

Abstract
Pertactin (PRN) is an autotransporter protein produced by all members of the Bordetella bronchiseptica cluster, which includes B. pertussis, B. parapertussis, and B. bronchiseptica. It is a primary component of acellular pertussis vaccines, and anti-PRN antibody titers correlate with protection. In vitro studies have suggested that PRN functions as an adhesin and that an RGD motif located in the center of the passenger domain is important for this function. Two regions of PRN that contain sequence repeats (region 1 [R1] and R2) show polymorphisms among strains and have been implicated in vaccine-driven evolution. We investigated the role of PRN in pathogenesis using B. bronchiseptica and natural-host animal models. A Δprn mutant did not differ from wild-type B. bronchiseptica in its ability to adhere to epithelial and macrophage-like cells in vitro or to establish respiratory infection in rats but was cleared much faster than wild-type bacteria in a mouse lung inflammation model. Unlike wild-type B. bronchiseptica, the Δprn mutant was unable to cause a lethal infection in SCID-Bg mice, but, like wild-type bacteria, it was lethal for neutropenic mice. These results suggest that PRN plays a critical role in allowing Bordetella to resist neutrophil-mediated clearance. Mutants producing PRN proteins in which the RGD motif was replaced with RGE or in which R1 and R2 were deleted were indistinguishable from wild-type bacteria in all assays, suggesting that these sequences do not contribute to PRN function.
Bordetella pertussis and Bordetella bronchiseptica cause respiratory infections in mammals. These two closely related bacterial subspecies display a high level of biochemical and genomic similarity and produce a similar set of virulence factors that are regulated at the transcriptional level by a highly conserved two-component signaling system called BvgAS (8, 46, 50, 65). However, they differ in symptomatic manifestations and host ranges: B. pertussis infects only humans and causes the acute disease whooping cough, while B. bronchiseptica has been isolated from nearly all mammals and typically causes chronic asymptomatic infections (41, 52). Nevertheless, some crucial factors are functionally interchangeable, such as filamentous hemagglutinin (FHA) (30) and the BvgAS signaling system (40), suggesting that studying B. bronchiseptica can reveal insights into the function of B. pertussis factors and vice versa. B. bronchiseptica and its natural-host animal models are therefore valuable tools for exploring molecular mechanisms of Bordetella pathogenesis.
Pertactin (PRN) is a putative Bordetella virulence factor belonging to the autotransporter (AT) family. It was discovered as a surface-localized immunogen of B. bronchiseptica in 1985 (49) and was found to have highly conserved homologs in B. pertussis and B. parapertussis shortly thereafter (5, 38). Many subsequent studies focused on the immunogenic and protective properties of PRN, leading to its inclusion in acellular pertussis vaccines (34, 48), and efficacy trials have shown that high anti-PRN antibody titers correlate with protection (6, 63).
Like all ATs, the nascent PRN polypeptide has a distinct central passenger domain flanked by an N-terminal signal sequence and a C-terminal porin domain (22). The signal sequence directs the polypeptide into the periplasm through the Sec system and is cleaved during translocation. The C-terminal porin domain, conserved among classical (monomeric) ATs, forms a channel in the outer membrane and is required for the export of the passenger domain to the cell surface (22, 23). Recent evidence indicates that the passenger domain of PRN crosses the outer membrane in a C- to N-terminal direction, with the N terminus being ultimately located distally from the cell surface (31). On the cell surface, the passenger domain folds into a predominantly right-handed β-helix with the individual rungs connected by loops of various lengths (14). The longest loop (residues 260 to 294 in PRN of B. pertussis Tohama I and residues 257 to 296 in PRN of B. bronchiseptica RB50), designated region 1 (R1), contains several Gly-Gly-Xaa-Xaa-Pro (GGXXP) repeats and an Arg-Gly-Asp (RGD) motif (45). RGD motifs have been shown to be important integrin binding sites for several mammalian proteins (57), and RGD motifs in some bacterial proteins, including PRN, were reported to function similarly (27-29, 37), although recent reports suggested otherwise (30, 66). Another loop of PRN (residues 563 to 614 of PRN in B. pertussis Tohama I and residues 564 to 608 of PRN in B. bronchiseptica RB50), designated region 2 (R2), is located at the C terminus of the passenger domain and contains Pro-Gln-Pro (PQP) repeats (45). Several studies have identified amino acid sequence variation within R1 and R2 of PRN among B. pertussis and B. bronchiseptica strains (2, 4, 44, 53, 54, 59). This information, together with evidence suggesting that R1 and R2 are immunodominant (21, 24, 25, 33, 61), led to the hypothesis that vaccination with a single PRN type has resulted in the selection of strains that produce antigenically distinct PRN proteins (i.e., that B. pertussis strains are undergoing vaccine-driven evolution) (4, 17, 45, 67).
Relatively few investigations have focused on PRN function. Based on in vitro studies with B. pertussis or PRN purified from B. pertussis, PRN was reported to function as an adhesin and an invasin that is dependent on the RGD motif (36, 37). However, two different studies failed to reveal a role for PRN in facilitating B. pertussis attachment to mammalian cells (15, 64). B. bronchiseptica PRN was also reported to function as an adhesin (13, 47). In vivo studies are limited to three reports: Khelef et al. and Roberts et al. observed no difference between the abilities of wild-type and pertactin-deficient B. pertussis strains to colonize or grow in the murine respiratory tract (32, 55), while Nicholson et al. found that a B. bronchiseptica prn mutant displayed reduced lung colonization in pigs at day 56 postinoculation (but not at earlier time points) compared with wild-type bacteria (47). Thus, despite 25 years of study and the commitment to include PRN in acellular pertussis vaccines, knowledge of how PRN contributes to Bordetella pathogenesis is rudimentary.
We report here an investigation of PRN function using B. bronchiseptica and natural-host animal models. Our results show that PRN, but neither its RGD motif nor R1 and R2, plays an important role in overcoming neutrophil-mediated clearance during the first week postinoculation.
MATERIALS AND METHODS
Bacterial strains.
The bacterial strains used in this study are listed in Table 1. B. bronchiseptica strains were maintained on Bordet-Gengou (BG) agar (BD Biosciences, San Jose, CA) supplemented with 7.5% defibrinated sheep blood (Mission Laboratories, Diamond Bar, CA) for 48 to 72 h at 37°C. To prepare B. bronchiseptica cells for adherence assays and infection experiments, bacteria were grown in Stainer-Scholte broth (60) at 37°C with shaking. Escherichia coli strains were cultured on Luria-Bertani (LB) agar or in LB broth. When appropriate, culture media were supplemented with kanamycin (Km; 50 μg/ml), streptomycin (Sm; 25 μg/ml), gentamicin (Gm; 20 μg/ml), or ampicillin (Ap; 100 μg/ml).
