

Abstract
Periodic outbreaks of highly pathogenic avian H5N1 influenza viruses and the current H1N1 pandemic highlight the need for a more detailed understanding of influenza virus pathogenesis. To investigate the host transcriptional response induced by pathogenic influenza viruses, we used a functional-genomics approach to compare gene expression profiles in lungs from 129S6/SvEv mice infected with either the fully reconstructed H1N1 1918 pandemic virus (1918) or the highly pathogenic avian H5N1 virus Vietnam/1203/04 (VN/1203). Although the viruses reached similar titers in the lung and caused lethal infections, the mean time of death was 6 days for VN/1203-infected animals and 9 days for mice infected with the 1918 virus. VN/1203-infected animals also exhibited an earlier and more potent inflammatory response. This response included induction of genes encoding components of the inflammasome. VN/1203 was also able to disseminate to multiple organs, including the brain, which correlated with changes in the expression of genes associated with hematological functions and lipoxin biogenesis and signaling. Both viruses elicited expression of type I interferon (IFN)-regulated genes in wild-type mice and to a lesser extent in mice lacking the type I IFN receptor, suggesting alternative or redundant pathways for IFN signaling. Our findings suggest that VN/1203 is more pathogenic in mice as a consequence of several factors, including the early and sustained induction of the inflammatory response, the additive or synergistic effects of upregulated components of the immune response, and inhibition of lipoxin-mediated anti-inflammatory responses, which correlated with the ability of VN/1203 to disseminate to extrapulmonary organs.
The World Health Organization officially declared on 11 June 2009 that spread of a new swine origin H1N1 influenza virus had reached the level of a global pandemic (29). Although less virulent than the H1N1 virus responsible for the 1918 influenza pandemic, initial studies indicate that this virus is more pathogenic than seasonal H1N1 influenza viruses (10). The continued emergence of new influenza viruses highlights the need to better understand influenza virus-host interactions and mechanisms of pathogenicity. Such an understanding is necessary to facilitate the development of safe and effective therapeutics and vaccines, critical aspects of preparedness for the current and future pandemics.
A clear reminder of the lethal potential of influenza virus infection is the 1918 pandemic, which resulted in over 50 million deaths worldwide (14). In addition, highly pathogenic avian H5N1 influenza viruses continue to circulate in diverse parts of the world (31, 45), and with human infections resulting in a greater than 50% mortality rate, there is considerable concern over the potential for a deadly new pandemic. Here, we sought to compare the host transcriptional responses to the reconstructed 1918 virus and the avian H5N1 virus Vietnam/1203/04 (VN/1203), with the goal of gaining insights into the underlying mechanisms that make these viruses so lethal.
Recently, we reported a study comparing the host response to 1918 and VN/1203 influenza viruses in a macaque model of infection (8). In that study, all 1918-infected macaques died while all VN/1203-infected animals recovered. Interestingly, only the 1918 virus induced the expression of genes encoding components of the inflammasome early during infection, pointing out different transcriptional mechanisms during infection with highly pathogenic H5N1 and H1N1 influenza viruses. Results from an independent study using mouse infection models suggest there are also differences in lethality and disease progression between the avian H5N1 influenza strains A/Hong Kong/483/97 and A/Hong Kong/486/97 (40). In that study, alpha/beta interferon receptor (IFN-α/βR)-deficient mice succumbed faster than wild-type mice to either H5N1 virus. In addition, there was systemic infection of IFN-α/β receptor-deficient mice with both viruses.
In the present study, we examined the host responses to the 1918 virus and VN/1203 in wild-type 129S6/SvEv mice; additional findings were obtained from IFN-α/β receptor-deficient mice and mouse embryonic fibroblasts derived from these animals. The VN/1203 virus was more pathogenic than the 1918 virus and was able to disseminate to extrapulmonary organs. Global gene expression profiling data revealed possible mechanisms underlying the greater pathogenicity of the VN/1203 virus.
MATERIALS AND METHODS
Viruses.
The reconstructed 1918 H1N1 virus (42) (1918), possessing the A/South Carolina/1/18 hemagglutinin (HA), and VN/1203 were previously shown to be highly virulent for both mice and ferrets (23, 42-43). The 1918 virus was generated utilizing the 12-plasmid reverse-genetics system in a mixture of Madin-Darby canine kidney (MDCK) (ATCC, Manassas, VA) and 293T (ATCC) cells as previously described (42). The titers of virus stocks were determined by plaque assay on MDCK cells, and stocks were maintained in Dulbecco's modified Eagle's medium (DMEM) (Gibco, Grand Island, NY) supplemented with 10% fetal calf serum (FCS) (HyClone, Logan, UT) and 1% penicillin/streptomycin (Gibco). The reconstructed 1918 and VN/1203 viruses were grown as previously described (33). All virus challenge experiments were performed under the guidance of the U.S. National Select Agent Program in negative-pressure HEPA-filtered biosafety level 3 enhanced (BSL-3+) laboratories and with the use of a battery-powered Racal HEPA filter respirator (Racal Health and Safety Inc., Frederick, MD) according to Biomedical Microbiological and Biomedical Laboratory procedures (35).
Mouse experiments.
All animal research was conducted according to the guidance of CDC's Institutional Animal Care and Use Committee in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. IFN-α/βR-deficient (IFNR1−/−) mice on the 129 background were obtained from Herbert Virgin at Washington University, St. Louis, MO. Wild-type SvEv129 (129S6/SvEv) mice (44) were used as age-matched controls in these experiments (Taconic, Bar Harbor, ME). Similar to the majority of inbred mouse strains, 129S6/SvEv animals carry defective alleles of the Mx1 gene (38). Eight-to-10-week-old female 129S6/SvEv and IFNR1−/− mice (on a 129 background) were anesthetized by intraperitoneal injection of 0.2 ml of 2,2,2-tribromoethanol in tert-amylalcohol (Avertin; Sigma-Aldrich, Milwaukee, WI). Ten times the 50% lethal dose (LD50), 3.2 × 104 PFU (1918) or 7 × 103 PFU (VN/1203), in 50 μl of infectious virus diluted in phosphate-buffered saline (PBS) was inoculated intranasally (i.n.). Fifty percent mouse infectious dose (MID50) and LD50 titers were determined by inoculating groups of seven mice i.n. with serial 10-fold dilutions of virus. For MID50 determination, three mice from each group were euthanized on day 4 postinoculation (p.i.), and lungs were collected and homogenized in 1 ml of cold PBS. The homogenates were frozen at −70°C and later thawed before centrifugation and titration for virus infectivity in eggs. The four remaining mice in each group were checked daily for disease signs and death for 14 days p.i. MID50 and LD50 titers were calculated by the method of Reed and Muench (34) and were expressed as the 50% egg infective dose (EID50) value corresponding to 1 MID50 or LD50.
