

Abstract
Although propagation of Saccharomyces cerevisiae prions requires Hsp104 protein disaggregating activity, overproducing Hsp104 “cures” cells of [PSI+] prions. Earlier evidence suggests that the Hsp70 mutant Ssa1-21 impairs [PSI+] by a related mechanism. Here, we confirm this link by finding that deletion of STI1 both suppresses Ssa1-21 impairment of [PSI+] and blocks Hsp104 curing of [PSI+]. Hsp104's tetratricopeptide repeat (TPR) interaction motif was dispensable for curing; however, cells expressing Sti1 defective in Hsp70 or Hsp90 interaction cured less efficiently, and the Hsp90 inhibitor radicicol abolished curing, implying that Sti1 acts in curing through Hsp70 and Hsp90 interactions. Accordingly, strains lacking constitutive or inducible Hsp90 isoforms cured at reduced rates. We confirm an earlier finding that elevating free ubiquitin levels enhances curing, but it did not overcome inhibition of curing caused by Hsp90 defects, suggesting that Hsp90 machinery is important for the contribution of ubiquitin to curing. We also find curing associated with cell division. Our findings point to crucial roles of Hsp70, Sti1, and Hsp90 for efficient curing by overexpressed Hsp104 and provide evidence supporting the earlier suggestion that destruction of prions by protein disaggregation does not adequately explain the curing.
Saccharomyces cerevisiae prions are self-replicating misfolded forms of normal cellular proteins. They are believed to propagate as amyloid, which is a highly ordered fibrous aggregate. What triggers prion formation is uncertain, but in order to be maintained in an expanding yeast population, prions must grow, replicate, and be transmitted to daughter cells during cell division. Growth occurs when soluble protein joins the fiber ends and is converted into the prion form (30, 52, 58). Replication is associated with fragmentation of prion polymers, which generates new prions from preexisting material (37, 50). Transmission is believed to occur by passive diffusion of prions with cytoplasm (57).
Although it is uncertain to what extent cellular factors influence growth or transmission of prions, it is clear that the Hsp104 disaggregation machinery is necessary for prion replication (10, 17, 55, 70). Hsp104 is a hexameric AAA+ chaperone that protects cells from a variety of stresses by resolubilizing proteins from aggregates (24, 25, 53). With help from Hsp70 and Hsp40, it extracts monomers from aggregates and extrudes them through its central pore (24, 41, 68). This machinery could act in prion replication by extracting monomers from amyloid fibers (29, 68), which would destabilize the fibers, causing them to break into more numerous pieces that each can continue to propagate the prion.
Paradoxically, overexpressing Hsp104 very efficiently “cures” cells of the [PSI+] prion, which is composed of the translation termination factor Sup35 (10). A widely held view of this curing is that elevating the cellular protein disaggregation activity causes complete destruction of prions. However, elevating Hsp104 has little or no effect on most other amyloidogenic prions (15, 16, 38, 47, 54, 66), although it can be inferred to cure [MCA] prions in cells also propagating a prion of an Mca1-Sup35 fusion (49). Together, these results suggest that prions of Sup35, and perhaps those of Mca1, are particularly sensitive to Hsp104 disaggregation activity. Alternatively, something in addition to or other than a simple increase in protein disaggregation is involved in the curing.
Although protein disaggregation activity of Hsp104 is required for both thermotolerance and prion propagation, we and others have identified mutations in Hsp104 that affect these processes separately (27, 32, 39, 60). The ability of Hsp104 to thread proteins through its central pore, however, is required for both processes (29, 41, 68), so this distinction in Hsp104 function could be due to differences in how Hsp104 interacts with amorphous aggregates of thermally denatured proteins and highly ordered prion aggregates or with cofactors that interact with the different prions as substrates. In any scenario, efficiency and specificity of Hsp104 function are affected by interactions with other components of the disaggregation machinery, in particular the Hsp70s and Hsp40s, which are believed to interact first with substrates to facilitate action of Hsp100 family disaggregases (2, 71, 72).
Increasing expression of either ubiquitin (Ub) or Ssb, an Hsp70 that has roles in protein translation and proteasome degradation, enhances Hsp104 curing of [PSI+] (3, 11, 12). Predictably, reducing expression of either of them reduces curing efficiency. The mechanisms underlying these effects are unknown, but the combined effects of Ssb and Ub are additive, suggesting that they act in different pathways. The role of Ub is indirect, as Sup35 is neither ubiquitylated nor degraded during curing. Whether other chaperones are involved in the effects of Ub on curing has not been investigated.
Earlier we isolated a mutant of the Hsp70 Ssa1, designated Ssa1-21, that weakens and destabilizes [PSI+] propagation (33). We later isolated several Hsp104 mutants that suppress this antiprion effect (29). The Hsp104 mutants retain normal functions in thermotolerance, protein disaggregation, and prion propagation, but when overexpressed, they are unable to cure [PSI+], even in wild-type cells. These findings argue against a specific hypersensitivity of [PSI+] to disaggregation and support the notion that something distinct from or in addition to complete destruction of prions is involved in the curing. They also imply that Ssa1-21 and elevated Hsp104 inhibit [PSI+] prions by similar mechanisms. A prediction from this conclusion is that other suppressors of Ssa1-21 will also inhibit curing of [PSI+] by overexpressed Hsp104. Indeed, we find here that alterations that suppress Ssa1-21 inhibition of [PSI+] do interfere with curing of [PSI+] by overexpressed Hsp104. We also provide evidence that Hsp90 has a critical role in this curing and that the ability of Ub to enhance curing depends on proper function of Hsp90 machinery.
MATERIALS AND METHODS
Strains, media, and plasmids.
Yeast strains are all derived from 779-6A (mata kar1-1 SUQ5 ade2-1 his3Δ202 leu2Δ1 trp1Δ63 ura3-52) (34). Deletions of HSP104, HSP82, HSC82, and STI1 were created by transforming strain 779-6A by using KanMX disruption cassettes that were PCR amplified from yeast deletion strains (ATCC) (31). 1/2YPD (0.5% yeast extract, 2% peptone, 2% glucose) is a rich medium that contains a limiting but undefined amount of adenine. YPAD is similar but contains 1% yeast extract and excess (400 mg/liter) adenine. SD (synthetic defined) medium contains 2% glucose, 7 g/liter yeast nitrogen base (YNB; Difco), and the appropriate nutritional supplements to maintain selection for plasmids and prions. Cells were grown at 30°C unless indicated otherwise.
Plasmids pRS313, pRS314, pRS315, and pRS316 are single-copy HIS3-, TRP1-, LEU2-, and URA3-based vectors, respectively (65). Plasmid pMR26, for overexpressing Hsp104, is pRS314 with HSP104 driven by the CUP1 promoter. It was constructed by inserting a BamHI-SacI fragment containing HSP104 plus a 130-bp 3′ sequence from pGCH17 (29) into p316CupNGMC digested by the same enzymes. Plasmid p316CupNGMC is pRS316 with a SUP35-GFP gene encoding the fusion protein NGMC under the control of the CUP1 promoter (62). Plasmid pMR80 (PCUP1::HSP104ΔN147) was constructed similarly by the use of pGCH23 (29) as the source of HSP104ΔN147. Plasmid pMR81 (PCUP1::HSP104ΔC4) was constructed by performing site-directed mutagenesis on pMR26 to change Hsp104 codons 905 and 906 to TAA. Plasmid pMR40 is pRS314 with Hsp104ΔC4 under the control of its own promoter. Plasmid pMR115 is pRS316 with SSE1 and 500 bp of 5′- and 3′-flanking DNA on a BamHI fragment. Plasmid pC210 for expressing Ssa1 was described earlier (61).
Plasmids containing STI1, sti1(C49Y), sti1ΔTPR1, sti1(G325D), sti1(T526I), and sti1ΔTPR2 were described previously (67). Plasmid pMR86 is pRS416 containing the GPD promoter, a His6 tag for N-terminal fusion, and the CYC1 terminator. It was constructed by first annealing oligonucleotides to create a 6HIS sequence flanked by SpeI and BamHI. This was inserted into XbaI/BamHI-digested p416-GPD (48). STI1 and the sti1(C49Y), sti1(G325D), and sti1(T526I) mutant genes were PCR amplified and inserted into BamHI-XhoI-digested pMR86 to give pMR87 through pMR90, respectively. Plasmid pMR29 is a multicopy HIS3-based vector (pRS423 [65]) containing UBI4 plus a 500-bp 5′ and 3′ sequence inserted in the BamHI site.
Monitoring [PSI+].
The [PSI+] prion propagates as self-replicating aggregates of the translation termination factor Sup35. Our ade2-1 strains cannot grow without exogenous adenine and are red when adenine is limiting due to the accumulation of a metabolite of the adenine biosynthetic pathway. When [PSI+] is present, depletion of Sup35 into prion aggregates causes reduced translation termination efficiency, allowing suppression of ade2-1 by the SUQ5 tRNA in our strains (14), which confers adenine prototrophy and white colony color. When [PSI+] propagation is weakened, less Sup35 is depleted, and cells display an intermediate (pink) colony color and weak or temperature-sensitive growth without adenine. Unstable [PSI+] propagation (i.e., loss of [PSI+] during mitosis) is seen as an appearance of red [psi−] clones in a population of [PSI+] cells.
Prion curing by Hsp104 overexpression.
The most commonly used method of overexpressing Hsp104 is galactose induction, but deleting Sti1 delays galactose-induced transcription (20). We therefore used copper (100 μM CuSO4) to activate the CUP1 promoter for Hsp104 overexpression. Copper induced Hsp104 efficiently and to similar degrees in all our strains (see Results). Hsp104, Hsp104ΔN147, and Hsp104ΔC4 were overexpressed using plasmids pMR26, pMR80, and pMR81, respectively. Cells containing the overexpression plasmid were first grown overnight in SD (made with copper-free YNB) without tryptophan or adenine to select for the plasmid and for [PSI+], which safeguards against potential leakiness of the CUP1 promoter. Starter cultures were diluted into SD lacking tryptophan and containing 400 mg/liter adenine and 100 μM CuSO4 to an optical density at 600 nm (OD600) of 0.05 and incubated at 30°C with shaking. Cells were diluted into fresh medium to maintain logarithmic growth. Growth of cultures was monitored by OD600s, and in all experiments, generations were defined as doublings of OD600. Periodically, culture aliquots were diluted and spread onto 1/2YPD to quantify culture density and monitor the appearance of [psi−] (entirely red) colonies. Most experiments were carried out through four to six generations because the interval where the rate of curing was most linear was between cell divisions 2 and 8.
