

Abstract
The peptide transporter from a cold-adapted bacterium has never been reported. In the present study, the dpp operon from the psychrophilic bacterium Pseudoalteromonas sp. strain SM9913 was cloned and analyzed. The dipeptide binding protein DppA of SM9913 was overexpressed in Escherichia coli, and its cold adaptation characteristics were studied. The recombinant DppA of SM9913 (PsDppA) displayed the highest ligand-binding affinity at 15°C, whereas the recombinant DppA of E. coli (EcDppA) displayed the highest ligand-binding affinity at 35°C. Thermal and guanidium hydrochloride unfolding analyses indicated that PsDppA has more structural instability than EcDppA. Six domain-exchanged mutants of PsDppA were expressed and purified. Analyses of these mutants indicated that domains III, I-2, and I-3 of PsDppA were less stable than those from EcDppA and that domains III and I-2 made a significant contribution to the high binding affinity of PsDppA at low temperatures. Structural and sequence analyses suggested that the state transition-involved regions in domain III and the α part of domain I-2 are the hot spots of optimization during cold adaptation and that decreasing the side-chain size in these regions is an important strategy for the cold adaptation of PsDppA.
The peptide transporter system of bacteria, which plays an important role in nutrition supply, has been extensively investigated, especially for Escherichia coli (9, 11, 20, 21), Salmonella enterica serovar Typhimurium (8, 12, 28, 29), and Lactococcus lactis (2, 13, 14, 25). At present, three peptide transporters—oligopeptide permease (Opp), dipeptide permease (Dpp), and dipeptide and tripeptide permease (Tpp or Dtp)—have been found in bacteria. Dpp proteins are transporters belonging to the ATP-binding cassette (ABC) superfamily and are composed of five subunits: the two integral membrane proteins DppB and DppC forming a permease for substrates, the two cytoplasmic proteins DppD and DppF in charge of ATP hydrolysis, and a periplasmic peptide-binding protein named DppA. At the genetic level, the five genes encoding these five proteins are always organized in an operon named dpp. Dpp has a preference for dipeptides and also transports some tripeptides (25, 27).
During peptide translocation, DppA performs identification and binding of substrates and determines the specificity and overall transport parameters for Dpp (27). The crystal structure of DppA from E. coli has been determined, which shows that DppA is composed of three domains (6). The ligand-binding cleft is located between domain I and domain III that are connected by two strands functioning as a hinge. After binding a ligand, the “open” protein turns into a “closed” form, and the ligand is completely buried (6). The tertiary structure of DppA is quite similar to that of OppA (2, 6, 29). It is reported that domain II in OppA is not involved in ligand binding directly, and its role has not been studied in detail (18, 28).
Cold-adapted microorganisms are a diverse group living in cold ecosystems, such as the polar and alpine regions and deep sea. Survival in these extreme environments requires the microorganisms to evolve a complex suite of structural and functional adaptations of all cellular constituents, such as membrane, enzymes, energy-generating systems, components responsible for nutrient uptake, and so on (16). There are extensive investigations on modification of membrane lipid composition and adjustment of cold-adapted enzymes (7). However, there has been no report on the peptide transport system from a cold-adapted microorganism. Thus, it is yet unclear how the peptide transport system of a cold-adapted microorganism is adapted to a cold environment.
Pseudoalteromonas sp. strain SM9913 is a psychrophilic bacterium isolated from deep-sea sediment, which secretes a large quantity of exopolysaccharide and proteases (5, 23). Our previous studies showed that the exopolysaccharide secreted by strain SM9913 may help the strain enrich the proteinaceous particles and the trace metal ions in the deep-sea environment, and the cold-adapted protease secreted by this strain can degrade various soluble and insoluble proteins to provide nutrients (4, 32). Efficient transport of peptides and amino acids in the deep-sea cold environment is very important for the growth of strain SM9913. In order to elucidate how the peptide transport system of strain SM9913 is adapted to a deep-sea cold environment, the dpp operon of strain SM9913 was cloned and analyzed in the present study. The periplasmic dipeptide-binding protein DppA of Pseudoalteromonas sp. strain SM9913 (PsDppA) was overexpressed in E. coli, and its ligand-binding affinity and structural stability were studied and compared to those of DppA of E. coli (EcDppA). Moreover, by domain-exchanged mutation, the cold adaptation mechanism of PsDppA at the domain level was studied.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Pseudoalteromonas sp. strain SM9913 was cultured with shaking at 15°C in modified 2216E marine medium (0.5% peptone, 0.1% yeast extract, seawater [pH 7.5]). E. coli strains DH5α, BL21(DE3), and OrigamiB(DE3) were grown at 37°C in Luria-Bertani (LB) medium, and appropriate antibiotics were supplemented when carrying plasmid vectors.
Cloning of the dpp operon.
Genomic DNA of strain SM9913 was prepared as previously described (4). Based on the conserved regions of several microbial DppAs, two degenerate primers, AP-F and AP-R, were designed, and a 1.2-kb DNA fragment, a part of the dppA gene of strain SM9913, was amplified from the genomic DNA by PCR with these two primers. Based on the 3′-end sequence of this DNA fragment, primer AD-F was designed. Then, a 3-kb DNA fragment comprising the downstream portion of dppA, the full dppB, the full dppC, and the upstream portion of dppD was amplified with the primers AD-F and AD-R, which were designed based on a conserved region of microbial DppD. Similarly, based on the 3′-end sequence of this DNA sequence, the primer DF-F was designed, and the remaining sequence of dppD and the upstream sequence of dppF were obtained by PCR with the primers DF-F and DF-R, which were designed based on a conserved region of microbial DppF. Finally, chromosome walking was used to amplify the 5′-end sequence of dppA and the 3′-end sequence of dppF by thermal asymmetric interlaced PCR (TAIL-PCR) with six specific primers (NA1, NA2, and NA3 and CF1, CF2, and CF3) and two general primers (NP and CP). Through assembly, a 5,276-bp sequence containing five open reading frames (genes dppABCDF) and the upstream promoter sequence were obtained. To verify the sequences of these genes, each of the five genes was amplified alone by PCR from the genomic DNA of strain SM9913 and sequenced. The sequences of all of the primers used above are shown in Table S1 in the supplemental material.