Replication of 1918 and H5N1 viruses in mice (3 animals/group/time point) was examined by determining the virus titers in lung (days 1, 3, and 4), spleen, liver, kidney, and brain (days 3 and 5) following i.n. inoculation with 106 EID50 of virus. Clarified homogenates were titrated for virus infectivity in eggs from an initial dilution of 1:10 (lung), making the limit of virus detection 101.2 EID50/ml. Additional inoculated mice (6 animals/group/virus, making a total of 24 animals) were followed for morbidity and mortality. For RNA isolation, lungs (2 animals/group/time point) were frozen in individual tubes and stored in solution D (4 M guanidinium thiocyanate, 25 mM sodium citrate, 0.5% sarcosyl, 0.1 M β-mercaptoethanol) as previously described (7). In addition, groups of uninfected 129S6/SvEv (6 animals) and IFNR1−/− (6 animals) mice were intranasally treated with 10,000 units of recombinant human IFN-α A/D (R&D Systems, Minneapolis, MN) and euthanized 8 and 24 h posttreatment.
Infection of MEFs.
Mouse embryonic fibroblasts (MEFs) derived from wild-type 129S6/SvEv mice or IFNR1−/− mice generated on the 129S6/SvEv background (27) were grown as monolayers in high-glucose Dulbecco's modified Eagle's medium (hgDMEM) supplemented to contain 10% heat-inactivated fetal calf serum (HyClone Laboratories), 2 mM l-glutamine, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 10 μM 2-mercaptoethanol, penicillin G (50 units/ml), and streptomycin sulfate (50 μg/ml). MDCK cells were grown as monolayers in hgDMEM supplemented to contain 10% heat-inactivated fetal calf serum, 2 mM l-glutamine, penicillin G (50 units/ml), and streptomycin sulfate (50 μg/ml). Near-confluent monolayers of cells were mock infected with PBS alone or infected with influenza virus diluted in infection medium (hgDMEM supplemented to contain 2% heat-inactivated calf serum, 2 mM l-glutamine, penicillin G [50 units/ml], streptomycin sulfate [50 μg/ml], and 50 mM HEPES) to the indicated multiplicity of infection (MOI). After 45 min of adsorption at 4°C, virus and medium were removed. Fresh infection medium was added to the cells, and the infections were allowed to proceed at 37°C until the indicated time postinfection.
Histopathology and immunohistochemistry (IHC).
Collected tissues were fixed by submersion in 10% neutral buffered formalin, routinely processed, and embedded in paraffin. Sections were cut to 5 μm and stained with hematoxylin and eosin (HE). Methods for immunohistochemical detection of influenza A viral antigen have been previously described (32). A duplicate 5-μm section was immunohistochemically stained to demonstrate influenza A virus nucleoprotein by first microwaving the sections in Antigen Retrieval Citra Solution (Biogenex, San Ramon, CA) for antigen exposure. A 1:2,000 dilution of a mouse-derived monoclonal antibody (P13C11) specific for a type A influenza virus nucleoprotein was applied and allowed to incubate for 12 h at 4°C. The primary antibody was then detected by the application of biotinylated goat anti-mouse IgG secondary antibody using the Mouse on Mouse System (M.O.M. kit; Vector Laboratories, Inc., Burlingame, CA) according to the manufacturer's instructions. The AEC-Peroxidase Substrate Kit (Vector Laboratories Inc.) was used as the substrate chromogen, and hematoxylin was used as a counterstain.
Microarray analysis and bioinformatics.
For all gene expression analyses, separate microarrays were run for each experimental sample. This included 2 animals/time point for 1918 virus-infected mice (24 animals total) or 3 animals/time point for VN/1203-infected mice (36 animals total). Equal masses of total RNA isolated from lungs collected from infected mice were amplified with a Low RNA Input Linear Amplification Kit (Agilent Technologies, Santa Clara, CA) according to the manufacturer's instructions. Global gene expression in infected lungs was compared to genetic-match mock control RNA for each group prepared from a pool of equal masses of total RNA from lung tissue of three uninfected mice. Probe labeling and microarray slide hybridization were performed as described elsewhere (16). Briefly, probes were hybridized on Agilent mouse oligonucleotide microarray slides (approximately 20,000 unique mouse genes). The slides were scanned with an Agilent DNA microarray scanner, and image data were processed using Agilent Feature Extractor (AFE) version 8.1.1.1. The AFE software was used to perform image analysis, including significance of signal and spatial detrending, and to apply a universal error model (for these hybridizations, the most conservative error model was applied). To correct for dye bias, AFE software was also used to perform linear and lowess (intensity dependent) dye normalization. The raw data were then loaded into a custom-designed laboratory information management system (LIMS). The raw data and associated sample information were subsequently loaded into Rosetta Resolver 7.2 (Rosetta Biosoftware, Seattle, WA). Each single microarray experiment incorporated reverse dye labeling techniques and resulted in two measurements for each gene (n = 2), allowing the calculation of a mean ratio between expression levels, standard deviations, and P values within the Rosetta Resolver System (Rosetta Biosoftware, Seattle, WA).
The Resolver system performs a squeeze operation that creates ratio profiles by combining replicates while applying error weighting. The error weighting consists of adjusting for additive and multiplicative noise. A P value is generated that represents the probability that a gene is differentially expressed. In this study, a threshold P value of 0.01 was used to identify genes that were significantly differentially expressed. The Resolver system then combines ratio profiles to create ratio experiments using an error-weighted average as described previously (38a). Spotfire Decision Site 9.1.1 (Spotfire, Somerville, MA) and Ingenuity Pathways Analysis (IPA) 8.0 (Ingenuity Systems, Redwood city, CA) were also used for data analysis and mining. Primary gene expression data are available at http://viromics.washington.edu, in accordance with proposed minimum information about a microarray experiment (MIAME) standards.
Functional and network analyses of statistically significant gene expression changes were performed using IPA. Analysis considered all genes from the data set that met the 2-fold (P < 0.01) change cutoff and that were associated with biological functions in the Ingenuity Pathways Knowledge Base. For all analyses, Fisher's exact test was used to determine the probability that each biological function assigned to the genes within each data set was due to chance alone.
qRT-PCR.
Reverse transcription (RT)-PCR was performed on samples from individual animals to validate specific cellular gene expression changes detected by microarray. The QuantiTect reverse transcription kit (Qiagen Inc., Valencia, CA) was used to generate cDNA. Quantitative real-time RT-PCR (qRT-PCR) was run on an ABI 7500 PCR system, using TaqMan chemistry (Applied Biosystems, Foster City, CA). Gene expression assays specific to mouse cellular genes were purchased from Applied Biosystems. Differences in gene expression are represented as log10 relative quantification to a calibrator and normalized to a reference, using the 2−ΔΔCT method (20).
RESULTS
VN/1203 is more pathogenic than the 1918 virus in mice.
Our previous studies of macaques infected with either VN/1203 or the 1918 virus indicated that the 1918 virus is more pathogenic in this animal model (8). However, of the 498 confirmed human cases of avian H5N1 influenza reported to the World Health Organization as of May 2010, 294 had resulted in death, representing a case fatality rate approaching 60%. Although there may be mild infections that go undiagnosed, H5N1 viruses appear to be significantly more pathogenic than the 1918 virus in humans (28). Although the genetic, anatomical, and physiological similarities between macaques and humans suggest that macaques provide a highly relevant animal model, the mouse infection model is widely used because it provides an approximation of human disease; larger numbers of animals can be used to improve statistical significance; and, importantly, a wide variety of gene knockout animals are available. Therefore, to gain additional insights into the host response to these viruses, we used a mouse infection model that included wild-type mice and mice lacking the type I IFN receptor, a critical component of the innate immune response. When inoculated into wild-type 129S6/SvEv mice, both VN/1203 and the 1918 virus caused lethal infections; however, the mean time of death was 6 days for VN/1203-infected animals and 9 days for mice infected with the 1918 virus (Fig. 1A). Animals were sacrificed according to weight loss euthanasia criteria (see Fig. S1 in the supplemental material). Moreover, VN/1203 disseminated to extrapulmonary organs, as demonstrated by detection of virus in brain and spleen (Fig. 1B), whereas the 1918 virus was not detected outside the respiratory tract. The observed differences in viral dissemination and time to death could not be attributed to differences in viral titers in the lungs, as the two viruses were present at similar levels at all time points examined (Fig. 1C).
