

Abstract
Autophagy is one of two major degradation systems in eukaryotic cells. The degradation mechanism of autophagy is required to maintain the balance between the biosynthetic and catabolic processes and also contributes to defense against invading pathogens. Recent studies suggest that a number of viruses can evade or subvert the host cell autophagic pathway to enhance their own replication. Here, we investigated the effect of autophagy on the KSHV (Kaposi's sarcoma-associated herpesvirus) life cycle. We found that the inhibition of autophagy reduces KSHV lytic reactivation from latency, and an enhancement of autophagy can be detected during KSHV lytic replication. In addition, RTA (replication and transcription activator), an essential viral protein for KSHV lytic reactivation, is able to enhance the autophagic process, leading to an increase in the number of autophagic vacuoles, an increase in the level of the lipidated LC3 protein, and the formation of autolysosomes. Moreover, the inhibition of autophagy affects RTA-mediated lytic gene expression and viral DNA replication. These results suggest that RTA increases autophagy activation to facilitate KSHV lytic replication. This is the first report demonstrating that autophagy is involved in the lytic reactivation of KSHV.
Autophagy is an intracellular catabolic mechanism and is principally responsible for the degradation of long-lived cellular proteins and damaged organelles. The degraded products are recycled primarily to supply nutrients when cells are undergoing nutrient deficiency. The hallmark of autophagy is the formation of double-membrane cytosolic vacuoles, called autophagosomes, which sequester entire organelles and large protein aggregates. Eventually, autophagosomes fuse with the lysosomes to generate single-membrane vacuoles, termed autolysosomes, where the contents subsequently are degraded and/or recycled by lysosomal hydrolases (34). This intracellular degradation system is tightly regulated by a family of genes, known as autophagy-related genes (Atg), which were described initially in yeast (27, 62). In higher eukaryotes, a number of Atg genes homologous to yeast Atg have been shown to be essential for autophagy formation in various eukaryotic systems (34). In addition to the classical homeostatic function, autophagy plays an important role in multiple biological processes, including differentiation, development, anti-aging, and cell death, and aberrant autophagy is implicated in a number of human diseases, such as cancer, neurodegeneration, and certain muscular myopathies (22, 35, 49).
The function of autophagy in cellular defense is to remove invading pathogens, including viruses; however, some pathogens have developed strategies to adopt the host autophagic machinery for their own survival and replication (26, 33). Infection by the single-stranded DNA virus B19 parvovirus induces autophagy to prolong the survival of the infected cells (42). The infection of the positive-stranded RNA viruses, such as poliovirus, coxsackievirus, and dengue virus, induces double-membrane vesicles resembling autophagosomes to increase viral RNA replication (21, 32, 53, 61). The induction of autophagosomes by poliovirus also is proposed to play a role in the nonlytic mechanism for poliovirus release (21, 25). Autophagy is also an important innate immunity mechanism that some viruses can evade by subverting or hijacking the autophagy process. For example, herpes simplex virus type 1 (HSV-1) ICP34.5 targets the mammalian autophagy protein Beclin 1 to block the host autophagy machinery to induce neurovirulence (43). Also, ICP34.5 can block autophagy through interference with the phosphorylation of eIF2α (eukaryotic translation initiation factor 2 alpha) by PKR (54). Epstein-Barr virus (EBV) latent membrane protein 1 (LMP1), which is required for the proliferation of infected B cells, utilizes autophagic degradation to limit its accumulation in EBV-infected B cells (30). The human immunodeficiency virus type I (HIV-1) envelope glycoprotein-mediated killing of uninfected CD4 T cells was found to be dependent on the autophagy machinery and may be involved in the pathogenesis of AIDS (14, 15). Overall, these studies present a complex picture defining the role of autophagy in viral pathogenesis, but this effect is both virus and host strain specific.
Kaposi's sarcoma-associated herpesvirus (KSHV) belongs to the gammaherpesvirus family and is associated with Kaposi's sarcoma and other malignancies, such as primary effusion lymphoma (PEL) and multicentric Castleman's disease (MCD). As in all herpesviruses, KSHV exhibits two phases in its replication cycle, latent and lytic phases. During latency, KSHV is capable of evading the immune surveillance to persist in host cells without viral production; however, infectious viral particles can be produced and released after the induction of lytic reactivation from latency as a result of stress or chemical stimuli, such as phorbol esters or sodium butyrate (4, 5, 39, 40, 46). In addition to in vitro stimuli, an immediate-early KSHV gene, ORF50, encodes the replication and transcription activator (RTA), which has the ability to initiate the entire lytic reactivation cascade (38). RTA is a typical transcriptional factor containing an N-terminal DNA-binding domain and a C-terminal activation domain. RTA can trigger KSHV lytic reactivation via the transcriptional activation of a number of viral lytic promoters (2, 6, 8, 11-13). However, for the virus to regulate between lytic replication and latency, the transactivation function of RTA can be suppressed by various viral and cellular repressors to limit the extent of lytic replication to return to latency (3, 17, 18, 20, 29, 58, 63-65). To overcome this suppression to facilitate lytic reactivation, viral RTA has the ability to promote the degradation of the repressors by modulating the ubiquitin-proteasome pathway (16, 66, 67). Since a cellular regulatory mechanism is adopted by KSHV to switch between the lytic cycle and latency and various viruses have adopted the autophagy pathway to regulate their replication, it will be of interest to determine whether KSHV replication also involves the autophagy pathway. In this study, we found that the activation of autophagy is enhanced during KSHV lytic reactivation, and the viral lytic replication inducer RTA is involved in this activation.
MATERIALS AND METHODS
Cells, viruses, plasmids, and reagents.
Human 293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad CA) supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT), 100 μg/ml penicillin-streptomycin (Mediatech) at 37°C with 5% CO2. For starvation experiments, the cells were washed with 1× phosphate-buffered saline (PBS) three times and incubated in 1× Earle's balanced solution (Invitrogen) for 90 min. BCBL-1 is a KSHV-positive primary effusion lymphoma cell line, and BJAB is a KSHV-negative cell line derived from Burkitt's lymphoma. TRExBCBL1-RTA and TRExBJAB-RTA, both carrying a tetracycline-inducible RTA gene, were provided by Jae Jung (University of Southern California, Los Angeles, CA) (41). These B cells were grown in RPMI 1640 medium (Gibco BRL) supplemented with 10% FBS and 100 μg/ml penicillin-streptomycin at 37°C with 5% CO2. Vero cells infected by rKSHV.219 virus were provided by Jeffrey Vieira (University of Washington, Seattle, WA) (57). The cells were grown in DMEM supplemented with 10% FBS, 100 μg/ml penicillin-streptomycin, and 6 μg/ml puromycin (Sigma) at 37°C with 5% CO2. Bac50, a recombinant baculovirus expressing KSHV RTA, also was provided by Jeffrey Vieira for the induction of KSHV lytic replication as described earlier (57). To establish the knockdown cell line, Beclin 1 (BECN 1) and scrambled negative control (Scramble) shRNA plasmids were purchased from Origene and were transfected into TRExBCBL1-RTA cells using the nucleofector system (Lonza), and the transfected cells were selected by puromycin.
