

Abstract
Airway macrophages provide a first line of host defense against a range of airborne pathogens, including influenza virus. In this study, we show that influenza viruses differ markedly in their abilities to infect murine macrophages in vitro and that infection of macrophages is nonproductive and no infectious virus is released. Virus strain BJx109 (H3N2) infected macrophages with high efficiency and was associated with mild disease following intranasal infection of mice. In contrast, virus strain PR8 (H1N1) was poor in its ability to infect macrophages and highly virulent for mice. Depletion of airway macrophages by clodronate-loaded liposomes led to the development of severe viral pneumonia in BJx109-infected mice but did not modulate disease severity in PR8-infected mice. The severe disease observed in macrophage-depleted mice infected with BJx109 was associated with exacerbated virus replication in the airways, leading to severe airway inflammation, pulmonary edema, and vascular leakage, indicative of lung injury. Thymic atrophy, lymphopenia, and dysregulated cytokine and chemokine production were additional systemic manifestations associated with severe disease. Thus, airway macrophages play a critical role in limiting lung injury and associated disease caused by BJx109. Furthermore, the inability of PR8 to infect airway macrophages may be a critical factor contributing to its virulence for mice.
Airway macrophages (Mφ) (AM), located at the interphase between air and lung tissue, provide the first line of defense following inhalation of airborne pathogens, including influenza viruses. In addition to phagocytosis of virions and virus-infected cells (16, 24), infection of AM represents an early event in recognition of the virus by the innate immune system. Following intranasal (i.n.) infection of mice, influenza virus replicates productively in type II epithelial cells lining the respiratory tract. Murine Mφ are also susceptible to influenza virus infection and viral proteins are produced, but replication is abortive and infectious progeny are not released (52, 69), although recent studies suggest limited release from mouse Mφ exposed to highly pathogenic H1N1 and H5N1 viruses (44). Following exposure to influenza virus, Mφ synthesize and release proinflammatory cytokines and alpha/beta interferon (26, 27, 45, 55), which further limit viral replication and spread within the respiratory tract. Inflammatory responses in the airways must be tightly regulated to ensure rapid virus clearance while avoiding excessive or chronic inflammation that may damage the delicate tissue-air interface.
Liposome-encapsulated dichloromethylene diphosphonate (clodronate or CL2-MDP) is taken up by phagocytic Mφ and accumulates in the cytosol, resulting in Mφ death and depletion (66). Administration of clodronate liposomes (CL-LIP) has been widely used to selectively deplete airway Mφ in mouse models without affecting circulating monocytes (for examples, see references 4, 6, 8, 36, 47, 64, and 71). While CL-LIP has been used predominantly in rodent models of infection, it is noteworthy that CL-LIP treatment of pigs, a natural host of influenza virus, resulted in enhanced morbidity and mortality following infection with a human H1N1 subtype virus (30). In murine studies, depletion of airway Mφ prior to influenza virus infection led to increased cytotoxic CD8+ T-cell responses in the lungs of virus-infected mice (71); however, treatment with CL-LIP 48 h after infection was associated with impaired CD8+ T-cell responses (41). Furthermore, CL-LIP treatment prior to intranasal infection of mice with a recombinant virus bearing the surface glycoproteins of the 1918 pandemic strain led to exacerbated disease and mortality (64). Treatment of mice with CL-LIP has been associated with enhanced virus replication (41, 64, 71); however, the mechanisms by which airway Mφ initiate and modulate inflammatory responses and disease after influenza virus infection have not been fully elucidated.