Homology modeling.
Homology models for PsDppA in an open conformation and a closed conformation were modeled by using Modeler 9v7 (24) with crystal structures of EcDppA as the templates (1DPE.pdb for the open conformation and 1DPP.pdb for the closed conformation). For each conformation, 1,000 models were created. The models with favorite objective function scores and DOPE scores were further validated by using the SAVES server (nihserver.mbi.ucla.edu/SAVES/) and PSQS server (www1.jcsg.org/psqs/). Ultimately, one model with high-quality scores for each conformation was selected.
Overexpression of PsDppA and EcDppA.
Our preparatory experiment showed that the signal peptide sequence of PsDppA could not be recognized by E. coli BL21(DE3). Substitution of the PsDppA signal peptide with one recognized by E. coli BL21(DE3) also failed to obtain the recombinant PsDppA in an active form in E. coli BL21(DE3). Homology modeling of PsDppA suggested that it contains two disulfide bonds: C-15—C-243 and C-431—C-444. Since E. coli OrigamiB(DE3) was reported to enhance cytoplasmic disulfide bond formation as an expression host (1, 22), PsDppA without its putative signal peptide was overexpressed in OrigamiB(DE3). The dppA gene of SM9913 (PsdppA) without the nucleic acids encoding the signal peptide sequence was cloned from the genomic DNA of strain SM9913 by PCR and then ligated into the NdeI/XhoI-digested pET-22b(+) plasmid for expression. The constructed vector pET-22b-PsdppA was transformed into OrigamiB(DE3) cells. The strain carrying pET-22b-PsdppA was grown at 37°C in LB medium with 50 μg of ampicillin/ml, 12.5 μg of tetracycline/ml, and 15 μg of kanamycin/ml to an optical density at 600 nm (OD600) of 0.7 to 0.8 and then was induced with 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside) at 15°C for 16 h.
To express EcDppA, the complete dppA gene (GenBank accession number NC010473) of E. coli (EcdppA) was cloned from the genomic DNA of E. coli BL21 and ligated into the NdeI/XhoI-digested pET-22b(+) plasmid. The constructed vector pET-22b-EcdppA was transformed into E. coli BL21(DE3) cells, and the strain was cultured at 37°C in LB medium supplemented with 50 μg of ampicillin/ml. When the OD600 reached ∼0.8, 0.5 mM IPTG was added, and the cells were grown at 20°C for 10 h.
Construction of domain-exchanged mutants.
The domain-exchanged mutants were constructed by replacing the PsDppA domain with the corresponding sequence from EcDppA. The sequence of domain II in PsDppA (D43 to T191 [D43-T191]) was replaced by the corresponding sequence of domain II in EcDppA (N34-T182) to construct mutant PsE2. Similarly, the sequence of domain III in PsDppA (L271-G487) was replaced by that of domain III in EcDppA (L262-S478) to construct mutant PsE3; the sequences of the three segments of domain I (I-1, I-2, and I-3) in PsDppA (K1-Y42 for I-1, G192-T269 for I-2, and M488-K516 for I-3) were replaced by the corresponding sequences of domain I in EcDppA (K1-Y33 for I-1, G183-P260 for I-2, and T479-E507 for I-3) to construct mutants PsE1-1, PsE1-2, and PsE1-3, respectively. Furthermore, the first 36 residues of domain I-2 in PsDppA (G192-I227) were replaced by the corresponding domain I sequence of EcDppA (G183-I218) to construct mutant PsE1-2β, and the last 42 residues (T228-T269) were replaced by the corresponding sequence of EcDppA (T219-P260) to construct mutant PsE1-2α. The chimeric PsE2, PsE3, PsE1-1, PsE1-2, PsE1-3, PsE1-2β, PsE1-2α genes were all generated by using gene splicing by overlap-extension PCR as described by Wurch et al. (31). Each of the above seven genes was ligated into the NdeI/XhoI-digested pET-22b(+) plasmid and was checked by DNA sequencing. The constructed vectors were then transformed into OrigamiB(DE3) cells to express the mutant proteins with an IPTG concentration of 0.2 mM and an inducing condition of 15°C for 16 h.
Protein purification.
PsDppA, EcDppA, and the domain-exchanged mutants were all expressed as C-terminally His6-tagged proteins for purification. Except for mutant PsE1-1, all of the expressed proteins were soluble and were purified with Novagen His-Bind resin at 4°C according to the manufacturer's instructions. Briefly, the harvested cell pellets were washed twice with binding buffer (25 mM phosphate buffer [pH 7.5], supplemented with 10 mM imidazole and 300 mM NaCl) and resuspended in the same buffer. After sonication on ice, the supernatant was collected by centrifugation at 13,000 rpm and then loaded on the column preequilibrated with 3 column volumes of binding buffer. The column was orderly washed with 10 column volumes of binding buffer and 6 column volumes of 25 mM phosphate buffer (pH 7.5), supplemented with 60 mM imidazole and 300 mM NaCl. Finally, the recombinant proteins were eluted with a gradient of increasing imidazole concentrations (80, 100, 150, and 200 mM). The purified proteins were analyzed by SDS-PAGE. The purification yield was ∼5 mg for PsDppA, 6 mg for EcDppA and 3 to 4 mg for each mutant per liter of cell culture (OD660 ≈ 2.5), with a purity of ≥95%. Endogenous ligands that might have been bound to the purified EcDppA were removed by using the previously described procedures with modification (25, 30). Guanidinium hydrochloride (GdmHCl) was added to a final concentration of 3 M to the EcDppA protein solution. The protein was then dialyzed against solutions with progressively decreasing concentrations of GdmHCl and finally dialyzed against 25 mM phosphate buffer (pH 7.5). The protein concentration was determined by using a bicinchoninic acid (BCA) protein assay kit (Bioteke, China), with bovine serum albumin as the standard.