Analysis of Sti1-interacting proteins.
Sti1-containing complexes were isolated as described previously (21). Briefly, sti1Δ cells carrying His6-tagged STI1 wild-type or mutant alleles on plasmids pMR87 to pMR90 were grown overnight in SD medium lacking uracil to an OD600 of 2. Cells were harvested, suspended in lysis buffer, and broken by agitation with glass beads. Equal amounts of total protein, determined using the bicinchoninic acid (BCA) assay (Pierce), were incubated with Ni-nitrilotriacetic acid (NTA) resin and washed first with lysis buffer and then extensively with lysis buffer plus 0.1% Tween 20 and 35 mM imidazole. Sti1-containing complexes were eluted with lysis buffer plus 500 mM imidazole and analyzed by Coomassie staining/SDS-PAGE and Western blotting for Sti1 and Hsp90.
Western analysis.
Cells collected by centrifugation were suspended in 100 mM Tris-HCl (pH 6.8), 4% SDS, and 10% glycerol and broken by agitation with glass beads for 1 min. Lysates were then boiled for 10 min in a water bath and cleared by centrifugation, and total protein concentrations were determined using the BCA assay (Pierce). Ten milligrams of total protein was separated on 4 to 20% Tris-HCl Criterion SDS-PAGE gels (Bio-Rad) and transferred to polyvinylidene difluoride (PVDF) membranes, which were probed with antibodies to Sti1, a gift from David Toft (Mayo Clinic, MN), or Hsp104, a gift from John Glover (University of Toronto). Hsp90 was detected using a C-terminal-specific antibody (a gift from David Toft), which reacts with both Hsc82 and Hsp82, and Stressgen antibody SPA-840, which reacts only with Hsp82. Hsp70 antibody was from Stressgen (SPA-822), and Sse1 antibodies were gifts from Jeff Brodsky (University of Pittsburgh) (28) and Bernd Bukau (University of Heidelberg) (56).
RESULTS
Mutations that suppress Ssa1-21 inhibition of [PSI+] also suppress Hsp104 curing of [PSI+].
We monitor [PSI+] in our strains by red/white colony color on media with limiting adenine (see Materials and Methods). In this system, [psi−] cells are red, and [PSI+] cells are white. Transient overexpression of Hsp104 causes cells to lose [PSI+] so that red [psi−] colonies appear in [PSI+] populations (10) (Fig. 1A). The Hsp70 mutant Ssa1-21 inhibits [PSI+] propagation, causing a weak and unstable prion phenotype, seen as a pink color of [PSI+] colonies, and frequent appearance of red [psi−] colonies (33) (Fig. 1A, lower center panel). We previously identified several mutations in the amino-terminal domain of Hsp104 that do not affect its normal functions in prion propagation or protein disaggregation but restore wild-type [PSI+] propagation in cells expressing Ssa1-21 (29). These Hsp104 mutants are also defective in curing [PSI+] when overexpressed, suggesting that Ssa1-21 impairs [PSI+] by a mechanism related to that caused by elevating Hsp104. To test this interpretation, we determined if other alterations that suppress prion inhibition by Ssa1-21 also interfere with the curing of [PSI+] by overexpression of Hsp104.
FIG. 1.
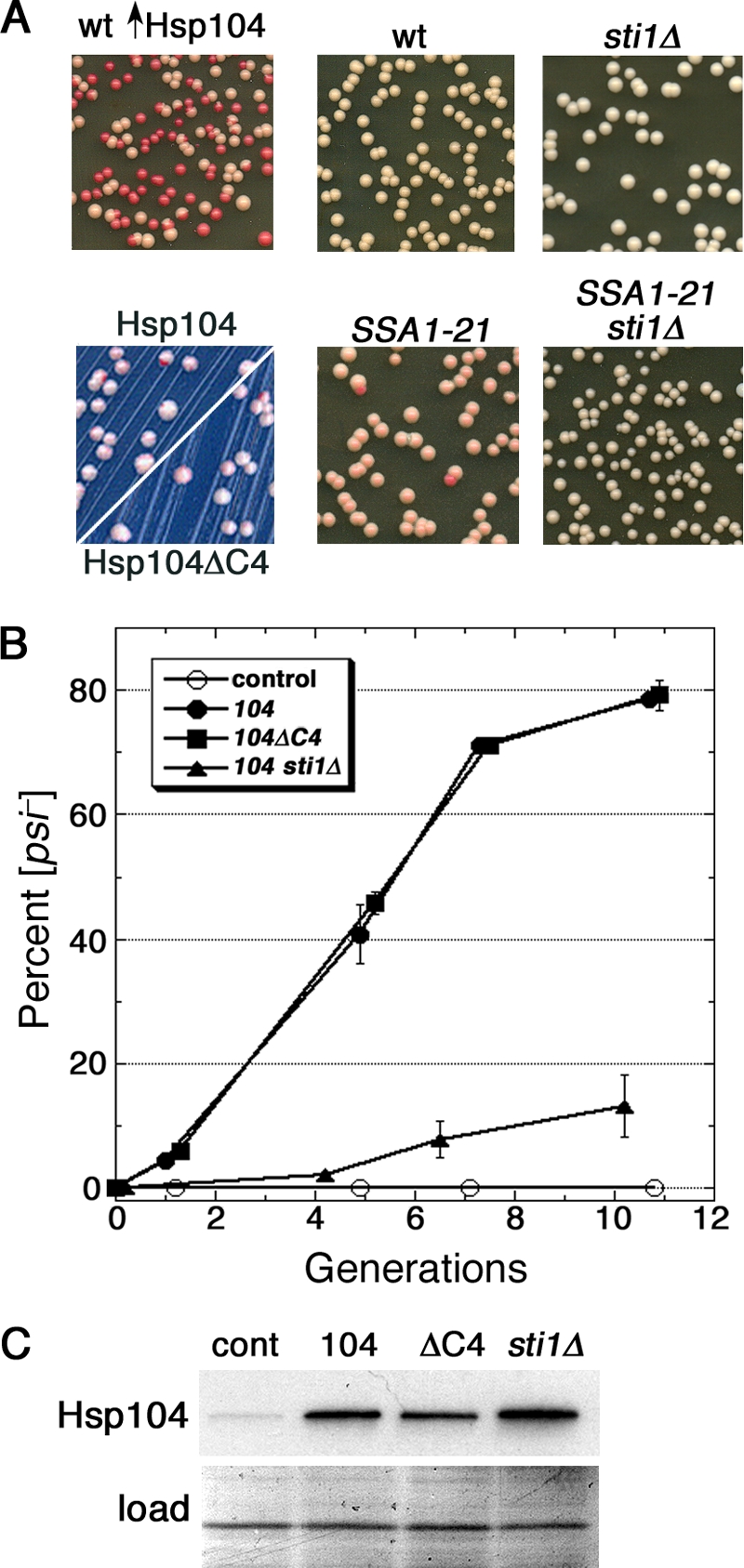
Sti1 mediates Ssa1-21 inhibition of [PSI+] and is critical for curing of [PSI+] by Hsp104 overexpression. (A) The upper left panel shows wild-type (wt) cells that overexpressed Hsp104 for ∼5 generations and then were spread onto 1/2YPD plates and grown for 2 to 3 days at 30°C. White colonies are [PSI+], and red colonies arose from cells that lost [PSI+]. The lower left panel shows hsp104Δ cells expressing Hsp104 or Hsp104ΔC4 (as indicated) from a plasmid, which produces roughly 3-fold more Hsp104 than chromosomally expressed Hsp104, grown similarly but on defined medium with limiting (8 mg/liter) adenine. Instability of [PSI+] caused by the modestly elevated Hsp104 is detectable as red sectoring in otherwise white colonies. Overall color and sectoring frequency of cells expressing the different Hsp104 proteins are indistinguishable. Wild-type and SSA1-21 mutant cells (center panels) or the same combined with deletion of STI1 (sti1Δ; right panels) were spread onto 1/2YPD and incubated as described above to illustrate [PSI+] phenotypes. All panels are representative 2.5-cm squares from plates, except those on the lower left, which are magnified ×2 relative to others. Images were made by scanning plates by the use of an HP ScanJet 6300C document scanner. Color and contrast of the assembly of images were optimized using Adobe Photoshop 7.0 software. (B) Appearance of [psi−] cells in growing cultures is shown as a function of generations (monitored as doubling of OD600). All cultures were grown in medium containing 100 μM CuSO4 to induce expression from the CUP1 promoter and then spread onto 1/2YPD plates and incubated as described above. Wild-type cells carried plasmids without HSP104 (control), with wild-type HSP104 (104), or with HSP104ΔC4 (104ΔC4). The sti1Δ cells over-express wild-type HSP104. To maintain continuous growth, cultures were diluted into fresh medium when the OD600 reached 1 to 2. No [psi−] colonies appeared among over 900 colonies from the control cultures at any of the time points. Values are averages of the results for three independent experiments ± standard error of the mean (SEM). (C) Western analysis of the abundance of Hsp104 and Hsp104ΔC4 in cells used for the experiment in panel B from cultures grown for roughly 10 generations. Bottom panel (load) shows a portion of the membrane used for the blot stained by amido black as a loading and transfer control.