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Description | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| DH5α | Molecular cloning strain | BRL, Gaithersburg, MD |
| SM10λpir | Conjugation strain | 43 |
| B. bronchiseptica | ||
| RB50 | Wild-type B. bronchiseptica | 9 |
| SP5 | RB50 containing an in-frame deletion of codons 227-756 of prn | 13 |
| RBXQ7 | RB50 in which codon 265 of prn was changed to encode glutamic acid instead of aspartic acid (PRN D265E) | This study |
| RB545 | RB50 containing deletions of codons 269-288 and 578-606 in prn (PRNΔR1ΔR2) | This study |
| SP5::pVC4 | SP5 with plasmid pVC4 integrated into the chromosome as shown in Fig. 1A | This study |
| RB50::PRN-NT-HA | RB50 with plasmid pPRN-NT-HA integrated into the chromosome | This study |
| RB545::PRN-NT-HA | RB545 with plasmid pPRN-NT-HA integrated into the chromosome | This study |
| RBXQ7::PRN-NT-HA | RBXQ7 with plasmid pPRN-NT-HA integrated into the chromosome | This study |
| RBX9 | RB50 containing an in-frame deletion of codons 5-3647 in fhaB | 11 |
| RBX26 | RB50 containing an in-frame deletion of codons 227-756 of prn and an in-frame deletion of codons 5-3647 in fhaB | This study |
| Plasmids | ||
| pEG7 | pBR322-based suicide plasmid (Apr and Gmr) | 10 |
| pSS4245 | pBR322-based allelic exchange plasmid (see Fig. 1 and text) | This study |
| pVC1 | pSS4245 containing a 1-kb DNA fragment from prn with codon 265 changed to encode glutamic acid instead of aspartic acid | This study |
| pCI78a | pSS4245 containing a 1-kb DNA fragment from prn with codons 578-606 deleted from the center | This study |
| pXQ4 | pSS4245 containing a 1-kb DNA fragment from prn with codons 269-288 deleted from the center | This study |
| pVC4 | pEG7 containing the entire prn gene and its promoter region | This study |
| pPRN-NT-HA | pEG7 containing a 0.6-kb fragment corresponding to the 5′ end of prn with nucleotides encoding an HA epitope inserted between codons 38 and 39 (corresponding to the first and second amino acids of the mature PRN protein following cleavage of the signal peptide) | This study |
Construction of B. bronchiseptica prn mutants.
Derivatives of plasmid pSS4245 (see Results for details) were constructed and used to generate B. bronchiseptica prn mutants. The DNA fragments used to construct plasmids pVC1, pCI78a, and pXQ4 (to ultimately create B. bronchiseptica strains RBXQ7 and RB545) were generated by using a two-step, overlap PCR method (58) and cloned into pSS4245. Each DNA fragment contained ∼1,000-bp DNA fragments with the sequence conferring the mutation or deletion in the center. For the complementation of the Δprn mutation, we constructed plasmid pVC4, which contains the entire prn gene and its promoter region, by plasmid rescue (10) (Fig. 1 and Table 1). To insert a hemagglutinin (HA) epitope into PRN for visualization by immunoblotting, we constructed plasmid pPRN-NT-HA, a pEG7 derivative containing a 0.6-kb DNA fragment encompassing the 5′ end of prn with nucleotides encoding the HA epitope inserted between codons 38 and 39 of prn. When pPRN-NT-HA integrates into the chromosome by homologous recombination in the region 3′ of the nucleotides encoding the HA epitope on the plasmid (confirmed by PCR), the resulting strains produce PRN containing an HA epitope between the first and second amino acids of the mature protein (after cleavage of the signal peptide). All plasmid constructs were confirmed by DNA sequence analysis.
FIG. 1.

Construction of prn mutants. (A) SP5, the Δprn mutant, was created using allelic exchange to delete codons 227 to 756 of prn from the RB50 chromosome. The Δprn mutation was complemented with plasmid pVC4, a suicide plasmid containing the wild-type (wt) prn gene and its entire promoter region. The integration of plasmid pVC4 in the region 5′ of the deletion in SP5 (as shown) leaves the region 3′ of the deletion in SP5 unaltered. RBXQ7 and RB545 were constructed by allelic exchange using pSS4245 as described in the text. (B) pSS4245 is an allelic exchange plasmid for use in Bordetella species. The positions of antibiotic resistance genes, the I-SceI restriction endonuclease-encoding gene (I-SceI CDS), the I-SceI recognition site, the ptx promoter, transcription terminators (TrrnB), and the origin of replication (oriV) are shown. (C) Whole-cell lysates of wild-type and prn mutants producing PRN proteins with HA epitopes at their N termini were probed with anti-HA antibody. The position of the 75-kDa molecular mass marker is shown.
Bacterial conjugations.
Matings between B. bronchiseptica strains and E. coli strain SM10λpir were conducted as described previously, with minor variations (3). Briefly, B. bronchiseptica cells grown under Bvg−-phase conditions were coincubated with SM10λpir cells carrying an allelic exchange plasmid (Table 1) on BG agar containing 50 mM MgSO4 (Bvg−-phase conditions) for 4 h. Cointegrants were selected and purified on BG-Sm-Km agar containing 50 mM MgSO4. Cointegrants (confirmed by PCR) were then streaked onto BG plates without added MgSO4 (Bvg+-phase conditions) and incubated for 2 days at 37°C, allowing for the selection of bacteria that had undergone a second recombination event and a loss of the allelic exchange plasmid. Colonies were screened for the presence of the correct mutation (deletion or nucleotide substitution) by PCR and DNA sequencing. To generate the complemented strain, E. coli strain SM10λpir harboring pVC4 (Table 1) was mated with the Δprn mutant, and cointegrants were selected on BG agar containing Gm and Sm. The integration of pVC4 at the correct location was confirmed by PCR.
Immunoblot analysis.
Cultures grown overnight were used for all bacterial cell preparations. Whole-cell lysates were prepared as described previously (40). SDS-PAGE was performed according to the method of Laemmli (35) using denaturing 8% SDS-polyacrylamide gels. Proteins separated by SDS-PAGE were transferred onto nitrocellulose membranes (Schleicher and Schuell BioScience, Dassel, Germany) and then probed with anti-His antibody (diluted 1:2,000) followed by incubation with IR800-conjugated secondary antibody (diluted 1:5,000) (Rockland, Gilbertsville, PA). Antigen-antibody complexes were visualized by using the Odyssey infrared imaging system (Li-Cor Biosciences, Lincoln, NE).
Adherence assay.
Adherence assays with rat lung epithelial cells (L2; ATCC CCL-149) and mouse alveolar macrophages (MH-S; ATCC CRL-2019) were performed as described previously (11, 26), with minor modifications. L2 cells were grown in Ham's F12K nutrient mixture (Gibco, Carlsbad, CA) containing 10% fetal calf serum in standard 12-well tissue culture plates. L2 cells were used when they reached 50 to 80% confluence. The culture medium was removed and replaced with Stainer-Scholte broth containing 107 CFU of a tested bacterial strain (multiplicity of infection [MOI] of 50); the plates were spun at 200 × g for 5 min and then incubated for 5 min at 37°C. The cells were then washed four times with Hanks' balanced salts solution, fixed with methanol, stained with Giemsa stain, and visualized by light microscopy. Adherence to MH-S cells was performed similarly except that bacteria were stained and visualized by using immunofluorescence as described previously (26). Adherence was quantified by counting the number of bacteria associated with seven groups of mammalian cells (5 L2 cells or 10 MH-S cells per group).