RTA expression plasmid (pCMVtagORF50), which encodes Flag-tagged full-length RTA, has been described previously (59). mRTA contains truncated RTA (amino acid 1 to 527), which was cloned in the pCMVtag-2A vector. Plasmid pGFP-LC3, encoding the green fluorescent protein (GFP)-tagged rat LC3 gene, was obtained from N. Mizushima (Tokyo Medical and Dental University, Japan) (23).
Bafilomycin A1, 3-methyladenine (3-MA), sodium butyrate (NaB), and 12-O-tetradecanoylphorbol-13-acetate (TPA) were purchased from Sigma.
Flow-cytometric analysis and cell viability assay.
The rKSHV.219-infected Vero cells were infected with Bac50 virus for 4 h and treated with sodium butyrate (Sigma) for 24 h and/or 3-methyladenine (Sigma) at the indicated time points. The cells were harvested and analyzed with a FACSCalibur flow cytometer using the Cell Quest software (BD Biosciences). rKSHV.219-infected Vero cells were treated with 3-MA for 24 and 40 h, and their viability was measured by a cell viability analyzer (Vi-cell; Beckman).
Western blot analysis.
Cells were harvested, and the cell pellets were resuspended in M-PER buffer (Pierce) containing protease inhibitor cocktail (Pierce) at 4°C for 20 min, followed by centrifugation at 10,000 rpm for 5 min at 4°C. Cell lysates were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to a polyvinylidene difluoride (PVDF) membrane (GE Health Care), followed by incubation with specific antibodies as described previously (65). LC3 antibody was purchased from Abcam Inc., and BECN1 antibody against human Beclin 1 was purchased from Santa Cruz Biotechnology. For protein loading controls, anti-tubulin antibody and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody were purchased from Santa Cruz Biotechnology. For the detection of His- and Flag-tagged proteins, the horseradish peroxidase (HRP)-conjugated anti-6×His antibody and mouse anti-Flag M2 monoclonal antibody were purchased from Clontech and Stratagene. Anti-K8a antibody was purchased from Novous, and anti-RTA antibody was provided by Luwen Zhang (University of Nebraska-Lincoln). The band intensities were measured by using the NIH image software ImageJ.
Confocal microscopy and LysoTracker red and 4′,6′-diamidino-2-phenylindole (DAPI) staining.
Quantitative GFP-LC3 autophagy assays were performed in 293T and TRExBJAB-RTA cells transfected with GFP-LC3 expression plasmids. The 293T cells were grown in 35-mm coverslip-bottom dishes (BD Biocoat) and cultured under the conditions indicated. To express RTA proteins in cells, RTA expression plasmid (pCMVtagORF50) was cotransfected into 293T cells along with GFP-LC3 expression plasmids. To induce RTA expression in TRExBJAB-RTA cells, the cells were treated with doxycycline after the transfection of GFP-LC3. For TRExBJAB-RTA, cells were fixed using 4% paraformaldehyde in PBS for 15 min at room temperature and then washed with PBS twice and deposited onto a slide for confocal microscopy visualization. A series of optical images were obtained with an Olympus FV500 confocal system on an inverted microscope, using the 488-nm laser line (522-nm emission) for GFP detection.
Cells containing ≥3 GFP-LC3 dots were defined as autophagy-positive cells. The number of cells with GFP-LC3 punctate dots relative to all GFP-LC3-positive cells was counted (a minimum of 200 GFP-LC3-positive cells were counted in total for each experiment) and is presented as percentages.
For LysoTracker red staining, the cells were treated with 1 μM LysoTracker Red DND-99 (Invitrogen) at 37°C for 30 min. Depending on the experiments, 293T cells were starved in Earle's balanced salts solution (EBSS; Gibco-BRL) for 90 min in the presence of LysoTracker red dye. Cells then were fixed in 3.7% formaldehyde, and nuclei were stained with DAPI (Calbiochem) for 10 min. Three-channel, optical images (DAPI, GFP, and Lysotracker red) were collected using the sequential scanning mode (405-, 488-, and 543-nm excitation; 450-, 522-, and 595-nm emission, respectively) of the Olympus FV500 confocal system.
Transmission electron microscopy (TEM).
293T cells were transfected with pCMVtagORF50 or pCMVtag (control) plasmids and harvested at 24 h posttransfection. Cell pellets were washed with 1× PBS once and fixed with 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) for 1 h at room temperature. Fixed cells were dehydrated through a graduated ethanol series and embedded in Epon 812 (Electron Microscopic Sciences, Fort Washington, PA). Thin sections (60 to 80 nm) were stained with uranyl acetate and lead citrate and observed under a transmission electron microscope (Hitachi H7500-I). A series of ultrastructural images were collected with a bottom-mount digital camera for the confirmational analysis of autophagy.
Real-time RT-PCR.
Total RNA was isolated using an RNA mini kit (Qiagen) using the protocol recommended by the supplier. RNA samples were digested with DNase (Invitrogen) to remove residual DNA. Real-time reverse transcription-PCR (RT-PCR) was carried out using an iScript one-step RT-PCR kit with SYBR green. The primers used for the mRNA quantitation were ORF57 (5′-GCATATTTGGTAGCGATGGG-3′ [forward] and 5′-GGGATAGTTAGGACAAAGGC-3′[reverse]) and K8.1 (5′-CGCTCCTAATCCTATGCCTT-3′ [forward] and 5′-CTGATAAACCTGTCCACTCC-3′ [reverse]). All reactions were performed in duplicate. For the calculation of the relative mRNA amount from quantitative real-time PCR, the CT (threshold cycle) value of each viral gene was normalized by the CT value of GAPDH, and the normalized CT values from samples were compared to those of the control samples (untreated).
Viral DNA copy number.
The intracellular viral DNA was extracted from TRExBCBL1-RTA cells using a genomic DNA purification kit (Gentra). The viral DNA was quantified by real-time PCR using iQ SYBR green supermix (Bio-Rad). To generate a standard curve for cycle thresholds versus genomic copy numbers, the pCMVtagORF50 plasmid was serially diluted to known concentrations. Primers for the amplification of the ORF50 gene were 5′-CAAACCCCATCCCAACAT-3′ and 5′-AGTAATCACGGCCCCTT-3′. The DNA copy number was calculated using Bio-Rad iCycler software (version 3.1).
RESULTS
An autophagy inhibitor suppresses KSHV lytic reactivation.
