

Abstract
Rap1GAP expression is decreased in human tumors. The significance of its downregulation is unknown. We show that Rap1GAP expression is decreased in primary colorectal carcinomas. To elucidate the advantages conferred on tumor cells by loss of Rap1GAP, Rap1GAP expression was silenced in human colon carcinoma cells. Suppressing Rap1GAP induced profound alterations in cell adhesion. Rap1GAP-depleted cells exhibited defects in cell/cell adhesion that included an aberrant distribution of adherens junction proteins. Depletion of Rap1GAP enhanced adhesion and spreading on collagen. Silencing of Rap expression normalized spreading and restored E-cadherin, β-catenin, and p120-catenin to cell/cell contacts, indicating that unrestrained Rap activity underlies the alterations in cell adhesion. The defects in adherens junction protein distribution required integrin signaling as E-cadherin and p120-catenin were restored at cell/cell contacts when cells were plated on poly-l-lysine. Unexpectedly, Src activity was increased in Rap1GAP-depleted cells. Inhibition of Src impaired spreading and restored E-cadherin at cell/cell contacts. These findings provide the first evidence that Rap1GAP contributes to cell/cell adhesion and highlight a role for Rap1GAP in regulating cell/matrix and cell/cell adhesion. The frequent downregulation of Rap1GAP in epithelial tumors where alterations in cell/cell and cell/matrix adhesion are early steps in tumor dissemination supports a role for Rap1GAP depletion in tumor progression.
Mammalian Rap proteins Rap1a/b and Rap2a/b/c are members of the Ras superfamily of small GTPases. Rap proteins are active when bound to GTP and inactive when bound to GDP. Cellular Rap activity is regulated by the concerted action of guanine nucleotide exchange factors that activate Rap (RapGEFs) and Rap-specific GTPase-activating proteins (RapGAPs) that inactivate Rap (reviewed in reference 10). The Rap1GAP family is composed of several members, including Rap1GAP, Rap1GAPII, Spa-1/SIPA1, and E6TP1/SIPA1L1. Several lines of evidence suggest that RapGAPs function as tumor and/or invasion suppressors. Downregulation of E6TP1 by human papillomavirus protein E6 contributes to cervical cancer (20, 21), and Spa-1 deficiency in mice induces a spectrum of myelodysplastic disorders similar to chronic myelogenous leukemia (26). The SPA1 gene was identified as a candidate for the metastasis efficiency modifier locus in mice (38). Although the relevance of this observation to humans is not yet clear, single-nucleotide polymorphisms in the SPA1 gene in human breast tumors have been associated with lymph node involvement and poor survival (15). Intriguingly, Spa-1 interacts with Brd4 (18) and Rrp-1b (13), the protein products of genes associated with patterns of extracellular matrix protein gene expression characteristic of metastatic tumors (14).
The RAP1GAP gene maps to 1p35-36, a chromosomal region subject to copy number alterations in human tumors (36, 49). Rap1GAP protein levels are decreased in pancreatic adenocarcinomas (53), papillary thyroid carcinomas (37, 47, 57), and melanomas (56). Rap1GAP downregulation has been shown to arise as a consequence of proteasomal degradation (46), loss of heterozygosity (37, 53), and promoter methylation (56, 57). Mutations of unknown significance in RAP1GAP have been identified in breast cancer (42). Although downregulation of Rap1GAP is frequent in human tumors, the functional significance of decreased Rap1GAP expression is unknown. Up to now, studies assessing the role of Rap1GAP in tumor cells have relied exclusively on overexpression experiments. Overexpression of Rap1GAP in oropharyngeal squamous cell (54) and pancreatic (53) carcinoma lines impaired tumor formation in mouse xenograft models. In vitro, overexpression of Rap1GAP impaired tumor cell proliferation (34, 47, 53, 54, 56) and enhanced apoptosis (34, 53, 56). In some instances, overexpression of Rap1GAP inhibited tumor cell migration and invasion (3, 47, 53, 56), while in others, it enhanced invasion (34). While these studies provide insight into cellular processes that can be deregulated by overexpression, they do not assess the significance of depletion of endogenous Rap1GAP in human tumors.
Colorectal cancer (CRC) is one of the leading causes of cancer deaths worldwide. The majority of CRC deaths arise as a consequence of distant metastases, most frequently to the liver. While the genetic basis of CRC is well understood (19, 48), less is known about the events that trigger the transition to metastatic disease. We report that Rap1GAP is highly expressed in normal colonic epithelium and that its expression is profoundly decreased in primary colorectal carcinomas. As one strategy to assess the significance of Rap1GAP depletion, the expression of Rap1GAP was silenced in human colon carcinoma cells. Silencing of Rap1GAP induced marked increases in Rap1 and Rap2 activity, the first evidence that Rap1GAP is an essential negative regulator of Rap GTPases in colon cancer. Rap1 regulates inside-out signaling through integrins (reviewed in references 8, 9, and 11) and is a target of outside-in signaling via cadherins (reviewed in reference 30). Downregulation of Rap1GAP induced profound alterations in cell/matrix and cell/cell adhesion. Suppressing Rap1GAP expression enhanced adhesion and spreading on collagen. Unexpectedly, based on the role of Rap1 in promoting cell/cell adhesion, silencing of Rap1GAP impaired cell/cell adhesion. These findings demonstrate a requirement for regulated Rap activity in the maintenance of epithelial cell structure and demonstrate a heretofore unappreciated role for Rap1GAP in the regulation of cell/cell adhesion. As the dissemination of tumor cells requires the weakening of cell/cell adhesion and an enhanced ability to adhere to collagen-rich interstitial matrices, our studies identify a potential mechanism through which loss of Rap1GAP contributes to tumor progression.
MATERIALS AND METHODS
Antibodies.
FAK (610087), E-cadherin (610404), Rap2 (610215), and p120 (610134) antibodies were purchased from BD Transduction Labs. FAK-pY576 (44652G) and FAK-pY397 (44624G) antibodies were from Invitrogen. Src (2109) and Src-pY416 (2101) antibodies were from Cell Signaling Technology. Rap1 (sc-65), β-catenin (sc-7199), Rap1GAP (sc-28189), and actin (sc-1615) antibodies were purchased from Santa Cruz.
Immunohistochemistry.
Tissue blocks from patients diagnosed with colon cancer were provided by Yu Lv, Beijing Chaoyang Hospital, Capital University of Medical Sciences, Beijing, China. Blocks containing tumor and adjacent normal tissues in the same sections were selected for analysis. Fresh hematoxylin-and-eosin-stained sections were made and reviewed by trained pathologists at the Hospital of the University of Pennsylvania. Freshly cut 5-μm sections were used for immunohistochemical staining. Sections were processed and stained for Rap1GAP as previously described (37). Slides were scanned on an Aperio Scanscope CS (Aperio Inc., Vista, CA) virtual slide scanner using ×40 magnification (0.5-μm/pixel resolution). The virtual slides were managed and manipulated using Imagescope software (Aperio Inc.). Single fields were extracted at ×400 magnification. Rap1GAP staining was quantified by H score as described in reference 23. In brief, the H score is calculated by adding 1+ events × number of events expressed as a percentage, 2+ events × number of events expressed as a percentage, and 3+ events × number of events expressed as a percentage. The highest possible H score is 300.
Cell lines.