FIG. 1.

VN/1203 infection is more pathogenic than 1918 infection in a mouse model of infection. (A) Mortality data from a total of 12 mice (6 mice/virus). (B) Viral dissemination data from a total of 12 mice (3 mice/virus/time point). d, day(s). (C) Lung virus titer data from a total of 18 wild-type mice (3 mice/virus/time point) infected with 1918 or VN/1203 virus. The limit of detection in this assay is indicated by black and grey bars at 1.2 × 101 EID50/ml.
Histopathology results indicated that two of six animals infected with the 1918 virus had mild epithelial necrosis in bronchioles and minimal peribronchiolar alveolitis at day 1 p.i. By day 3 p.i., three of the animals still lacked lesions and three presented mild to severe necrotizing bronchiolitis with associated neutrophilic casts in large bronchioles, peribronchiolar edema and inflammation, and moderate peribronchiolar neutrophilic-to-histiocytic alveolitis. By 4 days p.i., pneumonia and bronchiolitis were consistent in all animals and were slightly more severe than those observed at day 3 p.i. No lesions were observed in heart, spleen, brain, kidney, or liver in 1918-infected animals (see Fig. S2A and B in the supplemental material). Animals infected with VN/1203 presented at day 1 p.i. with moderate necrotizing bronchiolitis with neutrophilic inflammation and minimal peribronchiolar alveolitis. By days 3 and 4 p.i., the airway lesions were accompanied by minimal to mild peribronchiolar alveolitis. VN/1203-infected mice presented no lesions in extrapulmonary organs (see Fig. S2C and D in the supplemental material), but virus was detected in brain and spleen. Viral antigen was commonly present in bronchiolar epithelial cells and less commonly in alveolar epithelial cells and macrophages (see Fig. S2E and F in the supplemental material). Staining was more intense and more widely distributed in VN/1203-infected mice than in 1918 virus-infected mice. In summary, both viruses caused lethal infections, with death occurring approximately 2 days sooner among VN/1203-infected mice. VN/1203 disseminated to brain and spleen by 5 days p.i., but without noticeable tissue damage to those organs. The absence of pathology in extrapulmonary organs may be due to detection of recent virus replication that had not yet resulted in observable lesions. Alternatively, lesions may have been random and focal and not present on the samples examined.
VN/1203 elicits an earlier and more robust host transcriptional response.
In order to discover aspects of the host response that could explain differences in pathogenicity and dissemination, we used a functional-genomics approach to investigate the host transcriptional response elicited in the lungs of mice infected with VN/1203 or the 1918 virus. When analyzing the global transcriptional response, we found that VN/1203 infection induced the differential expression of numerous genes at all time points analyzed. Whereas the day 1 transcriptional response was attenuated in animals infected with the 1918 virus relative to the response induced by VN/1203, at later time points, the host responses to the two viruses were more similar (Fig. 2). This more robust transcriptional response to VN/1203 correlates with our observations that all VN/1203-infected animals showed moderate airway lesions at day 1 p.i., although we have not directly evaluated the extent to which specific gene expression changes are responsible for lesion development.
FIG. 2.

Global transcriptional responses to highly pathogenic 1918 and VN/1203 viruses. Shown is microarray analysis of whole lung tissue from 1918 and VN/1203 virus-infected wild-type mice at 1, 3, and 4 days p.i. The heat map illustrates the global transcriptional profile of 3,470 genes activated within cutoff values of ≥2-fold change and P ≤ 0.01. The genes shown in yellow were upregulated and those in blue were downregulated in infected relative to mock-infected animals. The box outlined in red indicates the attenuated transcriptional response in 1918 virus-infected animals.
The VN/1203 and 1918 viruses differentially regulate key cellular response pathways.
We next used Ingenuity Pathways Analysis to identify functional categories of differentially expressed genes. Our analyses showed that many of these genes were related to the inflammatory response (Fig. 3A) and included the upregulation of inflammasome genes in VN/1203-infected mice (but not in 1918-infected mice) at day 1 p.i. The upregulation of inflammasome genes in VN/1203-infected mice is also illustrated in the functional network shown in Fig. 3B. In this network, genes depicted in blue were upregulated by VN/1203 but not by the 1918 virus, whereas genes depicted in orange were upregulated by VN/1203 virus but downregulated by the 1918 virus. Of particular note, the key inflammasome components CASP1 (caspase 1), interleukin 1β (IL-1β), and NLRP3 (nucleotide-binding domain and leucine-rich-repeat-containing protein 3) were upregulated in response to VN/1203 infection.
FIG. 3.

VN/1203 virus differentially regulates the expression of inflammatory response genes. (A) Heat map illustrating ANOVA results for 142 inflammatory genes differentially transcribed within cutoff values of ≥2-fold change and ANOVA P ≤ 0.01 in wild-type mouse lungs infected with 1918 or VN/1203 virus. (B) Biological-network analysis of the top functional category inflammatory response (P = 4.22E−16) determined by Ingenuity Pathway Analysis. This analysis highlights a subset of genes that were upregulated during VN/1203 infection but not during 1918 infection (blue shading). Another subset of genes was anti-coregulated, that is, upregulated during VN/1203 infection but downregulated during 1918 infection (orange shading). A third subset of genes was upregulated by both viruses (green shading). The boxed genes highlight the differential regulation of the inflammasome components CASP1, IL-1β, and NLRP3.
In addition, VN/1203 also upregulated the expression of tumor necrosis factor alpha (TNF-α), a potent inflammatory molecule; IFN-γ; eukaryotic initiation factor 2 AK2 (eIF2AK2); protein kinase RNA activated (PKR); and additional chemokines and inflammation-related genes. Quantitative RT-PCR analyses were performed to verify the expression of specific transcripts, including IL-1β, NLRP3, CASP1, and TNF-α. The results from this method correlated well with the microarray results (see Fig. S3 in the supplemental material). Later during infection (days 3 and 4 p.i.), the inflammatory responses induced by the two viruses were still different but did not involve the inflammasome, and the numbers of genes differentially regulated were fewer than observed on day 1. This striking difference in the quality of the early inflammatory responses to two highly pathogenic influenza viruses correlates with the severity of disease.
We also observed that at day 1 p.i., VN/1203 infection induced the differential expression of genes associated with viral sensing, neutrophil activation, NF-κB signaling, and chemokine signaling (Fig. 4). Quantitative RT-PCR analyses were performed to verify the expression of specific transcripts, including IFN-β1 and IFN-γ. The results from this method correlated well with the microarray results (see Fig. S4A and B in the supplemental material). Because the products of many of these genes play important roles in regulating cellular gene expression, the activation of these genes may also be associated with the earlier and more robust host transcriptional and inflammatory response observed in VN/1203-infected animals.