To investigate whether autophagy affects KSHV lytic reactivation, a specific autophagy inhibitor, 3-methyladenine (3-MA), was used to suppress the autophagic pathway in rKSHV.219 latently infected Vero cells, and cells were examined by microscopy and flow cytometry (48). Vero cells were latently infected by a recombinant green fluorescent protein (GFP)-red fluorescent protein (RFP) double-labeled rKSHV.219 virus, which constitutively expresses GFP. Upon the induction of lytic reactivation, cells will express RFP under the control of the lytic PAN promoter (57). It had been shown that the highest number of cells expressing lytic proteins can be induced by treatment with sodium butyrate (NaB) and the infection of RTA expressing baculovirus Bac50 (57). Therefore, we followed this protocol for the induction of KSHV lytic reactivation in rKSHV.219-infected Vero cells. Figure 1 A shows that the number of cells harboring reactivated KSHV at 40 h postinduction was higher than that at 24 h postinduction. However, in the presence of the autophagy inhibitor 3-MA, the number of KSHV-reactivated cells was reduced compared to that of the untreated control. There was a 60% reduction of RFP-expressing cells at 24 h after 3-MA treatment and a 33% reduction at 40 h, as determined by flow cytometry (Fig. 1B). The cells expressing GFP were not affected by 3-MA treatment due to the similar percentage of cells expressing GFP in the absence and presence of 3-MA (89.2 and 90.2%, respectively; data not shown). In addition, similar cell viabilities were observed in both 3-MA-treated and untreated cells, indicating that the reduction in viral reactivation was not due to the toxic effect of 3-MA (Fig. 1C). These results demonstrated that 3-MA reduces KSHV lytic reactivation, this reduction likely is due to its effect on autophagy, and the effect is most prominent during the early phase of lytic reactivation.
FIG. 1.

Defective autophagy reduces KSHV lytic reactivation. (A) rKSHV.219 latently infected Vero cells were infected with Bac50 virus and treated with sodium butyrate (NaB), and cells with lytic replication were detected by RFP expression observed under a fluorescence microscope. Cells were also either untreated or treated with 3-methyladenine (3-MA) for 24 or 40 h. The GFP fluorescence and phase-contrast micrographs are showed in parallel. (B) The RFP signal expressed from cells as described for panel A were measured by flow cytometry. The results shown are based on the averages from three separate experiments. Results are expressed as means ± standard deviations. Asterisks indicate P < 0.05 (Student's t test). (C) The autophagic inhibitor 3-MA is not toxic to cells. A cell viability analyzer (Vi-cell; Beckman) was used to measure cell viability.
The activation of autophagy is enhanced during KSHV lytic replication.
The conversion of microtubule-associated protein 1 light chain 3 (LC3) is a hallmark of autophagy and has been used to detect autophagy activation. In quiescent cells, LC3 protein is expressed in the cytoplasm as a precursor protein known as LC3-I (18-kDa). When autophagy is activated, LC3-I can be conjugated to phosphatidylethanolamine (PE) by a ubiquitination-like reaction to generate a lipidated species termed LC3-II (16-kDa) that is associated with inner and outer membranes of autophagosomes (19, 24). Therefore, to examine the status of the autophagy activation during KSHV lytic replication, LC3 proteins were examined by the Western blot analysis of KSHV-positive cells. We observed that more LC3-II molecules were accumulated in BCBL-1 cells after 12-O-tetradecanoylphorbol-13-acetate (TPA) and NaB treatments. In addition, a time course study analyzing the cells at different time points after chemical stimulation showed that there was an increase in the amount of the endogenous autophagic protein Beclin 1, which is known to be responsible for the initiation of the autophagic pathway (Fig. 2 A). Therefore, the increases in the expression levels of two autophagic proteins as well as the presence of KSHV lytic gene product K8 after TPA and NaB stimulation indicate that the activation of autophagy is enhanced during KSHV lytic replication.
FIG. 2.
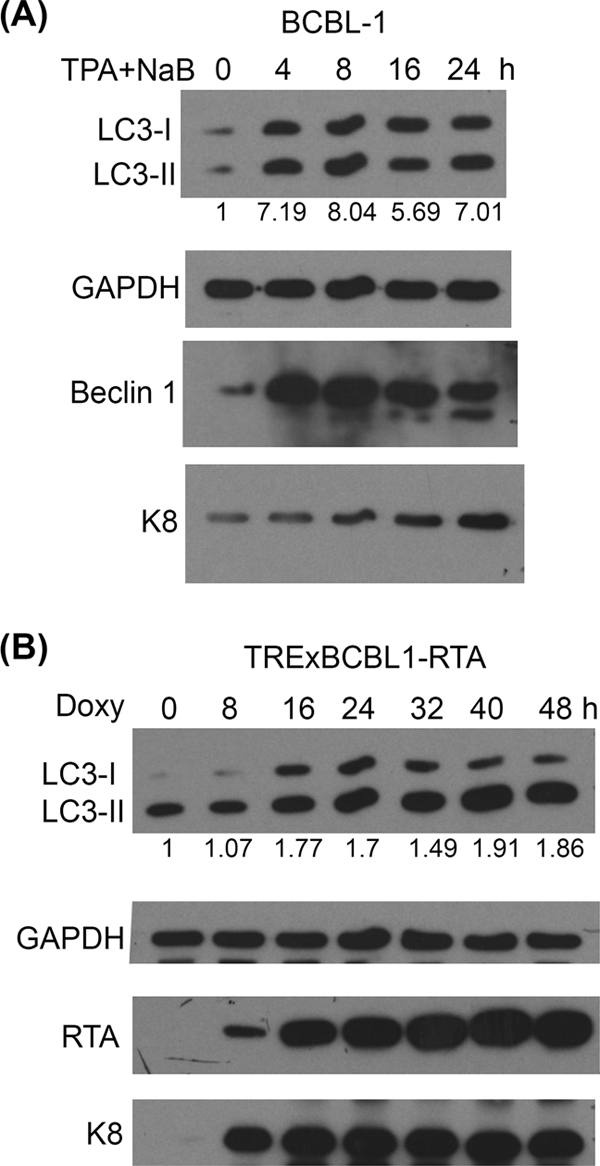
Autophagy is enhanced during KSHV lytic reactivation. (A) BCBL-1 cells were treated with 20 ng/ml TPA and 3 mM NaB for the indicated time. (B) TRExBCBL1-RTA cells were treated with 1 μg/ml doxycycline (Doxy) for the indicated time to induce RTA expression. Western blot analysis was performed using specific antibodies as indicated. The numbers below the blots indicate the relative amounts of LC3-II protein that were normalized by GAPDH protein. The exposure time of the Western blots for K8 shown in panel B is 10× less than that for panel A due to the much more efficient induction of the lytic reactivation of the TRExBCBL1-RTA cells.
The KSHV immediate-early gene product RTA is considered to be necessary and sufficient for KSHV lytic reactivation (38). Therefore, to determine whether the activation of autophagy also is increased during RTA-induced lytic replication, LC3 conversion was analyzed in RTA-inducible BCBL-1 cells (TRExBCBL1-RTA). In this cell line, RTA is integrated into the genome of BCBL-1 cells, and its expression is regulated by doxycycline (41). We demonstrated that the overexpression of RTA in TRExBCBL1-RTA efficiently triggers lytic replication, as shown by the enhancement of the expression of an early gene product, K8. Simultaneously, the accumulation of LC3-II was observed upon the overexpression of RTA in TRExBCBL1-RTA cells (Fig. 2B). These results indicate that there is an enhancement of autophagic process during KSHV lytic reactivation upon RTA overexpression or treatment by chemicals.