We observed in a previous study that influenza A virus strains show marked differences in their abilities to infect murine Mφ in vitro and implicated the Mφ mannose receptor (MMR) (CD206), a C-type lectin (46), in infectious virus entry (48). Virus strain BJx109, a reassortant virus bearing the surface glycoproteins of A/Beijing/353/89 (H3N2) and internal components derived from A/PR/8/34 (H1N1) (PR8) bears a highly glycosylated hemagglutinin (HA) molecule and was shown to infect Mφ to high levels, while the HA of PR8 is poorly glycosylated and the virus infected Mφ very poorly. In the current study, we demonstrate that intranasal infection of mice with BJx109 leads to mild clinical disease, while infection with an equivalent dose of PR8 leads to severe disease and rapid death. Depletion of airway Mφ by intranasal administration of CL-LIP prior to and during infection with influenza viruses had little effect on the course of PR8 infection; however, BJx109 infection of Mφ-depleted animals led to severe disease and death. Severe disease was associated with enhanced virus replication, severe airway inflammation, and pulmonary edema and vascular leakage, indicative of lung injury. Together, these data demonstrate that airway Mφ play a critical role in moderating disease severity during BJx109 infection. Furthermore, they suggest that the ability of PR8 to evade infectious uptake by airway Mφ is likely to be an important factor contributing to its virulence for mice.
MATERIALS AND METHODS
Mice and viruses.
C57BL/6 (B6) mice were bred and housed under specific-pathogen-free conditions at the Department of Microbiology and Immunology, University of Melbourne, Melbourne, Australia. Six- to 8-week-old male mice were used in all experiments. The influenza A virus strains used in this study were A/PR/8/34 (H1N1), as well as BJx109 (H3N2) and Phil/82-X (H3N2) which are high-yielding reassortants of PR8 that bear the surface glycoproteins of A/Beijing/353/89 (H3N2) and A/Philippines/2/82 (H3N2), respectively. Additional wild-type (i.e., nonreassortant) viruses used were A/New York/55/2004 (H3N2), A/Brisbane/9/2007 (H3N2), A/New Caledonia/20/1999 (H1N1), and A/Solomon Islands/3/2006 (H1N1). The viruses were grown in 10-day embryonated hen's eggs by standard procedures and titrated on Madin-Darby canine kidney (MDCK) cells as described previously (1).
The reassortant influenza viruses used in this study were generated by eight-plasmid reverse genetics as previously described (43). The viruses were 7:1 reassortants consisting of either the PR8 (H1N1) backbone with the HA or neuraminidase (NA) gene from BJx109 (H3N2; PR8-BJx109 HA and PR8-BJx109 NA, respectively). The rescued viruses were recovered after 3 days and amplified in the allantoic cavities of 10-day-old embryonated hens' eggs.
Infection of mouse Mφ and epithelial cells.
Resident peritoneal exudate cell (PEC) Mφ and bronchoalveolar lavage (BAL) Mφ were obtained from B6 mice as previously described (48). PEC Mφ (2.5 × 105 cells/well), BAL Mφ (2.5 × 105 cells/well), MDCK cells (1 × 104 cells/well), or the LA-4 lung epithelial cell line (3 × 104 cells/well) were seeded into 8-well glass chamber slides (LabTek; Nun) and incubated overnight at 37°C. The cell density for seeding was chosen to achieve similar densities of adherent cells from the different cell populations. Cell monolayers were washed with serum-free medium and infected with 106 PFU of influenza virus as described previously (48). At 7 to 9 h postinfection, the slides were washed in phosphate-buffered saline (PBS), fixed in acetone, and stained with monoclonal antibody (MAb) clone MP3.10G2.IC7 (WHO Collaborating Centre for Reference and Research on Influenza, Melbourne, Australia), specific for the nucleoprotein of type A influenza viruses, followed by fluorescein isothiocyanate (FITC)-conjugated sheep anti-mouse immunoglobulin (Silenus, Melbourne, Australia) and viewed under ×40 magnification. The percentage of fluorescent cells was determined in a minimum of 4 random fields with a minimum of 200 cells counted for each sample. To determine the number of adherent cells remaining after exposure to influenza virus, chamber slides were incubated in 80% (vol/vol) acetone in water for 2 min at room temperature and then stained with 10 μg/ml propidium iodide (PI). Nuclear morphology was assessed, intact nuclei were counted in 4 or more independent fields, and these data were used to determine the percentage of viable cells.