HCT 116, LoVo, Caco-2, DLD1, COLO 205, and COLO DM colon carcinoma cell lines were kindly provided by John Lynch (Department of Medicine, University of Pennsylvania). The variant of HT29 cells used in this study was originally characterized as a thyroid carcinoma cell line (WRO). DNA profiling using the Identifier kit from Applied Biosystems confirmed that this cell line is a pure line derived from the same individual as HT29 colon carcinoma cells profiled by the American Type Culture Collection (data not shown). HT29, DLD1, and COLO 205 cells were maintained in RPMI medium supplemented with 10% fetal bovine serum (FBS), HCT 116 and LoVo cells were maintained in McCoy's 5A medium containing 10% FBS, and Caco-2 cells were maintained in Dulbecco's modified Eagle's medium containing 20% FBS. SMARTvector human Rap1GAP and scrambled short hairpin RNA (shRNA) lentiviruses (S-005000-01) were purchased from Thermo Scientific Dharmacon (Boulder, CO) and used according to the manufacturer's recommendations. Viruses expressing two different Rap1GAP-directed shRNAs (shRNA2, TTGGTGTGTGAAGACGTCA; shRNA3, TCTTCTCACTCAAGTACGA) were used. Briefly, 0.5 × 105 to 1 × 105 HT29 cells were infected with viruses at multiplicities of infection of 5 to 20. After 48 h, cells were subcultured and selected in puromycin (0.5 to 5.0 mg/ml) and surviving cells were cloned by limiting dilution. The E11 and C10 cell lines were isolated from cells infected with Rap1GAPshRNA3; the C4 and F3 cell lines were isolated from cells infected with Rap1GAPshRNA2.
Transient transfection with siRNAs.
Small interfering RNAs (siRNAs) were delivered into HT29 cells by Amaxa-mediated electroporation. In brief, 1.5 × 106 cells were transfected with 200 nM siRNA. Rap2b (RAP2BHSS143582) and Rap2c (RAP2CHSS126791) siRNAs were purchased from Invitrogen. Rap1GAP (SI01737050 [CAGCGCCATTGGCATCGAGAA]), Rap1b (SI00111762), and scrambled (1027280) siRNAs were obtained from Qiagen.
Trypsin sensitivity assays.
Cells were treated with 0.01% trypsin in 2 mM CaCl2 or 2 mM EGTA for 30 min and lysed in radioimmunoprecipitation assay buffer, and cell lysates were analyzed by Western blotting. Control cells were lysed in the absence of trypsin treatment.
Adhesion assays.
Cells were released by trypsinization, resuspended in growth medium at 37°C for 60 min, and plated into 24-well plates (105 cells/well) coated with collagen IV (2 μg/ml) or poly-l-lysine (PLL; 0.01%) for 10 min. Nonadherent cells were removed by washing, and adherent cells were fixed in 3.7% formaldehyde and stained with 0.1% violet blue. After washing, stain was eluted with 1% deoxycholate and the absorbance at 590 nm was read. The number of total adherent cells was determined at 2 h postplating, and the percentage of adherent cells was plotted.
Cell dissociation assays.
Cells (1 × 106) were plated in 60-mm dishes for 48 h and trypsinized in the presence of 2 mM EGTA or 2 mM CaCl2 for 10 min at 37°C. After being pipetted 20 times, cells were plated and random images were acquired using a Nikon Eclipse TE2000 microscope, a 20× objective, and Northern Eclipse (v. 6) software (Empix Imaging). Cell dissociation is presented as the Np/Nt ratio, where Np is the total number of particles and Nt is the total number of cells.
Immunostaining.
Cells were fixed in methanol-acetone (1:1) for 15 min at room temperature. Cells were stained with primary antibodies diluted in 5 mg/ml bovine serum albumin-phosphate-buffered saline-0.2% Triton X-100 for 1 h at 37°C, followed by staining with Alexa Fluor-conjugated secondary antibodies for 1 h at 37°C. Images were acquired on a Zeiss Axiophot fluorescence microscope or a Zeiss Axiovert 100M confocal microscope. All images for a given antibody were acquired for the same times. Cell area was analyzed using Zeiss Axiovision software.
Rescue experiments.
Rap1GAP-depleted cell lines were infected with adenoviruses expressing HA-Rap1GAP or β-galactosidase as a control. At 2 days postinfection, cells were plated on coverslips, starved overnight, fixed, and stained for E-cadherin and p120-catenin as indicated above.
Statistical analysis.
All experiments were repeated three or more times unless otherwise noted. The bar graphs shown represent the combined results from at least three experiments. Statistical significance was determined by Student's t test.
RESULTS
Expression of Rap1GAP is decreased in primary colon carcinomas.
Tissue blocks from patients diagnosed with colon cancer were selected on the basis of containing tumor and adjacent normal tissues and stained with a highly specific Rap1GAP antibody (37, 47). Prominent staining for Rap1GAP was detected in areas of normal colonic epithelium (Fig. 1A). As previously reported (37, 47, 53), stromal cells did not stain for Rap1GAP. Rap1GAP staining was decreased in the majority (12/15) of the tumors analyzed (Fig. 1A). Semiquantitative staining according to H score (23) revealed that Rap1GAP expression was significantly decreased in tumor tissue compared to that in the adjacent normal epithelium (Fig. 1B). These data demonstrate that Rap1GAP is a frequent target for downregulation in CRCs and add colon cancer to an expanding list of cancers in which Rap1GAP expression is suppressed.
FIG. 1.

Decreased expression of Rap1GAP in primary colorectal carcinomas. (A) Tissue sections containing adjacent normal colonic epithelium and tumor tissue were stained for Rap1GAP as previously described (37, 47). (B) Rap1GAP staining was quantified by H score (23). The H scores in tumor versus normal tissue were compared using the nonparametric Wilcoxon signed-rank test and found to be significantly different (P = 0.0007).
Silencing of Rap1GAP expression increases Rap activity.
To elucidate the potential advantages conferred on cells by Rap1GAP downregulation, we set out to silence Rap1GAP expression in tumor cells. To identify a cell model for these studies, Rap1GAP expression was analyzed in a panel of human colon carcinoma cell lines. Rap1GAP expression was highest in HT29 cells, which exhibit an epithelial morphology and express E-cadherin (Fig. 2A). Hence, these cells were selected for gene silencing experiments. Proof-of-principle experiments were conducted to determine whether there are functional consequences associated with silencing of Rap1GAP expression. Both Rap1 and Rap2 activities were increased in HT29 cells transfected with Rap1GAP-directed siRNAs (Fig. 2B and C). Based on this, we proceeded to isolate cell lines in which Rap1GAP expression was chronically suppressed, a situation that more closely resembles that in human tumors. HT29 cells were infected with lentiviruses expressing Rap1GAP-directed or scrambled shRNAs, and drug-resistant cells were cloned by limiting dilution. Rap1GAP expression was decreased in all of the cell lines exposed to Rap1GAP shRNAs and unchanged in cells exposed to scrambled shRNAs (six lines of each are shown in Fig. 2D). The two cell lines with the greatest reduction in Rap1GAP protein levels (E11 and C10) and a control cell line (Scr and A2) were selected for analysis.
FIG. 2.

Rap1GAP is an essential negative regulator of Rap activity in colon carcinoma cells. (A) Rap1GAP expression was analyzed in a panel of human colon carcinoma cell lines by Western blotting. With the exception of COLO DM, E-cadherin was expressed in all of the lines. Western blotting for actin confirmed equal protein loading. (B and C) HT29 cells were transiently transfected with Rap1GAP-directed or scrambled (scr) siRNAs, and Rap1/2 activity (46) was assessed in growing cells. Total cell lysates were analyzed by Western blotting for Rap and Rap1GAP expression. Rap1 and Rap2 activities were increased in HT29 cells transfected with Rap1GAP siRNAs. Rap activity is presented as n-fold over that in control transfected cells (scr), which was set to 1. (D) Rap1GAP expression in cell lines selected to express Rap1GAP versus scrambled shRNAs was analyzed. The two cell lines with the lowest levels of Rap1GAP (E11 and C10) and a control cell line (Scr and A2) were selected for further analysis. Rap1 (E) and Rap2 (F) activities were analyzed in serum-starved cells. The increase in Rap activity in both Rap1GAP-depleted cell lines was statistically significant (P < 0.05). Basal levels of Rap1 and Rap2 activities were similar in Scr and HT29 cells (data not shown).