RTA is able to enhance the autophagic process in 293T cells.
To investigate whether RTA is able to induce the autophagic process, the presence of autophagic vacuoles was monitored after the cotransfection of RTA and GFP-tagged LC3 expression plasmids in 293T cells. Since LC3-II is lipidated and associated with the inner and outer membrane of autophagic vacuoles, the green fluorescent signal from GFP-LC3 can be shifted from a diffuse cytosolic/nuclear staining to a punctate pattern outlining the autophagic vacuoles in the cells if autophagy is triggered. In cells transfected with the control GFP vector and starved in salt buffer, no obvious green punctate dots were observed (Fig. 3 A). In contrast, green punctate dots of GFP-LC3 were very distinct and found to be distributed in the cytosol after the starvation and/or transfection of RTA expression plasmids. Very few GFP punctate dots were seen in GFP-LC3-transfected cells in nutrient-rich media. The percentage of GFP-LC3-positive cells containing green punctate dots then was quantified, and the results are shown in Fig. 3B. Very few cells (2%) transfected with pCMVtag vector were found to contain GFP-LC3 dots, and as expected, the nutrition deprivation of these cells showed an increase in cells containing GFP-LC3 dots to about 14%. Consistently with previous reports, we also observed that the transfection of Beclin 1, which is one of autophagic proteins that is important for autophagic nucleation (36), moderately increased the number of GFP-LC3 dot-containing cells to about 4.2%. The number of Beclin 1-transfected cells containing GFP-LC3 punctate dots markedly increases to about 19% upon starvation. Interestingly, the overexpression of RTA in the cells by the transient transfection of the RTA expression plasmid dramatically increased the percentage of GFP-LC3 dot-containing cells (19.9%), which was further increased to about 35% upon starvation. The increased number of cells containing GFP-LC3 punctate dots was found to be dependent on the presence of RTA in a dose-dependent manner (Fig. 3C). In addition, the ultrastructural analysis of transfected 293T cells by electron microscopy revealed an increased prevalence of double membrane-autophagic vacuoles in the cytoplasm of the RTA-transfected cells (Fig. 4 B) compared to the vector-transfected 293T cells (Fig. 4A). To demonstrate that the RTA-mediated induction of autophagy is not an overexpression phenotype, a carboxyl-terminal truncated RTA plasmid (mRTA) was overexpressed in the 293T cells for GFP dot analysis. The expression level of mRTA was much higher than that of the wild-type RTA (Fig. 3D, left) but was not sufficient to enhance autophagy, as indicated by the number of cells containing GFP-LC3 dots (Fig. 3D, right). To confirm that the increase in GFP-LC3 dots by RTA is due to the induction of autophagy, 3-MA was added to determine whether the presence of GFP-LC3 dots can be inhibited. Indeed, 3-MA was able to reduce GFP-LC3 dot expression even with the highest concentration of RTA added. There was a reduction of GFP-LC3 dot-expressing cells from 25 to 10% (Fig. 3C). To further confirm the ability of RTA to induce the activation of autophagy, LC3 conversion was analyzed by Western blotting. As expected, LC3-II accumulation was increased either in the presence of RTA or upon starvation (Fig. 5 A). Less accumulation of LC3-II was observed with the transfection of vector alone. Taken together, these results indicate that RTA is capable of inducing autophagic vacuole formation without the involvement of other viral proteins.
FIG. 3.

Induction of autophagic vacuoles by overexpression of RTA. (A) Confocal microscopy of autophagic vacuoles. GFP or GFP-LC3 plasmid was cotransfected with pCMVtag or pCMVtagORF50 (RTA) expression plasmid into 293T cells. At 22 h after transfection, the cells were cultured either in regular medium (nutrient rich) or buffered saline (1× Earle's balanced solution) (starvation) for another 90 min. The phase-contrast micrograph is shown in parallel. Scale bars = 10 μm. (B) Quantitation of the number of cells containing GFP punctate dots in transfected 293T cells. GFP-LC3 was cotransfected with pCMVtag, Beclin 1, or pCMVtagORF50 (RTA) into 293T cells, and the cells were cultured in regular medium (nutrient rich) or buffered saline (starvation). (C) Induction of autophagy by RTA in a dose-dependent manner. An increasing concentration of RTA expression was cotransfected with GFP-LC3 into 293T cells. An autophagic inhibitor, 3-MA, was added to the transfected cells for 3 h. The number of cells containing green punctate dots then was quantified. (D) The increase in the autophagy by RTA is not due to an overexpression phenotype. GFP-LC3 was cotransfected with pCMVtag, pCMVtagORF50 (RTA), or mutant RTA (mRTA) into 293T cells, and the cells were cultured in regular medium. The protein expression levels of RTA and mRTA are detected by Western blotting with anti-flag antibody as shown in the left panel. The number of cells containing green dots is shown in the right panel. All of the results are based on averages from three independent repeats. Results are expressed as means ± standard deviations.
FIG. 4.

TEM ultrastructural confirmation of autophagic vacuoles. 293T cells were transfected with pCMVtag (A) as a control or with pCMVtagORF50 (RTA) (B) and were cultured under nutrient-rich conditions. Arrows indicate autophagic vacuoles. N, nucleus. Panels B1, B2, and B3 are magnified views of autophagic vacuole structures from the boxed regions shown in panel B. M, mitochondria. Scale bars = 1 μm.
FIG. 5.

Induction of autolysosomes by RTA. (A) Western blot analysis of LC3 in 293T cells transfected with pCMVtag or pCMVtafORF50 (RTA) and treated with bafilomycin A1 (as indicated by the letter B) or dimethylsulfoxide (DMSO) (control) for 4 h or salt buffer (as indicated by the letter S) for 90 min. The numbers indicate the relative amounts of LC3-II protein that were normalized with the GAPDH protein. (B) Confocal microscopy of autolysosomes. The plasmids transfected into 293T cells were as indicated. For starvation, the transfected cells were starved for 90 min in salt buffer. Lysosomes were stained by LysoTracker red dye (red), autophagosomes were stained by GFP-LC3 (green), and autolysosomes were stained by LysoTracker red and GFP-LC3 showing a yellow signal. The nuclei were stained with DAPI (blue). Scale bars = 10 μm.
RTA is capable of inducing the formation of autolysosomes.