We showed earlier that deleting the tetratricopeptide repeat (TPR)-containing Hsp70/Hsp90 cochaperone Sti1 (Hop1 in mammals) has little effect on [PSI+] in wild-type cells but restores a normal [PSI+] phenotype in SSA1-21 cells, a finding confirmed here (Fig. 1A, right panels). Sti1 interacts with both Hsp70 and Hsp90 to bridge them and facilitates substrate transfer from Hsp70 to Hsp90 in a folding pathway for a variety of client proteins (9, 67). Here, we find that deleting STI1 caused a considerable inhibition in the curing of [PSI+] by overexpressed Hsp104 (Fig. 1B and Table 1). The reduced curing of sti1Δ cells was not due to reduced Hsp104 expression (Fig. 1C and 2 C). These data show that Sti1 is required for efficient prion curing.
TABLE 1.
Rates of Hsp104 curing of [PSI+]a
| Strain | % [psi−] cells/generation |
|---|---|
| Wild type plus empty vector | <0.001 |
| Wild type plus Hsp104 | 9.0 ± 0.3 |
| Wild type plus Hsp104ΔC4 | 8.9 ± 0.6 |
| sti1Δ mutant plus Hsp104 | 0.2 ± 0.2 |
| hsc82Δ mutant plus Hsp104 | 0.3 ± 0.2 |
| hsp82Δ mutant plus Hsp104 | 4.1 ± 0.2 |
Hsp104 (or Hsp104ΔC4 where indicated) proteins were overexpressed from copper-inducible alleles on pMR26 and pMR81, respectively. Cells removed periodically from cultures grown for four to six generations were spread onto 1/2YPD to determine the proportion of [psi−] (entirely red) colonies. At each time point, 500 to 1,500 colonies were assessed. Values are averages of the results for at least three experiments ± standard deviation (n = 3 to 19). Generations were measured as doublings of culture OD600. No [psi−] colonies were detected in cultures that did not overexpress Hsp104 (first row).
FIG. 2.
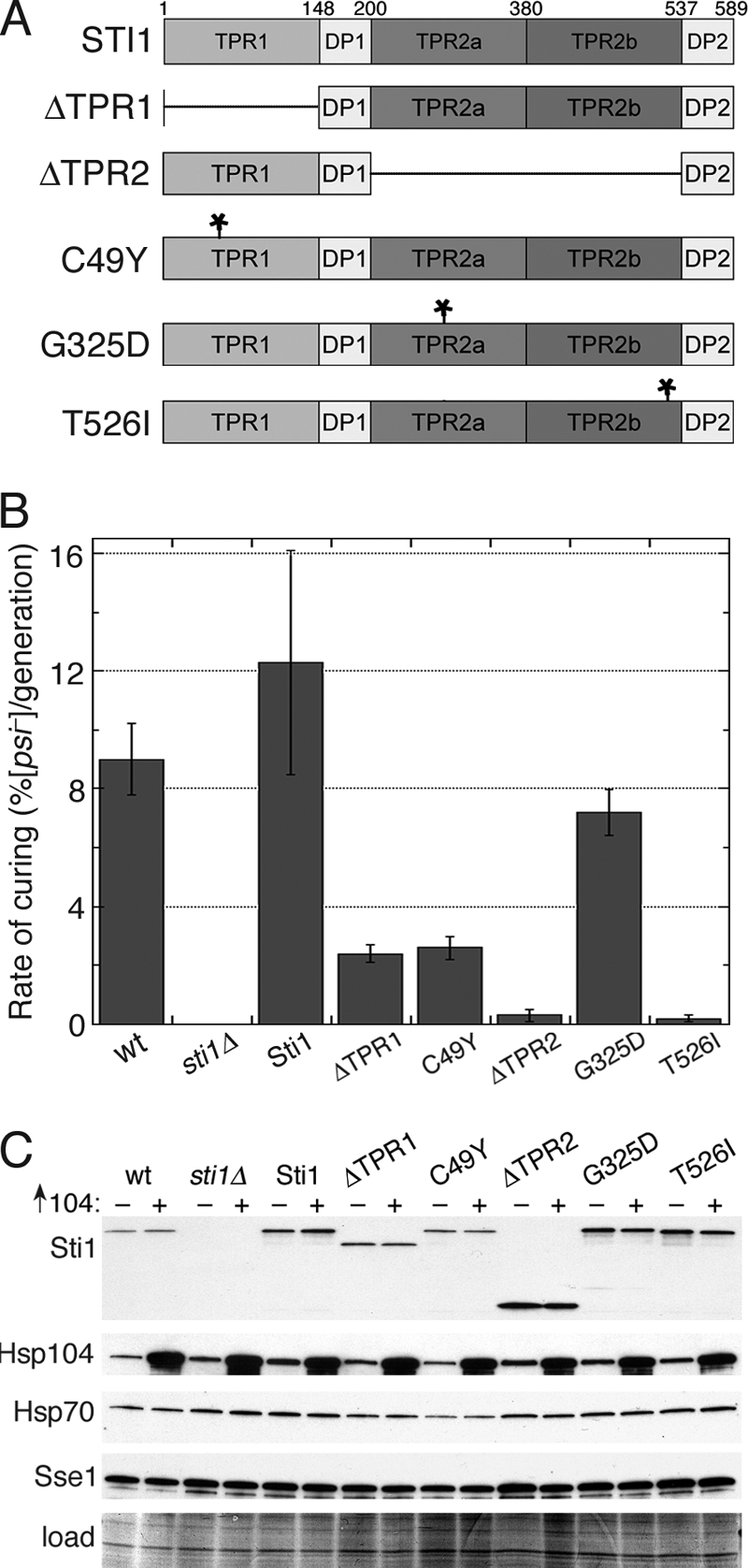
Both TPR domains of Sti1 are important for prion curing by overexpressed Hsp104. (A) Domain structure of Sti1 and mutants used in this study. Numbers above the diagram indicate the amino acid residue position; asterisks indicate the locations of point mutations indicated on the left. Deleted regions are indicated with horizontal lines. DP, aspartate-proline region. (B) Rates of prion curing were determined in cultures overexpressing Hsp104 for four or five generations. Strains to the right of the wild type (wt; carries empty vector) are sti1Δ with an empty vector or a vector encoding the indicated Sti1 protein. Values are averages of the results for three independent experiments ± standard deviation. (C) Western analysis of abundance of proteins indicated on the left in cells from experiments shown in panel B that were grown for four generations. All strains were exposed to copper and carry either an empty vector (−) or pMR26 for expression of Hsp104 (+), as indicated above each lane. Lower panel (load) is a portion of the blotted membrane stained by amido black as a loading and transfer control.
While our paper was in revision, a paper reporting a similar conclusion was published (46). There are two differences between our two studies. Although the strains they used originated in our laboratory and are isogenic to ours, their sti1Δ deletion strain carried a disruption allele that apparently was constructed separately. Additionally, that study used galactose induction for overexpressing Hsp104. We used copper induction because galactose-induced transcription from the galactose promoter is delayed in sti1Δ cells (20). Apparently, this delay does not significantly influence prion curing, and depleting Sti1 inhibits curing similarly when Hsp104 expression is induced with galactose (46). Together, our independent findings corroborate the conclusion that Sti1 plays an important role in curing of [PSI+] by Hsp104 overexpression.
The TPR interaction motif of Hsp104 is dispensable for [PSI+] curing.
A conserved carboxy-terminal acidic motif (EEVD) on Hsp70 and Hsp90 mediates their physical interactions with TPR-containing cochaperones (59). Hsp104 contains a similar carboxy-terminal motif (DDLD) that mediates physical interactions with Sti1 and other TPR-containing Hsp90 cochaperones (1) but is dispensable for its function in thermotolerance (42). To determine if the effects of TPR proteins on the curing were through direct interactions with Hsp104, we overexpressed Hsp104 that lacks the DDLD motif, designated Hsp104ΔC4. Hsp104ΔC4 cured [PSI+] like wild-type Hsp104 did (Fig. 1B and Table 1), indicating that a direct interaction between the Hsp104 C-terminal motif and TPR cochaperones is not required for, nor detectably influences, the curing of [PSI+]. This was not surprising since the interaction of Hsp104 with Sti1 and other TPR-containing cochaperones in vivo is detected only when cells are grown on a nonfermentable carbon source (i.e., conditions requiring respiration [1]), which we do not use here. We further found that cells lacking chromosomal HSP104 and expressing wild-type Hsp104 or Hsp104ΔC4 from the HSP104 promoter on a single-copy plasmid propagated [PSI+] similarly (Fig. 1A, lower left panel), which indicates that the C-terminal motif is not necessary for Hsp104 function in prion propagation and implies that Hsp104ΔC4 cures prions by the same mechanism as that used by Hsp104 rather than by inactivating endogenous Hsp104.
Both TPR domains of Sti1 are required for efficient curing of [PSI+] by excess Hsp104.
Fig. 2A shows a diagram of the domain structure of Sti1, which has different TPR domains that mediate interactions with different chaperones. TPR1 interacts with Ssa1 and Hsp104, and the longer TPR2 domain, which is subdivided into TPR2a and TPR2b, mediates physical and functional interactions primarily with Hsp90 (1, 21, 22, 67). By assessing various domain deletions and point mutations of Sti1, we earlier showed that Sti1 can regulate Hsp70 and Hsp90 separately in vivo (67). In this system, Sti1 lacking TPR2 (Sti1ΔTPR2) can regulate Ssa1 effects on [PSI+], but it cannot regulate essential Hsp90 functions. In contrast, Sti1 lacking TPR1 (Sti1ΔTPR1) does not regulate Ssa1 effects on [PSI+] but can regulate essential functions of Hsp90. This earlier work shows that it is possible to alter TPR1 and TPR2 of Sti1 in ways that specifically affect functions of Ssa1 and Hsp90, respectively.