We first investigated whether chronic suppression of Rap1GAP results in sustained increases in Rap activity. Basal levels of Rap1 (Fig. 2E) and Rap2 (Fig. 2F) activity were significantly increased in both Rap1GAP-depleted cell lines. The Rap1GAP-directed shRNAs target different sequences on Rap1GAP than the siRNAs used in the transient-knockdown experiments, confirming that increased Rap activity is not an off-target effect. These findings identify Rap1GAP as an essential regulator of Rap activity in human colon carcinoma cells and suggest that downregulation of Rap1GAP in human tumors is associated with increased Rap activity.
Rap1GAP is required for cell/cell adhesion.
Rap1GAP-depleted cells exhibited dramatic alterations in cell morphology. Unlike HT29 cells, which form rounded colonies with abundant cell/cell contacts, Rap1GAP-depleted cells were more dispersed. To explore the molecular mechanism underlying the alterations in cell morphology, we first examined whether silencing of Rap1GAP alters the expression of adherens junction (AJ) proteins. Steady-state E-cadherin, β-catenin, and p120-catenin protein levels were largely unchanged in Rap1GAP-depleted cells, although we consistently noted a slight decrease in E-cadherin protein levels in E11 cells (Fig. 3A). Strikingly, however, the distribution of all three proteins was altered. E-cadherin and β-catenin were enriched at sites of cell/cell contact in control cells (Fig. 3B, Scr). The accumulation of E-cadherin and β-catenin at cell/cell junctions was reduced in the Rap1GAP-depleted cell lines. p120-catenin was found at cell/cell contacts and at lower levels in the cytoplasm of control cells. The accumulation of p120-catenin at cell/cell contacts was reduced in Rap1GAP-depleted cells, where an increased proportion of p120-catenin appeared to be stranded in the cytoplasm. Similar effects were observed in two additional cells lines that expressed Rap1GAP-directed shRNAs that target different sequences on Rap1GAP (see Fig. S1 in the supplemental material). Furthermore, transient silencing of Rap1GAP expression in HT29 cells using siRNAs that target regions on Rap1GAP distinct from the shRNAs induced similar alterations in the distribution of E-cadherin (Fig. 4). The distribution of E-cadherin in control siRNA-transfected HT29 cells and that in the Scr cell line were similar (compare Fig. 3B and 4).
FIG. 3.

Downregulation of Rap1GAP alters the distribution of AJ proteins. (A) Rap1GAP-depleted cells were analyzed for E-cadherin, β-catenin, and p120-catenin expression by Western blotting. Rap1GAP expression is also shown. Actin was used to document equal protein loading. (B) Cells (E11, C10, and Scr) were serum starved overnight, fixed, stained for E-cadherin, β-catenin, and p120-catenin, and analyzed by confocal microscopy. Images for each antibody were acquired for the same times. (C) Cells were lysed directly on plastic (total) or trypsinized in the presence of calcium (T/Ca) or EDTA (T/E). Cell lysates (40 μg) were fractionated by SDS-PAGE and subjected to Western blotting for E-cadherin and actin as a loading control. (D) Summary of the results obtained from three independent trypsin sensitivity experiments. (E) Soluble fractions from Rap1GAP-depleted and control cells were subjected to Western blotting for p120-catenin. Partitioning of p120-catenin to the soluble fraction was significantly increased (P < 0.05) in Rap1GAP-depleted cells. Steady-state (total) levels of p120-catenin were not altered by downregulation of Rap1GAP.
FIG. 4.

Acute silencing of Rap1GAP impairs the accumulation of E-cadherin at cell/cell contacts. HT29 cells were transiently transfected with Rap1GAP-directed or scrambled siRNAs that target sequences distinct from those targeted by the Rap1GAP and scrambled shRNAs. After 48 h, cells were serum starved overnight, fixed, and stained for E-cadherin and Rap1GAP. DAPI staining was included to label cell nuclei. Examples of Rap1GAP siRNA-transfected cells that lack E-cadherin at cell/cell junctions are indicated with arrows. Examples of control transfected cells where E-cadherin accumulates at cell/cell junctions are also indicated with arrows.
We next explored whether restoring Rap1GAP expression rescues the defects in AJ protein distribution. Rap1GAP was overexpressed in the E11 and C10 Rap1GAP-deficient cell lines using an adenovirus. Cells expressing Rap1GAP were more rounded and compact than control infected cells. Importantly, the accumulation of E-cadherin and p120-catenin at cell/cell contacts was increased in Rap1GAP-expressing cells (see Fig. S2 to S4 in the supplemental material). Taken together, these data demonstrate that the effects of Rap1GAP depletion on AJ proteins are not due to clonal heterogeneity or off-target effects.
Cell fractionation experiments failed to reveal consistent differences in the partitioning of E-cadherin and β-catenin between soluble and insoluble fractions in Rap1GAP-depleted versus control cells (data not shown). To assess whether cell surface E-cadherin was reduced in Rap1GAP-depleted cells, trypsin sensitivity experiments were conducted. These studies revealed that the proportion of E-cadherin on the cell surface was not reduced in Rap1GAP-depleted cells (Fig. 3C and D). These data suggest that loss of Rap1GAP alters the distribution of membrane E-cadherin and β-catenin, rather than their association with membranes. On the other hand, p120-catenin was significantly increased in the soluble fraction of Rap1GAP-silenced cells (Fig. 3E), consistent with the enhanced cytoplasmic staining for p120-catenin observed in these cells.
To explore if silencing of Rap1GAP has functional effects on cell/cell adhesion, cell dissociation assays were performed. Cells were trypsinized in calcium to maintain cell/cell adhesion or in EGTA to disrupt adhesion, and the proportions of particles (aggregates and single cells) were determined. As expected, calcium decreased the dissociation index in control cells (Fig. 5A). In contrast, E11 and C10 cells were dissociated in the presence of calcium. Collectively, these results demonstrate that Rap1GAP contributes to the maintenance of cell/cell adhesion in human colon cancer cells.
FIG. 5.
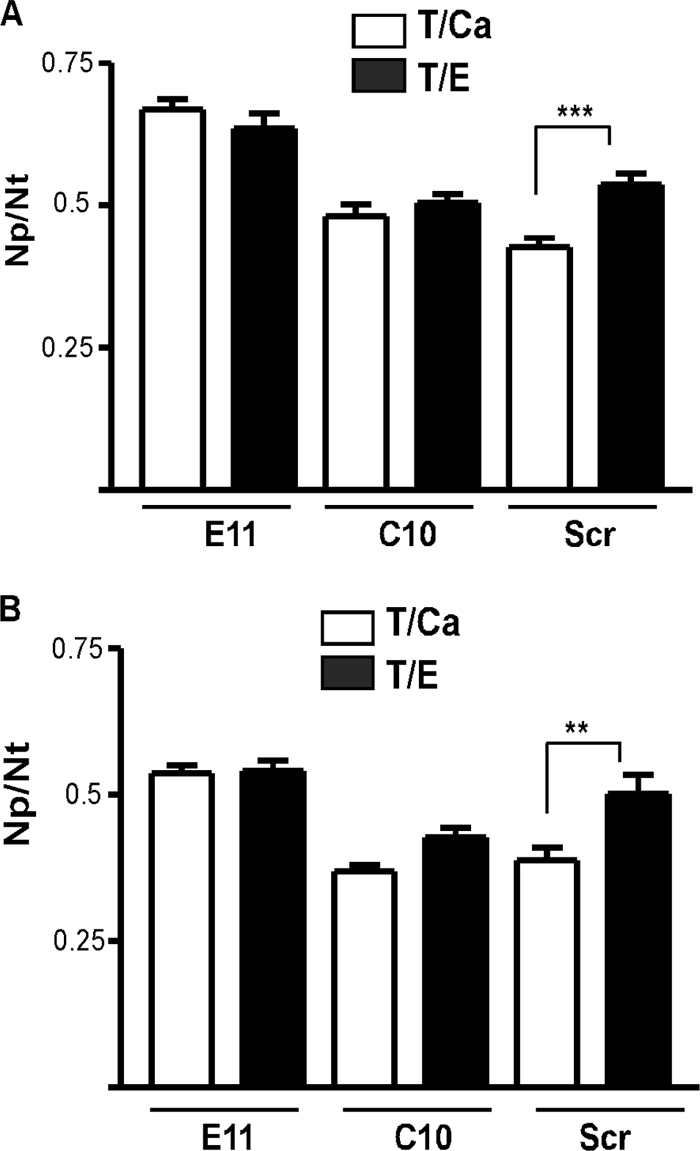
Silencing of Rap1GAP weakens cell/cell adhesion. (A) Cells were trypsinized in the presence of calcium or EGTA, and images were acquired immediately after plating. The graph shows the dissociation index as the Np/Nt ratio, where Np is the number of particles (aggregates and single cells) and Nt is the total number of cells in the presence of calcium (T/Ca) or EGTA (T/E). The dissociation index in the presence of calcium was significantly decreased in control cells but not in the Rap1GAP-depleted cell lines. (B) Dissociation assays were performed with cells plated on PLL for 24 h. Calcium significantly decreased the dissociation index in control cells but not in Rap1GAP-depleted cells.