Animal experiments. (i) Rat colonization experiments.
Female Wistar rats (3 to 4 weeks old) were inoculated with 1,000 CFU of Bordetella delivered in 10 μl to the external nares. Animals were euthanized at 14 and 28 days postinoculation, and colonization levels in the nasal cavity and trachea were determined as described previously (42).
(ii) Mouse lung inflammation experiments.
Groups of six to eight BALB/c mice (3 to 4 weeks old; Charles River Laboratories) were inoculated intranasally with 5 × 105 CFU of Bordetella in a 50-μl volume. At 1 h, 3 days, and 11 days postinoculation, three left lung lobes were recovered to enumerate bacterial CFU (42). Data shown are the results of two independent experiments.
(iii) Infection of immunodeficient and neutropenic mice.
SCID-beige mice (3 to 4 weeks old; Charles River Laboratories) were inoculated with 5 × 105 CFU of Bordetella delivered to the external nares in a 50-μl volume. Animals were monitored daily for signs of respiratory distress and euthanized when moribund. Neutropenic mice were obtained by intraperitoneally injecting BALB/c mice with 0.25 g cyclophosphamide per kg of body weight 4 days prior to inoculation. The reduction of neutrophils by more than 90% was confirmed by Diff-Quick staining of blood smears and microscopic examination. Animal protocols were approved by the IACUC at the University of California, Santa Barbara (protocol 6-04-601). A Student's unpaired t test was used for statistical analyses.
RESULTS
Construction of prn mutant strains.
B. bronchiseptica strain SP5 is isogenic with B. bronchiseptica RB50 except for the deletion of codons 227 through 756 from the prn gene (Fig. 1A) (13). For complementation experiments, we constructed plasmid pVC4 by plasmid rescue. pVC4 contains the entire prn gene, including its promoter, from RB50 on suicide plasmid pEG7. The integration of pVC4 into the chromosome of SP5 by homologous recombination within the region 5′ of the deletion in prn (confirmed by PCR) (Fig. 1A) results in a merodiploid containing both the wild-type and deletion prn alleles. The organization of the region 3′ of the prn deletion in the merodiploid is the same as that in SP5, and therefore, if the deletion mutation exerts any polar effects on downstream genes (which is unlikely because the gene 3′ of prn [cysG] is convergently transcribed), it will also do so in the merodiploid.
To investigate the roles of the PRN RGD triplet and R1 and R2 in pathogenesis, we constructed mutants using a newly developed allelic exchange system. Allelic exchange plasmid pSS4245 (Fig. 1B) contains the origin of replication from pBR322 and does not replicate in Bordetella. The plasmid also contains genes encoding resistance to ampicillin (bla), streptomycin (Str), kanamycin (Kmr), bleomycin (bler), and tetracycline (Tetr). For counterselection, pSS4245 carries a gene for an intron-encoded restriction endonuclease, I-SceI, under the control of the pertussis toxin (ptx) promoter (which is expressed only under Bvg+-phase conditions) and an I-SceI cleavage site. We constructed a derivative of this plasmid containing a 1-kb DNA fragment corresponding to codons 99 to 431 of PRN but with the codon for aspartic acid 265 changed to encode glutamic acid to change the RGD in the passenger domain of PRN to RGE. This plasmid, pVC1, was introduced into RB50 by conjugation, and cointegrants were selected on BG agar containing Km (to select for the plasmid), Sm (to inhibit the growth of E. coli), and 50 mM MgSO4 (Bvg−-phase conditions). Cointegrants were restreaked once onto the same medium and then streaked onto BG agar without Gm or MgSO4. Under these Bvg+-phase conditions, the ptx promoter is activated, the gene encoding I-SceI is expressed, and the I-SceI that is produced cleaves at its recognition site, resulting in a double-stranded DNA break in the chromosome. Repair by homologous recombination between sequences that flank the integrated plasmid results in the loss of the plasmid from the chromosome and, if recombination occurs on the opposite side of the lesion from where the initial recombination event occurred, the transfer of the lesion to the chromosome. Colonies that grew on BG agar were screened by PCR and DNA sequencing to identify mutants containing the DNA changes conferring the Asp265Glu (D265E or RGD→RGE) substitution. One strain, called RBXQ7, was used for further studies. A similar approach was used in two successive steps to construct a strain, called RB545, in which codons 269 to 288 and 578 to 606 of the prn gene, corresponding to R1 and R2, respectively, were deleted (Fig. 1A).
To visualize the PRN proteins produced in the mutant strains, we constructed derivatives in which nucleotides encoding hemagglutinin (HA) epitopes were inserted between codons 38 and 39 of prn. RBXQ7 (the PRN D265E mutant) produced a PRN protein that was indistinguishable in size and amount from that produced by RB50, and RB545 (the ΔR1ΔR2 mutant) produced a slightly smaller PRN protein, as expected (Fig. 1C). Note that pro-Prn (the passenger domain plus the porin domain) runs with a mobility of ∼90 kDa and that the passenger domain alone runs at ∼69 kDa. Strains that produced PRN proteins lacking HA epitopes were used for all subsequent experiments (growth curves, adherence assays, and animals experiments) to avoid any possible detrimental effects from the HA epitopes.
To determine if any of the mutations that we introduced into B. bronchiseptica affected the growth rate, we compared the growth rates of wild-type and mutant strains incubated in Stainer-Scholte broth at 37°C (Bvg+-phase conditions). Growth curves for all strains were indistinguishable (data not shown), indicating that the mutations did not alter the ability of B. bronchiseptica to grow in vitro under conditions in which prn is expressed.
Pertactin does not appear to contribute to the adherence of B. bronchiseptica to epithelial or macrophage-like cell lines.
PRN was reported previously to contribute to Bordetella adherence to epithelial and/or macrophage-like cell lines (37). We compared wild-type and mutant strains of B. bronchiseptica for their abilities to adhere to rat lung epithelial L2 cells and mouse macrophage-like MH-S cells. SP5, the Δprn mutant, was indistinguishable from RB50 in its ability to adhere to these cells lines (Fig. 2). A ΔfhaB strain, RBX9, which is defective for adherence to several cell lines (11), is shown for comparison. As expected, a ΔfhaB Δprn double mutant strain, RBX26, behaved similarly to the ΔfhaB strain (Fig. 2). Therefore, PRN is not required for B. bronchiseptica strain RB50 to adhere to mammalian cells, at least those tested here.
FIG. 2.

Adherence of B. bronchiseptica strains to L2 (rat lung epithelial) and MH-S (murine alveolar macrophage-like) cell lines. Data from at least two independent experiments were averaged, and the standard deviations are shown (MOI = 50). Asterisks indicate a statistically significant difference compared with RB50.
PRN is not required for respiratory tract colonization in rats.
We used a rat model to determine the contribution of PRN to the colonization of the respiratory tract. Female Wistar rats were inoculated with 1,000 CFU of either wild-type RB50 or the Δprn mutant delivered in a small volume (10 μl) to the nares. Colonization levels in tracheas and nasal septa were determined at days 14 and 28 postinoculation. The Δprn mutant was recovered at levels similar to those of RB50 at both time points in both tissues (data not shown), indicating that prn is not required for B. bronchiseptica to colonize the respiratory tracts of rats.