Dipeptide binding.
The dipeptide binding of DppA proteins was measured by intrinsic fluorescence assay as described by Sanz et al. (25) on an FP-6500 spectrofluorometer (Jasco, Japan) equipped with a Peltier-type thermostat-controlled cell holder (Jasco ETC-273). Excitation was performed at 280 nm with a 3-nm bandwidth, and emission was scanned from 300 to 400 nm with a 5-nm bandwidth. Spectra were determined with DppA proteins at a concentration of ∼100 μg/ml in 25 mM phosphate buffer (pH 7.5) in the absence or presence of saturating dipeptides.
Ligand-binding affinity.
In order to analyze the ligand-binding affinity of DppA proteins at different temperatures, the dissociation constant (Kd) of DppA proteins to dipeptide Ala-Phe was determined by titration of intrinsic fluorescence of DppA with different ligand concentrations. The dissociation constants (Kd) were determined by nonlinear fitting of the data to the equations as described by Lanfermeijer et al. (14).
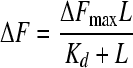 |
(1) |
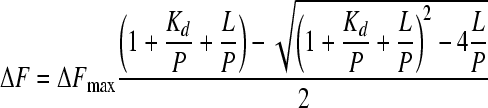 |
(2) |
In the above equations, Kd is the dissociation constant, ΔF is the measured fluorescence change, ΔFmax is the maximum fluorescence change, and L is the total ligand concentration. The saturation equation (equation 1) was used when the dissociation constants were at least 3-fold higher than the protein concentration, and the general equilibrium equation (equation 2) was applied when the dissociation constants and the protein concentration were in the same range. Nonlinear least-squares regression was performed by using the Origin program (OriginLab).
Thermal and GdmHCl unfolding.
Thermal unfolding of DppA proteins was measured by both circular dichroism (CD) spectroscopy and differential scanning calorimetry (DSC). CD spectra of DppA proteins with the same concentration (3.3 to 3.4 μM) were collected from 260 to 190 nm at a scan speed of 200 nm/min on a Jasco J-810 spectropolarimeter with a Julabo computer-controlled thermostat (Japan). All data are averages of three scans. The ellipticity at 222 nm was recorded as the temperature increased from 25 to 85°C at a rate of 1°C/min. DSC was performed on a Microcal VP-DSC instrument at a scan rate of 60°C/h, and the protein concentration was 1 mg/ml. GdmHCl unfolding of PsDppA and EcDppA was monitored by CD spectroscopy. Native PsDppA and EcDppA were incubated at room temperature for 1 h with various concentrations of GdmHCl (0 to 5 M), and then the CD spectra were measured. All measurements were performed in 25 mM phosphate buffer (pH 7.5).
Nucleotide sequence accession numbers.
The five genes of strain SM9913 (dppABCDF) were deposited in GenBank under accession numbers EU121590, EU136168, EU136169, EU136170, and EU136171.
RESULTS
Cloning and sequence analysis of the dpp operon of Pseudoalteromonas sp. strain SM9913.
A 5,276-bp DNA fragment containing genes dppABCDF was cloned from the genomic DNA of strain SM9913 as described in Materials and Methods. Sequencing results showed that the five genes are organized in a typical five-gene dpp operon (20). The first open reading frame, gene dppA, starts with a GTG initiation codon and encodes a protein of 535 amino acid residues with a predicted N-terminal signal peptide of 19 amino acid residues. The deduced DppB and DppC were predicted to be two integral membrane proteins, and the deduced DppD and DppF have high similarity to ATP-binding proteins, especially those from ABC-type transporters. Searches with BLASTP in the GenBank database showed that the Dpp proteins of strain SM9913 have the highest identities (85 to 94%) to those deduced from the genome sequence of the Antarctic psychrophilic strain Pseudoalteromonas haloplanktis TAC125 (17), relatively high identities to those from the same-genus mesophilic strains P. tunicata D2 (65 to 79%) and P. atlantica T6c (45 to 51%), and low identities (40 to 43%) to those from mesophilic E. coli (3, 20) (see Table S2 in the supplemental material).
Sequence and structural analysis of PsDppA.
In the Dpp system, DppA is the most studied protein. The structure and function of DppA from E. coli has been well studied, and it is the only DppA protein whose structure has been determined (6, 19). There are two major conformations of DppA: the ligand-free open conformation (19) and the ligand-bound closed conformation (6). PsDppA structures in the two conformations were modeled (see Fig. S1 in the supplemental material for the sequence alignment, Fig. 1 for the models, and Fig. S2 in the supplemental material for structure superposition). Quality assessment indicated that the obtained models were reliable (see Table S3 in the supplemental material). As shown in Fig. 1, PsDppA is composed of three domains: domain I (K1-Y42 for I-1, G192-T269 for I-2, and M488-K516 for I-3), domain II (D43-T191), and domain III (N270-G487).
FIG. 1.

Three-dimensional structures of modeled PsDppA in an open conformation (A) and a closed conformation (B) with crystal structures of EcDppA as the templates (1DPE.pdb and 1DPP.pdb). Domains are represented in different colors: the first segment of domain I (K1-Y42) in cyan, the second segment of domain I (G192-T269) in blue, the third segment of domain I (M488-K516) in yellow, domain II (D43-T191) in green, and domain III (L271-G487) in red. Domain I-2β (β8 to β10 in Fig. S1 in the supplemental material) is labeled “β,” and domain I-2α (α9, α10, and β11 in Fig. S1 in the supplemental material) is labeled “α.”