FIG. 4.
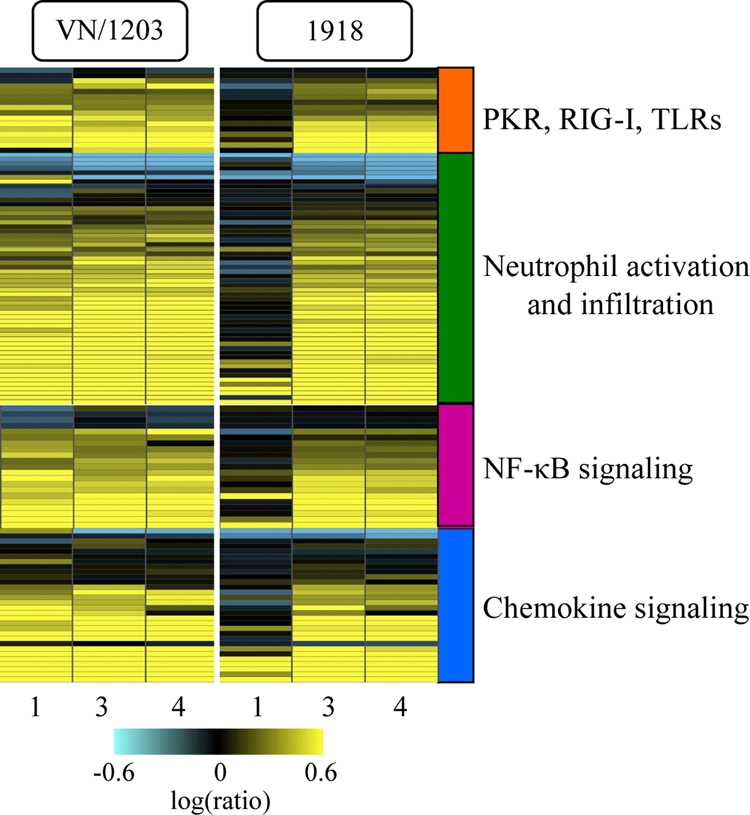
The VN/1203 and 1918 viruses differentially regulate key cellular signaling pathways. Shown is ANOVA of microarray data from lung samples using cutoff values of ≥2-fold change and ANOVA P ≤ 0.01 comparing the infections of wild-type animals with 1918 and VN/1203 viruses. The results were uploaded into IPA for functional analysis. Selected canonical pathways were analyzed, and the results are presented as heat maps illustrating the expression of genes associated with viral sensing, neutrophil activation, NF-κB signaling, and chemokine signaling to 1918 and VN/1203 viruses. Yellow indicates upregulated genes; blue indicates downregulated genes.
VN/1203 dissemination correlates with sustained activation of the inflammatory response and altered hematological function and lipoxin signaling.
To attempt to gain insight into the ability of VN/1203 to disseminate to the brain and spleen, we also devised an analysis strategy to identify gene expression changes that correlated with tissue dissemination. Briefly, gene expression data from all time points were stratified into four groups: wild-type mice infected by the 1918 virus, wild-type mice infected by VN/1203, IFNR1−/− mice infected by the 1918 virus, and IFNR1−/− mice infected by VN/1203. Among these groups, only wild-type mice infected by the 1918 virus showed no dissemination during infection. Differentially expressed genes in each group compared to mock infection were identified (≥2.0-fold change), and pathway analysis was carried out using Ingenuity Pathway Analysis and GeneGo MetaCore. Two functional categories, which included genes associated with hematological development and function and genes associated with the inhibitory effects of lipoxin on inflammation, leukocyte trafficking, and neutrophil-mediated tissue injury, showed statistically significant changes in the groups associated with virus dissemination but not the group not associated (wild-type mice infected by 1918). Many of the genes in the hematological development and function category are associated with proinflammatory responses and were strongly upregulated during VN/1203 infection (Fig. 5A). Lipoxins, in contrast, are eicosanoids with potent anti-inflammatory properties (37). The gene responsible for lipoxin biogenesis, Alox5, was downregulated during VN/1203 infection (Fig. 5B), as was the gene encoding suppressor of cytokine signaling 2 (Socs2), the expression of which can be induced by lipoxins to control proinflammatory responses (22). These finding suggest that not only does VN/1203 infection result in the induction of numerous proinflammatory genes, but lipoxin-mediated anti-inflammatory responses are also impaired. No change in the expression of Alox5, Socs2, or Fprl1 (the primary receptor for lipoxins) was observed in response to infection with the 1918 virus.
FIG. 5.

Association of hematological function and lipoxin signaling with VN/1203 virus dissemination. (A) Differential regulation of hematological system development- and function-related genes. (B) Differential regulation of lipoxin signaling. Shown is ANOVA of lung gene expression data using cutoff values of ≥2-fold change and ANOVA P ≤ 0.01 comparing the infection of wild-type animals with VN/1203 or the 1918 virus. The results were uploaded into IPA for functional analysis. Selected biological functions were analyzed, and the results are presented as heat maps. Yellow indicates upregulated genes; blue indicates downregulated genes. WT, wild type.
We also investigated the observed dissemination of the 1918 virus to brain and spleen during infection of IFNR1−/− mice. Our results indicated that the host responses elicited by 1918 virus infection were distinctly different in wild-type and immune-deficient animals. While the activation of important host functions, such as immune cell trafficking, inflammatory response, and cell death, were almost at similar levels in both mouse strains by day 1 p.i., these functions changed dramatically in wild-type mice, which responded to 1918 infection by an steady increase in the expression of genes associated with these functions by days 3 and 4 p.i. However, far fewer genes in these categories were transcriptionally activated in IFNR1−/− mice by days 3 and 4 p.i. (see Fig. S5 in the supplemental material).
VN/1203 and 1918 viruses induce the activation of type I IFN-regulated genes even in the absence of the IFN-α/β receptor.
Activation of IFN signaling is an important component of the innate antiviral response, and our previous studies of the 1918 virus in mice and VN/1203 in macaques suggested differences in IFN signaling in response to these viruses (6, 17). We therefore sought to directly compare the IFN response to these viruses in the mouse infection model. To identify IFN-regulated genes, we treated uninfected mice with IFN and identified the resulting differential expression of genes in the lungs. We also took advantage of mouse genetics and used IFNR1−/− mice to determine the importance of an intact IFN response to the host response to these viruses.
To identify IFN-regulated genes, we treated wild-type or IFNR1−/− mice with 10,000 U of recombinant human IFN-α A/D and harvested lung tissue at 8 and 24 h after treatment. These samples were then used to generate a set of 854 type I IFN-regulated genes, which was used as a common gene set for clustering and analysis of variance (ANOVA) of gene expression data from virus-infected animals. We found that activation of type I IFN-regulated genes was stronger in VN/1203-infected mice than in mice infected with the 1918 virus and that VN/1203 elicited steady and sustained activation of these genes even in animals lacking the IFN-α/β receptor (Fig. 6A). In animals infected with the 1918 virus, there was little activation of the IFN-regulated genes at day 1 p.i., but by days 3 and 4, IFN-regulated gene expression reached almost the same level as that induced by VN/1203. Since it was possible that IFNR1−/− mice compensated for the absence of IFNR1 with increased expression of IFN-λ3 (also known as IL28B) (4, 18), we used quantitative RT-PCR to detect IL28B in lung samples at 1, 3, and 4 days p.i. We found that IL28B was not overexpressed in the IFNR1−/− mice and that expression correlated with the temporal regulation of IFN-β1 and, to a lesser extent, IFN-γ (see Fig. S4C in the supplemental material).