We surmised that the aberrant distribution of E-cadherin, β-catenin, and p120-catenin in Rap1GAP-depleted cells was due to unrestrained Rap activity. To test this, we investigated whether acute depletion of Rap can reverse these effects. Silencing of Rap1 and Rap2 expression (Fig. 6A and B) in Rap1GAP-depleted cells elicited dramatic effects on cell morphology. Cells depleted of Rap were rounder and more compact and exhibited an increase in cell/cell contacts compared to control siRNA-transfected cells. Moreover, silencing of Rap expression restored the accumulation of E-cadherin (Fig. 6A) and p120-catenin (Fig. 6B) at cell/cell contacts. Similar results were obtained with C10 cells and with both Rap1GAP-deficient cell lines using independent sets of Rap1- and Rap2-directed siRNAs (data not shown). These results confirm that unrestrained Rap activity underlies the defects in cell/cell adhesion and that, remarkably, silencing of Rap expression is sufficient to restore cell/cell adhesion in cells chronically depleted of Rap1GAP.
FIG. 6.

The effects of silencing of Rap1GAP on AJ proteins are Rap dependent. Rap1GAP-depleted (E11) and control (Scr) cells were transiently transfected with Rap1- and Rap2-directed (+) versus scrambled (−) siRNAs. After 48 h, cells were fixed and stained for E-cadherin (A) or p120-catenin (B). Note the accumulation of p120-catenin at cell/cell contacts in Rap1/2-depleted cells (indicated by arrows). Total cell lysates prepared from replicate plates were analyzed for Rap expression.
Downregulation of Rap1GAP enhances cell/substrate adhesion.
In addition to being more dispersed, Rap1GAP-depleted cells appeared flatter and more spread than control cells. Measurements of cell size revealed that acute (Fig. 7A) or chronic (Fig. 7B) silencing of Rap1GAP increased cell area. The increase in area was reversed by silencing of Rap1 and Rap2 expression, documenting that enhanced spreading was a consequence of increased Rap activity. Given the widespread role of Rap1 in regulating integrin activation (reviewed in references 9 and 11), we explored whether silencing of Rap1GAP enhances integrin-mediated adhesion. Downregulation of Rap1GAP increased adhesion (Fig. 8A) and spreading (Fig. 8B and C) on collagen IV. In contrast, there was no difference in the ability of Rap1GAP-depleted versus control cells to adhere or spread on PLL. The localization of active FAK to adhesive structures, a marker of integrin activation, was markedly increased in Rap1GAP-depleted cells plated on collagen IV (Fig. 8C).
FIG. 7.

Downregulation of Rap1GAP increases cell spreading. (A) The areas of HT29 cells transiently transfected with Rap1GAP-directed or scrambled siRNAs were compared. Downregulation of Rap1GAP significantly (P < 0.05) increased cell area. (B) The area of cells in which Rap1GAP expression was chronically suppressed (E11, C10) was significantly increased (P < 0.0005) compared to that of control (Scr) cells. Silencing of Rap1 and Rap2 expression significantly reduced the area of Rap1GAP-depleted cells (P < 0.05 for E11; P < 0.005 for C10).
FIG. 8.

Silencing of Rap1GAP enhances integrin-dependent adhesion and spreading. (A) Rap1GAP-depleted and control cells were plated on collagen IV (open bars) or PLL (filled bars) for 10 min. Silencing of Rap1GAP significantly increased adhesion to collagen (P < 0.0005 for E11, P < 0.005 for C10). (B) The area of Rap1GAP-depleted versus control cells was measured following plating on collagen IV (open bars) or PLL (filled bars) for the times (minutes) indicated. Depletion of Rap1GAP significantly increased cell spreading on collagen IV (P < 0.001 for E11; P < 0.01 for C10 at 60 min). (C) Cells plated on collagen IV or PLL for 60 min were fixed and stained for autophosphorylated FAK (FAKY397). Images were acquired for the same times. (D) Plating of Rap1GAP-depleted cells on PLL (for 16 h) restored the accumulation of E-cadherin and p120-catenin at cell/cell contacts (for examples, see arrowed cells).
We next explored whether the aberrant distribution of AJ proteins in Rap1GAP-depleted cells required matrix adhesion. When Rap1GAP-depleted cells were plated on PLL, the accumulation of E-cadherin and p120-catenin at cell/cell contacts was restored (Fig. 8D). To assess whether impaired integrin signaling restores the functional defects in cell/cell adhesion, the dissociation indices of cells in the absence (Fig. 5A) and presence (Fig. 5B) of PLL were compared. Calcium decreased the dissociation index in control cells but not in Rap1GAP-depleted cells under both conditions. Therefore, although impairing integrin-mediated adhesion restored the accumulation of AJ proteins at cell/cell contacts, it was not sufficient to restore the functional defect in cell/cell adhesion in Rap1GAP-depleted cells.
Downregulation of Rap1GAP increases Src activity.
The profound alterations in cell morphology observed in Rap1GAP-depleted cells were reminiscent of those of Src activation in colon cancer cells. Overexpression of activated Src or activation of endogenous Src disrupted cell/cell adhesion through enhanced cell/matrix adhesion (2, 40). We reasoned that the increase in matrix adhesion conferred by silencing of Rap1GAP could be associated with increased Src activity. To test this, Rap1GAP-depleted and control cells were plated on collagen IV for various times and Src autophosphorylation at Y416 was analyzed as one indicator of Src kinase activity. These experiments were conducted in the absence of serum to selectively monitor adhesion-induced signaling. Autophosphorylation of Src was consistently increased in Rap1GAP-depleted compared to control cells (Fig. 9A and B). Src-mediated phosphorylation of FAK at Y576 was also enhanced in Rap1GAP-depleted cells (Fig. 9A and C). Both phosphorylation events were dependent upon matrix adhesion, as neither protein was phosphorylated in suspended cells (data not shown).
FIG. 9.