PRN is required for B. bronchiseptica to grow and/or resist inflammatory clearance in the lungs of mice.
We have used a mouse model to investigate the roles of specific virulence genes in overcoming inflammation-mediated clearance in the lower respiratory tract (18, 19, 26, 30). In this model, animals were inoculated with 5 × 105 CFU of bacteria in 50 μl. The number of bacteria recovered from the lungs of animals inoculated with wild-type B. bronchiseptica typically increases to about 5 × 106 during the first 7 days of infection and then decreases slowly, and the bacteria are eventually cleared from the lungs by about 30 days postinoculation (26). Days 3 and 11 postinoculation have proven to be the most informative time points, especially with regard to the ability of B. bronchiseptica to influence and/or resist the inflammatory response (22, 26). We inoculated 3- to 4-week-old female BALB/c mice with wild-type B. bronchiseptica RB50, SP5 (the Δprn mutant), and SP5::pVC4 (the complemented strain) and determined the number of CFU in the lungs at 3 and 11 days postinoculation. Additional animals were sacrificed at 1 to 3 h postinoculation (day 0) to confirm that similar numbers of bacteria were delivered to each site initially (Fig. 3). The Δprn mutant was recovered from the lungs in much smaller numbers than wild-type bacteria at both 3 and 11 days postinoculation (Fig. 3). The complemented strain was recovered at levels similar to those of wild-type bacteria at all time points, indicating that the defect displayed by the Δprn strain was due specifically to the deletion of the prn gene (Fig. 3). The gross appearance of lung tissues recovered from all mice was healthy and no different from that of lungs harvested from uninfected mice. These results indicate that PRN is required for B. bronchiseptica to grow and/or resist inflammation-mediated clearance in the lungs during the first week postinoculation.
FIG. 3.

Colonization of mouse lungs. Shown are data for colonization of mouse lungs at 1 to 3 h (day 0), 3 days, and 11 days postinoculation with 5 × 105 CFU of wild-type or prn mutant strains. Each symbol indicates the number of CFU from a single animal. The horizontal bar indicates the geometric mean. For animals inoculated with the ΔfhaB strain, the means for the moribund animals (∼108 CFU) and healthy animals (∼106 CFU) were calculated separately. The dashed line represents the lower limit of detection. *, P < 0.05 compared with RB50.
PRN is required for the ΔfhaB strain to cause a bimodal or hyperinflammatory response in mice.
Intranasal inoculation of BALB/c mice with ΔfhaB B. bronchiseptica results in a hyperinflammatory response compared with inoculation with wild-type B. bronchiseptica (26, 30). At a dose of 5 × 105 CFU, this hyperinflammatory response leads to a rapid clearance of the bacteria in about half of the animals and inflammation-mediated pulmonary damage that allows the growth of the bacteria to high numbers and, ultimately, death of the mice within about 4 days in the other half of the animals (26, 30). The gross appearance of lungs recovered from moribund mice is easily distinguishable from that of healthy animals (patches of white and dark red with obvious signs of consolidation and infarction), while the gross appearance of lungs from healthy animals was indistinguishable from that of uninfected mice. The microscopic appearance of hematoxylin- and eosin-stained histological sections corresponds to the macroscopic appearance: lungs from moribund mice were filled with inflammatory cells, predominantly neutrophils and lymphocytes, while lungs of healthy mice show only mild inflammation (24). We reasoned that if PRN plays a proinflammatory role, the hyperinflammation that is seen in response to infection with the ΔfhaB strain will not occur in animals inoculated with a Δprn ΔfhaB double mutant and that animals will not suffer inflammation-mediated lung damage (and death), but the bacteria may grow to high numbers in the lungs due to the lack of recruitment of inflammatory cells. Alternatively, if PRN plays a role specifically in resisting inflammation-mediated clearance, a ΔfhaB Δprn double mutant will be cleared faster than the Δprn single mutant due to the hyperrobust inflammatory response induced in the absence of FHA's immunomodulatory effects. The number of CFU recovered from mice inoculated with 5 × 105 CFU of the Δprn ΔfhaB mutant was lower than that recovered from mice inoculated with wild-type bacteria and the single mutants at day 3 postinoculation, and no bacteria were recovered from Δprn ΔfhaB-inoculated mice at day 11 postinoculation (Fig. 3). Moreover, none of the mice inoculated with the double mutant showed any signs of respiratory distress or illness at any time during the experiment, and all lungs that were removed from these animals appeared healthy, with no macroscopic signs of inflammation-mediated damage. PRN is therefore required for the ΔfhaB strain to cause a lethal infection (which occurs in about half of the mice at the dose used in this model), possibly by contributing to the ability of the bacteria to resist clearance by inflammatory cells, which consequently causes a sustained activation of inflammation.
Neither the RGD motif nor region 1 or region 2 is required for PRN to contribute to defense against inflammation-mediated clearance.
To determine if the RGD triplet or the repeats in regions 1 and 2 are important for PRN function in vivo, we compared the PRN D265E mutant and the PRNΔR1ΔR2 mutant with wild-type B. bronchiseptica in the mouse model. The numbers of CFU of both mutants recovered from the lungs of inoculated mice were similar to those for wild-type bacteria at all time points (Fig. 3), indicating that neither the RGD motif nor regions 1 and 2 are required for B. bronchiseptica to grow and/or resist inflammation-mediated clearance in mice.
PRN is required for B. bronchiseptica to overcome the innate immune response.
Our data indicate that PRN is important for the growth of B. bronchiseptica in the lungs during the first week postinoculation, suggesting that it plays a role in overcoming inflammation-mediated clearance. To investigate this possibility further, we used SCID-Bg mice, which lack B and T cells and have defective NK cells and are therefore dependent on innate immunity for protection against pathogens (12, 56). Consistent with previous results (12, 26, 56), wild-type B. bronchiseptica caused a lethal systemic infection in SCID-Bg mice (Fig. 4). In contrast, mice inoculated with the Δprn strain were healthy for the duration of the experiment (Fig. 4). PRN is therefore required for B. bronchiseptica to overcome innate immune mechanisms that are functional in SCID-Bg mice.
FIG. 4.
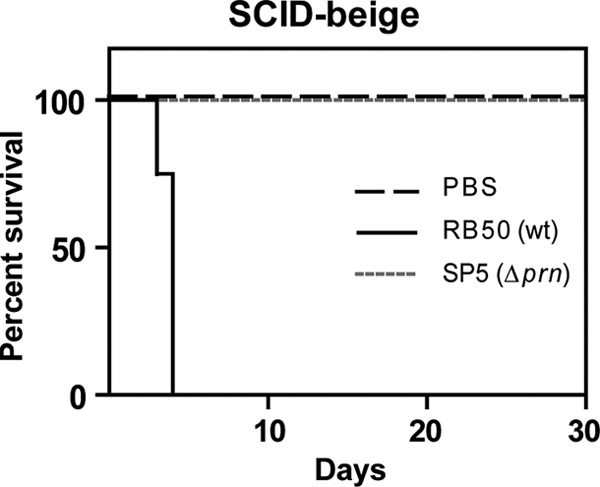
Infection of SCID-beige mice. Groups of six mice were infected intranasally with phosphate-buffered saline (PBS) or 500 CFU of RB50 or SP5 and monitored for signs of disease. Shown are the percentages of surviving mice over time.