FIG. 6.

Expression of interferon-regulated genes in response to interferon treatment or VN/1203 or 1918 infection. (A) Expression of type I interferon response genes in wild-type or IFNR1−/− mice infected with 1918 and VN/1203 viruses. Expression was determined by ANOVA of microarray data using cutoff values of ≥2-fold change and ANOVA P ≤ 0.01, respectively, from mock-infected wild-type mouse lung samples treated with interferon or untreated. This analysis generated a subset of 854 genes (right) that were used to cluster the mouse data. KO, knockout. (B) Expression of type I interferon response genes in MEFs infected with 1918 and VN/1203 viruses. Expression was determined by ANOVA analysis using cutoff values of ≥2-fold change and ANOVA P ≤ 0.01, respectively.
IFNR1−/− mice infected with either virus died sooner than infected wild-type animals, and IFNR1−/− mice infected with VN/1203 died approximately 3 days sooner than those infected with the 1918 virus (see Fig. S6A in the supplemental material). The percentage of survival was determined by the weight loss parameter, according to the CDC's Institutional Animal Care and Use Committee (see Fig. S1B in the supplemental material). VN/1203 disseminated to brain, liver, and spleen, and in contrast to wild-type animals, the 1918 virus was also detected in the brains and spleens of IFNR1−/− mice at day 5 p.i. (see Fig. S6B in the supplemental material). As in the case of wild-type mice, viral dissemination in the IFNR1−/− mice did not correlate with the viral titer (see Fig. S6C in the supplemental material). Even though the 1918 virus was detected in the brains and spleens of IFNR1−/− mice at day 5 p.i., histopathology revealed no lesions in these organs (Fig. 7A and B). Lesions in spleens were observed in VN/1203-infected IFNR1−/− mice (Fig. 7C and D). Staining for influenza virus antigen in the bronchus was more uniformly distributed in VN/1203-infected than 1918-infected mice, and staining was more uniform in bronchiolar epithelium than in alveolar cells (Fig. 7E and F).
FIG. 7.

Histopathology analysis of 1918 and VN/1203 infections in IFNR1−/− mice. (A) Normal spleen of an IFNR1−/− mouse infected by 1918 virus at day 1 p.i. (B) Normal liver of an IFNR1−/− mouse infected by 1918 virus at day 4 p.i. (C) Apoptotic spleen of an IFNR1−/− mouse infected by VN/1203 virus at day 1 p.i. (D) Lymphocyte depletion in the periarteriolar lymphatic sheath with heterophilic splenitis of an IFNR1−/− mouse infected by VN/1203 virus at day 1 p.i. (E) Common viral staining in bronchus of an IFNR1−/− mouse infected by 1918 virus at day 3 p.i. (F) Intense antigen staining in the bronchus of an IFNR1−/− mouse infected by VN/1203 virus at day 3 p.i.
To determine whether we could recapitulate our results using a simpler system, we performed in vitro 1918 and VN/1203 infection experiments using MEFs from wild-type and IFNR1−/− mice. Cells were harvested at 8 and 24 h p.i. for microarray analysis. Our analyses showed that wild-type and IFNR1−/− MEFs both exhibited a type I IFN response to VN/1203 infection, whereas the 1918 virus elicited weak activation of IFN-regulated genes. Interestingly, 1918 infection of MEFs recapitulated the delayed activation of the type I IFN response that was observed in mouse lungs (Fig. 6B). These results clearly indicate a significant difference in the host responses to the H1N1 1918 virus and the avian H5N1 VN/1203 virus and correlate well with our mouse microarray data. The combination of stronger inflammatory and type I IFN responses induced by VN/1203 virus likely contributes to the increased pathogenesis of this virus in the mouse model.
DISCUSSION
In the present study, we observed that the highly pathogenic H5N1 VN/1203 virus was able to disseminate to extrapulmonary organs regardless of the expression of the type I IFN receptor. However, whereas the 1918 virus did not disseminate in wild-type mice (Fig. 1B), dissemination to brain and spleen was observed in the absence of the type I IFN receptor (see Fig. S6B in the supplemental material), although no damage to these tissues was apparent. These results clearly indicate a role for the IFN response in the control of influenza infection, as has been previously described (11, 40). Moreover, systemic disease and tissue damage, as suggested by necrosis of spleen and liver tissue, was observed only in IFNR1−/− mice infected with VN/1203 (Fig. 7). However, IFN signaling was not sufficient to prevent a lethal outcome, nor could it control the spread of VN/1203 to extrapulmonary organs (Fig. 1A and B). The observed dissemination to extrapulmonary organs could not be explained by differences in viral loads, since the viruses exhibited similar titers in the lungs at the measured time points (Fig. 1C; see Fig. S6C in the supplemental material).
VN/1203 induced an earlier and stronger host response.
An additional distinction between the viruses used in this study was the time to death following infection. In wild-type and IFNR1−/− mice, VN/1203-infected animals died sooner than 1918-infected mice. In addition, IFNR1−/− animals infected with either VN/1203 or the 1918 virus died sooner than infected immunocompetent wild-type mice. Similar results have recently been reported by Szretter et al. during infection of wild-type and IFNR1−/− mice with A/Hong Kong/483/97, a highly pathogenic H5N1 influenza virus (40).
We used global gene expression profiling to determine whether differences in the host transcriptional response to infection would provide clues to differences in disease outcome. Because the mouse LD50 for VN/1203 was lower than that of the 1918 virus, we normalized the experimental design by using the LD50 of each virus to determine the inoculum. Although this resulted in infection with different amounts of virus, this approach allowed a more accurate comparison of the host response to these viruses, and viral titers were nearly equivalent at all measured time points. Mice infected with VN/1203 elicited a strong transcriptional activation of the innate response at 1 day p.i. that persisted throughout infection (Fig. 2). In contrast, the host transcriptional response was delayed in 1918 virus-infected mice. Extensive functional-genomics analysis using Ingenuity Pathways Analysis showed that the inflammatory response was one of the most significant functional categories differentially regulated by the VN/1203 and 1918 viruses (Fig. 3A). In particular, we found that at day 1 p.i., the VN/1203 virus upregulated genes associated with the inflammatory response, including the inflammasome components NLRP3, IL-1β, and CASP1, whose products are important for the innate immune response to influenza virus infection (Fig. 3B) (3, 13, 41).