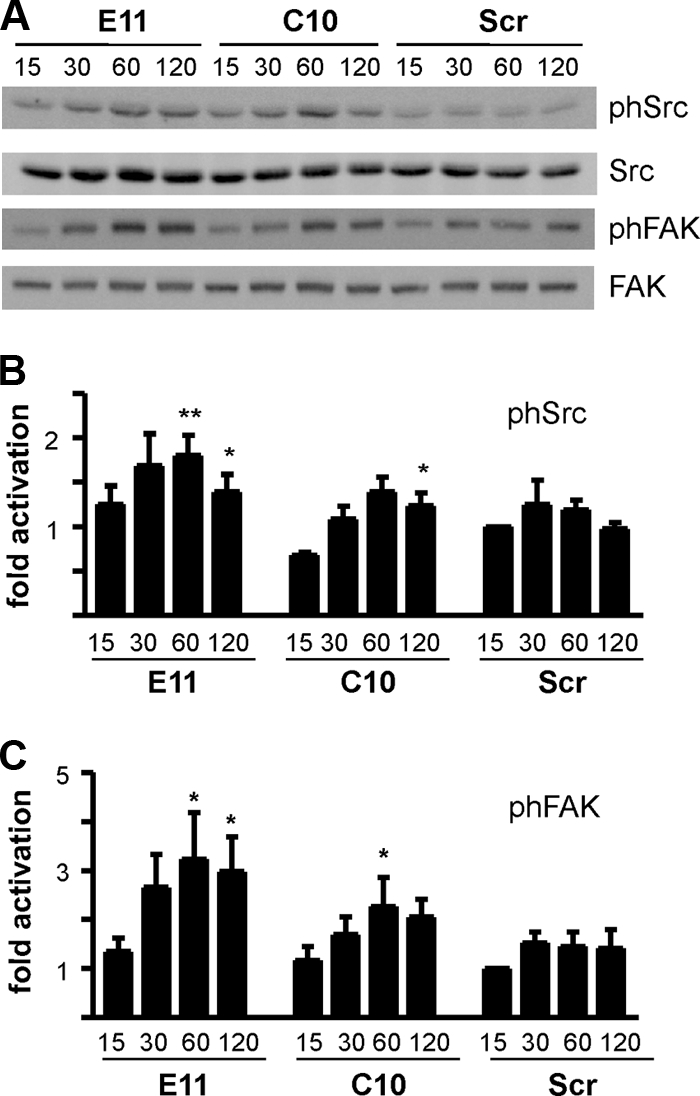
Src activity is increased in Rap1GAP-depleted cells. (A) Rap1GAP-depleted and control cells were plated on collagen IV for the times (minutes) indicated, and total cell lysates were prepared and subjected to Western blotting for autophosphorylated Src (SrcY416 and pSrc), Src, phosphorylated FAK (FAKY576 and pFAK), and FAK. (B and C) The results from multiple experiments are summarized. Phosphorylated Src was normalized to total Src, and phosphorylated FAK was normalized to total FAK. Phosphorylation of Src (P < 0.0005 for E11; P < 0.05 for C10 at 60 min) and FAK (P < 0.0001 for E11; P < 0.05 for C10 at 60 min) was significantly increased in Rap1GAP-depleted cells.
The similarity between the effects of Src activation and silencing of Rap1GAP on cell adhesion suggested that some of the effects of Rap1GAP depletion could be mediated through Src. We confirmed that Src activity was increased in Rap1GAP-depleted cells in the absence of replating (Fig. 10A and B). To assess whether Src activity contributed to the aberrant distribution of AJ proteins, Rap1GAP-depleted cells were treated with the selective Src inhibitor SU6656. Inhibition of Src activity restored the accumulation of E-cadherin at cell/cell contacts in Rap1GAP-depleted cells (Fig. 10C). This was accompanied by an increase in the proportion of E-cadherin in the insoluble fraction, further evidence of increased cell/cell adhesion (data not shown). In contrast, inhibiting Src activity did not fully restore p120-catenin at cell/cell junctions, suggesting that depletion of Rap1GAP elicits Src-independent effects on the distribution of p120-catenin. Inhibition of Src activity decreased the area of Rap1GAP-depleted cells (Fig. 10D). Taken together, these data indicate that chronic suppression of Rap1GAP induces Src-dependent effects on cell/matrix adhesion and the accumulation of E-cadherin at cell/cell contacts.
FIG. 10.

Loss of Rap1GAP induces Src-dependent effects on E-cadherin. (A) Rap1GAP-depleted and control cells were starved overnight, and total cell lysates were prepared and subjected to Western blotting for autophosphorylated and total Src. (B) Summary of multiple experiments showing that Src autophosphorylation was significantly (P < 0.005 for E11; P < 0.05 for C10) increased in Rap1GAP-depleted cells. (C) Rap1GAP-depleted and control cells were exposed to SU6656 (10 μM), fixed, and stained for E-cadherin and p120-catenin. Exposure to SU6656 for 30 min was sufficient to restore membrane E-cadherin. SU6656 failed to fully restore membrane p120-catenin, even after 120 min (shown here). Images for each antibody were acquired for the same times. (D) Measurements of cell size revealed that SU6656 significantly reduced the area of Rap1GAP-depleted cells (P < 0.05 for E11; P < 0.0005 for C10).
DISCUSSION
Our findings provide the first insight into the significance of downregulation of Rap1GAP in human tumor cells. We previously reported that the expression of Rap1GAP is decreased in thyroid adenomas and papillary thyroid carcinomas (37, 47). We now show that Rap1GAP is highly expressed in colonic epithelium and that its expression is markedly decreased in primary colorectal carcinomas. The widespread downregulation of Rap1GAP in human tumors prompted us to explore the cellular consequences associated with the depletion of Rap1GAP. Silencing of Rap1GAP in human colon cancer cells enhanced adhesion to collagen, an important component of tumor stroma, and weakened cell/cell adhesion. These findings provide a rationale for the frequent downregulation of Rap1GAP in epithelial tumors where alterations in cell/matrix adhesion and the weakening of cell/cell contacts are important contributing factors in tumor dissemination.
Rap activation promotes cell/cell adhesion and junctional integrity (reviewed in reference 30). Moreover, activated Rap and RapGEFs have been shown to associate with components of cell/cell junctions (1, 7, 17, 24, 29, 41). Thus, we were initially surprised to find that Rap1GAP-depleted cells, which exhibit durable increases in Rap1 and Rap2 activity, formed fewer cell/cell contacts than control cells. Although steady-state levels of E-cadherin, β-catenin, and p120-catenin were unchanged in Rap1GAP-depleted cells, the intracellular distribution of all three proteins was altered. Most notably, the accumulation of E-cadherin, β-catenin, and p120-catenin at cell/cell contacts was reduced. This occurred in the absence of demonstrable changes in the partitioning of E-cadherin and β-catenin between soluble and insoluble fractions or in cell surface pools of E-cadherin, although p120-catenin was increased in the soluble fraction of Rap1GAP-depleted cells. p120-catenin plays an important role in retaining E-cadherin at the cell surface (reviewed in references 31 and 51). Its increased localization in the cytoplasm prompted us to explore if silencing of Rap1GAP elicited functional effects on cell/cell adhesion. Cell dissociation assays revealed that cell/cell adhesion was weakened in Rap1GAP-depleted cells. These findings are in agreement with reports that cadherin-mediated adhesion can be reduced in the absence of demonstrable changes in steady-state levels or cell surface localization of E-cadherin (52).
The defects in cell/cell adhesion in Rap1GAP-depleted cells were due to unrestrained Rap activity, as silencing of Rap expression increased the number of cell/cell contacts and the accumulation of E-cadherin, β-catenin, and p120-catenin at cell/cell junctions. Although Rap activation promotes cell/cell adhesion, similar to our findings, overexpression of activated Rap has been reported to disrupt cell/cell contacts (7, 24). Rap is activated when cell/cell junctions form (24, 39) and when cell/cell contacts are disrupted (1, 4). In the latter instance, trans ligation of E-cadherin was required to inactivate Rap, highlighting a requirement for Rap inactivation in the formation and/or maturation of cell/cell contacts. Collectively, these findings suggest that cell/cell adhesion requires the cycling of Rap between active and inactive states. The observation that sustained activation of endogenous Rap disrupts cell/cell adhesion in human colon cancer cells demonstrates a heretofore unappreciated role for Rap1GAP in the regulation of cell/cell adhesion. We surmise that Rap1GAP is required for the inactivation of Rap that precedes the formation of cell/cell junctions (1, 4). Interestingly, overexpression of Rap1GAP has been shown to impair the formation (24, 39) or maintenance (16, 41, 50) of cell/cell contacts. The similar effects of Rap1GAP overexpression and downregulation may indicate that Rap1GAP functions as part of a macromolecular complex, where alterations in Rap1GAP expression impact stoichiometry. Besides interacting with Rap, Rap1GAP binds to heterotrimeric G protein α subunits (27, 33, 35) and AF6 (43, 55), a protein that associates with activated Rap and localizes to cell/cell junctions (7).