We expressed wild-type and mutant STI1 alleles from plasmids in sti1Δ cells to determine if specifically inhibiting the ability of Sti1 to regulate Hsp70 or Hsp90 affected curing. Cells expressing Sti1ΔTPR1 or Sti1(C49Y), which have impaired regulation of Hsp70, were cured by overexpressed Hsp104 at a rate 6-fold lower than were cells expressing wild-type Sti1 (Fig. 2B). The rate of curing of [PSI+] in cells expressing Sti1ΔTPR2, which interacts normally with Hsp70 with regard to [PSI+] propagation, was reduced 40-fold (Fig. 2B), pointing to an even more prominent role for Hsp90 in the curing. All of the Sti1 mutants were expressed at levels similar to or above that of Sti1 in wild-type cells, and Hsp104 was induced to similar levels in all the strains (Fig. 2C). To address the role of TPR2 in more detail, we tested curing of cells expressing Sti1 mutants Sti1(G325D) (in TPR2a) and Sti1(T526I) (in TPR2b), which have no effect on [PSI+] but disrupt the ability of Sti1 to regulate Hsp90 function (67). Cells expressing Sti1(G325D) and Sti1(T526I) were cured roughly 2- and 40-fold less efficiently, respectively, than those expressing wild-type Sti1 (Fig. 2B). These results are consistent with our earlier data showing that G325D and T526I had the same proportional effect on essential Hsp90 function (67). They also indicate that TPR2 function with regard to curing is destroyed by the T526I substitution and show that while Sti1-Hsp90 interaction is dispensable for [PSI+] propagation, it is crucial for the curing of [PSI+] by Hsp104.
Elevating abundance of a variety of chaperones and cochaperones can weaken or destabilize prions, but only Sse1 (an Hsp70 regulator), and to a lesser extent the Hsp70s Ssa1 to Ssa4, inhibit Hsp104 curing of [PSI+] (4, 19, 51, 56). We compared the amounts of Sse1 and overall Hsp70 expressed in our strains to determine if any of the inhibitory effects we observed might be due to induction of high levels of these chaperones. The levels of these proteins were similar to that of the wild type, although they seemed to vary somewhat (Fig. 2C). Since it was reported that a roughly 50% increase in Ssa1 reduced curing in typical [PSI+] cultures from 43% cured to 16% cured (51), even small variations could be meaningful. Some differences in abundance, however, were opposite of what would be predicted on the basis of effects on curing. For example, although wild-type cells looked to have more of both Hsp70 and Sse1 than did cells expressing Sti1(C49Y), they cured more efficiently. Thus, although increases in abundance of Sse1 or Hsp70 probably contributed to some of the reduction in curing, the large reductions in curing we see do not appear to be explained adequately just by differences in expression of these factors known to inhibit Hsp104 curing of [PSI+].
With regard to effects on curing, it is difficult to know how meaningful the small differences we see in the abundances of Hsp70 and Sse1 might be, because none of the earlier studies monitoring Hsp104 curing present similar blotting data to indicate the degree to which induced Sse1 or Ssa1 were elevated. To get a better idea of how levels of Ssa1 and Sse1 correlate with inhibition of curing, we tested curing of cells coexpressing Sse1 and Ssa1. Cells coexpressing Ssa1 had an increase in Hsp70 abundance that was clearly higher (Fig. 3) than that of cells lacking Sti1 or expressing mutated Sti1 (Fig. 2C). Nevertheless, these cells had only a 3-fold reduction in curing rate (Fig. 3). Similarly, the roughly 10-fold reduction in curing of the cells coexpressing Sse1 was accompanied by an increase in Sse1 abundance that also was clearly greater than that seen for any of the Sti1 mutants. The Sse1-overexpressing cells also had a level of Ssa1 comparable to that of cells overexpressing Ssa1. Thus, the reduction in the curing of Sti1 mutants is not explained simply by increases in Ssa1 or Sse1. These data are in line with our interpretation that Sti1 regulation of Hsp70 and Hsp90 plays a significant role in the curing of [PSI+] by overexpressed Hsp104.
FIG. 3.
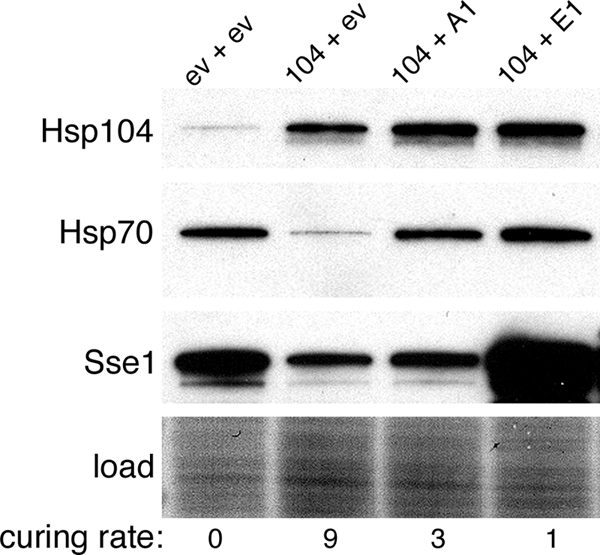
Curing of cells coexpressing Ssa1 or Sse1. Curing was done as described in the legend for Fig. 1, using cells carrying plasmids with HSP104 (104), SSA1 (A1), and SSE1 (E1) or the corresponding empty vectors (ev). Western analysis probing for the chaperones indicated on the left was done using cultures grown for four generations. Numbers below the image indicate rates of prion curing (%[psi−] per generation) for the strains labeled directly above.
Another observation from this experiment was that in the absence of Ssa1 or Sse1 coexpression, overexpressing Hsp104 caused a reduction in abundance of Ssa1 and Sse1. This effect was a general one that we noticed consistently in our experiments. Since Ssa1 and Sse1 promote [PSI+] appearance and propagation (19, 51), repressed expression of these factors might contribute to the curing mechanism.
Sti1 mutations affect Hsp90 interaction.
To determine if the Sti1 mutations affecting Hsp90 function altered physical interaction of Sti1 with Hsp90, we evaluated the amount of Hsp90 that copurified with Sti1 (Fig. 4 A). In line with the genetic results here and in the earlier study (67), both the G325D and T526I mutations reduced Sti1-Hsp90 physical interaction. Unexpectedly, although the T526I mutation caused a greater inhibition of curing than did G325D, it did not cause a greater reduction of the Sti1-Hsp90 interaction. This discrepancy might mean that T526I is more important than G325D for functional consequences of the Sti1 interaction or that it is involved in an Hsp90-independent Sti1 function important for the curing. Together, these data show that these Sti1 mutations disrupted physical interactions with Hsp90.
FIG. 4.
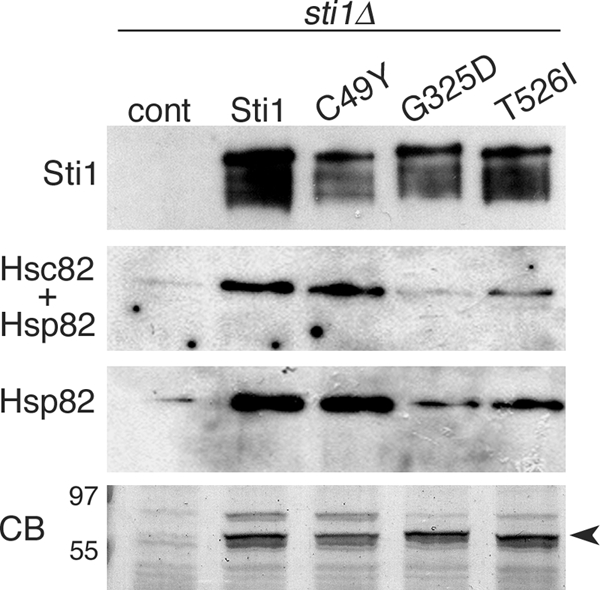
Sti1 mutations that suppress Hsp104-mediated curing affect interaction with Hsp90. Cells lacking chromosomal STI1 and carrying empty vector (cont) or plasmids expressing His6-tagged Sti1, Sti1(C49Y), Sti1(G325D), or Sti1(T526I) were grown as indicated in Materials and Methods. Sti1-containing complexes were purified from lysates by using metal affinity chromatography. Complexes were separated by SDS-PAGE, blotted, and probed with antibodies specific to Sti1 (top panel), an antibody raised against the C terminus of yeast Hsp90 that reacts with both Hsc82 and Hsp82 (Hsc82 + Hsp82) and a commercially available antibody raised against human Hsp90α that recognizes yeast Hsp82 only (Hsp82). The bottom panel (CB) shows a portion of a Coomassie brilliant blue-stained gel of the purified complexes, with molecular size markers indicated. Identical volumes of samples were loaded in this gel and in the gels used for the blots. The arrowhead indicates the position of Sti1.
We were unable to detect interactions between Sti1 and Hsp70 by this method so we used the alternative approach of purifying Ssa1 and assessing the amount of wild-type or mutant Sti1 that copurified. This method also was unsuccessful. Apparently, Sti1-Hsp70 interactions in our strains were not strong enough to survive the purification conditions. Although our results do not allow us to conclude whether the C49Y substitution influences the physical interaction of Sti1 with Hsp70, this mutation did not affect Hsp90 interaction (Fig. 4), and our previous data demonstrate that it greatly disrupts essentially all TPR1-mediated functional interaction between Sti1 and Hsp70 in vivo (67).
Inhibiting Hsp90 blocks Hsp104 curing of [PSI+].
Although reducing Hsp90 abundance does not noticeably affect [PSI+] propagation (31, 67) and overexpressing Hsp90 does not affect [PSI+] propagation or curing of [PSI+] by overexpressed Hsp104 (51), evidence that Hsp90 is important for eliminating [PSI+] when Hsp104 is overexpressed is lacking. Since depleting both constitutive (Hsc82) and inducible (Hsp82) isoforms is lethal, we used radicicol, a selective inhibitor of Hsp90 ATPase activity, to test for such an involvement. Exposing [PSI+] cells to radicicol at concentrations ranging from 1 to 200 μM has no effect on [PSI+] stability (31). At a concentration of 25 μg/ml (70 μM), radicicol has little effect on the growth of our wild-type cells but markedly reduces growth of cells with compromised Hsp90 machinery function, such as those lacking Sti1 (Fig. 5 A).
FIG. 5.