PsDppA and its mesophilic homolog EcDppA were expressed and purified to study the cold adaptation characteristics of PsDppA. SDS-PAGE analysis showed that their relative molecular masses correspond to those predicted from the amino acid sequences (58.8 kDa for PsDppA and 57.4 kDa for EcDppA) (Fig. 2 A). Far-UV CD spectra of PsDppA and EcDppA showed they have similar secondary structures (Fig. 2B).
FIG. 2.

(A) SDS-PAGE analysis of recombinant PsDppA and EcDppA purified by nickel affinity chromatography. Protein samples were separated by SDS-PAGE (12.5%) and stained by Coomassie brilliant blue G250. Approximately 4 μg of EcDppA and 2.2 μg of PsDppA samples were loaded. (B) CD spectra of PsDppA (solid line) and EcDppA (broken line) with a concentration of 3.4 μM at 15°C.
Ligand-binding affinity of PsDppA.
The binding mechanism of DppA is referred to as the “Venus's flytrap” mechanism (15). Upon ligand binding, DppA undergoes a large conformational change, which results in closure of the cleft between domain I and domain III and leads to a change in its intrinsic fluorescence. Thus, fluorescence spectroscopy is often used to detect the ligand binding of the DppA proteins, and fluorescence titration is applied to measure the binding affinity (10, 25, 27). The emission spectrum of the purified PsDppA showed a maximum at 337 nm, and it displayed a blue shift of 3 to 5 nm with an overall fluorescence decrease after saturating Ala-Phe was added (Fig. 3 A). In the assay of EcDppA, after saturating Ala-Phe was added, an overall fluorescence decrease with a blue shift of 1 to 2 nm in the fluorescence spectrum was observed (Fig. 3B), a finding consistent with the result described by Smith et al. (27).
FIG. 3.

Intrinsic fluorescence change of PsDppA (A) and EcDppA (B) induced by the addition of saturating dipeptide Ala-Phe and fluorescence titration of PsDppA at 15°C (C) and EcDppA at 35°C (D). In an intrinsic fluorescence change assay (left), the emission spectra of DppA proteins with a concentration of ∼100 μg/ml in the absence (solid lines) or presence (broken lines) of saturating Ala-Phe were recorded at 15°C. In fluorescence titration (right), the concentration of DppA proteins was 0.86 μM. The solid line represents the best fitting of the data to equation 2 in Materials and Methods. The intersection point of the dotted lines corresponds to the binding stoichiometry. All of the experiments were performed in 25 mM phosphate buffer (pH 7.5). Each experiment was repeated five times, and similar results were obtained each time.
In fluorescence titration, the fluorescence changes at 360 nm for PsDppA and at 350 nm for EcDppA were shown to be ligand concentration dependent and were used in peptide binding analysis. The binding stoichiometry was determined to be 1.1 ± 0.2 dipeptide/protein molecule for PsDppA and 0.95 ± 0.1 for EcDppA (Fig. 3C and D). The lowest Kd of PsDppA to Ala-Phe was observed at 15°C in the temperature range of 10 to 30°C (Table 1), showing that the optimal ligand-binding temperature for PsDppA was 15°C. In contrast, the optimal ligand-binding temperature for EcDppA was 35°C (Table 1). These results suggested that, compared to EcDppA, PsDppA is adapted to efficiently bind peptides at a low temperature.
TABLE 1.
Dissociation constants and Tm values of EcDppA, PsDppA, and mutants
| Protein |
Kd (μM)a |
Tm (°C) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| 10°C | 15°C | 20°C | 25°C | 30°C | 35°C | 40°C | DSC | CD | |
| PsDppA | 0.11 ± 0.02 | 0.04 ± 0.02 | 0.18 ± 0.03 | 0.29 ± 0.03 | 0.40 ± 0.03 | ND | ND | 43 | 49 |
| EcDppA | ND | ND | 0.12 ± 0.02 | 0.09 ± 0.02 | 0.05 ± 0.02 | 0.02 ± 0.01 | 0.04 ± 0.01 | 71 | 65 |
| PsE2 | 8.2 ± 1.6 | 5.6 ± 0.6 | 10.3 ± 0.8 | 12.8 ± 1.9 | ND | ND | ND | 43 | ND |
| PsE3 | ND | ND | 13.2 ± 1.5 | 9.4 ± 0.9 | 5.8 ± 0.7 | 12.6 ± 0.5 | ND | 43.5, 65 | 61 |
| PsE1-2 | ND | ND | ND | 6.0 ± 0.8 | 3.9 ± 0.4 | 1.5 ± 0.2 | 4.5 ± 0.1 | 46 | 51 |
| PsE1-3 | 8.1 ± 0.6 | 5.9 ± 0.2 | 8.3 ± 0.6 | 13.2 ± 1.4 | ND | ND | ND | 48 | 55 |
| PsE1-2β | 5.9 ± 0.5 | 4.7 ± 0.7 | 7.1 ± 1.1 | 9.1 ± 1.5 | ND | ND | ND | ND | ND |
| PsE1-2α | ND | ND | 0.17 ± 0.02 | 0.13 ± 0.02 | 0.09 ± 0.03 | 0.12 ± 0.03 | ND | ND | ND |
ND, not determined. The lowest Kd value for each protein is indicated in boldface.
Stability of PsDppA.
The unfolding of PsDppA induced by temperature and GdmHCl was monitored and compared to those of EcDppA. The Tm value of PsDppA determined by CD is 49°C, 16°C lower than that of EcDppA (Fig. 4 A). According to DSC results, the Tm value of PsDppA is 43 ± 0.3°C, 28°C lower than that of EcDppA (Fig. 4B). The GdmHCl unfolding transition of PsDppA and EcDppA was determined by monitoring the loss of the secondary structure with the increase of GdmHCl concentration by using CD (Fig. 4C). Complete secondary structure loss was observed when the GdmHCl concentration was 2.5 M for PsDppA and 3.5 M for EcDppA. All of these results suggested that PsDppA has lower stability in structure than EcDppA.