It is well established that the fusion of autophagosomes with lysosomes to generate autolysosomes is a critical step in the autophagy degradation process. Moreover, autolysosomes contain various lysosomal enzymes that are able to digest sequestered material, including LC3-II, which is associated with the inner membrane of autophagosomes. Thus, to further determine whether RTA has the ability to stimulate the maturation of autophagic vacuoles into degradative organelles, LC3-II accumulation was analyzed by Western blotting in the presence of an inhibitor to prevent LC3-II degradation by lysosomal enzymes. To prevent degradation, cells were treated with bafilomycin A1, a specific inhibitor of vacuolar proton ATPases. As expected, cells transfected with the control plasmid pCMVtag showed an increase in LC3-II due to the effect of bafilomycin to prevent LC3-II degradation. However, the increase in the control cells is less than that of cells transfected with RTA. This indicates that fusion between the autophagosomes and lysosomes was enhanced in the presence of RTA (Fig. 5A). To further confirm this result, we assessed the presence of autolysosomes in cells by using GFP-LC3 and LysoTracker red staining, which stains for acidic organelles such as lysosomes. Clearly, in the RTA-expressing or starved cells, LC3 was found to be colocalized with LysoTracker red, suggesting the formation of the autolysosomes (Fig. 5B). Taken together, these results indicated that RTA is able to induce LC3 conversion and the formation of autophagosomes and autolysosomes to mediate the induction of the autophagy degradation pathway.
The enhancement of autophagy activation by RTA is cell type independent.
To further demonstrate whether RTA-induced autophagic activation can be observed in lymphoma cells, the autophagic vacuole formation and LC3 molecular conversion were estimated in an RTA-inducible BJAB cell line, TRExBJAB-RTA. In this cell line, the RTA gene is integrated into the chromosomal DNA under the control of a tetracycline-inducible promoter. TRExBJAB-RTA and control TRExBJAB cells first were transfected with the GFP-LC3 plasmid, and GFP-LC3 punctate dots were analyzed at the indicated time points after doxycycline treatment. Before the addition of doxycycline (at 0 h), both cell lines have similar basal numbers of cells containing GFP-LC3 dots, and doxycycline is unable to enhance the formation of GFP-LC3 punctate dots in the control TRExBJAB cells without the integrated RTA gene. In contrast, the percentage of cells containing GFP-LC3 punctate dots was increased upon the induction of RTA expression in the TRExBJAB-RTA cells (Fig. 6 A). In addition, the levels of the processed LC3-II proteins were found to be enhanced upon RTA induction in the TRExBJAB-RTA cells but remained constant in control TRExBJAB cells (Fig. 6B). Consistently with our results obtained with 293T cells, the increase in the autophagy activation by RTA likely is cell type independent.
FIG. 6.

Enhancement of autophagy by RTA in B cells. (A) Induction of autophagic vacuoles by RTA. After transfection with GFP-LC3 plasmids, TRExBJAB and TRExBJAB-RTA cells were treated with doxycycline for the indicated time. The number of cells containing green punctate dots then was quantified. The results are from the averages from three independent experiments and are expressed as means ± standard deviations. (B) LC3 was analyzed by the Western blot analysis of TRExBJAB and TRExBJAB-RTA cells treated with doxycycline for the indicated time. The numbers shown below the blots indicate the relative amounts of LC3-II protein, which were normalized with the GAPDH protein.
Inhibition of autophagy affects RTA-mediated lytic replication.
Our findings suggest that the autophagic process can be enhanced by RTA expression, and the inhibition of autophagy affects KSHV lytic reactivation in infected Vero cells. Therefore, the inhibition of autophagy may affect RTA-induced KSHV lytic gene expression. To test this possibility, we measured the mRNA levels of early and late genes (ORF57 and K8.1) in RTA-inducible BCBL-1 cells (TRExBCBL1-RTA) in the presence or absence of an autophagy inhibitor (3-MA). In the reverse transcription quantitative PCR analysis, the induction of RTA by doxycycline was able to elevate the expression of ORF57 and K8.1 mRNA, indicating that lytic replication is triggered. However, the suppression of autophagy by 3-MA treatment leads to a reduction in the RTA-induced lytic gene mRNA expression of ORF57 and K8.1 (62 and 72% reduction, respectively) (Fig. 7 A). To specifically inhibit autophagy, we employed a gene knockdown approach using shRNA against Beclin 1, which is required for the initiation of autophagy (36). After the transfection of shRNA plasmid in TRExBCBL-RTA cells and puromycin selection, the reduction of Beclin 1 mRNA and protein was confirmed by reverse transcription-quantitative PCR and Western blotting (Fig. 7B). We found that the RTA-mediated mRNA expression of ORF57 and K8.1 were decreased by 53 and 42% reduction, respectively, in the Beclin 1 knockdown cells (Fig. 7C). It is well known that RTA triggers lytic gene expression to facilitate KSHV lytic replication. Thus, we also measured the viral DNA copy number in the TRExBCBL-RTA cells with and without 3-MA treatment. Figure 7D shows that the inhibition of autophagy by 3-MA reduces RTA-mediated viral DNA replication (29% inhibition). Taken together, these results indicate that autophagy positively regulates RTA-mediated lytic replication.
FIG. 7.

Defective autophagy reduces RTA-mediated lytic replication. (A) Inhibition of autophagy by 3-MA affects RTA-mediated lytic gene expression. TRExBCBL1-RTA cells were treated with different combinations of doxycycline (Doxy) and 3-MA. ORF57 and K8.1 mRNA were quantified by real-time PCR. (B) Quantitative real-time PCR and Western blot analysis detected the mRNA and protein levels of Beclin 1 from scrambled negative (Scramble) and Beclin 1 (BECN 1) knockdown BCBL1-RTA cells. (C) Inhibition of autophagy by the knockdown of Beclin 1 reduces RTA-mediated lytic gene expression. ORF57 and K8.1 mRNA levels from scrambled negative (Scramble) and Beclin 1 (BECN 1) knockdown BCBL1-RTA cells with or without doxycycline (Doxy) treatment were quantified by real-time PCR. (D) Inhibition of autophagy reduces viral DNA replication. Cellular viral DNA copy number from TRExBCBL1-RTA cells with different combinations of 3-MA and doxycycline (Doxy) was quantified by real-time PCR. All of the results are averages from three independent experiments. Results are expressed as means ± standard deviations.
DISCUSSION
Autophagy has been implicated as an antiviral immune defense, and a number of viruses were found to develop various strategies to block autophagy to overcome the antiviral function of autophagy (28). However, several viruses have been found to utilize autophagy to enhance viral replication. In our present study, autophagy is enhanced during KSHV lytic replication and positively regulates KSHV lytic replication. It has been shown that infections by RNA viruses, such as poliovirus, coxsackievirus, influenza A virus, hepatitis C virus, and dengue virus, can induce autophagy, and this induction increases viral RNA replication and viral yield (1, 21, 32, 61, 68). Since positive-stranded RNA virus replications are associated with cytoplasmic membranes of infected cells (47), autophagosome-like structures may serve as the membrane scaffold for viral RNA replication based on the localization of their viral proteins and viral RNA genome in the autophagosomes (21, 45, 60). In contrast to most RNA viruses, KSHV DNA replication occurs in the nucleus. Therefore, it is unlikely that the autophagic vacuoles in the cytoplasm can serve as sites for KSHV replication. It has been demonstrated that KSHV adopts various cellular machineries and recruits different cellular proteins to maintain latency or to initiate lytic replication (10, 66); therefore, autophagy could be one of the cellular mechanisms utilized by KSHV to establish an adequate environment for lytic reactivation.