To determine the titer of infectious virus present in supernatants from influenza virus-infected LA-4 epithelial cells or PEC Mφ, cell monolayers were incubated with 106 PFU of BJx109 or PR8 virus for 60 min, washed three times to remove free virus, and cultured at 37°C. At 2 and 24 h postinfection, the supernatants were removed and the presence of infectious virus was determined by standard plaque assay on MDCK cell monolayers in the presence of trypsin.
Infection and treatment of mice.
Mice were anesthetized and infected with 105 PFU of influenza virus via the i.n. route in 50 μl of PBS. The mice were weighed daily and assessed for visual signs of clinical disease, including inactivity, ruffled fur, labored breathing, and huddling behavior. Animals that had lost ≥25% of their original body weight and/or displayed evidence of pneumonia were euthanized. All research complied with the University of Melbourne's Animal Experimentation Ethics guidelines and policies. At various times after infection, mice were euthanized, and the lungs, nasal tissues, brain, and heart were removed, homogenized in PBS, and clarified by centrifugation. Titers of infectious virus in the tissue homogenates were determined by standard plaque assay on MDCK cells.
For depletion of airway Mφ, mice were treated i.n. with 100 μl of CL-LIP while under light anesthesia with isoflurane. Clodronate was a kind gift of Roche Diagnostics (Mannheim, Germany). It was encapsulated in liposomes as described previously (65). Mice were treated 48 h prior to infection and every 72 h thereafter, unless otherwise stated. Control animals received an equivalent volume of saline-loaded liposomes (SL-LIP) or PBS alone. In some experiments, mice were treated with CL-LIP diluted to 30% in PBS. Depletion of airway Mφ in the BAL fluid and lung parenchyma was monitored by flow cytometry as described below.
Recovery of leukocytes from mice.
BAL cells and heparinized blood were obtained as described previously (61). Lung cell suspensions were prepared by incubating minced lung tissue with 2 mg/ml collagenase A (Roche Diagnostics, Germany) for 30 min at 37°C and then pressing it through a fine wire mesh. Samples were treated with Tris-NH4Cl (0.14 M NH4Cl in 17 mM Tris, adjusted to pH 7.2) to lyse erythrocytes and washed in RPMI 1640 medium supplemented with 10% fetal calf serum (FCS) (RF10). Cell numbers and cell viability were assessed via trypan blue exclusion using a hemocytometer.
Differential leukocyte counts and flow cytometry.
For flow cytometry analysis, single-cell suspensions prepared from the blood, BAL fluid, and lung were incubated on ice for 20 min with supernatants from hybridoma 2.4G2 to block Fc receptors and then stained with appropriate combinations of FITC, phycoerythrin (PE), allophycocyanin (APC), or biotinylated monoclonal antibodies to Ly6G (1A8), Gr-1 (RB6-8C5), CD45.2 (104), CD8a (53-6.7), CD4 (GK1.5), B220 (RA3-6B2), NK1.1 (PK136), major histocompatibility complex (MHC) class II (I-Ab; AF6-120.1), CD11b (M1/70), and CD11c (HL3) (all from BD PharMingen) and F4/80 (BM8; Caltag Laboratory). Isotype controls were included as appropriate to facilitate gating of leukocyte populations. Living cells were analyzed by the addition of 10 μg/ml PI to each sample, and the cells were analyzed on a FACSCalibur flow cytometer. A minimum of 50,000 live cells (PI−) were collected. Airway Mφ were identified as CD11chigh MHC class IIint cells in lung cell suspensions and BAL fluids, while pulmonary dendritic cells (pulDC) were identified as CD11chigh MHC class IIhigh (31).
Pulmonary histopathology.
Lungs were inflated, fixed in 4% formaldehyde, and processed in paraffin wax as previously described (62). Airway inflammation of hematoxylin and eosin (H&E)-stained lung sections was evaluated on a subjective scale of 0, 1, 2, 3, 4, or 5 (corresponding to none, very mild, mild, moderate, marked, and severe inflammation, respectively) on randomized, blinded sections by three independent readers (59). Tissues were graded for peribronchiolar inflammation (around 3 to 5 small airways per section) and alveolitis in multiple random fields per section. Immunoperoxidase staining of paraffin-embedded lung sections was performed using polyclonal goat-anti influenza A virus antibody (AbD Serotec, United Kingdom) and the goat Vectastain ABC kit (Vector Laboratories) as previously described (62). Lung sections were viewed on a Leica DMI3000 B microscope and photographed at ×10 magnification with a Leica DFC 490 camera running from the Leica application software.