FIG. 4.

Thermal unfolding and GdmHCl unfolding of PsDppA and EcDppA. (A) Thermal unfolding curves of PsDppA (▪) and EcDppA (•) detected by CD. CD spectra of DppA proteins with the same concentration (3.3 to 3.4 μM) were collected from 260 to 190 nm. The ellipticity at 222 nm was recorded as the temperature increased from 25 to 85°C at a rate of 1°C/min. (B) Thermal unfolding curves of PsDppA (solid line) and EcDppA (broken line) detected by DSC at a scan rate of 60°C/h with a protein concentration of ∼1 mg/ml. (C) GdmHCl unfolding of PsDppA (▪, solid line) and EcDppA (•, dashed line) detected by CD. Native PsDppA and EcDppA (3.4 μM) were incubated at room temperature for 1 h with various concentrations of GdmHCl (0 to 5 M), and then the CD spectra were measured by recording the ellipticity at 222 nm. Experiments were all performed in 25 mM phosphate buffer at pH 7.5. All of the these experiments were repeated three times.
Stability of domain-exchanged mutants.
Since homology modeling showed that PsDppA has three highly similar domains to EcDppA, domain-exchanged mutations were performed to investigate the contribution of each domain to the structural instability of PsDppA protein. The domain-exchanged mutants (PsE2, PsE3, PsE1-2, PsE1-3, PsE1-2β, and PsE1-2α) were expressed and purified (Fig. 5). The secondary structures of the purified mutant proteins were similar to those of PsDppA and EcDppA (see Fig. S3 in the supplemental material), and all exhibited an ability to bind to dipeptide Ala-Phe (see Fig. S4 in the supplemental material), suggesting that all of the mutants fold correctly. The structural stability of the mutants was determined as shown in Table 1 and Fig. 6.
FIG. 5.

(A) Schematic representation of the structures of PsDppA, EcDppA, and domain-exchanged mutants. The sequences from PsDppA are indicated in gray, and those from EcDppA are indicated in white. (B) SDS-PAGE analysis of the mutant proteins purified by nickel affinity chromatography.
FIG. 6.
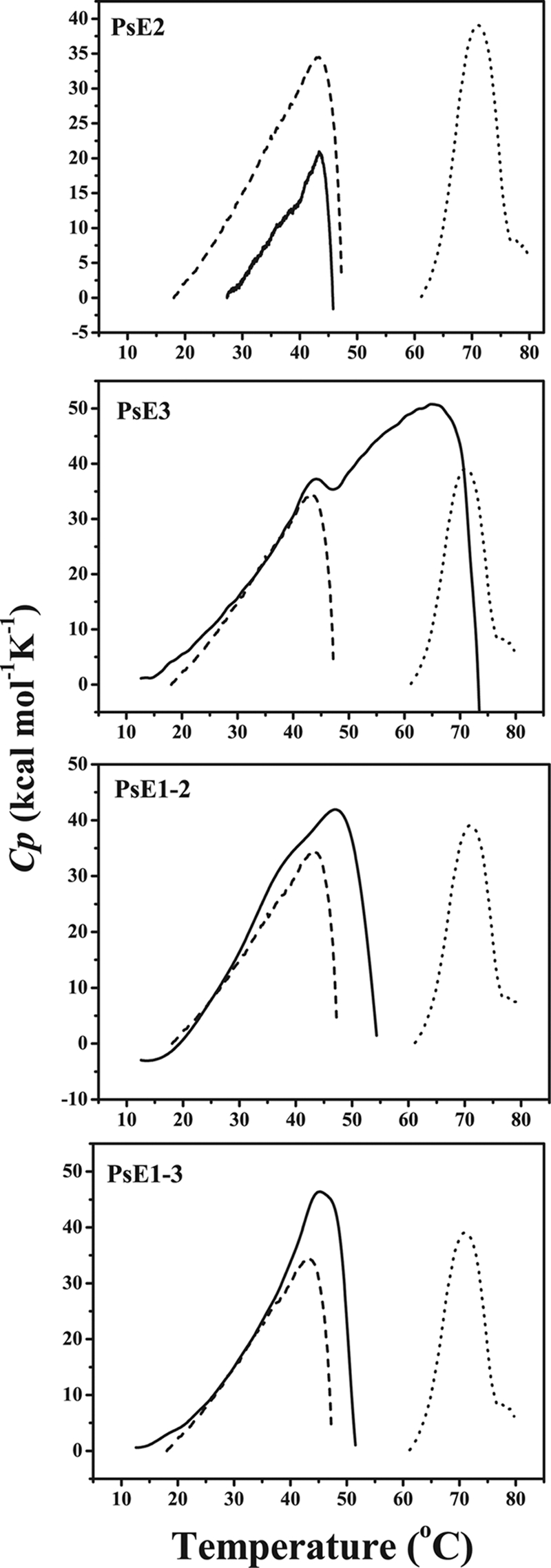
Thermal unfolding of domain-exchanged mutants. Thermal unfolding curves of the mutants PsE2, PsE3, PsE1-2, and PsE1-3 (solid lines) detected by DSC with a protein concentration of ∼1 mg/ml in 25 mM phosphate buffer (pH 7.5) compared to those of PsDppA (dashed line) and EcDppA (dotted line). Each sample was evaluated three times, and the same result was obtained each time.