Rap1GAP-depleted cells were more spread than control cells. Acute plating experiments revealed that silencing of Rap1GAP enhanced adhesion and spreading on collagen IV, as well as the formation or stabilization of structures containing activated FAK. The concomitant effects of Rap1GAP depletion on cell/cell and cell/matrix adhesion prompted us to assess whether these effects are related. Impairing integrin-dependent cell spreading restored the accumulation of E-cadherin, β-catenin, and p120-catenin at cell/cell contacts in Rap1GAP-depleted cells, suggesting that defects in AJ protein localization are due to enhanced interaction of cells with the extracellular matrix. However, cell/cell adhesion remained weakened in Rap1GAP-depleted cells plated on PLL. This suggests that downregulation of Rap1GAP elicits integrin-dependent and -independent effects on cell/cell adhesion. Balzac et al. reported that the disassembly of cadherin-mediated cell/cell junctions activates Rap and that Rap activation was required for the subsequent formation of integrin-dependent focal contacts (4). These findings position Rap as an important site of integration between cadherin- and integrin-mediated signals. Our findings further support this notion by showing that the presence or absence of Rap1GAP is an important determinant of the balance of cell/cell and cell/substrate adhesion.
The expression and activity of Src are increased in CRC with a trend toward increasing activity with disease progression (6, 12, 25, 44). Overexpression of activated Src in KM12C colon cancer cells stimulated the formation of integrin-mediated focal contacts and delayed the recruitment of E-cadherin to cell/cell contacts (2). Inhibitory antibodies to αv or β1 integrins restored the accumulation of E-cadherin at cell/cell contacts, linking the defects in cell/cell adhesion to enhanced integrin signaling. Similarly, increasing endogenous Src activity in HT29 cells enhanced integrin activation and disrupted cell/cell contacts (40). Because silencing of Rap1GAP exhibited effects similar to those of Src activation, we examined whether Src activity is altered by Rap1GAP depletion. Rap1GAP-depleted cells exhibited increased Src activity, as monitored by activating phosphorylation of Src or Src-dependent phosphorylation of FAK. These results are consistent with a report that overexpression of Rap1GAP inhibited Src phosphorylation in melanoma cell lines (56). Intriguingly, inhibition of Src kinase activity restored cell/cell contact localization of E-cadherin but not of p120-catenin. Src inhibition also impaired cell spreading, suggesting that at least some of the effects of Rap1GAP depletion on cell adhesion are mediated through Src. Although Rap is typically activated downstream from Src, our findings suggest that downregulation of Rap1GAP acts upstream from Src in deregulating cell adhesion.
The significance of the effects of Rap1GAP depletion in vitro to those in human tumors remains to be established. E-cadherin and p120-catenin are membranous in normal colonic epithelium, and there is a trend toward the increased cytoplasmic localization of both proteins in colorectal tumors (5, 22, 32). We observed cytoplasmic staining for E-cadherin and p120-catenin in primary colon tumors where Rap1GAP expression was decreased (unpublished data). Because downregulation of Rap1GAP and alterations in the localization of E-cadherin and p120-catenin are frequent events, the analysis of a large number of tumors is required to determine if these effects are correlated. Remarkably, silencing of Rap expression restored cell/cell adhesion in cells where Rap1GAP expression was chronically suppressed. This implies that the defects in Rap1GAP-depleted cells are potentially reversible and that strategies to restore Rap1GAP expression or to inhibit Rap activity should be evaluated for their effects on cell/cell adhesion in human tumors.
The signals that trigger the downregulation of Rap1GAP in human tumors remain to be identified. Acute expression of activated Ras decreased Rap1GAP expression in thyroid epithelial cells (47). Ras mutations are prevalent in tumors of the pancreas, thyroid, and colon, all of which exhibit downregulation of Rap1GAP (37, 53). However, Rap1GAP expression was decreased in papillary thyroid carcinomas, which rarely exhibit Ras mutations. Wnt signaling has been shown to target SIPAL1 for proteasomal turnover (45). Whether the same is true for Rap1GAP remains to be determined. Other signals regulate Rap1GAP protein stability (28), suggesting that multiple signals impinge upon RapGAPs. Investigation of the regulation of Rap1GAP expression and activity, together with the identification of its protein partners, promises to provide further insight into the roles of Rap signaling in the control of cell architecture.
Supplementary Material
Acknowledgments
We are grateful to John Lynch for providing cell lines and to Margaret Chou and Richard Assoian for reviewing the manuscript.
This work was supported by Public Health Service grant CA127986 awarded to J.M.
We have no conflicts of interest to disclose.
Footnotes
Published ahead of print on 3 May 2010.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Asuri, S., J. Yan, N. C. Paranavitana, and L. A. Quilliam. 2008. E-cadherin dis-engagement activates the Rap1 GTPase. J. Cell. Biochem. 105:1027-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avizienyte, E., A. W. Wyke, R. J. Jones, G. W. McLean, M. A. Westhoff, V. G. Brunton, and M. C. Frame. 2002. Src-induced de-regulation of E-cadherin in colon cancer cells requires integrin signalling. Nat. Cell Biol. 4:632-638. [DOI] [PubMed] [Google Scholar]
- 3.Bailey, C. L., P. Kelly, and P. J. Casey. 2009. Activation of Rap1 promotes prostate cancer metastasis. Cancer Res. 69:4962-4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balzac, F., M. Avolio, S. Degani, I. Kaverina, M. Torti, L. Silengo, J. V. Small, and S. F. Retta. 2005. E-cadherin endocytosis regulates the activity of Rap1: a traffic light GTPase at the crossroads between cadherin and integrin function. J. Cell Sci. 118:4765-4783. [DOI] [PubMed] [Google Scholar]
- 5.Bellovin, D. I., R. C. Bates, A. Muzikansky, D. L. Rimm, and A. M. Mercurio. 2005. Altered localization of p120 catenin during epithelial to mesenchymal transition of colon carcinoma is prognostic for aggressive disease. Cancer Res. 65:10938-10945. [DOI] [PubMed] [Google Scholar]
- 6.Biscardi, J. S., D. A. Tice, and S. J. Parsons. 1999. c-Src, receptor tyrosine kinases, and human cancer. Adv. Cancer Res. 76:61-119. [DOI] [PubMed] [Google Scholar]