Our data indicate that as early as day 1 p.i., VN/1203 is able to exacerbate the inflammatory response and trigger the type I IFN response. These responses are sustained throughout infection, which likely favors a “cytokine storm” response that is detrimental to the host (30). In-depth analysis of the transcriptional profiles suggested that the delayed host response during 1918 infection (Fig. 2) was a consequence of the inability of the host immune response to activate the transcription of critical cell signaling pathways involved in the classical response to viral pathogens (Fig. 4). For example, the transcriptional activation of the pattern recognition receptors (PRRs) toll-like receptors (TLRs) and RIG-I helicase were almost nonexistent at 1 day p.i. Furthermore, the activation of genes encoding proteins that modulate the immune response, such as chemokines and NF-κB, was also delayed. The activation of the effector components PKR, inflammatory response, and neutrophil activation and infiltration were also unchanged. There was activation of these cell signaling categories by 3 and 4 days p.i., although to a lesser extent than during VN/1203 infection.
Dissemination of VN/1203 in wild-type mice correlates with sustained activation of the inflammatory response and altered hematological function and lipoxin signaling.
Our results indicated that VN/1203 infection induced strong activation of the inflammatory response (Fig. 3A), including inflammasome genes, at day 1 p.i. (Fig. 3B). Virus dissemination and an intense inflammatory response have also been reported as features of H5N1 pathogenesis in humans (9). The physiological consequence of acute inflammation is the transvasation of immune cells from the blood to the infected tissues and the production of chemokines, cytokines, and vasoactive amines. Activated neutrophils release reactive oxygen and nitrogen species that act on pathogen and host targets in an indiscriminate manner (25).
We used a bioinformatics approach to attempt to gain insight into why VN/1203 was able to disseminate during infection of wild-type mice. This analysis revealed two biological categories associated with dissemination. The first category was the differential regulation of hematological system development and function (Fig. 5A), which was steadily activated at all time points analyzed. These same genes were activated only later and to a lesser extent during 1918 virus infection. The second identified category was the inhibitory effect of lipoxin signaling (Fig. 5B). These results depict a scenario where VN/1203 elicits a strong and steady upregulation of genes involved in the hematological function, a critical physiological response to acute inflammation, to trigger the resolution and healing processes at the sites of inflammation. Recently, it has been reported that people with severe cases of human swine H1N1 influenza infection are at risk to develop blood clots in the lungs (1), suggesting that dysregulation of the hematological system is likely important for the severity of disease.
Using an independent analytical tool for functional analysis (GeneGo), we also found a small set of genes related to hematological function whose expression was differentially regulated by the two viruses. These genes include fibrinogen α, β, and γ (FGA, FGB, and FGG); thrombomodulin (F2); and coagulation factor 3 (F3), which are components of the blood coagulation pathway (data not shown). Interestingly, it has been reported that disseminated intravascular coagulation generated by strong activation of the coagulation system that results in microvascular thrombosis (24) may be a contributing factor for multisystem organ failure during Ebola virus infection (12, 36). These results may suggest that the robust expression of hematological-function genes related to proinflammatory responses and blood coagulation system genes may contribute to VN/1203 virus dissemination.
In addition, we found that loss of the inhibitory action of lipoxin on neutrophil function was also associated with dissemination of VN/1203 in wild-type mice. Lipoxins play a role in the modulation of immune responses during microbial infection (21) and have potent anti-inflammatory properties in diseases such as tuberculosis, toxoplasmosis, arthritis, and cystic fibrosis (2, 5, 15, 21, 39). Studies of Toxoplasma gondii and Mycobacterium tuberculosis infection indicate that lipoxin-dependent inhibition of proinflammatory type 1 responses favor transmission and propagation of these pathogens (21). In particular, lipoxins inhibit important defense mechanisms, such as leukocyte migration, translocation of NF-κB, leukotriene function, TNF-induced chemokine production, natural killer cell function, and expression of chemokine receptor and adhesion molecules. The differential transcriptional regulation of lipoxin signaling and its inhibitory action on neutrophil functions may be important factors in differences in VN/1203 and 1918 virus pathogenesis.
Another avenue that we investigated was the detected dissemination of the 1918 virus in IFNR1−/− animals. Our data indicate that infection of wild-type mice with the 1918 virus resulted in the activation of important host functions, such as the inflammatory response, immune cell trafficking, and cell death, which at day 1 p.i. were at levels equivalent to those of the response elicited in IFNR1−/− mice. However, while wild-type animals produced a robust and steady increase in the activation of genes associated with these functions at days 3 and 4 p.i., this response was weak in IFNR1−/− animals (see Fig. S5 in the supplemental material). These results suggest that an intact immune response is necessary to control dissemination of the 1918 virus. However, this response must be controlled, since the exacerbated inflammatory response elicited by VN/1203 was also correlated with virus dissemination.
VN/1203 and the 1918 virus induce the activation of type I IFN-regulated genes even in the absence of the IFN-α/β receptor.
Gene expression data from 1918 and VN/1203 infections indicated that both viruses induced the activation of type I IFN-regulated genes in wild-type mice. Interestingly, the absence of IFNR1 did not completely abrogate the expression of type I IFN-related genes in response to either VN/1203 or the 1918 virus, and VN/1203 in particular elicited a strong IFN response (Fig. 6A). Our results suggest that the response to VN/1203 infection may be due in part to differences in the activation of TLRs and NF-κB signaling cascades. Recently, it has been shown that IFN-λ is important for controlling influenza replication in mice (4, 26). Therefore, a formal possibility in our experiments was that some of the observed transcriptional activation changes were due in part to the compensatory effects of IFN-λ expression. However, our quantitative RT-PCR results suggest that this was not the case (see Fig. S4C in the supplemental material).
The mouse model is a complex system in which cell infiltration is a landmark of the host response. Using MEFs, a system in which there is no cell infiltration, we were able to recapitulate our in vivo results. MEFs also exhibited a delay in the transcriptional activation of the innate response to 1918 infection relative to that elicited by VN/1203 (Fig. 6B). These results suggest that in mice or in mouse-derived cells, VN/1203 elicits a stronger IFN response that correlates with disease outcome. These data illustrate the complex and delicate homeostatic balance of the host response against highly pathogenic influenza infections.
Taken together, our results suggest that VN/1203-infected mice likely died faster as a consequence of several factors: (i) the early and sustained induction of an inflammatory response that may be detrimental rather than protective to the host; (ii) the additive and/or synergistic effects of the virus-sensing (TLRs and RIG-I), modulator (chemokines, NF-κB, and IFN), and effector (PKR, neutrophil infiltration, and inflammation) components of the immune response, which were transcriptionally induced in a steady manner throughout infection; (iii) the inhibition of lipoxin-mediated anti-inflammatory responses; and (iv) the ability of VN/1203 to disseminate to extrapulmonary organs.
Interestingly, our findings using the mouse model show intriguing differences and similarities compared with our findings using a cynomolgus macaque model of influenza virus infection (8). We previously reported that all 1918 virus-infected macaques succumbed to infection, whereas, despite the fact that there were initial signs of tissue damage, all VN/1203-infected macaques recovered. Interestingly, in macaques, it was the 1918 virus that upregulated key inflammasome components, whereas these genes were downregulated during VN/1203 infection. Consolidation of our data from macaque (8) and mouse infection studies suggests that the early and sustained upregulation of the inflammatory response is detrimental to the host (Fig. 8). It is also worth noting that in both animal models upregulation of the inflammatory response took place at 24 h p.i. (in macaques by the 1918 virus and in mice by VN/1203). It is also possible that differences in cellular microRNA expression between mouse and macaque may affect disease outcome, as we have recently reported that cellular microRNAs influence the inflammatory response to influenza virus infection (19). Together, our findings highlight the importance of the inflammatory response and suggest new lines of experimentation, including the infection of inflammasome knockout mice and specific targeting of the Alox5 gene, responsible for lipoxin biogenesis. The inflammatory response is also an attractive target for drug intervention, and the development of new drugs that target the inflammatory response may prevent severe tissue damage.
