andresson. 2000. Clostridium perfringens beta-toxin forms multimeric transmembrane pores in human endothelial cells. Microb. Pathog. 28:45-50. [DOI] [PubMed] [Google Scholar]
- 45.Stevens, D. L. 1997. Necrotizing clostridial soft tissue infections, p. 141-151. In J. I. Rood, B. A. McClaine, J. G. Songer, and R. W. Titball (ed.), The clostridia: molecular biology and pathogenesis. Academic Press, San Diego, CA.
- 46.Tsai, S. H., Y. W. Liu, W. C. Tang, Z. W. Zhou, C. Y. Hwang, G. Y. Hwang, B. R. Ou, C. P. Hu, V. C. Yang, and J. K. Chen. 2007. Characterization of porcine arterial endothelial cells cultured on amniotic membrane, a potential matrix for vascular tissue engineering. Biochem. Biophys. Res. Commun. 357:984-990. [DOI] [PubMed] [Google Scholar]
- 47.Uzal, F. A., J. Saputo, S. Sayeed, J. E. Vidal, D. J. Fisher, R. Poon, V. Adams, M. E. Fernandez-Miyakawa, J. I. Rood, and B. A. McClane. 2009. Development and application of new mouse models to study the pathogenesis of Clostridium perfringens type C enterotoxemias. Infect. Immun. 77:5291-5299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Hinsbergh, V. W. 2001. The endothelium: vascular control of haemostasis. Eur. J. Obstet. Gynecol. Reprod. Biol. 95:198-201. [DOI] [PubMed] [Google Scholar]
- 49.Vidal, J. E., B. A. McClane, J. Saputo, J. Parker, and F. A. Uzal. 2008. Effects of Clostridium perfringens beta-toxin on the rabbit small intestine and colon. Infect. Immun. 76:4396-4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vidal, J. E., K. Ohtani, T. Shimizu, and B. A. McClane. 2009. Contact with enterocyte-like Caco-2 cells induces rapid upregulation of toxin production by Clostridium perfringens type C isolates. Cell. Microbiol. 11:1306-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Watson, D. A., J. H. Andrew, S. Banting, J. R. Mackay, R. G. Stillwell, and M. Merrett. 1991. Pig-bel but no pig: enteritis necroticans acquired in Australia. Med. J. Aust. 155:47-50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.