Hsp90 function is important for curing of [PSI+] by overexpressed Hsp104. (A) Sensitivity of strains (all are [PSI+]) to the Hsp90-specific inhibitor radicicol. Overnight YPAD cultures were diluted to an OD600 of 1.0, and 5-fold serial dilutions were spotted onto plates without and with (+rad) 25 μg/ml radicicol. Plates were scanned after incubation for 3 days at 30°C. (B) Radicicol blocks curing of [PSI+] by overexpressed Hsp104. Wild-type cells with plasmid for copper-inducible overexpression of Hsp104 were grown under inducing conditions (100 μM CuSO4) for 1.5 generations, at which time (“add” arrow) the overexpressing culture was split. One culture (closed circles) was untreated, while radicicol (25 μg/ml final concentration) was added to the other (closed triangles). After growing for another 3.5 generations (“remove” arrow), the radicicol-treated culture was washed, suspended in fresh radicicol-free medium, and grown for another 2.5 generations. Cultures grown without CuSO4 and either lacking (open circles) or containing (open triangles) radicicol were monitored in parallel. Values are averages of the results for three independent experiments ± standard deviation. (C) Western analysis of chaperones (indicated on left) in cells used in a replicate of the experiment shown in panel B. The “load” image shows a portion of the blotted membrane stained by amido black. (D) Western analysis of Hsp90 and other chaperones in hsc82Δ and hsp82Δ cells used in curing experiments. The blot in the top image is probed with an antibody that recognizes both isoforms of Hsp90 (Hsc82 + Hsp82), while the panel beneath it is an identical blot probed with a different antibody reactive only to the inducible Hsp82 isoform (see also Fig. 3). The lower panels show blots probed for Hsp104, Hsp70, and Sse1, as indicated. All cultures contained 100 μM CuSO4; control cells (cont) carry the empty vector.
When radicicol was included at this concentration in wild-type cultures overexpressing Hsp104, cell growth was minimally affected, indicating that elevating Hsp104 does not alter radicicol sensitivity, but curing of [PSI+] was completely blocked (Fig. 5B). The effect was fully reversible, as curing was rapidly restored to a normal rate when cells were subsequently transferred to medium without radicicol (Fig. 5B, “remove” arrow). The inhibitory effect of radicicol was not due to reduced expression of Hsp104 (Fig. 5C). Radicicol caused an increase in expression of Hsp104, Hsp70, and Sse1 (Fig. 5C, lane 2), but again, overexpressing Hsp104 moderated this effect somewhat, so the level of Sse1 was not as high when radicicol was present in Hsp104-overexpressing cells (Fig. 5C, compare lanes 3 and 4). The radicicol-induced increase was clearly less than that of cells overexpressing Sse1 from a plasmid (Fig. 3) from which [PSI+] was cured more efficiently. Thus, even if the increased amount of Sse1 caused by radicicol contributed to the inhibition of curing, it does not explain the complete inhibition caused by radicicol. These results also are in line with our other data that indicate that Hsp90 plays a role in the curing of [PSI+] by overexpression of Hsp104.
As in mammalian cells, S. cerevisiae cells bear both constitutive (Hsc82) and inducible (Hsp82) Hsp90 isoforms. Deleting HSC82 causes a greater reduction in overall abundance of Hsp90 than does deleting HSP82 (8), an effect we see in our strains (Fig. 5D, upper panel). Even though hsc82Δ cells had less overall Hsp90 (as Hsp82) than did wild-type cells, they grew better on plates containing radicicol (Fig. 5A). Thus, cells expressing only Hsc82 are more sensitive to radicicol toxicity than those expressing only Hsp82. A similar difference in sensitivity of cells expressing Hsc82 or Hsp82 was observed in a screen for mutants hypersensitive to growth inhibitory effects of macbecin II, another Hsp90 inhibitor (43). This functional difference between Hsp90 isoforms appears to be conserved, as the human stress-inducible Hsp90α is reported to confer less resistance to radicicol than does the constitutive isoform Hsp90β (45).
Our results suggest that much, if not all, of the Hsp82 was still active in the radicicol-treated wild-type cells that failed to cure, which implies that Hsc82 is more important than Hsp82 for the curing of [PSI+] by overexpressed Hsp104. As a test of this hypothesis, we overexpressed Hsp104 in the hsc82Δ and hsp82Δ strains. Hsp104 overexpression cured hsc82Δ cells of [PSI+] very inefficiently (17-fold reduction) but cured hsp82Δ cells only 2-fold less well than did wild-type cells (Table 1). Although the smaller overall amount of Hsp90 in hsc82Δ cells might have contributed to the reduced efficiency of curing of these cells, the difference in Hsp90 abundance between the hsc82Δ and hsp82Δ strains was much smaller than the difference in curing efficiency. These data are consistent with Hsc82 having a more significant role in the curing than Hsp82.
Additionally, even though hsp82Δ cells had more Sse1 than did hsc82Δ cells (Fig. 5D), they cured more efficiently. Moreover, the absolute level of Sse1 was much greater in hsp82Δ cells than in cells exposed to radicicol or those expressing the TPR2 mutants (compare Fig. 5D with Fig. 5C and 2C), but again, the hsp82Δ cells cured much more efficiently. These data show that abundance of Sse1 had less to do with the reduction of curing than the alteration of Hsp90 machinery and suggest that the inhibitory effects of Sse1 on curing depend on certain functions of this machinery.
Hsp90 machinery mediates Ub effects on curing.
The Ub-proteasome system (UPS) mutations that reduce levels of free Ub, such as depletion of the Ub recycling enzyme Ubp6, partially inhibit the curing of [PSI+] by overexpressed Hsp104 (3, 12). Introducing the Ub gene UBI4 on a high-copy-number plasmid restores the curing of such mutant cells, indicating that the inhibition of curing is caused by the reduction in free Ub. In wild-type cells, elevating Ub increases curing efficiency and strengthens nonsense suppression by [PSI+] in general. Thus, Ub influences both Hsp104 curing and [PSI+] propagation (3, 12).
We looked for a link between the UPS and Hsp90 in the curing by testing if elevating free Ub influenced the curing of cells with Hsp90 system defects. As expected, high-copy-number UBI4 increased efficiency of curing of wild-type cells (Fig. 6). However, it did not significantly affect the curing of sti1Δ, hsc82Δ, or hsp82Δ cells. These results suggest that the positive effect that Ub has on prion curing by Hsp104 overexpression requires normal function of the Hsp90 machinery.
FIG. 6.
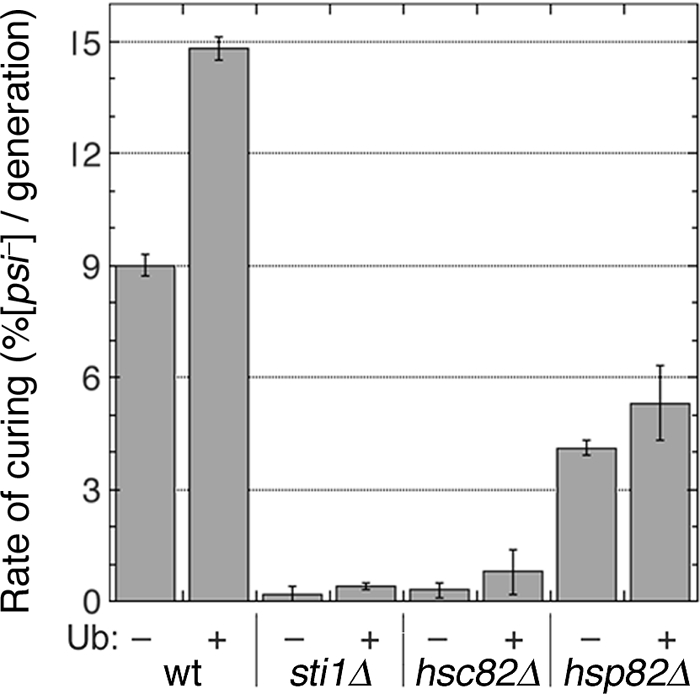
Ub effects on curing of [PSI+] by Hsp104 overexpression. Rates of curing of cultures grown for four to six generations were determined as described in the legend for Fig. 1. Strains (indicated at bottom) carry empty vector (−) or the same plasmid with UBI4 (+), which encodes Ub. Values are averages of the results for three independent experiments ± standard deviation. Student's t test values are as follows: wild type, P = 0.0014; others, P = 0.1.
Curing of [PSI+] by excess Hsp104 is associated with cell division.
A widely held view to explain the curing of [PSI+] by excess Hsp104 is that increasing Hsp104 disaggregation activity destroys prion templates through a direct mass action effect. If excess Hsp104 cures [PSI+] by this mechanism in vivo, then it should be possible to cure nondividing cells. We tested if curing could proceed in nondividing cells by monitoring the curing of cells in a culture depleted of a nutrient required for growth (Fig. 7). After inducing overexpression of Hsp104 in wild-type cells for 5 h, the culture was harvested by centrifugation, washed, suspended in the same medium but lacking leucine, and then split into two equal portions. Leucine was added to one of these cultures, and both were incubated for another 20 h, during which the culture containing leucine went through five generations and the leucine-depleted culture doubled only once. Leucine was then added to the leucine-depleted culture, whereupon it immediately resumed logarithmic growth at the normal rate, and both cultures were grown another 10 h. The appearance of [psi−] cells in both cultures correlated with the growth of the cultures, showing a clear association between curing and cell division (Fig. 7A and B). Similar results were obtained when histidine was depleted. Starved cells overexpressed Hsp104 to similar levels as nonstarved cells (Fig. 7C). These results are consistent with the notion that effective curing of [PSI+] by overexpressing Hsp104 depends on cell division and argue against curing being caused solely by direct wholesale destruction of prions.
FIG. 7.