Assessment of lung edema.
The lung wet-to-dry weight ratio was used as an index of lung water accumulation during influenza virus infection. After euthanasia of mice, the lungs were surgically dissected, blotted dry, and weighed immediately (wet weight). The lung tissue was then dried in an oven at 60°C for 72 h and reweighed as dry weight. The ratio of wet to dry weight was calculated for each animal to assess tissue edema as previously described (2, 70). The concentration of protein in cell-free BAL supernatant was measured by adding Bradford protein dye. A standard curve using bovine serum albumin (BSA) was constructed, and the optical density (OD) was determined at 595 nm.
Cytokine bead array for the detection of inflammatory mediators.
The levels of gamma interferon (IFN-γ), tumor necrosis factor alpha (TNF-α), interleukin 6 (IL-6), IL-10, IL-12.p70, and monocyte chemoattractant protein 1 (MCP-1) in BAL supernatants and serum were determined with the use of a cytokine bead array mouse inflammation kit (Becton Dickinson) according to the manufacturer's instructions. The detection limit for the assay was 20 pg/ml for all cytokines tested.
Statistical analysis.
For the comparison of two sets of values, Student's t test (two-tailed, two-sample equal variance) was used. When comparing three or more sets of values, data were analyzed by one-way analysis of variance (ANOVA), followed by posthoc analysis using Tukey's multiple-comparison test. For the analysis of histopathological data, a Kruskal-Wallis test (nonparametric) was used, followed by the Dunn's posthoc test. Survival proportions were compared using the Mantel-Cox log rank test. A P value of ≤0.05 was considered statistically significant.
RESULTS
Influenza viruses differ in their abilities to infect murine Mφ, but not murine epithelial cells, in vitro.
Initial studies compared the infectivities of influenza virus strains BJx109 and PR8 for Mφ and epithelial cells using primary cells and representative cell lines. Consistent with our previous findings (50), primary Mφ isolated from PECs or BAL fluids via adherence to glass chamber slides were highly susceptible to infection by BJx109, but not by PR8 (Fig. 1 A). In contrast, BJx109 and PR8 infected epithelial cells, including a mouse airway epithelial cell line (LA-4), to equivalent levels, indicating there is no defect in the ability of PR8 to infect mammalian cells per se (Fig. 1A). As both HA and NA glycoproteins can modulate the efficiency of virus infection of target cells (67), we used engineered PR8 viruses expressing either the HA (PR8-BJx109 HA) or NA (PR8-BJx109 NA) of BJx109 to demonstrate that efficient infection of airway Mφ was associated with expression of the BJx109 HA, not the NA (Fig. 1B). In contrast, expression of either PR8 or BJx109 HA did not alter the ability of engineered viruses to infect LA-4 airway epithelial cells.
FIG. 1.