PRN is required for defense of B. bronchiseptica against neutrophils.
SCID-Bg mice are at least partially dependent on neutrophils to control bacterial infection, and neutrophils are required for mice to control B. bronchiseptica infection (12, 19, 56). If PRN is specifically required for B. bronchiseptica to resist neutrophil-mediated clearance, then both wild-type and Δprn bacteria should cause a lethal infection in neutropenic mice. If PRN is required to overcome a neutrophil-independent innate immune clearance mechanism, then neutropenic mice should be able to control infection by the Δprn mutant. Mice rendered neutropenic by treatment with cyclophosphamide were inoculated with wild-type B. bronchiseptica or the Δprn mutant and quickly succumbed to infection by both strains (Fig. 5). Together with the results obtained with the SCID-Bg mice, these data indicate that PRN is required for B. bronchiseptica to resist neutrophil-mediated clearance.
FIG. 5.
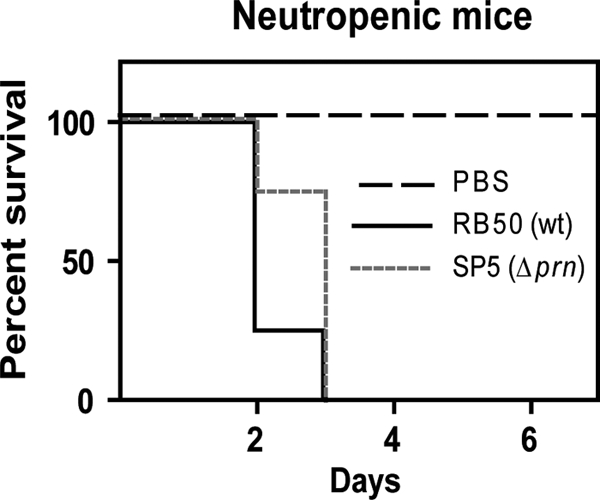
Infections of neutropenic mice. Groups of six BALB/c mice rendered neutropenic by cyclophosphamide treatment were inoculated with PBS or 5 × 105 CFU of either RB50 or SP5. Shown are the percentages of surviving mice over time.
DISCUSSION
Although pertactin is typically referred to as a Bordetella adhesin, previously reported data supporting this designation are inconclusive. Only three studies compared the abilities of wild-type and PRN-deficient B. pertussis strains to adhere to mammalian cells in vitro: in two of these studies, the PRN-deficient mutants were reported to adhere less efficiently to CHO cells, HeLa cells (37), and monocytes (20) than wild-type bacteria, while the third study reported no difference in adherence to NCI-H292 and HEp-2 cells between wild-type and mutant bacteria (64). Those studies all used the same strain of B. pertussis in which the prn gene was disrupted by the insertion of a kanamycin resistance gene. Whether the contradictory results are due to differences in the host cells used or adherence assay protocols is unknown. Two studies compared wild-type B. bronchiseptica with derivatives containing in-frame deletion mutations in prn for their abilities to mediate adherence: Nicholson et al. reported that a PRN-deficient mutant was less efficient at adhering to L2 cells and PK-15 cells than wild-type B. bronchiseptica strain KM22 (47), and Edwards et al. observed a decreased adherence of a PRN-deficient derivative of B. bronchiseptica strain RB50 (SP5, the same strain used in our study) to ciliated rabbit tracheal explant cells compared with that of wild-type bacteria (13). We found no difference between the abilities of RB50 and SP5 to adhere to L2 cells and MH-S cells. We do not know if our results differ from those of Nicholson et al. because of differences in the bacterial strains used or differences in adherence assay protocols. For B. bronchiseptica strain RB50, however, we can conclude that while PRN may contribute to adherence to ciliated epithelial cells in the trachea (13), it appears not to contribute to adherence to epithelial and macrophage cell lines in vitro. Whether PRN functions as a Bordetella “adhesin” therefore remains unresolved and may remain so until its host cell receptor, if one exists, is identified.
Although it is unlikely to mimic the natural course of infection for any respiratory pathogen, the murine lung inflammation model is useful for investigating the ability of bacteria to resist and/or overcome the innate immune responses of their mammalian hosts. In this model, wild-type B. bronchiseptica strain RB50 typically increases in number in the lungs by about 1 log during the first week of infection and is then gradually cleared from the lungs over the next ∼30 days (19, 26, 30). The Δprn mutant, in contrast, was reduced in number by 2 logs during the first 3 days postinoculation compared with wild-type bacteria and was maintained at that level over the next week. The inability of the Δprn mutant to grow in the lungs of mice during the first week postinoculation is similar what was previously observed for a B. bronchiseptica mutant that is unable to produce adenylate cyclase toxin (ACT) (19). Also, similar to the phenotype of ACT-deficient B. bronchiseptica (19), the Δprn mutant was unable to cause a lethal infection in SCID-Bg mice but was capable of causing a lethal infection in neutropenic mice. These results indicate that, like ACT, PRN is required to resist neutrophil-mediated clearance. The fact that the ΔfhaB Δprn double mutant was cleared from the lungs faster than the Δprn or ΔfhaB single mutant is also consistent with a role for PRN in resisting neutrophils. Our previous results indicate that inoculation with FHA-deficient B. bronchiseptica induces a more robust inflammatory response in the lungs of mice than does inoculation with FHA-producing B. bronchiseptica (26, 30). Armed with ACT and PRN (as well as other virulence factors), the ΔfhaB mutant is able to resist complete clearance by the hyperinflammatory response at day 11 postinoculation. Without PRN (shown here) or ACT (C. S. Inatsuka et al., manuscript in preparation), the bacteria are unable to resist the hyperinflammatory response and are completely cleared from the lungs by day 11. ACT was shown previously to inhibit bactericidal activities of phagocytic cells (7, 16, 51, 68). Our data suggest that Prn may be required for this ability, perhaps by allowing interactions between the bacteria and phagocytic cells so that ACT can be efficiently delivered and/or by affecting signaling events in the host cells that make them susceptible to ACT-mediated inhibition. We are currently developing in vitro assays using human peripheral blood neutrophils to explore this possibility.
Based on inhibition by RGD-containing peptides, the RGD motif in the passenger domain of PRN was previously proposed to mediate adherence to CHO cells and the invasion of HeLa cells by B. pertussis (36, 37). Everest et al., however, who compared strains of E. coli and Salmonella enterica serovar Typhimurium producing wild-type PRN or a PRN RGE mutant, found no evidence of a role for the PRN RGD in adherence or invasion (15). Consistent with the results of Everest et al., we found that a B. bronchiseptica strain producing a PRN protein with a D265E substitution was indistinguishable from wild-type B. bronchiseptica in its ability to grow and/or resist inflammation-mediated clearance in the lungs of mice, suggesting that the RGD motif at positions 263 to 265 of PRN does not contribute to PRN function in vivo. We showed recently that the RGD motif in the mature FHA protein was not required for B. bronchiseptica to colonize the tracheas of rats or to modulate the inflammatory response in the lungs of mice (30), and Waters et al. showed a few years ago that the RGD motif of the aggregation substance was not responsible for the interaction of Enterococcus faecalis with host cells (66). At present, to our knowledge, there is no published report showing a role for an RGD triplet in a bacterial protein in a bacterium-host interaction in vivo. Whether such motifs contribute to any interactions between pathogens or symbionts and their eukaryotic hosts therefore remains undetermined.