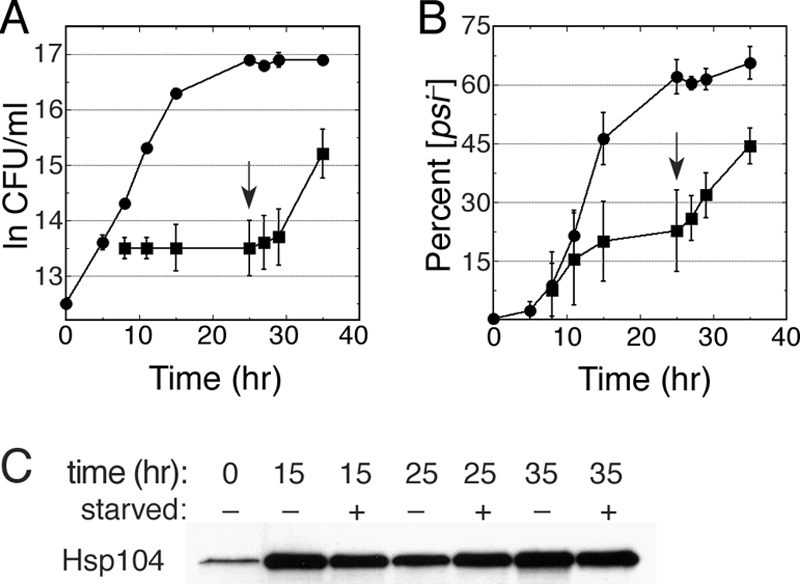
Curing of [PSI+] by Hsp104 overexpression is associated with cell division. Curing of a wild-type culture was done as described in the legend for Fig. 1 except that the culture was harvested 5 h after inducing Hsp104 expression and split into two flasks, one containing identical medium (control; circles) and the other into a similar medium lacking a required nutrient (leucine or histidine; squares). After incubation for another 20 h, the depleted nutrient was added back to the second culture (arrow). (A) Growth of cultures is shown as log of CFU per ml as a function of time. (B) The proportion of [psi−] colonies in the same culture aliquots used to monitor growth is shown. Values in panels A and B are averages from the results for three independent experiments, (two with leucine depleted, one with histidine depleted) ± standard deviation. (C) Western analysis of Hsp104 abundance in cells of the same aliquots of starved (+) and unstarved (−) cultures removed at the indicated time points from the experiment shown in panels A and B.
Hsp90 machinery does not affect curing of [PSI+] by Hsp104 inactivation.
Inactivating Hsp104 by adding millimolar guanidine to growth medium arrests prion seed replication so that the seeds present at the time of guanidine addition become diluted among the cells in a growing culture, eventually giving rise to [psi−] cells when the number of cells becomes greater than the number of preexisting prions. We tested the possibility that the Hsp90 machinery affects curing caused by Hsp104 inactivation by growing sti1Δ, hsc82Δ, and hsp82Δ cells in medium containing three millimolar guanidine-HCl, which inactivates Hsp104 ATPase (24, 26). [PSI+] cells grown in YPAD were diluted into YPAD containing 3 mM guanidine-HCl, and the proportion of [psi−] cells in the cultures was monitored periodically for roughly 12 generations. All of the mutant strains cured like wild-type cells, having similar lags before [psi−] cells appeared and similar rates of subsequent curing (Fig. 8). These results show that prion replication was arrested equally well by guanidine in all the strains and that all strains had similar numbers of starting seeds. Thus, the Sti1 and Hsp90 mutations do not affect the normal function of Hsp104 in prion propagation or the dependency of [PSI+] on Hsp104 activity.
FIG. 8.
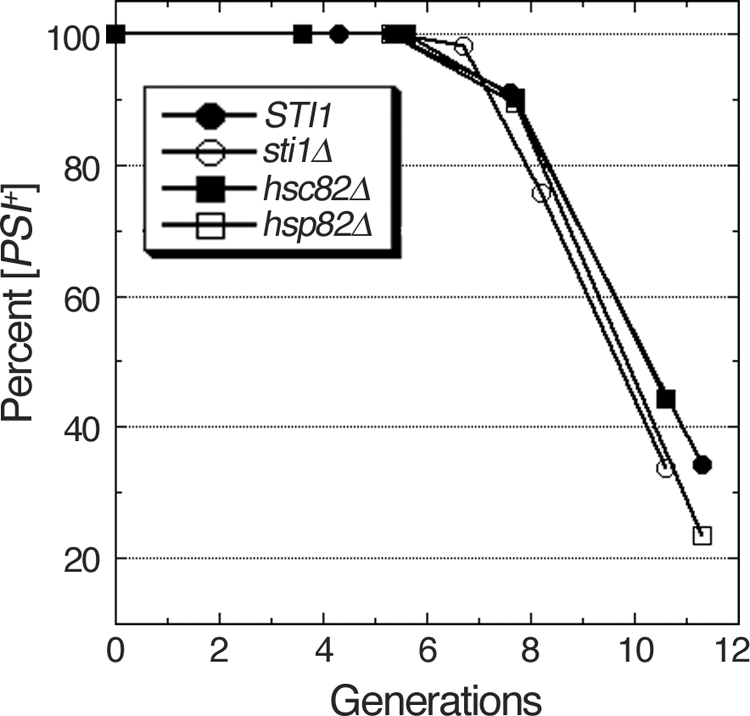
Curing of [PSI+] by guanidine inactivation of Hsp104. Cells grown in YPAD were diluted into similar medium containing 3 mM guanidine hydrochloride and grown continuously for roughly 12 generations. Aliquots of cells were removed periodically to determine the proportion of [PSI+] cells as described in the legend for Fig. 1. The proportion of [PSI+] cells remaining is shown as a function of generations.
DISCUSSION
We previously showed that antiprion effects of Ssa1-21 require a curing-competent Hsp104, and we show here that an extragenic suppressor of Ssa1-21 (sti1Δ) also suppresses curing of [PSI+] by overexpressed Hsp104 in wild-type cells, a result also found by others (46). Our data confirm an important role for this Hsp70/90 cofactor in the curing and suggest that Ssa1-21 induces an Hsp104-like prion curing activity even when Hsp104 is present in low abundance. This explanation strengthens the link between Ssa1-21 and Hsp104 curing activities and is consistent with the dominant impairment of [PSI+] propagation by Ssa1-21.
We demonstrate that the role of Sti1 in curing by overexpressed Hsp104 is indirect, through functional interactions of Sti1 with Hsp70 and, more prominently, Hsp90. Although mutations in Sti1 that disrupt physical and functional interactions of Sti1 with Hsp90 do not affect [PSI+] propagation, they severely inhibit curing of [PSI+] by overexpressed Hsp104. These results highlight the distinction in the way Hsp104 functions in prion replication and in prion curing and point to Sti1 and Hsp90 as major determinants of this distinction in Hsp104 function with regard to the curing. Our finding that hsp82Δ cells are more sensitive than hsc82Δ cells to radicicol resembles that of previous work showing a similar difference in sensitivity to the structurally unrelated Hsp90 inhibitor macbecin II (43). These and other results that we show here uncover isoform-specific differences between Hsc82 and Hsp82 with regard to effects on prions and point to Hsc82 as having a more important role than Hsp82 in curing by overexpressed Hsp104. Although these data point to exquisite specificity in functions important for the curing, the expanding number of chaperone factors that influence the curing speaks to the complexity of the curing mechanism and suggests that overexpressed Hsp104 does not act alone to disrupt [PSI+] propagation.
The involvement of Ub adds another level of complexity. Although alterations that reduce levels of free Ub also adversely affect [PSI+] propagation (12; M. Reidy and D. C. Masison, unpublished results), a substantial effort failed to detect ubiquitylated Sup35 or differences in Sup35 stability, implying that any effect on the prion due to protein turnover is indirect and not by degradation of Sup35 (3). Since ubiquitylation can regulate protein activity as well as signal protein degradation (36), the degradation or regulation of an unknown factor might contribute to the curing mechanism. Alternatively, reducing Ub might simply slow proteasomal degradation in general, which could lead to higher levels of misfolded protein substrates that titrate chaperones involved in curing. Ub is also important for endosome/lysosome trafficking, and a link between these processes and aggregation of prions and polyglutamine has been reported (44). The extent to which the influence of Ub on curing might involve these processes has not been investigated in detail. Although increasing Ub enhanced the curing of wild-type cells, it did not significantly affect the curing of cells depleted of Sti1 or Hsp90, which uncovers a link between Ub and the Hsp90 machinery in curing and shows that Sti1 and Hsp90 are important for mediating the contribution of Ub to the curing.
The mechanism of prion curing by overexpressing Hsp104 has remained elusive, but a widely held view to explain it is that increasing Hsp104 dismantles prions until no functional templates remain to propagate the prion state. This view is based on the logical assumption that increasing Hsp104 abundance corresponds to increased disaggregation activity in the cell due to simple mass action effects, and it is supported by biochemical evidence showing that Hsp104 can break down infectious Sup35 amyloid into noninfectious material (63, 64). Our in vivo data presented here, however, are inconsistent with this mechanism as the sole explanation for the curing. Although the factors we identify here that are important for the curing are involved in protein folding, they have not been implicated as having a direct role in, or even influencing, Hsp104 protein disaggregation. Additionally, our earlier findings with an amino-terminally altered Hsp104 showed that Hsp104's ability to confer thermotolerance, to resolubilize proteins from aggregates, and to act in prion replication are together insufficient for overexpressed Hsp104 to cure cells of the [PSI+] prion (29). The findings by Moosavi et al. (46) that Sti1 is required for curing by overexpression of wild-type Hsp104 but not by overexpression of a dominant negative form of Hsp104, which cures by the different mechanism of Hsp104 inactivation, led them to promote the hypothesis that [PSI+] elimination by Hsp104 overexpression is not simply a consequence of complete dissolution of prion aggregates. They further suggest that [PSI+] curing is through a mechanism distinct from the remodeling activity of Hsp104.
Since Hsp104 lacking its amino-terminal domain actually enhances [PSI+] propagation somewhat, it is unlikely that this domain is important for direct interactions of Hsp104 with Sup35 as a prion substrate. The conserved amino-terminal domain of Hsp100 family proteins of prokaryotic species is also dispensable for function in thermotolerance (7, 13), suggesting that this domain has a function beyond stress protection in other species as well. Prion curing appears to require another Hsp104 function, dependent upon its amino-terminal domain, that is active only when Hsp104 abundance is elevated under conditions where it is not normally induced.