Mutant PsE2 had the same Tm value (43 ± 0.3°C, determined by DSC) as PsDppA, indicating that domain II of PsDppA has similar structural stability to that of EcDppA. The DSC profile of PsE3 showed that PsE3 mutant is composed of two different calorimetric units, a heat-labile one with a Tm of 43.5°C, similar to that of PsDppA, and a heat-stable one with a Tm of 65°C, which may correspond to domain III from EcDppA. The Tm value of PsE3 measured by CD was 61°C (see Fig. S5A in the supplemental material), 12°C higher than that of PsDppA. These results indicated that domain III of PsDppA has lower structural stability than that of EcDppA, which would make a significant contribution to the high thermolability of PsDppA protein. The Tm values of PsE1-2 (46 ± 0.3°C) and PsE1-3 (48 ± 0.3°C) measured by DSC were both higher than that of PsDppA (43 ± 0.3°C), indicating that domain I in PsDppA is also a heat-labile domain compared to that in EcDppA. The CD measurement also showed that PsE1-2 (Tm of 51°C; see Fig. S5B in the supplemental material) and PsE1-3 (Tm of 55°C; see Fig. S5C in the supplemental material) had higher Tm values than PsDppA. Therefore, it could be deduced that domain I also makes a contribution to the high thermolability of PsDppA, although the replacement of domain I-1 could not produce soluble protein.
Ligand-binding affinity of domain-exchanged mutants.
The Kd values for domain-exchanged mutants at different temperatures were shown in Table 1. Mutants PsE2 and PsE1-3 both displayed the highest binding affinity at 15°C, like PsDppA, suggesting that domains II and I-3 in PsDppA contribute little to the high binding affinity of PsDppA at low temperatures. In contrast, PsE3 showed its highest binding affinity at 30°C, and PsE1-2 showed its highest binding affinity at 35°C. These results indicated that domains III and I-2 contribute significantly to the high binding affinity of PsDppA at low temperatures. In addition, domain I-2 is composed of two distinct parts: the α part (42 residues, named I-2α) containing two α-helices and one strand and the β part (36 residues, named I-2β) containing three-stranded β-sheets. Mutants PsE1-2α and PsE1-2β were expressed to replace these two parts, respectively, in PsDppA with those from EcDppA. PsE1-2α displayed the highest binding affinity at 30°C, while PsE1-2β showed no difference from PsDppA, which suggested that domain I-2α contributes significantly to the high binding affinity of PsDppA at low temperatures. Therefore, it was concluded that domains III and I-2α, located at the entrance of the binding cleft of PsDppA, play an important role in keeping the high ligand-binding affinity of PsDppA at low temperatures.
DISCUSSION
As a protein from a cold-adapted bacterium, PsDppA has a much lower optimal temperature for ligand binding (15°C) than its mesophilic homolog, EcDppA (35°C). PsDppA also has a much less stable structure than EcDppA. These results suggest that PsDppA might employ a cold adaptation strategy similar to that reported in many cold-adapted enzymes, i.e., increasing structural flexibility with decreasing structural stability as a tradeoff (7). Domain-exchanged mutation showed that a more stable domain is related to a higher optimal ligand-binding temperature. However, there is an exception in that PsE1-3 has a more stable domain I-3 than PsDppA but the same optimal ligand-binding temperature as PsDppA, suggesting that some structural optimization might be used to promote the kinetic binding rate at low temperature rather than the binding affinity.
The binding mechanism of DppA is referred to as the “Venus's flytrap” mechanism (15). Upon ligand binding, DppA turns from an open conformation into a closed conformation. The major conformational change during this state transition is that domain III bends toward domain I, with the two strands (around L262 and T479 in EcDppA) between domains III and I-2α as the hinge. As shown in Fig. 7 (thin line), this major conformational change is accompanied by conformational changes in many other flexible regions. It is speculated here that further flexibility optimization of these state transition-involved regions should contribute to the cold adaptation of DppA.
FIG. 7.

Local conformational change of EcDppA during state transition (thin line) and the residue size difference between PsDppA and EcDppA (thick line). PyMol (http://www.pymol.org/) was used to superpose the local structures of the open conformation (1DPE.pdb chain A) and the closed conformation (1DPP.pdb chain A). The residue size difference between PsDppA and EcDppA (ΔMw) was calculated as the average relative molecular masses of PsDppA residues minus those of EcDppA residues. A ΔMw of ±14 Da corresponds to one -CH2. Both local structural superposition and relative molecular mass were calculated by using a sliding window of 21 residues. EcDppA numbering was used. r.m.s.d., root mean square deviation.
As shown in some cold-adapted enzymes, small residues will promote the structural flexibility (26). To find out whether the residue size is favorable for the cold adaptation of PsDppA, PsDppA and EcDppA were compared by evaluating the relative molecular masses of the peptide segments with a sliding window of 21 residues. As shown in Fig. 7 (thick line), in domain III of PsDppA, the state transition-involved regions have smaller residue sizes, suggesting that these regions have more structural flexibility compared to those in EcDppA. In contrast, in other domains of PsDppA, the residues in the state transition-involved regions are not always smaller and, moreover, some regions not involved in the state transition have smaller residues, such as I-2α. Although the strand between I-2α and III (around L262 in EcDppA) undergoes a large conformational change during state transition, I-2α itself has very similar local conformations in both open and closed states. As shown in Fig. 7, I-2α has smaller residues in PsDppA compared to that in EcDppA. Comparison of the sequences and molecular masses of domain I-2α from different bacteria also showed that the overall relative molecular mass of I-2α correlates well with the optimal growth temperature of its source bacterium (see Fig. S6 in the supplemental material). I-2α from a bacterium with a lower optimal growth temperature has a lower relative molecular mass (see Fig. S6 in the supplemental material). In addition to decreasing residue size, other strategies are also used to further improve the flexibility of this domain. Analysis of amino acid composition showed that I-2α has fewer prolines in PsDppA (two prolines) than in EcDppA (five prolines). Comparison of the PsDppA model and the EcDppA crystal structure showed that I-2α has fewer salt bridges in PsDppA (one salt bridge [R260-D262]) than in EcDppA (three salt bridges [K228-E233, D244-R247, and E258-K249]). These structural characteristics favor the improvement of I-2α structural flexibility. Therefore, I-2α is a hotspot of optimization during the cold adaptation of PsDppA.