Our study also failed to reveal a role for R1 and R2 of PRN in the ability of B. bronchiseptica to infect rats or mice. This result was unexpected given the plethora of reports indicating that these regions are variable and immunogenic and that vaccine-driven evolution is selecting for strains carrying prn alleles that are antigenically distinct from the alleles used to produce proteins for acellular pertussis vaccines (4, 17, 39, 45, 61, 67). If these regions are not required for PRN function but represent important immunogenic targets, why are they maintained? Our study focused only on the contribution of PRN to the establishment of colonization (in rats) and the ability to resist inflammation-mediated clearance (in mice), and it is possible that PRN, and domains within PRN, contributes to other aspects of Bordetella pathogenesis, such as persistence and circulation within immune populations.
We also report here a new allelic exchange system for use in Bordetella. This system uses an intron-encoded restriction endonuclease, I-SceI, under the control of the pertussis toxin (ptx) promoter and an I-SceI cleavage site to select for the loss of the plasmid from cointegrants. Previous allelic exchange systems used for B. pertussis relied on the rpsL gene, conferring sensitivity to streptomycin, for counterselection, which required the isolation and use of streptomycin-resistant derivatives of wild-type B. pertussis strains (62). The new system can be used with wild-type B. pertussis strains, alleviating concerns that any phenotypes displayed by the resulting mutants are due to the mutation conferring streptomycin sensitivity. Allelic exchange systems used for B. bronchiseptica relied on the Bacillus subtilis sacB gene for counterselection (1). We and others have observed that the frequency of obtaining B. bronchiseptica cointegrants is much lower for a suicide plasmid containing the sacB gene than for plasmids that do not contain the sacB gene and that those cointegrants that are obtained are often sucrose resistant, suggesting the selection of mutations in the sacB gene. We obtained cointegrants of pSS4245 at a high frequency, and after counterselection by growing the bacteria under Bvg+-phase conditions, 100% of the colonies obtained had lost the plasmid. The pSS4245 allelic exchange system is therefore superior to those that have been used previously for B. pertussis and B. bronchiseptica.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (grant AI43986) to P.A.C.
Editor: A. J. Bäumler
Footnotes
Published ahead of print on 26 April 2010.
REFERENCES
- 1.Akerley, B. J., P. A. Cotter, and J. F. Miller. 1995. Ectopic expression of the flagellar regulon alters development of the Bordetella-host interaction. Cell 80:611-620. [DOI] [PubMed] [Google Scholar]
- 2.Boursaux-Eude, C., and N. Guiso. 2000. Polymorphism of repeated regions of pertactin in Bordetella pertussis, Bordetella parapertussis, and Bordetella bronchiseptica. Infect. Immun. 68:4815-4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buboltz, A. M., T. L. Nicholson, L. S. Weyrich, and E. T. Harvill. 2009. Role of the type III secretion system in a hypervirulent lineage of Bordetella bronchiseptica. Infect. Immun. 77:3969-3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cassiday, P., G. Sanden, K. Heuvelman, F. Mooi, K. M. Bisgard, and T. Popovic. 2000. Polymorphism in Bordetella pertussis pertactin and pertussis toxin virulence factors in the United States, 1935-1999. J. Infect. Dis. 182:1402-1408. [DOI] [PubMed] [Google Scholar]
- 5.Charles, I. G., G. Dougan, D. Pickard, S. Chatfield, M. Smith, P. Novotny, P. Morrissey, and N. F. Fairweather. 1989. Molecular cloning and characterization of protective outer membrane protein P.69 from Bordetella pertussis. Proc. Natl. Acad. Sci. U. S. A. 86:3554-3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cherry, J. D., J. Gornbein, U. Heininger, and K. Stehr. 1998. A search for serologic correlates of immunity to Bordetella pertussis cough illnesses. Vaccine 16:1901-1906. [DOI] [PubMed] [Google Scholar]
- 7.Confer, D. L., and J. W. Eaton. 1982. Phagocyte impotence caused by an invasive bacterial adenylate cyclase. Science 217:948-950. [DOI] [PubMed] [Google Scholar]
- 8.Cotter, P. A., and A. M. Jones. 2003. Phosphorelay control of virulence gene expression in Bordetella. Trends Microbiol. 11:367-373. [DOI] [PubMed] [Google Scholar]
- 9.Cotter, P. A., and J. F. Miller. 1994. BvgAS-mediated signal transduction: analysis of phase-locked regulatory mutants of Bordetella bronchiseptica in a rabbit model. Infect. Immun. 62:3381-3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cotter, P. A., and J. F. Miller. 1997. A mutation in the Bordetella bronchiseptica bvgS gene results in reduced virulence and increased resistance to starvation, and identifies a new class of Bvg-regulated antigens. Mol. Microbiol. 24:671-685. [DOI] [PubMed] [Google Scholar]
- 11.Cotter, P. A., M. H. Yuk, S. Mattoo, B. J. Akerley, J. Boschwitz, D. A. Relman, and J. F. Miller. 1998. The filamentous hemagglutinin (FHA) of Bordetella bronchiseptica is required for efficient establishment of tracheal colonization. Infect. Immun. 66:5921-5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dorshkind, K., G. M. Keller, R. A. Phillips, R. G. Miller, G. C. Bosma, M. O'Toole, and M. J. Bosma. 1984. Functional status of cells from lymphoid and myeloid tissues in mice with severe combined immunodeficiency disease. J. Immunol. 132:1804-1808. [PubMed] [Google Scholar]
- 13.Edwards, J. A., N. A. Groathouse, and S. Boitano. 2005. Bordetella bronchiseptica adherence to cilia is mediated by multiple adhesin factors and blocked by surfactant protein A. Infect. Immun. 73:3618-3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Emsley, P., I. G. Charles, N. F. Fairweather, and N. W. Isaacs. 1996. Structure of Bordetella pertussis virulence factor P.69 pertactin. Nature 381:90-92. [DOI] [PubMed] [Google Scholar]
- 15.Everest, P., J. Li, G. Douce, I. Charles, J. De Azavedo, S. Chatfield, G. Dougan, and M. Roberts. 1996. Role of the Bordetella pertussis P.69/pertactin protein and the P.69/pertactin RGD motif in the adherence to and invasion of mammalian cells. Microbiology 142:3261-3268. [DOI] [PubMed] [Google Scholar]