Possibilities for this function of Hsp104 include indirect effects through an interaction with a specific substrate or cofactor that is involved in a process that [PSI+] prions rely on to be efficiently replicated or transmitted during cell division or with other factors that interact specifically with Sup35. Sup35 interacts with components involved in endosome/vacuole dynamics and cortical actin organization, and exposing cells to the actin depolymerizing compound latrunculin A can cure them of [PSI+] (5, 6, 23, 44). The interaction between Sup35 and cytoskeletal components affects the aggregation of Sup35, and Hsp104 seems to be important for this interaction (5, 23). Hsp104 is also involved in actin organization and has a role in linking actin cytoskeleton and polarisome dynamics with retention of damaged proteins in mother cells during cell division (18, 40, 69). Hsp104 also partitions between cytosolic protein quality control compartments and can influence the recovery of proteins from them (35). Hsp104's amino-terminal domain might be important for [PSI+] curing by directing interactions of Hsp104 with factors acting in any of these pathways in a way that interferes specifically with replication or transmission of [PSI+] prions or that promotes or perturbs interactions of Sup35 with components involved in these processes. It is also possible that Hsp104 undergoes posttranslational modifications influenced by its expression level that might differentially regulate its activity.
Although curing of [PSI+] by overexpressed Hsp104 was tied to cell division, it occurred soon after induction of Hsp104 and at a low and roughly linear rate (about 10%/cell division). With 90% or so of cells escaping curing each generation, the curing effect seems relatively inefficient. Whether this pattern reflects an inherent inefficiency of a particular activity or an efficient process occurring in only a specific subpopulation of cells in the culture, such as those of a particular replicative age, remains to be determined.
Acknowledgments
We thank Jeff Brodsky (University of Pittsburgh), Bernd Bukau (University of Heidelberg), John Glover (University of Toronto), and David Toft (Mayo Clinic, Minnesota) for antibodies, and Andy Golden and other colleagues for helpful comments on the manuscript.
This research was supported by the intramural program of the National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health.
Footnotes
Published ahead of print on 17 May 2010.
REFERENCES
- 1.Abbas-Terki, T., O. Donze, P. A. Briand, and D. Picard. 2001. Hsp104 interacts with Hsp90 cochaperones in respiring yeast. Mol. Cell. Biol. 21:7569-7575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Acebron, S. P., I. Martin, U. del Castillo, F. Moro, and A. Muga. 2009. DnaK-mediated association of ClpB to protein aggregates. A bichaperone network at the aggregate surface. FEBS Lett. 583:2991-2996. [DOI] [PubMed] [Google Scholar]
- 3.Allen, K. D., T. A. Chernova, E. P. Tennant, K. D. Wilkinson, and Y. O. Chernoff. 2007. Effects of ubiquitin system alterations on the formation and loss of a yeast prion. J. Biol. Chem. 282:3004-3013. [DOI] [PubMed] [Google Scholar]
- 4.Allen, K. D., R. D. Wegrzyn, T. A. Chernova, S. Muller, G. P. Newnam, P. A. Winslett, K. B. Wittich, K. D. Wilkinson, and Y. O. Chernoff. 2005. Hsp70 chaperones as modulators of prion life cycle: novel effects of Ssa and Ssb on the Saccharomyces cerevisiae prion [PSI+]. Genetics 169:1227-1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bailleul, P. A., G. P. Newnam, J. N. Steenbergen, and Y. O. Chernoff. 1999. Genetic study of interactions between the cytoskeletal assembly protein Sla1 and prion-forming domain of the release factor Sup35 (eRF3) in Saccharomyces cerevisiae. Genetics 153:81-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bailleul-Winslett, P. A., G. P. Newnam, R. D. Wegrzyn, and Y. O. Chernoff. 2000. An antiprion effect of the anticytoskeletal drug latrunculin A in yeast. Gene Expr. 9:145-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beinker, P., S. Schlee, Y. Groemping, R. Seidel, and J. Reinstein. 2002. The N terminus of ClpB from Thermus thermophilus is not essential for the chaperone activity. J. Biol. Chem. 277:47160-47166. [DOI] [PubMed] [Google Scholar]
- 8.Borkovich, K. A., F. W. Farrelly, D. B. Finkelstein, J. Taulien, and S. Lindquist. 1989. Hsp82 is an essential protein that is required in higher concentrations for growth of cells at higher temperatures. Mol. Cell. Biol. 9:3919-3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen, S., V. Prapapanich, R. A. Rimerman, B. Honore, and D. F. Smith. 1996. Interactions of p60, a mediator of progesterone receptor assembly, with heat shock proteins Hsp90 and Hsp70. Mol. Endocrinol. 10:682-693. [DOI] [PubMed] [Google Scholar]
- 10.Chernoff, Y. O., S. L. Lindquist, B. Ono, S. G. Inge-Vechtomov, and S. W. Liebman. 1995. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 268:880-884. [DOI] [PubMed] [Google Scholar]
- 11.Chernoff, Y. O., G. P. Newnam, J. Kumar, K. Allen, and A. D. Zink. 1999. Evidence for a protein mutator in yeast: role of the Hsp70-related chaperone Ssb in formation, stability, and toxicity of the [PSI] prion. Mol. Cell. Biol. 19:8103-8112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chernova, T. A., K. D. Allen, L. M. Wesoloski, J. R. Shanks, Y. O. Chernoff, and K. D. Wilkinson. 2003. Pleiotropic effects of Ubp6 loss on drug sensitivities and yeast prion are due to depletion of the free ubiquitin pool. J. Biol. Chem. 278:52102-52115. [DOI] [PubMed] [Google Scholar]
- 13.Clarke, A. K., and M. J. Eriksson. 2000. The truncated form of the bacterial heat shock protein ClpB/HSP100 contributes to development of thermotolerance in the cyanobacterium Synechococcus sp. strain PCC 7942. J. Bacteriol. 182:7092-7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cox, B. S. 1965. “PSI” a cytoplasmic suppressor of super-suppressor in yeast. Heredity 20:505-521. [Google Scholar]
- 15.Derkatch, I. L., M. E. Bradley, P. Zhou, Y. O. Chernoff, and S. W. Liebman. 1997. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics 147:507-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Du, Z., K. W. Park, H. Yu, Q. Fan, and L. Li. 2008. Newly identified prion linked to the chromatin-remodeling factor Swi1 in Saccharomyces cerevisiae. Nat. Genet. 40:460-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eaglestone, S. S., L. W. Ruddock, B. S. Cox, and M. F. Tuite. 2000. Guanidine hydrochloride blocks a critical step in the propagation of the prion-like determinant [PSI+] of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A. 97:240-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Erjavec, N., L. Larsson, J. Grantham, and T. Nystrom. 2007. Accelerated aging and failure to segregate damaged proteins in Sir2 mutants can be suppressed by overproducing the protein aggregation-remodeling factor Hsp104p. Genes Dev. 21:2410-2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan, Q., K. W. Park, Z. Du, K. A. Morano, and L. Li. 2007. The role of Sse1 in the de novo formation and variant determination of the [PSI+] prion. Genetics 177:1583-1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Floer, M., G. O. Bryant, and M. Ptashne. 2008. HSP90/70 chaperones are required for rapid nucleosome removal upon induction of the GAL genes of yeast. Proc. Natl. Acad. Sci. U. S. A. 105:2975-2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flom, G., R. H. Behal, L. Rosen, D. G. Cole, and J. L. Johnson. 2007. Definition of the minimal fragments of Sti1 required for dimerization, interaction with Hsp70 and Hsp90 and in vivo functions. Biochem. J. 404:159-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flom, G., J. Weekes, J. J. Williams, and J. L. Johnson. 2006. Effect of mutation of the tetratricopeptide repeat and aspartate-proline 2 domains of Sti1 on Hsp90 signaling and interaction in Saccharomyces cerevisiae. Genetics 172:41-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ganusova, E. E., L. N. Ozolins, S. Bhagat, G. P. Newnam, R. D. Wegrzyn, M. Y. Sherman, and Y. O. Chernoff. 2006. Modulation of prion formation, aggregation, and toxicity by the actin cytoskeleton in yeast. Mol. Cell. Biol. 26:617-629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glover, J. R., and S. Lindquist. 1998. Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell 94:73-82. [DOI] [PubMed] [Google Scholar]
- 25.Glover, J. R., and R. Lum. 2009. Remodeling of protein aggregates by Hsp104. Protein Pept. Lett. 16:587-597. [DOI] [PubMed] [Google Scholar]
- 26.Grimminger, V., K. Richter, A. Imhof, J. Buchner, and S. Walter. 2004. The prion curing agent guanidinium chloride specifically inhibits ATP hydrolysis by Hsp104. J. Biol. Chem. 279:7378-7383. [DOI] [PubMed] [Google Scholar]
- 27.Hattendorf, D. A., and S. L. Lindquist. 2002. Cooperative kinetics of both Hsp104 ATPase domains and interdomain communication revealed by AAA sensor-1 mutants. EMBO J. 21:12-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hrizo, S. L., V. Gusarova, D. M. Habiel, J. L. Goeckeler, E. A. Fisher, and J. L. Brodsky. 2007. The Hsp110 molecular chaperone stabilizes apolipoprotein B from endoplasmic reticulum-associated degradation (ERAD). J. Biol. Chem. 282:32665-32675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hung, G. C., and D. C. Masison. 2006. N-terminal domain of yeast Hsp104 chaperone is dispensable for thermotolerance and prion propagation but necessary for curing prions by Hsp104 overexpression. Genetics 173:611-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Inoue, Y., A. Kishimoto, J. Hirao, M. Yoshida, and H. Taguchi. 2001. Strong growth polarity of yeast prion fiber revealed by single fiber imaging. J. Biol. Chem. 276:35227-35230. [DOI] [PubMed] [Google Scholar]