FIG. 8.

Model illustrating the consolidation of data obtained from macaques and mice infected with highly pathogenic 1918 and VN/1203 viruses. Provided is an illustration depicting our current understanding of influenza virus infections in macaques and mice. The network diagrams for macaques (8) and mice (Fig. 3B) emphasize the differential regulation of the inflammatory response and activation of inflammasome components that was found during 1918 and VN/1203 virus infections, respectively.
Supplementary Material
Acknowledgments
This study was funded in part by National Institute of Allergy and Infectious Diseases grants P01 AI058113 and 2P01 AI058113 (to M.G.K.) and Current Research Information Systems Project 6612-32000-049-00D (to D.E.S.).
We thank Sarah Belisle, Jenny Tisoncik, and Angela Rasmussen (Department of Microbiology, University of Washington) for helpful discussions and for reviewing the manuscript.
The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the funding agency.
Footnotes
Published ahead of print on 26 May 2010.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Agarwal, P. P., S. Cinti, and E. A. Kazerooni. 2009. Chest radiographic and CT findings in novel swine-origin influenza A (H1N1) virus (S-OIV) infection. Am. J. Roentgenol. 193:1488-1493. [DOI] [PubMed] [Google Scholar]
- 2.Aliberti, J., C. Serhan, and A. Sher. 2002. Parasite-induced lipoxin A4 is an endogenous regulator of IL-12 production and immunopathology in Toxoplasma gondii infection. J. Exp. Med. 196:1253-1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allen, I. C., M. A. Scull, C. B. Moore, E. K. Holl, E. McElvania-TeKippe, D. J. Taxman, E. H. Guthrie, R. J. Pickles, and J. P. Ting. 2009. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30:556-565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ank, N., M. B. Iversen, C. Bartholdy, P. Staeheli, R. Hartmann, U. B. Jensen, F. Dagnaes-Hansen, A. R. Thomsen, Z. Chen, H. Haugen, K. Klucher, and S. R. Paludan. 2008. An important role for type III interferon (IFN-lambda/IL-28) in TLR-induced antiviral activity. J. Immunol. 180:2474-2485. [DOI] [PubMed] [Google Scholar]
- 5.Bafica, A., C. A. Scanga, C. Serhan, F. Machado, S. White, A. Sher, and J. Aliberti. 2005. Host control of Mycobacterium tuberculosis is regulated by 5-lipoxygenase-dependent lipoxin production. J. Clin. Invest. 115:1601-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baskin, C. R., H. Bielefeldt-Ohmann, T. M. Tumpey, P. J. Sabourin, J. P. Long, A. Garcia-Sastre, A. E. Tolnay, R. Albrecht, J. A. Pyles, P. H. Olson, L. D. Aicher, E. R. Rosenzweig, K. Murali-Krishna, E. A. Clark, M. S. Kotur, J. L. Fornek, S. Proll, R. E. Palermo, C. L. Sabourin, and M. G. Katze. 2009. Early and sustained innate immune response defines pathology and death in nonhuman primates infected by highly pathogenic influenza virus. Proc. Natl. Acad. Sci. U. S. A. 106:3455-3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chomczynski, P., and N. Sacchi. 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162:156-159. [DOI] [PubMed] [Google Scholar]
- 8.Cilloniz, C., K. Shinya, X. Peng, M. J. Korth, S. C. Proll, L. D. Aicher, V. S. Carter, J. H. Chang, D. Kobasa, F. Feldmann, J. E. Strong, H. Feldmann, Y. Kawaoka, and M. G. Katze. 2009. Lethal influenza virus infection in macaques is associated with early dysregulation of inflammatory related genes. PLoS Pathog. 5:e1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Jong, M. D. 2008. H5N1 transmission and disease: observations from the frontlines. Pediatr. Infect. Dis. J. 27:S54-S56. [DOI] [PubMed] [Google Scholar]
- 10.Fraser, C., C. A. Donnelly, S. Cauchemez, W. P. Hanage, M. D. Van Kerkhove, T. D. Hollingsworth, J. Griffin, R. F. Baggaley, H. E. Jenkins, E. J. Lyons, T. Jombart, W. R. Hinsley, N. C. Grassly, F. Balloux, A. C. Ghani, N. M. Ferguson, A. Rambaut, O. G. Pybus, H. Lopez-Gatell, C. M. Apluche-Aranda, I. B. Chapela, E. P. Zavala, D. M. Guevara, F. Checchi, E. Garcia, S. Hugonnet, and C. Roth. 2009. Pandemic potential of a strain of influenza A (H1N1): early findings. Science 324:1557-1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Sastre, A., A. Egorov, D. Matassov, S. Brandt, D. E. Levy, J. E. Durbin, P. Palese, and T. Muster. 1998. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 252:324-330. [DOI] [PubMed] [Google Scholar]
- 12.Geisbert, T. W., H. A. Young, P. B. Jahrling, K. J. Davis, E. Kagan, and L. E. Hensley. 2003. Mechanisms underlying coagulation abnormalities in Ebola hemorrhagic fever: overexpression of tissue factor in primate monocytes/macrophages is a key event. J. Infect. Dis. 188:1618-1629. [DOI] [PubMed] [Google Scholar]
- 13.Ichinohe, T., H. K. Lee, Y. Ogura, R. Flavell, and A. Iwasaki. 2009. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 206:79-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson, N. P., and J. Mueller. 2002. Updating the accounts: global mortality of the 1918-1920 “Spanish” influenza pandemic. Bull. Hist Med. 76:105-115. [DOI] [PubMed] [Google Scholar]
- 15.Karp, C. L., L. M. Flick, R. Yang, J. Uddin, and N. A. Petasis. 2005. Cystic fibrosis and lipoxins. Prostaglandins Leukot. Essent. Fatty Acids 73:263-270. [DOI] [PubMed] [Google Scholar]
- 16.Kash, J. C., T. M. Tumpey, S. C. Proll, V. Carter, O. Perwitasari, M. J. Thomas, C. F. Basler, P. Palese, J. K. Taubenberger, A. Garcia-Sastre, D. E. Swayne, and M. G. Katze. 2006. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature 443:578-581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobasa, D., S. M. Jones, K. Shinya, J. C. Kash, J. Copps, H. Ebihara, Y. Hatta, J. H. Kim, P. Halfmann, M. Hatta, F. Feldmann, J. B. Alimonti, L. Fernando, Y. Li, M. G. Katze, H. Feldmann, and Y. Kawaoka. 2007. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature 445:319-323. [DOI] [PubMed] [Google Scholar]
- 18.Li, W., A. Lewis-Antes, J. Huang, M. Balan, and S. V. Kotenko. 2008. Regulation of apoptosis by type III interferons. Cell Prolif. 41:960-979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li, Y., E. Y. Chan, J. Li, C. Ni, X. Peng, E. Rosenzweig, T. M. Tumpey, and M. G. Katze. 2010. MicroRNA expression and virulence in pandemic influenza virus-infected mice. J. Virol. 84:3023-3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar]