- 16.Friedman, R. L., R. L. Fiederlein, L. Glasser, and J. N. Galgiani. 1987. Bordetella pertussis adenylate cyclase: effects of affinity-purified adenylate cyclase on human polymorphonuclear leukocyte functions. Infect. Immun. 55:135-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fry, N. K., S. Neal, T. G. Harrison, E. Miller, R. Matthews, and R. C. George. 2001. Genotypic variation in the Bordetella pertussis virulence factors pertactin and pertussis toxin in historical and recent clinical isolates in the United Kingdom. Infect. Immun. 69:5520-5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harvill, E. T., P. A. Cotter, and J. F. Miller. 1999. Pregenomic comparative analysis between Bordetella bronchiseptica RB50 and Bordetella pertussis Tohama I in murine models of respiratory tract infection. Infect. Immun. 67:6109-6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harvill, E. T., P. A. Cotter, and J. F. Miller. 1999. Probing the function of a bacterial virulence factor by manipulating host immunity. Infect. Immun. 67:1493-1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hazenbos, W. L., B. M. van den Berg, J. W. van't Wout, F. R. Mooi, and R. van Furth. 1994. Virulence factors determine attachment and ingestion of nonopsonized and opsonized Bordetella pertussis by human monocytes. Infect. Immun. 62:4818-4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He, Q., J. Makinen, G. Berbers, F. R. Mooi, M. K. Viljanen, H. Arvilommi, and J. Mertsola. 2003. Bordetella pertussis protein pertactin induces type-specific antibodies: one possible explanation for the emergence of antigenic variants? J. Infect. Dis. 187:1200-1205. [DOI] [PubMed] [Google Scholar]
- 22.Henderson, I. R., F. Navarro-Garcia, M. Desvaux, R. C. Fernandez, and D. Ala'Aldeen. 2004. Type V protein secretion pathway: the autotransporter story. Microbiol. Mol. Biol. Rev. 68:692-744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henderson, I. R., F. Navarro-Garcia, and J. P. Nataro. 1998. The great escape: structure and function of the autotransporter proteins. Trends Microbiol. 6:370-378. [DOI] [PubMed] [Google Scholar]
- 24.Hijnen, M., R. de Voer, F. R. Mooi, R. Schepp, E. E. Moret, P. van Gageldonk, G. Smits, and G. A. Berbers. 2007. The role of peptide loops of the Bordetella pertussis protein P.69 pertactin in antibody recognition. Vaccine 25:5902-5914. [DOI] [PubMed] [Google Scholar]
- 25.Hijnen, M., F. R. Mooi, P. G. van Gageldonk, P. Hoogerhout, A. J. King, and G. A. Berbers. 2004. Epitope structure of the Bordetella pertussis protein P.69 pertactin, a major vaccine component and protective antigen. Infect. Immun. 72:3716-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inatsuka, C. S., S. M. Julio, and P. A. Cotter. 2005. Bordetella filamentous hemagglutinin plays a critical role in immunomodulation, suggesting a mechanism for host specificity. Proc. Natl. Acad. Sci. U. S. A. 102:18578-18583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ishibashi, Y., S. Claus, and D. A. Relman. 1994. Bordetella pertussis filamentous hemagglutinin interacts with a leukocyte signal transduction complex and stimulates bacterial adherence to monocyte CR3 (CD11b/CD18). J. Exp. Med. 180:1225-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishibashi, Y., and A. Nishikawa. 2002. Bordetella pertussis infection of human respiratory epithelial cells up-regulates intercellular adhesion molecule-1 expression: role of filamentous hemagglutinin and pertussis toxin. Microb. Pathog. 33:115. [DOI] [PubMed] [Google Scholar]
- 29.Ishibashi, Y., and A. Nishikawa. 2003. Role of nuclear factor-kappa B in the regulation of intercellular adhesion molecule 1 after infection of human bronchial epithelial cells by Bordetella pertussis. Microb. Pathog. 35:169-177. [DOI] [PubMed] [Google Scholar]
- 30.Julio, S. M., C. S. Inatsuka, J. Mazar, C. Dieterich, D. A. Relman, and P. A. Cotter. 2009. Natural-host animal models indicate functional interchangeability between the filamentous haemagglutinins of Bordetella pertussis and Bordetella bronchiseptica and reveal a role for the mature C-terminal domain, but not the RGD motif, during infection. Mol. Microbiol. 71:1574-1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Junker, M., R. N. Besingi, and P. L. Clark. 2009. Vectorial transport and folding of an autotransporter virulence protein during outer membrane secretion. Mol. Microbiol. 71:1323-1332. [DOI] [PubMed] [Google Scholar]
- 32.Khelef, N., C. M. Bachelet, B. B. Vargaftig, and N. Guiso. 1994. Characterization of murine lung inflammation after infection with parental Bordetella pertussis and mutants deficient in adhesins or toxins. Infect. Immun. 62:2893-2900. (Erratum, 62:5707.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.King, A. J., G. Berbers, H. F. van Oirschot, P. Hoogerhout, K. Knipping, and F. R. Mooi. 2001. Role of the polymorphic region 1 of the Bordetella pertussis protein pertactin in immunity. Microbiology 147:2885-2895. [DOI] [PubMed] [Google Scholar]
- 34.Kobisch, M., and P. Novotny. 1990. Identification of a 68-kilodalton outer membrane protein as the major protective antigen of Bordetella bronchiseptica by using specific-pathogen-free piglets. Infect. Immun. 58:352-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 36.Leininger, E., C. A. Ewanowich, A. Bhargava, M. S. Peppler, J. G. Kenimer, and M. J. Brennan. 1992. Comparative roles of the Arg-Gly-Asp sequence present in the Bordetella pertussis adhesins pertactin and filamentous hemagglutinin. Infect. Immun. 60:2380-2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leininger, E., M. Roberts, J. G. Kenimer, I. G. Charles, N. Fairweather, P. Novotny, and M. J. Brennan. 1991. Pertactin, an Arg-Gly-Asp-containing Bordetella pertussis surface protein that promotes adherence of mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 88:345-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li, L. J., G. Dougan, P. Novotny, and I. G. Charles. 1991. P.70 pertactin, an outer-membrane protein from Bordetella parapertussis: cloning, nucleotide sequence and surface expression in Escherichia coli. Mol. Microbiol. 5:409-417. [DOI] [PubMed] [Google Scholar]
- 39.Litt, D. J., S. E. Neal, and N. K. Fry. 2009. Changes in genetic diversity of the Bordetella pertussis population in the United Kingdom between 1920 and 2006 reflect vaccination coverage and emergence of a single dominant clonal type. J. Clin. Microbiol. 47:680-688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martinez de Tejada, G., J. F. Miller, and P. A. Cotter. 1996. Comparative analysis of the virulence control systems of Bordetella pertussis and Bordetella bronchiseptica. Mol. Microbiol. 22:895-908. [DOI] [PubMed] [Google Scholar]