- 7.Boettner, B., E. E. Govek, J. Cross, and L. Van Aelst. 2000. The junctional multidomain protein AF-6 is a binding partner of the Rap1A GTPase and associates with the actin cytoskeletal regulator profilin. Proc. Natl. Acad. Sci. U. S. A. 97:9064-9069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bos, J. L. 2005. Linking Rap to cell adhesion. Curr. Opin. Cell Biol. 17:123-128. [DOI] [PubMed] [Google Scholar]
- 9.Bos, J. L., K. de Bruyn, J. Enserink, B. Kuiperij, S. Rangarajan, H. Rehmann, J. Riedl, J. de Rooij, F. van Mansfeld, and F. Zwartkruis. 2003. The role of Rap1 in integrin-mediated cell adhesion. Biochem. Soc. Trans. 31:83-86. [DOI] [PubMed] [Google Scholar]
- 10.Bos, J. L., H. Rehmann, and A. Wittinghofer. 2007. GEFs and GAPs: critical elements in the control of small G proteins. Cell 129:865-877. [DOI] [PubMed] [Google Scholar]
- 11.Caron, E. 2003. Cellular functions of the Rap1 GTP-binding protein: a pattern emerges. J. Cell Sci. 116:435-440. [DOI] [PubMed] [Google Scholar]
- 12.Cartwright, C. A., C. A. Coad, and B. M. Egbert. 1994. Elevated c-Src tyrosine kinase activity in premalignant epithelia of ulcerative colitis. J. Clin. Invest. 93:509-515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crawford, N. P., X. Qian, A. Ziogas, A. G. Papageorge, B. J. Boersma, R. C. Walker, L. Lukes, W. L. Rowe, J. Zhang, S. Ambs, D. R. Lowy, H. Anton-Culver, and K. W. Hunter. 2007. Rrp1b, a new candidate susceptibility gene for breast cancer progression and metastasis. PLoS Genet. 3:e214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crawford, N. P., R. C. Walker, L. Lukes, J. S. Officewala, R. W. Williams, and K. W. Hunter. 2008. The diasporin pathway: a tumor progression-related transcriptional network that predicts breast cancer survival. Clin. Exp. Metastasis 25:357-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crawford, N. P., A. Ziogas, D. J. Peel, J. Hess, H. Anton-Culver, and K. W. Hunter. 2006. Germline polymorphisms in SIPA1 are associated with metastasis and other indicators of poor prognosis in breast cancer. Breast Cancer Res. 8:R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cullere, X., S. K. Shaw, L. Andersson, J. Hirahashi, F. W. Luscinskas, and T. N. Mayadas. 2005. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood 105:1950-1955. [DOI] [PubMed] [Google Scholar]
- 17.Dubé, N., M. R. Kooistra, W. J. Pannekoek, M. J. Vliem, V. Oorschot, J. Klumperman, H. Rehmann, and J. L. Bos. 2008. The RapGEF PDZ-GEF2 is required for maturation of cell-cell junctions. Cell Signal. 20:1608-1615. [DOI] [PubMed] [Google Scholar]
- 18.Farina, A., M. Hattori, J. Qin, Y. Nakatani, N. Minato, and K. Ozato. 2004. Bromodomain protein Brd4 binds to GTPase-activating SPA-1, modulating its activity and subcellular localization. Mol. Cell. Biol. 24:9059-9069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fearon, E. R., and B. Vogelstein. 1990. A genetic model for colorectal tumorigenesis. Cell 61:759-767. [DOI] [PubMed] [Google Scholar]
- 20.Gao, Q., L. Singh, A. Kumar, S. Srinivasan, D. E. Wazer, and V. Band. 2001. Human papillomavirus type 16 E6-induced degradation of E6TP1 correlates with its ability to immortalize human mammary epithelial cells. J. Virol. 75:4459-4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao, Q., S. Srinivasan, S. N. Boyer, D. E. Wazer, and V. Band. 1999. The E6 oncoproteins of high-risk papillomaviruses bind to a novel putative GAP protein, E6TP1, and target it for degradation. Mol. Cell. Biol. 19:733-744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hahn-Strömberg, V., H. Edvardsson, L. Bodin, and L. Franzen. 2008. Disturbed expression of E-cadherin, beta-catenin and tight junction proteins in colon carcinoma is unrelated to growth pattern and genetic polymorphisms. APMIS 116:253-262. [DOI] [PubMed] [Google Scholar]
- 23.Hodi, Z., J. Chakrabarti, A. H. Lee, J. E. Ronan, C. W. Elston, K. L. Cheung, J. F. Robertson, and I. O. Ellis. 2007. The reliability of assessment of oestrogen receptor expression on needle core biopsy specimens of invasive carcinomas of the breast. J. Clin. Pathol. 60:299-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hogan, C., N. Serpente, P. Cogram, C. R. Hosking, C. U. Bialucha, S. M. Feller, V. M. Braga, W. Birchmeier, and Y. Fujita. 2004. Rap1 regulates the formation of E-cadherin-based cell-cell contacts. Mol. Cell. Biol. 24:6690-6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Irby, R. B., W. Mao, D. Coppola, J. Kang, J. M. Loubeau, W. Trudeau, R. Karl, D. J. Fujita, R. Jove, and T. J. Yeatman. 1999. Activating SRC mutation in a subset of advanced human colon cancers. Nat. Genet. 21:187-190. [DOI] [PubMed] [Google Scholar]
- 26.Ishida, D., K. Kometani, H. Yang, K. Kakugawa, K. Masuda, K. Iwai, M. Suzuki, S. Itohara, T. Nakahata, H. Hiai, H. Kawamoto, M. Hattori, and N. Minato. 2003. Myeloproliferative stem cell disorders by deregulated Rap1 activation in SPA-1-deficient mice. Cancer Cell 4:55-65. [DOI] [PubMed] [Google Scholar]
- 27.Jordan, J. D., K. D. Carey, P. J. Stork, and R. Iyengar. 1999. Modulation of rap activity by direct interaction of Galpha(o) with Rap1 GTPase-activating protein. J. Biol. Chem. 274:21507-21510. [DOI] [PubMed] [Google Scholar]
- 28.Jordan, J. D., J. C. He, N. J. Eungdamrong, I. Gomes, W. Ali, T. Nguyen, T. G. Bivona, M. R. Philips, L. A. Devi, and R. Iyengar. 2005. Cannabinoid receptor-induced neurite outgrowth is mediated by Rap1 activation through G(alpha)o/i-triggered proteasomal degradation of Rap1GAPII. J. Biol. Chem. 280:11413-11421. [DOI] [PubMed] [Google Scholar]
- 29.Kawajiri, A., N. Itoh, M. Fukata, M. Nakagawa, M. Yamaga, A. Iwamatsu, and K. Kaibuchi. 2000. Identification of a novel beta-catenin-interacting protein. Biochem. Biophys. Res. Commun. 273:712-717. [DOI] [PubMed] [Google Scholar]
- 30.Kooistra, M. R., N. Dubé, and J. L. Bos. 2007. Rap1: a key regulator in cell-cell junction formation. J. Cell Sci. 120:17-22. [DOI] [PubMed] [Google Scholar]
- 31.Kowalczyk, A. P., and A. B. Reynolds. 2004. Protecting your tail: regulation of cadherin degradation by p120-catenin. Curr. Opin. Cell Biol. 16:522-527. [DOI] [PubMed] [Google Scholar]
- 32.Lugli, A., I. Zlobec, P. Minoo, K. Baker, L. Tornillo, L. Terracciano, and J. R. Jass. 2007. Prognostic significance of the wnt signalling pathway molecules APC, beta-catenin and E-cadherin in colorectal cancer: a tissue microarray-based analysis. Histopathology 50:453-464. [DOI] [PubMed] [Google Scholar]
- 33.Meng, J., J. L. Glick, P. Polakis, and P. J. Casey. 1999. Functional interaction between Galpha(z) and Rap1GAP suggests a novel form of cellular cross-talk. J. Biol. Chem. 274:36663-36669. [DOI] [PubMed] [Google Scholar]