- 31.Jones, G., Y. Song, S. Chung, and D. C. Masison. 2004. Propagation of Saccharomyces cerevisiae [PSI+] prion is impaired by factors that regulate Hsp70 substrate binding. Mol. Cell. Biol. 24:3928-3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jung, G., G. Jones, and D. C. Masison. 2002. Amino acid residue 184 of yeast Hsp104 chaperone is critical for prion-curing by guanidine, prion propagation, and thermotolerance. Proc. Natl. Acad. Sci. U. S. A. 99:9936-9941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jung, G., G. Jones, R. D. Wegrzyn, and D. C. Masison. 2000. A role for cytosolic Hsp70 in yeast [PSI+] prion propagation and [PSI+] as a cellular stress. Genetics 156:559-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jung, G., and D. C. Masison. 2001. Guanidine hydrochloride inhibits Hsp104 activity in vivo: a possible explanation for its effect in curing yeast prions. Curr. Microbiol. 43:7-10. [DOI] [PubMed] [Google Scholar]
- 35.Kaganovich, D., R. Kopito, and J. Frydman. 2008. Misfolded proteins partition between two distinct quality control compartments. Nature 454:1088-1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Komander, D. 2009. The emerging complexity of protein ubiquitination. Biochem. Soc. Trans. 37:937-953. [DOI] [PubMed] [Google Scholar]
- 37.Kryndushkin, D. S., I. M. Alexandrov, M. D. Ter-Avanesyan, and V. V. Kushnirov. 2003. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J. Biol. Chem. 278:49636-49643. [DOI] [PubMed] [Google Scholar]
- 38.Kryndushkin, D. S., F. Shewmaker, and R. B. Wickner. 2008. Curing of the [URE3] prion by Btn2p, a Batten disease-related protein. EMBO J. 27:2725-2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurahashi, H., and Y. Nakamura. 2007. Channel mutations in Hsp104 hexamer distinctively affect thermotolerance and prion-specific propagation. Mol. Microbiol. 63:1669-1683. [DOI] [PubMed] [Google Scholar]
- 40.Liu, B., L. Larsson, A. Caballero, X. Hao, D. Oling, J. Grantham, and T. Nystrom. 2010. The polarisome is required for segregation and retrograde transport of protein aggregates. Cell 140:257-267. [DOI] [PubMed] [Google Scholar]
- 41.Lum, R., J. M. Tkach, E. Vierling, and J. R. Glover. 2004. Evidence for an unfolding/threading mechanism for protein disaggregation by Saccharomyces cerevisiae Hsp104. J. Biol. Chem. 279:29139-29146. [DOI] [PubMed] [Google Scholar]
- 42.Mackay, R. G., C. W. Helsen, J. M. Tkach, and J. R. Glover. 2008. The C-terminal extension of Saccharomyces cerevisiae Hsp104 plays a role in oligomer assembly. Biochemistry 47:1918-1927. [DOI] [PubMed] [Google Scholar]
- 43.McClellan, A. J., Y. Xia, A. M. Deutschbauer, R. W. Davis, M. Gerstein, and J. Frydman. 2007. Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell 131:121-135. [DOI] [PubMed] [Google Scholar]
- 44.Meriin, A. B., X. Zhang, N. B. Miliaras, A. Kazantsev, Y. O. Chernoff, J. M. McCaffery, B. Wendland, and M. Y. Sherman. 2003. Aggregation of expanded polyglutamine domain in yeast leads to defects in endocytosis. Mol. Cell. Biol. 23:7554-7565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Millson, S. H., A. W. Truman, A. Racz, B. Hu, B. Panaretou, J. Nuttall, M. Mollapour, C. Soti, and P. W. Piper. 2007. Expressed as the sole Hsp90 of yeast, the alpha and beta isoforms of human Hsp90 differ with regard to their capacities for activation of certain client proteins, whereas only Hsp90beta generates sensitivity to the Hsp90 inhibitor radicicol. FEBS J. 274:4453-4463. [DOI] [PubMed] [Google Scholar]
- 46.Moosavi, B., J. Wongwigkarn, and M. F. Tuite. 2010. Hsp70/Hsp90 co-chaperones are required for efficient Hsp104-mediated elimination of the yeast [PSI+] prion but not for prion propagation. Yeast 27:167-179. [DOI] [PubMed] [Google Scholar]
- 47.Moriyama, H., H. K. Edskes, and R. B. Wickner. 2000. [URE3] prion propagation in Saccharomyces cerevisiae: requirement for chaperone Hsp104 and curing by overexpressed chaperone Ydj1p. Mol. Cell. Biol. 20:8916-8922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mumberg, D., R. Muller, and M. Funk. 1995. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156:119-122. [DOI] [PubMed] [Google Scholar]
- 49.Nemecek, J., T. Nakayashiki, and R. B. Wickner. 2009. A prion of yeast metacaspase homolog (Mca1p) detected by a genetic screen. Proc. Natl. Acad. Sci. U. S. A. 106:1892-1896. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 50.Ness, F., P. Ferreira, B. S. Cox, and M. F. Tuite. 2002. Guanidine hydrochloride inhibits the generation of prion “seeds” but not prion protein aggregation in yeast. Mol. Cell. Biol. 22:5593-5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Newnam, G. P., R. D. Wegrzyn, S. L. Lindquist, and Y. O. Chernoff. 1999. Antagonistic interactions between yeast chaperones Hsp104 and Hsp70 in prion curing. Mol. Cell. Biol. 19:1325-1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Osherovich, L. Z., B. S. Cox, M. F. Tuite, and J. S. Weissman. 2004. Dissection and design of yeast prions. PLoS Biol. 2:E86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parsell, D. A., A. S. Kowal, M. A. Singer, and S. Lindquist. 1994. Protein disaggregation mediated by heat-shock protein Hsp104. Nature 372:475-478. [DOI] [PubMed] [Google Scholar]
- 54.Patel, B. K., J. Gavin-Smyth, and S. W. Liebman. 2009. The yeast global transcriptional co-repressor protein Cyc8 can propagate as a prion. Nat. Cell Biol. 11:344-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paushkin, S. V., V. V. Kushnirov, V. N. Smirnov, and M. D. Ter-Avanesyan. 1996. Propagation of the yeast prion-like [psi+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J. 15:3127-3134. [PMC free article] [PubMed] [Google Scholar]
- 56.Sadlish, H., H. Rampelt, J. Shorter, R. D. Wegrzyn, C. Andreasson, S. Lindquist, and B. Bukau. 2008. Hsp110 chaperones regulate prion formation and propagation in S. cerevisiae by two discrete activities. PLoS One 3:e1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Satpute-Krishnan, P., and T. R. Serio. 2005. Prion protein remodelling confers an immediate phenotypic switch. Nature 437:262-265. [DOI] [PubMed] [Google Scholar]
- 58.Scheibel, T., A. S. Kowal, J. D. Bloom, and S. L. Lindquist. 2001. Bidirectional amyloid fiber growth for a yeast prion determinant. Curr. Biol. 11:366-369. [DOI] [PubMed] [Google Scholar]
- 59.Scheufler, C., A. Brinker, G. Bourenkov, S. Pegoraro, L. Moroder, H. Bartunik, F. U. Hartl, and I. Moarefi. 2000. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell 101:199-210. [DOI] [PubMed] [Google Scholar]
- 60.Schirmer, E. C., O. R. Homann, A. S. Kowal, and S. Lindquist. 2004. Dominant gain-of-function mutations in Hsp104p reveal crucial roles for the middle region. Mol. Biol. Cell 15:2061-2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schwimmer, C., and D. C. Masison. 2002. Antagonistic interactions between yeast [PSI+] and [URE3] prions and curing of [URE3] by Hsp70 protein chaperone Ssa1p but not by Ssa2p. Mol. Cell. Biol. 22:3590-3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sharma, D., C. N. Martineau, M. T. Le Dall, M. Reidy, D. C. Masison, and M. Kabani. 2009. Function of Ssa subfamily of Hsp70 within and across species varies widely in complementing Saccharomyces cerevisiae cell growth and prion propagation. PLoS One 4:e6644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shorter, J., and S. Lindquist. 2004. Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science 304:1793-1797. [DOI] [PubMed] [Google Scholar]
- 64.Shorter, J., and S. Lindquist. 2008. Hsp104, Hsp70 and Hsp40 interplay regulates formation, growth and elimination of Sup35 prions. EMBO J. 27:2712-2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sikorski, R. S., and P. Hieter. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sondheimer, N., N. Lopez, E. A. Craig, and S. Lindquist. 2001. The role of Sis1 in the maintenance of the [RNQ+] prion. EMBO J. 20:2435-2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Song, Y., and D. C. Masison. 2005. Independent regulation of Hsp70 and Hsp90 chaperones by Hsp70/Hsp90-organizing protein Sti1 (Hop1). J. Biol. Chem. 280:34178-34185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tessarz, P., A. Mogk, and B. Bukau. 2008. Substrate threading through the central pore of the Hsp104 chaperone as a common mechanism for protein disaggregation and prion propagation. Mol. Microbiol. 68:87-97. [DOI] [PubMed] [Google Scholar]
- 69.Tessarz, P., M. Schwarz, A. Mogk, and B. Bukau. 2009. The yeast AAA+ chaperone Hsp104 is part of a network that links the actin cytoskeleton with the inheritance of damaged proteins. Mol. Cell. Biol. 29:3738-3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wegrzyn, R. D., K. Bapat, G. P. Newnam, A. D. Zink, and Y. O. Chernoff. 2001. Mechanism of prion loss after Hsp104 inactivation in yeast. Mol. Cell. Biol. 21:4656-4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zietkiewicz, S., J. Krzewska, and K. Liberek. 2004. Successive and synergistic action of the Hsp70 and Hsp100 chaperones in protein disaggregation. J. Biol. Chem. 279:44376-44383. [DOI] [PubMed] [Google Scholar]
- 72.Zietkiewicz, S., A. Lewandowska, P. Stocki, and K. Liberek. 2006. Hsp70 chaperone machine remodels protein aggregates at the initial step of Hsp70-Hsp100-dependent disaggregation. J. Biol. Chem. 281:7022-7029. [DOI] [PubMed] [Google Scholar]