To explore the possible mechanism and biological significance of the enhancement of autophagic activation during KSHV lytic replication, we tested the ability of a key viral protein, RTA, which is responsible for the initiation of KSHV lytic reactivation, in the induction of autophagy. We observed that RTA possesses the ability to increase the autophagic activation, and this regulation is independent of other KSHV viral proteins. Since RTA is a typical transcriptional factor located in the nucleus, it is not clear how RTA can affect this intracellular pathway, which occurs in the cytoplasm. It is known that RTA upregulates expression of a number of intracellular and viral genes (9, 37, 52, 56, 63); thus, it is possible that RTA promotes autophagic gene expression to trigger the autophagic pathway. A similar mechanism has been found for hepatitis B virus (HBV), where the HBV X protein can upregulate Beclin 1 mRNA and protein levels to activate autophagy in hepatocytes (55). Therefore, the relationship between RTA and autophagy-related protein expression needs to be further investigated.
In the classic autophagic pathway, portions of cytoplasm are sequestered to form the double-membrane vacuoles, autophagosomes. Subsequently, autophagosomes fused with lysosomes to become autolysosomes, where the degradation process occurs. However, there are two reports showing the blockage of autolysosomal maturation by hepatitis C virus and coxsackievirus B3, even though the induction of autophagosome-like structures can be observed during their infections, suggesting that the degradation function of autophagy is blocked by hepatitis C virus and coxsackievirus B3 (51, 61). From our results, we found that the overexpression of RTA can enhance both autophagosomes and autolysosome formation. There was an enhancement of GFP-LC3 and lysosomal staining in the presence of RTA, which are similar to those observed during starvation. In addition, the increase in LC3-II protein level can be observed in RTA-expressing cells treated with bafilomycin A1, suggesting that RTA is able to induce the autophagy degradation process. A number of cellular and viral proteins have been shown to interfere with the RTA-mediated transcription of lytic genes to maintain KSHV latency (3, 18, 20, 29, 58, 63-65); hence, the induction of autophagy by RTA to degrade these repressor proteins may be a mechanism to enable the virus to initiate lytic replication. Since RTA has been shown to promote protein degradation through the proteasome degradation pathway (16, 66, 67), it is possible that RTA also adopts the autophagy degradation machinery for the clearance of cellular or viral transcriptional repressors to lead to KSHV lytic replication. Therefore, further studies need to be conducted to demonstrate this possibility.
Currently, very little information is available regarding the roles of autophagy on herpesvirus infection. Autophagy can be an antiviral defense against HSV and HCMV (human cytomegalovirus) infection (7, 43, 44), but autophagy also could be an advantage for viral infection, as in the case of EBV, which uses autophagy to regulate viral LMP-1 expression (30). In our study, we have shown that KSHV RTA may play a role in inducing autophagy to enhance lytic replication; nevertheless, two other KSHV proteins, vFLIP (31) and vBcl2 (50), have been reported to possess antiautophagic function. It is possible that KSHV can either enhance or suppress autophagy during different phases of infection and is dependent upon the expression of various viral and cellular proteins involved. Thus, the roles of autophagy seem to differ among different viruses, and the regulation of autophagy may involve various viral and cellular proteins, as in the case of KSHV. How these various KSHV proteins function to regulate autophagy in infected cells during various phases of the viral replication cycle needs to be further determined. In conclusion, our study shows that the activation of autophagy is enhanced during KSHV lytic reactivation, induced either by chemical stimuli or by RTA. RTA is able to activate the entire process of autophagy. The inhibition of autophagy diminishes RTA-mediated lytic gene expression and reduces KSHV lytic replication, indicating that autophagy is involved in KSHV lytic replication.
Acknowledgments
We thank Han Chen and Terri Fangman at the microscopy core facility, University of Nebraska-Lincoln, for their assistance in TEM and confocal microscopy. We thank J. U. Jung at the University of Southern California for providing RTA-inducible BCBL1 and BJAB cell lines, Jeffrey Vieira at the University of Washington for providing the rKSHV.219-infected cell line and Bac50 virus, and Tamotsu Yoshimori at the National Institute of Genetics, Japan, and Noboru Mizushima at Tokyo Medical and Dental University for providing GFP-LC3 plasmid. We thank Pankaj Kumar for helpful discussions.
This study was supported in part by PHS grant CA75903 and NCRR COBRE grant RR15635 to C.W.
Footnotes
Published ahead of print on 19 May 2010.
REFERENCES
- 1.Ait-Goughoulte, M., T. Kanda, K. Meyer, J. S. Ryerse, R. B. Ray, and R. Ray. 2008. Hepatitis C virus genotype 1a growth and induction of autophagy. J. Virol. 82:2241-2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bowser, B. S., S. Morris, M. J. Song, R. Sun, and B. Damania. 2006. Characterization of Kaposi's sarcoma-associated herpesvirus (KSHV) K1 promoter activation by Rta. Virology 348:309-327. [DOI] [PubMed] [Google Scholar]
- 3.Brown, H. J., M. J. Song, H. Deng, T. T. Wu, G. Cheng, and R. Sun. 2003. NF-kappaB inhibits gammaherpesvirus lytic replication. J. Virol. 77:8532-8540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang, J., R. Renne, D. Dittmer, and D. Ganem. 2000. Inflammatory cytokines and the reactivation of Kaposi's sarcoma-associated herpesvirus lytic replication. Virology 266:17-25. [DOI] [PubMed] [Google Scholar]
- 5.Chang, M., H. J. Brown, A. Collado-Hidalgo, J. M. Arevalo, Z. Galic, T. L. Symensma, L. Tanaka, H. Deng, J. A. Zack, R. Sun, and S. W. Cole. 2005. Beta-adrenoreceptors reactivate Kaposi's sarcoma-associated herpesvirus lytic replication via PKA-dependent control of viral RTA. J. Virol. 79:13538-13547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang, P. J., D. Shedd, L. Gradoville, M. S. Cho, L. W. Chen, J. Chang, and G. Miller. 2002. Open reading frame 50 protein of Kaposi's sarcoma-associated herpesvirus directly activates the viral PAN and K12 genes by binding to related response elements. J. Virol. 76:3168-3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaumorcel, M., S. Souquere, G. Pierron, P. Codogno, and A. Esclatine. 2008. Human cytomegalovirus controls a new autophagy-dependent cellular antiviral defense mechanism. Autophagy 4:46-53. [DOI] [PubMed] [Google Scholar]