- 34.Mitra, R. S., M. Goto, J. S. Lee, D. Maldonado, J. M. Taylor, Q. Pan, T. E. Carey, C. R. Bradford, M. E. Prince, K. G. Cordell, K. L. Kirkwood, and N. J. D'Silva. 2008. Rap1GAP promotes invasion via induction of matrix metalloproteinase 9 secretion, which is associated with poor survival in low N-stage squamous cell carcinoma. Cancer Res. 68:3959-3969. [DOI] [PubMed] [Google Scholar]
- 35.Mochizuki, N., Y. Ohba, E. Kiyokawa, T. Kurata, T. Murakami, T. Ozaki, A. Kitabatake, K. Nagashima, and M. Matsuda. 1999. Activation of the ERK/MAPK pathway by an isoform of rap1GAP associated with G alpha. Nature 400:891-894. [DOI] [PubMed] [Google Scholar]
- 36.Nagai, H., M. Negrini, S. L. Carter, D. R. Gillum, A. L. Rosenberg, G. F. Schwartz, and C. M. Croce. 1995. Detection and cloning of a common region of loss of heterozygosity at chromosome 1p in breast cancer. Cancer Res. 55:1752-1757. [PubMed] [Google Scholar]
- 37.Nellore, A., K. Paziana, C. Ma, O. M. Tsygankova, Y. Wang, K. Puttaswamy, A. U. Iqbal, S. R. Franks, Y. Lv, A. B. Troxel, M. D. Feldman, J. L. Meinkoth, and M. S. Brose. 2009. Loss of Rap1GAP in papillary thyroid cancer. J. Clin. Endocrinol. Metab. 94:1026-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park, Y. G., X. Zhao, F. Lesueur, D. R. Lowy, M. Lancaster, P. Pharoah, X. Qian, and K. W. Hunter. 2005. Sipa1 is a candidate for underlying the metastasis efficiency modifier locus Mtes1. Nat. Genet. 37:1055-1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Price, L. S., A. Hajdo-Milasinovic, J. Zhao, F. J. Zwartkruis, J. G. Collard, and J. L. Bos. 2004. Rap1 regulates E-cadherin-mediated cell-cell adhesion. J. Biol. Chem. 279:35127-35132. [DOI] [PubMed] [Google Scholar]
- 40.Rengifo-Cam, W., A. Konishi, N. Morishita, H. Matsuoka, T. Yamori, S. Nada, and M. Okada. 2004. Csk defines the ability of integrin-mediated cell adhesion and migration in human colon cancer cells: implication for a potential role in cancer metastasis. Oncogene 23:289-297. [DOI] [PubMed] [Google Scholar]
- 41.Sakurai, A., S. Fukuhara, A. Yamagishi, K. Sako, Y. Kamioka, M. Masuda, Y. Nakaoka, and N. Mochizuki. 2006. MAGI-1 is required for Rap1 activation upon cell-cell contact and for enhancement of vascular endothelial cadherin-mediated cell adhesion. Mol. Biol. Cell 17:966-976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sjöblom, T., S. Jones, L. D. Wood, D. W. Parsons, J. Lin, T. D. Barber, D. Mandelker, R. J. Leary, J. Ptak, N. Silliman, S. Szabo, P. Buckhaults, C. Farrell, P. Meeh, S. D. Markowitz, J. Willis, D. Dawson, J. K. Willson, A. F. Gazdar, J. Hartigan, L. Wu, C. Liu, G. Parmigiani, B. H. Park, K. E. Bachman, N. Papadopoulos, B. Vogelstein, K. W. Kinzler, and V. E. Velculescu. 2006. The consensus coding sequences of human breast and colorectal cancers. Science 314:268-274. [DOI] [PubMed] [Google Scholar]
- 43.Su, L., M. Hattori, M. Moriyama, N. Murata, M. Harazaki, K. Kaibuchi, and N. Minato. 2003. AF-6 controls integrin-mediated cell adhesion by regulating Rap1 activation through the specific recruitment of Rap1GTP and SPA-1. J. Biol. Chem. 278:15232-15238. [DOI] [PubMed] [Google Scholar]
- 44.Talamonti, M. S., M. S. Roh, S. A. Curley, and G. E. Gallick. 1993. Increase in activity and level of pp60c-src in progressive stages of human colorectal cancer. J. Clin. Invest. 91:53-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsai, I. C., J. D. Amack, Z. H. Gao, V. Band, H. J. Yost, and D. M. Virshup. 2007. A Wnt-CKIvarepsilon-Rap1 pathway regulates gastrulation by modulating SIPA1L1, a Rap GTPase activating protein. Dev. Cell 12:335-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsygankova, O. M., E. Feshchenko, P. S. Klein, and J. L. Meinkoth. 2004. Thyroid-stimulating hormone/cAMP and glycogen synthase kinase 3{beta} elicit opposing effects on Rap1GAP stability. J. Biol. Chem. 279:5501-5507. [DOI] [PubMed] [Google Scholar]
- 47.Tsygankova, O. M., G. V. Prendergast, K. Puttaswamy, Y. Wang, M. D. Feldman, H. Wang, M. S. Brose, and J. L. Meinkoth. 2007. Downregulation of Rap1GAP contributes to Ras transformation. Mol. Cell. Biol. 27:6647-6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vogelstein, B., E. R. Fearon, S. R. Hamilton, S. E. Kern, A. C. Preisinger, M. Leppert, Y. Nakamura, R. White, A. M. Smits, and J. L. Bos. 1988. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 319:525-532. [DOI] [PubMed] [Google Scholar]
- 49.Williamson, C., A. A. Pannett, J. T. Pang, C. Wooding, M. McCarthy, M. N. Sheppard, J. Monson, R. N. Clayton, and R. V. Thakker. 1997. Localisation of a gene causing endocrine neoplasia to a 4 cM region on chromosome 1p35-p36. J. Med. Genet. 34:617-619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wittchen, E. S., R. A. Worthylake, P. Kelly, P. J. Casey, L. A. Quilliam, and K. Burridge. 2005. Rap1 GTPase inhibits leukocyte transmigration by promoting endothelial barrier function. J. Biol. Chem. 280:11675-11682. [DOI] [PubMed] [Google Scholar]
- 51.Xiao, K., R. G. Oas, C. M. Chiasson, and A. P. Kowalczyk. 2007. Role of p120-catenin in cadherin trafficking. Biochim. Biophys. Acta 1773:8-16. [DOI] [PubMed] [Google Scholar]
- 52.Yap, A. S., M. S. Crampton, and J. Hardin. 2007. Making and breaking contacts: the cellular biology of cadherin regulation. Curr. Opin. Cell Biol. 19:508-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang, L., L. Chenwei, R. Mahmood, K. van Golen, J. Greenson, G. Li, N. J. D'Silva, X. Li, C. F. Burant, C. D. Logsdon, and D. M. Simeone. 2006. Identification of a putative tumor suppressor gene Rap1GAP in pancreatic cancer. Cancer Res. 66:898-906. [DOI] [PubMed] [Google Scholar]
- 54.Zhang, Z., R. S. Mitra, B. S. Henson, N. S. Datta, L. K. McCauley, P. Kumar, J. S. Lee, T. E. Carey, and N. J. D'Silva. 2006. Rap1GAP inhibits tumor growth in oropharyngeal squamous cell carcinoma. Am. J. Pathol. 168:585-596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang, Z., H. Rehmann, L. S. Price, J. Riedl, and J. L. Bos. 2005. AF6 negatively regulates Rap1-induced cell adhesion. J. Biol. Chem. 280:33200-33205. [DOI] [PubMed] [Google Scholar]
- 56.Zheng, H., L. Gao, Y. Feng, L. Yuan, H. Zhao, and L. A. Cornelius. 2009. Down-regulation of Rap1GAP via promoter hypermethylation promotes melanoma cell proliferation, survival, and migration. Cancer Res. 69:449-457. [DOI] [PubMed] [Google Scholar]
- 57.Zuo, H., M. Gandhi, M. M. Edreira, D. Hochbaum, V. L. Nimgaonkar, P. Zhang, J. Dipaola, V. Evdokimova, D. L. Altschuler, and Y. E. Nikiforov. 2010. Downregulation of Rap1GAP through epigenetic silencing and loss of heterozygosity promotes invasion and progression of thyroid tumors. Cancer Res. 70:1389-1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.