- 41.Mattoo, S., and J. D. Cherry. 2005. Molecular pathogenesis, epidemiology, and clinical manifestations of respiratory infections due to Bordetella pertussis and other Bordetella subspecies. Clin. Microbiol. Rev. 18:326-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mattoo, S., J. F. Miller, and P. A. Cotter. 2000. Role of Bordetella bronchiseptica fimbria in tracheal colonization and development of a humoral immune response. Infect. Immun. 68:2024-2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller, V. L., and J. J. Mekalanos. 1988. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J. Bacteriol. 170:2575-2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mooi, F. R., H. Hallander, C. H. Wirsing von Konig, B. Hoet, and N. Guiso. 2000. Epidemiological typing of Bordetella pertussis isolates: recommendations for a standard methodology. Eur. J. Clin. Microbiol. Infect. Dis. 19:174-181. [DOI] [PubMed] [Google Scholar]
- 45.Mooi, F. R., H. van Oirschot, K. Heuvelman, H. G. van der Heide, W. Gaastra, and R. J. Willems. 1998. Polymorphism in the Bordetella pertussis virulence factors P.69/pertactin and pertussis toxin in the Netherlands: temporal trends and evidence for vaccine-driven evolution. Infect. Immun. 66:670-675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Musser, J. M., E. L. Hewlett, M. S. Peppler, and R. K. Selander. 1986. Genetic diversity and relationships in populations of Bordetella spp. J. Bacteriol. 166:230-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nicholson, T. L., S. L. Brockmeier, and C. L. Loving. 2009. Contribution of Bordetella bronchiseptica filamentous hemagglutinin and pertactin to respiratory disease in swine. Infect. Immun. 77:2136-2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Novotny, P., A. P. Chubb, K. Cownley, and I. G. Charles. 1991. Biologic and protective properties of the 69-kDa outer membrane protein of Bordetella pertussis: a novel formulation for an acellular pertussis vaccine. J. Infect. Dis. 164:114-122. [DOI] [PubMed] [Google Scholar]
- 49.Novotny, P., A. P. Chubb, K. Cownley, and J. A. Montaraz. 1985. Adenylate cyclase activity of a 68,000-molecular-weight protein isolated from the outer membrane of Bordetella bronchiseptica. Infect. Immun. 50:199-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parkhill, J., M. Sebaihia, A. Preston, L. D. Murphy, N. Thomson, D. E. Harris, M. T. Holden, C. M. Churcher, S. D. Bentley, K. L. Mungall, A. M. Cerdeno-Tarraga, L. Temple, K. James, B. Harris, M. A. Quail, M. Achtman, R. Atkin, S. Baker, D. Basham, N. Bason, I. Cherevach, T. Chillingworth, M. Collins, A. Cronin, P. Davis, J. Doggett, T. Feltwell, A. Goble, N. Hamlin, H. Hauser, S. Holroyd, K. Jagels, S. Leather, S. Moule, H. Norberczak, S. O'Neil, D. Ormond, C. Price, E. Rabbinowitsch, S. Rutter, M. Sanders, D. Saunders, K. Seeger, S. Sharp, M. Simmonds, J. Skelton, R. Squares, S. Squares, K. Stevens, L. Unwin, S. Whitehead, B. G. Barrell, and D. J. Maskell. 2003. Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat. Genet. 35:32-40. [DOI] [PubMed] [Google Scholar]
- 51.Pearson, R. D., P. Symes, M. Conboy, A. A. Weiss, and E. L. Hewlett. 1987. Inhibition of monocyte oxidative responses by Bordetella pertussis adenylate cyclase toxin. J. Immunol. 139:2749-2754. [PubMed] [Google Scholar]
- 52.Preston, A., J. Parkhill, and D. J. Maskell. 2004. The bordetellae: lessons from genomics. Nat. Rev. Microbiol. 2:379-390. [DOI] [PubMed] [Google Scholar]
- 53.Register, K. B. 2004. Comparative sequence analysis of Bordetella bronchiseptica pertactin gene (prn) repeat region variants in swine vaccines and field isolates. Vaccine 23:48-57. [DOI] [PubMed] [Google Scholar]
- 54.Register, K. B. 2001. Novel genetic and phenotypic heterogeneity in Bordetella bronchiseptica pertactin. Infect. Immun. 69:1917-1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roberts, M., N. F. Fairweather, E. Leininger, D. Pickard, E. L. Hewlett, A. Robinson, C. Hayward, G. Dougan, and I. G. Charles. 1991. Construction and characterization of Bordetella pertussis mutants lacking the vir-regulated P.69 outer membrane protein. Mol. Microbiol. 5:1393-1404. [DOI] [PubMed] [Google Scholar]
- 56.Roder, J., and A. Duwe. 1979. The beige mutation in the mouse selectively impairs natural killer cell function. Nature 278:451-453. [DOI] [PubMed] [Google Scholar]
- 57.Ruoslahti, E. 1996. RGD and other recognition sequences for integrins. Annu. Rev. Cell Dev. Biol. 12:697-715. [DOI] [PubMed] [Google Scholar]
- 58.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 59.Shin, E. K., R. Jung, and T. W. Hahn. 2007. Polymorphism of pertactin gene repeat regions in Bordetella bronchiseptica isolates from pigs. J. Vet. Med. Sci. 69:771-774. [DOI] [PubMed] [Google Scholar]
- 60.Stainer, D. W., and M. J. Scholte. 1970. A simple chemically defined medium for the production of phase I Bordetella pertussis. J. Gen. Microbiol. 63:211-220. [DOI] [PubMed] [Google Scholar]
- 61.Stenger, R. M., M. C. Poelen, E. E. Moret, B. Kuipers, S. C. Bruijns, P. Hoogerhout, M. Hijnen, A. J. King, F. R. Mooi, C. J. Boog, and C. A. van Els. 2009. Immunodominance in mouse and human CD4+ T-cell responses specific for the Bordetella pertussis virulence factor P.69 pertactin. Infect. Immun. 77:896-903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stibitz, S., W. Black, and S. Falkow. 1986. The construction of a cloning vector designed for gene replacement in Bordetella pertussis. Gene 50:133-140. [DOI] [PubMed] [Google Scholar]
- 63.Storsaeter, J., H. O. Hallander, L. Gustafsson, and P. Olin. 1998. Levels of anti-pertussis antibodies related to protection after household exposure to Bordetella pertussis. Vaccine 16:1907-1916. [DOI] [PubMed] [Google Scholar]
- 64.van den Berg, B. M., H. Beekhuizen, R. J. Willems, F. R. Mooi, and R. van Furth. 1999. Role of Bordetella pertussis virulence factors in adherence to epithelial cell lines derived from the human respiratory tract. Infect. Immun. 67:1056-1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van der Zee, A., F. Mooi, J. Van Embden, and J. Musser. 1997. Molecular evolution and host adaptation of Bordetella spp.: phylogenetic analysis using multilocus enzyme electrophoresis and typing with three insertion sequences. J. Bacteriol. 179:6609-6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Waters, C. M., C. L. Wells, and G. M. Dunny. 2003. The aggregation domain of aggregation substance, not the RGD motifs, is critical for efficient internalization by HT-29 enterocytes. Infect. Immun. 71:5682-5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weber, C., C. Boursaux-Eude, G. Coralie, V. Caro, and N. Guiso. 2001. Polymorphism of Bordetella pertussis isolates circulating for the last 10 years in France, where a single effective whole-cell vaccine has been used for more than 30 years. J. Clin. Microbiol. 39:4396-4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weingart, C. L., and A. A. Weiss. 2000. Bordetella pertussis virulence factors affect phagocytosis by human neutrophils. Infect. Immun. 68:1735-1739. [DOI] [PMC free article] [PubMed] [Google Scholar]