- 21.Machado, F. S., and J. Aliberti. 2008. Role of lipoxin in the modulation of immune response during infection. Int. Immunopharmacol. 8:1316-1319. [DOI] [PubMed] [Google Scholar]
- 22.Machado, F. S., J. E. Johndrow, L. Esper, A. Dias, A. Bafica, C. N. Serhan, and J. Aliberti. 2006. Anti-inflammatory actions of lipoxin A4 and aspirin-triggered lipoxin are SOCS-2 dependent. Nat. Med. 12:330-334. [DOI] [PubMed] [Google Scholar]
- 23.Maines, T. R., X. H. Lu, S. M. Erb, L. Edwards, J. Guarner, P. W. Greer, D. C. Nguyen, K. J. Szretter, L. M. Chen, P. Thawatsupha, M. Chittaganpitch, S. Waicharoen, D. T. Nguyen, T. Nguyen, H. H. Nguyen, J. H. Kim, L. T. Hoang, C. Kang, L. S. Phuong, W. Lim, S. Zaki, R. O. Donis, N. J. Cox, J. M. Katz, and T. M. Tumpey. 2005. Avian influenza (H5N1) viruses isolated from humans in Asia in 2004 exhibit increased virulence in mammals. J. Virol. 79:11788-11800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mammen, E. F. 2000. Disseminated intravascular coagulation (DIC). Clin. Lab. Sci. 13:239-245. [PubMed] [Google Scholar]
- 25.Medzhitov, R. 2008. Origin and physiological roles of inflammation. Nature 454:428-435. [DOI] [PubMed] [Google Scholar]
- 26.Mordstein, M., G. Kochs, L. Dumoutier, J. C. Renauld, S. R. Paludan, K. Klucher, and P. Staeheli. 2008. Interferon-lambda contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog. 4:e1000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muller, U., U. Steinhoff, L. F. Reis, S. Hemmi, J. Pavlovic, R. M. Zinkernagel, and M. Aguet. 1994. Functional role of type I and type II interferons in antiviral defense. Science 264:1918-1921. [DOI] [PubMed] [Google Scholar]
- 28.Neumann, G., T. Noda, and Y. Kawaoka. 2009. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459:931-939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Novel Swine Origin influenza A (H1N1) Virus Investigation Team. 2009. Emergence of a Novel Swine-Origin Influenza A (H1N1) Virus in Humans. N. Engl. J. Med. 360:2605-2615. [DOI] [PubMed] [Google Scholar]
- 30.Owen, D. M., and M. Gale, Jr. 2009. Fighting the flu with inflammasome signaling. Immunity 30:476-478. [DOI] [PubMed] [Google Scholar]
- 31.Peiris, J. S., W. C. Yu, C. W. Leung, C. Y. Cheung, W. F. Ng, J. M. Nicholls, T. K. Ng, K. H. Chan, S. T. Lai, W. L. Lim, K. Y. Yuen, and Y. Guan. 2004. Re-emergence of fatal human influenza A subtype H5N1 disease. Lancet 363:617-619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perkins, L. E., and D. E. Swayne. 2001. Pathobiology of A/chicken/Hong Kong/220/97 (H5N1) avian influenza virus in seven gallinaceous species. Vet. Pathol. 38:149-164. [DOI] [PubMed] [Google Scholar]
- 33.Perrone, L. A., A. Ahmad, V. Veguilla, X. Lu, G. Smith, J. M. Katz, P. Pushko, and T. M. Tumpey. 2009. Intranasal vaccination with 1918 influenza virus-like particles protects mice and ferrets from lethal 1918 and H5N1 influenza virus challenge. J. Virol. 83:5726-5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reed, L. J., and H. Muench. 1938. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 27:493-497. [Google Scholar]
- 35.Richmond, J., and R. McKinney. 2007. Laboratory biosafety level criteria, p. 41-71. In J. Richmond and R. McKinney (ed.), Biosafety in microbiological and biomedical laboratories. Centers for Disease Control and Prevention, Atlanta, GA.
- 36.Rubins, K. H., L. E. Hensley, V. Wahl-Jensen, K. M. Daddario DiCaprio, H. A. Young, D. S. Reed, P. B. Jahrling, P. O. Brown, D. A. Relman, and T. W. Geisbert. 2007. The temporal program of peripheral blood gene expression in the response of nonhuman primates to Ebola hemorrhagic fever. Genome Biol. 8:R174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Serhan, C. N., N. Chiang, and T. E. Van Dyke. 2008. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 8:349-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Staeheli, P., R. Grob, E. Meier, J. G. Sutcliffe, and O. Haller. 1988. Influenza virus-susceptible mice carry Mx genes with a large deletion or a nonsense mutation. Mol. Cell. Biol. 8:4518-4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38a.Stoughton, R., and H. Dai. February 2002. Statistical combining of cell expression profiles. U.S. patent 6,351,712.
- 39.Svensson, C. I., M. Zattoni, and C. N. Serhan. 2007. Lipoxins and aspirin-triggered lipoxin inhibit inflammatory pain processing. J. Exp. Med. 204:245-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Szretter, K. J., S. Gangappa, J. A. Belser, H. Zeng, H. Chen, Y. Matsuoka, S. Sambhara, D. E. Swayne, T. M. Tumpey, and J. M. Katz. 2009. Early control of H5N1 influenza virus replication by the type I interferon response in mice. J. Virol. 83:5825-5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas, P. G., P. Dash, J. R. Aldridge, Jr., A. H. Ellebedy, C. Reynolds, A. J. Funk, W. J. Martin, M. Lamkanfi, R. J. Webby, K. L. Boyd, P. C. Doherty, and T. D. Kanneganti. 2009. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 30:566-575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tumpey, T. M., C. F. Basler, P. V. Aguilar, H. Zeng, A. Solorzano, D. E. Swayne, N. J. Cox, J. M. Katz, J. K. Taubenberger, P. Palese, and A. Garcia-Sastre. 2005. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science 310:77-80. [DOI] [PubMed] [Google Scholar]
- 43.Tumpey, T. M., T. R. Maines, N. Van Hoeven, L. Glaser, A. Solorzano, C. Pappas, N. J. Cox, D. E. Swayne, P. Palese, J. M. Katz, and A. Garcia-Sastre. 2007. A two-amino acid change in the hemagglutinin of the 1918 influenza virus abolishes transmission. Science 315:655-659. [DOI] [PubMed] [Google Scholar]
- 44.van den Broek, M. F., U. Muller, S. Huang, M. Aguet, and R. M. Zinkernagel. 1995. Antiviral defense in mice lacking both alpha/beta and gamma interferon receptors. J. Virol. 69:4792-4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yuen, K. Y., P. K. Chan, M. Peiris, D. N. Tsang, T. L. Que, K. F. Shortridge, P. T. Cheung, W. K. To, E. T. Ho, R. Sung, and A. F. Cheng. 1998. Clinical features and rapid viral diagnosis of human disease associated with avian influenza A H5N1 virus. Lancet 351:467-471. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.