The mutation data could be explained by the distribution of residues with decreased size. The strong correlation between a decrease in residue size and conformational change in domain III suggests that domain III has been optimized to improve the flexibility of these regions for keeping the high binding affinity of PsDppA at a low temperature. This is in accordance with the result that PsE3 had a higher optimal ligand-binding temperature than PsDppA. The lack of such correlation in domain II is in agreement with the result that PsE2 had the same optimal ligand-binding temperature as PsDppA. Although the average residue size of domain I-3 of PsDppA is smaller than that of EcDppA, domain I-3 undergoes only small conformational changes during state transition and, therefore, PsE1-3 had the same optimal ligand-binding temperature as PsDppA. Domain I-2 is composed of two parts: I-2α and I-2β. The residue size in I-2α decreased, while that in I-2β increased, compared to that in EcDppA, suggesting that I-2α has been optimized to increase the flexibility of domain I-2. This analysis agreed with the result that PsE1-2α had a higher optimal ligand-binding temperature than PsDppA and that PsE1-2β the same optimal ligand-binding temperature as PsDppA.
Generally, the mutants had lower binding affinity than PsDppA, suggesting that the mutants are trapped in conformations suboptimal for the binding of peptide. This should be caused mainly by the suboptimal interactions at the interfaces between the EcDppA domain and the PsDppA domains in domain-exchanged mutants. In addition, as suggested by the residue size profile, the difference in side-chain size between PsDppA domain and EcDppA domain should play an important role. The PsE1-2α has a Kd similar to that of PsDppA, which might be a result of both the high flexibility of domain I-2α and the relatively large distance from the binding sites. It was also noted that domain III and domain I-2α function in a cooperative manner, and the mutant would not have a low optimal ligand-binding temperature if either domain in PsDppA was replaced by the corresponding domain from EcDppA.
Our results showed that the DppA from the deep-sea psychrophilic bacterium Pseudoalteromonas sp. strain SM9913 is a cold-adapted protein with low structural stability and high ligand-binding affinity at a low temperature, which can facilitate the dipeptide translocation for SM9913, surviving in the permanently cold deep sea. Domains III and I-2α at the entrance of the cleft have high structural flexibility and play an important role in efficient ligand binding of PsDppA at low temperatures. Sequence and structural analyses suggested that increasing the local flexibility by decreasing the residue size is an important strategy in the cold adaptation of PsDppA.
Supplementary Material
Acknowledgments
This study was supported by the National Natural Science Foundation of China (40876072, 30770040, and 30770062), the Hi-Tech Research and Development program of China (2007AA091903), and the COMRA Program (DYXM-115-02-2-6).
Footnotes
Published ahead of print on 7 May 2010.
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Aslund, F., M. Zheng, J. Beckwith, and G. Storz. 1999. Regulation of the OxyR transcription factor by hydrogen peroxide and the cellular thiol-disulfide status. Proc. Natl. Acad. Sci. U. S. A. 96:6161-6165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berntsson, R. P., M. K. Doeven, F. Fusetti, R. H. Duurkens, D. Sengupta, S. J. Marrink, A. M. Thunnissen, B. Poolman, and D. J. Slotboom. 2009. The structural basis for peptide selection by the transport receptor OppA. EMBO J. 28:1332-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blattner, F. R., G. Plunkett III, C. A. Bloch, N. T. Perna, V. Burland, M. Riley, J. Collado-Vides, J. D. Glasner, C. K. Rode, G. F. Mayhew, J. Gregor, N. W. Davis, H. A. Kirkpatrick, M. A. Goeden, D. J. Rose, B. Mau, and Y. Shao. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453-1462. [DOI] [PubMed] [Google Scholar]
- 4.Chen, X. L., B. B. Xie, J. T. Lu, H. L. He, and Y. Zhang. 2007. A novel type of subtilase from the psychrotolerant bacterium Pseudoalteromonas sp. SM9913: catalytic and structural properties of deseasin MCP-01. Microbiology 153:2116-2125. [DOI] [PubMed] [Google Scholar]
- 5.Chen, X. L., Y. Z. Zhang, P. J. Gao, and X. W. Luan. 2003. Two different proteases produced by a deep-sea psychrotrophic bacterial strain, Pseudoalteromonas sp. SM9913. Mar. Biol. 143:989-993. [Google Scholar]
- 6.Dunten, P., and S. L. Mowbray. 1995. Crystal structure of the dipeptide binding protein from Escherichia coli involved in active transport and chemotaxis. Protein Sci. 4:2327-2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feller, G., and C. Gerday. 2003. Psychrophilic enzymes: hot topics in cold adaptation. Nat. Rev. Microbiol. 1:200-208. [DOI] [PubMed] [Google Scholar]
- 8.Gibson, M. M., M. Price, and C. F. Higgins. 1984. Genetic characterization and molecular cloning of the tripeptide permease (tpp) genes of Salmonella typhimurium. J. Bacteriol. 160:122-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guyer, C. A., D. G. Morgan, N. Osheroff, and J. V. Staros. 1985. Purification and characterization of a periplasmic oligopeptide binding protein from Escherichia coli. J. Biol. Chem. 260:10812-10818. [PubMed] [Google Scholar]