- 8.Chen, J., K. Ueda, S. Sakakibara, T. Okuno, and K. Yamanishi. 2000. Transcriptional regulation of the Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor gene. J. Virol. 74:8623-8634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deng, H., J. T. Chu, M. B. Rettig, O. Martinez-Maza, and R. Sun. 2002. Rta of the human herpesvirus 8/Kaposi sarcoma-associated herpesvirus up-regulates human interleukin-6 gene expression. Blood 100:1919-1921. [DOI] [PubMed] [Google Scholar]
- 10.Deng, H., Y. Liang, and R. Sun. 2007. Regulation of KSHV lytic gene expression. Curr. Top. Microbiol. Immunol. 312:157-183. [DOI] [PubMed] [Google Scholar]
- 11.Deng, H., M. J. Song, J. T. Chu, and R. Sun. 2002. Transcriptional regulation of the interleukin-6 gene of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus). J. Virol. 76:8252-8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng, H., A. Young, and R. Sun. 2000. Auto-activation of the rta gene of human herpesvirus-8/Kaposi's sarcoma-associated herpesvirus. J. Gen. Virol. 81:3043-3048. [DOI] [PubMed] [Google Scholar]
- 13.Duan, W., S. Wang, S. Liu, and C. Wood. 2001. Characterization of Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8 ORF57 promoter. Arch. Virol. 146:403-413. [DOI] [PubMed] [Google Scholar]
- 14.Espert, L., and M. Biard-Piechaczyk. 2009. Autophagy in HIV-induced T cell death. Curr. Top. Microbiol. Immunol. 335:307-321. [DOI] [PubMed] [Google Scholar]
- 15.Espert, L., M. Denizot, M. Grimaldi, V. Robert-Hebmann, B. Gay, M. Varbanov, P. Codogno, and M. Biard-Piechaczyk. 2006. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J. Clin. Investig. 116:2161-2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gould, F., S. M. Harrison, E. W. Hewitt, and A. Whitehouse. 2009. Kaposi's sarcoma-associated herpesvirus RTA promotes degradation of the Hey1 repressor protein through the ubiquitin proteasome pathway. J. Virol. 83:6727-6738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gwack, Y., H. Byun, S. Hwang, C. Lim, and J. Choe. 2001. CREB-binding protein and histone deacetylase regulate the transcriptional activity of Kaposi's sarcoma-associated herpesvirus open reading frame 50. J. Virol. 75:1909-1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gwack, Y., H. Nakamura, S. H. Lee, J. Souvlis, J. T. Yustein, S. Gygi, H. J. Kung, and J. U. Jung. 2003. Poly(ADP-ribose) polymerase 1 and Ste20-like kinase hKFC act as transcriptional repressors for gamma-2 herpesvirus lytic replication. Mol. Cell. Biol. 23:8282-8294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ichimura, Y., T. Kirisako, T. Takao, Y. Satomi, Y. Shimonishi, N. Ishihara, N. Mizushima, I. Tanida, E. Kominami, M. Ohsumi, T. Noda, and Y. Ohsumi. 2000. A ubiquitin-like system mediates protein lipidation. Nature 408:488-492. [DOI] [PubMed] [Google Scholar]
- 20.Izumiya, Y., S. F. Lin, T. Ellison, L. Y. Chen, C. Izumiya, P. Luciw, and H. J. Kung. 2003. Kaposi's sarcoma-associated herpesvirus K-bZIP is a coregulator of K-Rta: physical association and promoter-dependent transcriptional repression. J. Virol. 77:1441-1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jackson, W. T., T. H. Giddings, Jr., M. P. Taylor, S. Mulinyawe, M. Rabinovitch, R. R. Kopito, and K. Kirkegaard. 2005. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 3:e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jin, S., and E. White. 2007. Role of autophagy in cancer: management of metabolic stress. Autophagy 3:28-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kabeya, Y., N. Mizushima, T. Ueno, A. Yamamoto, T. Kirisako, T. Noda, E. Kominami, Y. Ohsumi, and T. Yoshimori. 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19:5720-5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kirisako, T., Y. Ichimura, H. Okada, Y. Kabeya, N. Mizushima, T. Yoshimori, M. Ohsumi, T. Takao, T. Noda, and Y. Ohsumi. 2000. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J. Cell Biol. 151:263-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kirkegaard, K., and W. T. Jackson. 2005. Topology of double-membraned vesicles and the opportunity for non-lytic release of cytoplasm. Autophagy 1:182-184. [DOI] [PubMed] [Google Scholar]
- 26.Kirkegaard, K., M. P. Taylor, and W. T. Jackson. 2004. Cellular autophagy: surrender, avoidance and subversion by microorganisms. Nat. Rev. Microbiol. 2:301-314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klionsky, D. J., J. M. Cregg, W. A. Dunn, Jr., S. D. Emr, Y. Sakai, I. V. Sandoval, A. Sibirny, S. Subramani, M. Thumm, M. Veenhuis, and Y. Ohsumi. 2003. A unified nomenclature for yeast autophagy-related genes. Dev. Cell 5:539-545. [DOI] [PubMed] [Google Scholar]
- 28.Kudchodkar, S. B., and B. Levine. 2009. Viruses and autophagy. Rev. Med. Virol. 19:359-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lan, K., D. A. Kuppers, S. C. Verma, and E. S. Robertson. 2004. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen inhibits lytic replication by targeting Rta: a potential mechanism for virus-mediated control of latency. J. Virol. 78:6585-6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee, D. Y., and B. Sugden. 2008. The latent membrane protein 1 oncogene modifies B-cell physiology by regulating autophagy. Oncogene 27:2833-2842. [DOI] [PubMed] [Google Scholar]
- 31.Lee, J. S., Q. Li, J. Y. Lee, S. H. Lee, J. H. Jeong, H. R. Lee, H. Chang, F. C. Zhou, S. J. Gao, C. Liang, and J. U. Jung. 2009. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 11:1355-1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee, Y. R., H. Y. Lei, M. T. Liu, J. R. Wang, S. H. Chen, Y. F. Jiang-Shieh, Y. S. Lin, T. M. Yeh, C. C. Liu, and H. S. Liu. 2008. Autophagic machinery activated by dengue virus enhances virus replication. Virology 374:240-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levine, B. 2005. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell 120:159-162. [DOI] [PubMed] [Google Scholar]
- 34.Levine, B., and D. J. Klionsky. 2004. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell 6:463-477. [DOI] [PubMed] [Google Scholar]
- 35.Levine, B., and G. Kroemer. 2008. Autophagy in the pathogenesis of disease. Cell 132:27-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liang, X. H., S. Jackson, M. Seaman, K. Brown, B. Kempkes, H. Hibshoosh, and B. Levine. 1999. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402:672-676. [DOI] [PubMed] [Google Scholar]
- 37.Liu, Y., Y. Cao, D. Liang, Y. Gao, T. Xia, E. S. Robertson, and K. Lan. 2008. Kaposi's sarcoma-associated herpesvirus RTA activates the processivity factor ORF59 through interaction with RBP-Jkappa and a cis-acting RTA responsive element. Virology 380:264-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lukac, D. M., R. Renne, J. R. Kirshner, and D. Ganem. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252:304-312. [DOI] [PubMed] [Google Scholar]
- 39.Miller, G., L. Heston, E. Grogan, L. Gradoville, M. Rigsby, R. Sun, D. Shedd, V. M. Kushnaryov, S. Grossberg, and Y. Chang. 1997. Selective switch between latency and lytic replication of Kaposi's sarcoma herpesvirus and Epstein-Barr virus in dually infected body cavity lymphoma cells. J. Virol. 71:314-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller, G., M. O. Rigsby, L. Heston, E. Grogan, R. Sun, C. Metroka, J. A. Levy, S. J. Gao, Y. Chang, and P. Moore. 1996. Antibodies to butyrate-inducible antigens of Kaposi's sarcoma-associated herpesvirus in patients with HIV-1 infection. N. Engl. J. Med. 334:1292-1297. [DOI] [PubMed] [Google Scholar]
- 41.Nakamura, H., M. Lu, Y. Gwack, J. Souvlis, S. L. Zeichner, and J. U. Jung. 2003. Global changes in Kaposi's sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J. Virol. 77:4205-4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakashima, A., N. Tanaka, K. Tamai, M. Kyuuma, Y. Ishikawa, H. Sato, T. Yoshimori, S. Saito, and K. Sugamura. 2006. Survival of parvovirus B19-infected cells by cellular autophagy. Virology 349:254-263. [DOI] [PubMed] [Google Scholar]
- 43.Orvedahl, A., D. Alexander, Z. Talloczy, Q. Sun, Y. Wei, W. Zhang, D. Burns, D. A. Leib, and B. Levine. 2007. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1:23-35. [DOI] [PubMed] [Google Scholar]
- 44.Orvedahl, A., and B. Levine. 2008. Autophagy and viral neurovirulence. Cell Microbiol. 10:1747-1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Panyasrivanit, M., A. Khakpoor, N. Wikan, and D. R. Smith. 2009. Co-localization of constituents of the dengue virus translation and replication machinery with amphisomes. J. Gen. Virol. 90:448-456. [DOI] [PubMed] [Google Scholar]
- 46.Renne, R., W. Zhong, B. Herndier, M. McGrath, N. Abbey, D. Kedes, and D. Ganem. 1996. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 2:342-346. [DOI] [PubMed] [Google Scholar]
- 47.Salonen, A., T. Ahola, and L. Kaariainen. 2005. Viral RNA replication in association with cellular membranes. Curr. Top. Microbiol. Immunol. 285:139-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seglen, P. O., and P. B. Gordon. 1982. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. U. S. A. 79:1889-1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shintani, T., and D. J. Klionsky. 2004. Autophagy in health and disease: a double-edged sword. Science 306:990-995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sinha, S., C. L. Colbert, N. Becker, Y. Wei, and B. Levine. 2008. Molecular basis of the regulation of Beclin 1-dependent autophagy by the gamma-herpesvirus 68 Bcl-2 homolog M11. Autophagy 4:989-997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sir, D., W. L. Chen, J. Choi, T. Wakita, T. S. Yen, and J. H. Ou. 2008. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 48:1054-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song, M. J., H. J. Brown, T. T. Wu, and R. Sun. 2001. Transcription activation of polyadenylated nuclear RNA by RTA in human herpesvirus 8/Kaposi's sarcoma-associated herpesvirus. J. Virol. 75:3129-3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suhy, D. A., T. H. Giddings, Jr., and K. Kirkegaard. 2000. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J. Virol. 74:8953-8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tallóczy, Z., W. Jiang, H. W. T. Virgin, D. A. Leib, D. Scheuner, R. J. Kaufman, E. L. Eskelinen, and B. Levine. 2002. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl. Acad. Sci. U. S. A. 99:190-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tang, H., L. Da, Y. Mao, Y. Li, D. Li, Z. Xu, F. Li, Y. Wang, P. Tiollais, T. Li, and M. Zhao. 2009. Hepatitis B virus X protein sensitizes cells to starvation-induced autophagy via up-regulation of beclin 1 expression. Hepatology 49:60-71. [DOI] [PubMed] [Google Scholar]
- 56.Ueda, K., K. Ishikawa, K. Nishimura, S. Sakakibara, E. Do, and K. Yamanishi. 2002. Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) replication and transcription factor activates the K9 (vIRF) gene through two distinct cis elements by a non-DNA-binding mechanism. J. Virol. 76:12044-12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vieira, J., and P. M. O'Hearn. 2004. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology 325:225-240. [DOI] [PubMed] [Google Scholar]
- 58.Wang, J., J. Zhang, L. Zhang, W. Harrington, Jr., J. T. West, and C. Wood. 2005. Modulation of human herpesvirus 8/Kaposi's sarcoma-associated herpesvirus replication and transcription activator transactivation by interferon regulatory factor 7. J. Virol. 79:2420-2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang, S., S. Liu, M. Wu, Y. Geng, and C. Wood. 2001. Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8 ORF50 gene product contains a potent C-terminal activation domain which activates gene expression via a specific target sequence. Arch. Virol. 146:1415-1426. [DOI] [PubMed] [Google Scholar]
- 60.Wileman, T. 2006. Aggresomes and autophagy generate sites for virus replication. Science 312:875-878. [DOI] [PubMed] [Google Scholar]
- 61.Wong, J., J. Zhang, X. Si, G. Gao, I. Mao, B. M. McManus, and H. Luo. 2008. Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 82:9143-9153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xie, Z., and D. J. Klionsky. 2007. Autophagosome formation: core machinery and adaptations. Nat. Cell Biol. 9:1102-1109. [DOI] [PubMed] [Google Scholar]
- 63.Yada, K., E. Do, S. Sakakibara, E. Ohsaki, E. Ito, S. Watanabe, and K. Ueda. 2006. KSHV RTA induces a transcriptional repressor, HEY1, that represses rta promoter. Biochem. Biophys. Res. Commun. 345:410-418. [DOI] [PubMed] [Google Scholar]
- 64.Yang, Z., H. J. Wen, V. Minhas, and C. Wood. 2009. The zinc finger DNA-binding domain of K-RBP plays an important role in regulating Kaposi's sarcoma-associated herpesvirus RTA-mediated gene expression. Virology 391:221-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang, Z., and C. Wood. 2007. The transcriptional repressor K-RBP modulates RTA-mediated transactivation and lytic replication of Kaposi's sarcoma-associated herpesvirus. J. Virol. 81:6294-6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang, Z., Z. Yan, and C. Wood. 2008. Kaposi's sarcoma-associated herpesvirus transactivator RTA promotes degradation of the repressors to regulate viral lytic replication. J. Virol. 82:3590-3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu, Y., S. E. Wang, and G. S. Hayward. 2005. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 22:59-70. [DOI] [PubMed] [Google Scholar]
- 68.Zhou, Z., X. Jiang, D. Liu, Z. Fan, X. Hu, J. Yan, M. Wang, and G. F. Gao. 2009. Autophagy is involved in influenza A virus replication. Autophagy 5:321-328. [DOI] [PubMed] [Google Scholar]