- 10.Guyer, C. A., D. G. Morgan, and J. V. Staros. 1986. Binding specificity of the periplasmic oligopeptide-binding protein from Escherichia coli. J. Bacteriol. 168:775-779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harder, D., J. Stolz, F. Casagrande, P. Obrdlik, D. Weitz, D. Fotiadis, and H. Daniel. 2008. DtpB (YhiP) and DtpA (TppB, YdgR) are prototypical proton-dependent peptide transporters of Escherichia coli. FEBS J. 275:3290-3298. [DOI] [PubMed] [Google Scholar]
- 12.Higgins, C. F., and M. M. Hardie. 1983. Periplasmic protein associated with the oligopeptide permeases of Salmonella typhimurium and Escherichia coli. J. Bacteriol. 155:1434-1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lanfermeijer, F. C., F. J. Detmers, W. N. Konings, and B. Poolman. 2000. On the binding mechanism of the peptide receptor of the oligopeptide transport system of Lactococcus lactis. EMBO J. 19:3649-3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lanfermeijer, F. C., A. Picon, W. N. Konings, and B. Poolman. 1999. Kinetics and consequences of binding of nona- and dodecapeptides to the oligopeptide binding protein (OppA) of Lactococcus lactis. Biochemistry 38:14440-14450. [DOI] [PubMed] [Google Scholar]
- 15.Mao, B., M. R. Pear, J. A. McCammon, and F. A. Quiocho. 1982. Hinge-bending in l-arabinose-binding protein. The “Venus's-flytrap” model. J. Biol. Chem. 257:1131-1133. [PubMed] [Google Scholar]
- 16.Margesin, R., G. Neuner, and K. B. Storey. 2007. Cold-loving microbes, plants, and animals: fundamental and applied aspects. Naturwissenschaften 94:77-99. [DOI] [PubMed] [Google Scholar]
- 17.Médigue, C., E. Krin, G. Pascal, V. Barbe, A. Bernsel, P. N. Bertin, F. Cheung, S. Cruveiller, S. D'Amico, A. Duilio, G. Fang, G. Feller, C. Ho, S. Mangenot, G. Marino, J. Nilsson, E. Parrilli, E. P. Rocha, Z. Rouy, A. Sekowska, M. L. Tutino, D. Vallenet, G. von Heijne, and A. Danchin. 2005. Coping with cold: the genome of the versatile marine Antarctica bacterium Pseudoalteromonas haloplanktis TAC125. Genome Res. 15:1325-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Monnet, V. 2003. Bacterial oligopeptide-binding proteins. Cell. Mol. Life Sci. 60:2100-2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nickitenko, A. V., S. Trakhanov, and F. A. Quiocho. 1995. 2 Å resolution structure of DppA, a periplasmic dipeptide transport/chemosensory receptor. Biochemistry 34:16585-16595. [DOI] [PubMed] [Google Scholar]
- 20.Olson, E. R., D. S. Dunyak, L. M. Jurss, and R. A. Poorman. 1991. Identification and characterization of dppA, an Escherichia coli gene encoding a periplasmic dipeptide transport protein. J. Bacteriol. 173:234-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Payne, J. W., and M. W. Smith. 1994. Peptide transport by micro-organisms. Adv. Microb. Physiol. 36:1-80. [DOI] [PubMed] [Google Scholar]
- 22.Prinz, W. A., F. Aslund, A. Holmgren, and J. Beckwith. 1997. The role of the thioredoxin and glutaredoxin pathways in reducing protein disulfide bonds in the Escherichia coli cytoplasm. J. Biol. Chem. 272:15661-15667. [DOI] [PubMed] [Google Scholar]
- 23.Qin, G. K., L. Z. Zhu, X. L. Chen, P. G. Wang, and Y. Zhang. 2007. Structural characterization and ecological roles of a novel exopolysaccharide from the deep-sea psychrotolerant bacterium Pseudoalteromonas sp. SM9913. Microbiology 153:1566-1572. [DOI] [PubMed] [Google Scholar]
- 24.Sali, A., and T. L. Blundell. 1993. Comparative protein modeling by satisfaction of spatial restraints. J. Mol. Biol. 234:779-815. [DOI] [PubMed] [Google Scholar]
- 25.Sanz, Y., F. C. Lanfermeijer, W. N. Konings, and B. Poolman. 2000. Kinetics and structural requirements for the binding protein of the Di-tripeptide transport system of Lactococcus lactis. Biochemistry 39:4855-4862. [DOI] [PubMed] [Google Scholar]
- 26.Siddiqui, K. S., and R. Cavicchioli. 2006. Cold-adapted enzymes. Annu. Rev. Biochem. 75:403-433. [DOI] [PubMed] [Google Scholar]
- 27.Smith, M. W., D. R. Tyreman, G. M. Payne, N. J. Marshall, and J. W. Payne. 1999. Substrate specificity of the periplasmic dipeptide-binding protein from Escherichia coli: experimental basis for the design of peptide prodrugs. Microbiology 145:2891-2901. [DOI] [PubMed] [Google Scholar]
- 28.Tame, J. R., E. J. Dodson, G. Murshudov, C. F. Higgins, and A. J. Wilkinson. 1995. The crystal structures of the oligopeptide-binding protein OppA complexed with tripeptide and tetrapeptide ligands. Structure 3:1395-1406. [DOI] [PubMed] [Google Scholar]
- 29.Tame, J. R., G. N. Murshudov, E. J. Dodson, T. K. Neil, G. G. Dodson, C. F. Higgins, and A. J. Wilkinson. 1994. The structural basis of sequence-independent peptide binding by OppA protein. Science 264:1578-1581. [DOI] [PubMed] [Google Scholar]
- 30.Wang, X. G., J. M. Kidder, J. P. Scagliotti, M. S. Klempner, R. Noring, and L. T. Hu. 2004. Analysis of differences in the functional properties of the substrate binding proteins of the Borrelia burgdorferi oligopeptide permease (Opp) operon. J. Bacteriol. 186:51-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wurch, T., F. Lestienne, and P. J. Pauwels. 1998. A modified overlap extension PCR method to create chimeric genes in the absence of restriction enzymes. Biotechnol. Tech. 12:653-657. [Google Scholar]
- 32.Zhao, G. Y., X. L. Chen, H. L. Zhao, B. B. Xie, B. C. Zhou, and Y. Z. Zhang. 2008. Hydrolysis of insoluble collagen by deseasin MCP-01 from deep-sea Pseudoalteromonas sp. SM9913: collagenolytic characters, collagen-binding ability of C-terminal PKD domain and implication for its novel role in deep-sea sedimentary particulate organic nitrogen degradation. J. Biol. Chem. 283:36100-36107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.