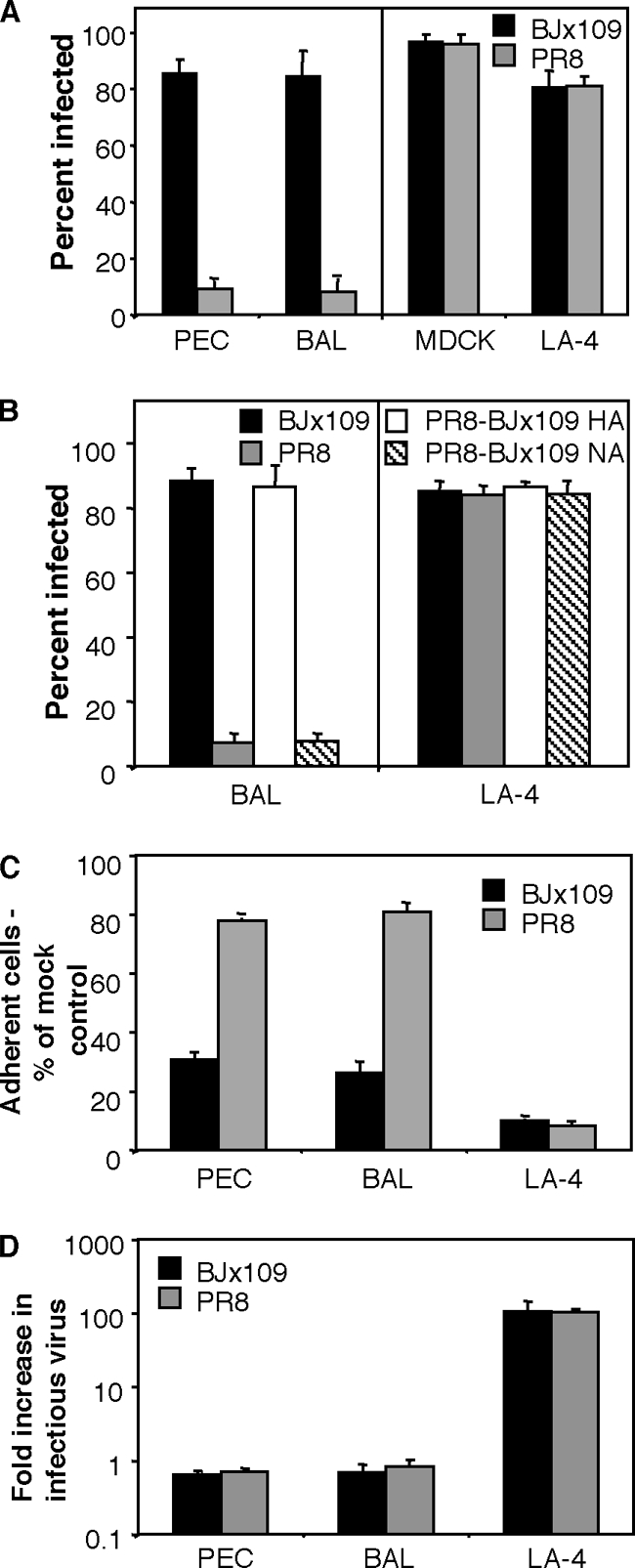
Influenza viruses BJx109 and PR8 differ in their abilities to infect Mφ, but not epithelial cells. (A) Monolayers of mouse PEC Mφ, BAL Mφ, MDCK cells, and LA-4 epithelial cells were infected in chamber slides with 106 PFU of BJx109 or PR8, as described in Materials and Methods. At 6 to 8 h postinfection, cells were fixed and stained via immunofluorescence for expression of influenza A virus nucleoprotein (NP). The mean percent infection (plus 1 standard deviation [SD]) from a minimum of 4 independent fields per chamber is shown for a representative experiment, and infection of Mφ by PR8 was significantly reduced compared to BJx109 in 4 independent experiments (P < 0.01; one-way ANOVA). (B) BAL Mφ or LA-4 epithelial cells were infected with 106 PFU of BJx109 or PR8 or with reverse-engineered viruses expressing 7 genes derived from PR8 with the HA (PR8-BJx109 HA) or the NA (PR8-BJx109 NA) of BJx109 and stained by immunofluorescence at 8 h postinfection. The data from a representative experiment are shown, and infection of Mφ with PR8 or PR8-BJx109 NA was significantly different from that with BJx109 in 3 independent experiments (P < 0.01; one-way ANOVA). (C) Numbers of adherent PEC Mφ and LA-4 cells 24 h after exposure to influenza viruses. Monolayers of PEC Mφ and LA-4 cells were infected in chamber slides with 106 PFU of BJx109 or PR8 or mock infected with medium alone. At 24 h postinfection, cells were fixed and stained via immunofluorescence for nucleic acids using PI. Nuclear morphology was assessed, and intact nuclei were counted in 4 independent fields. The data are expressed as a percentage of the number of nuclei counted in mock-infected controls. The error bars represent 1 SD. The data from a representative experiment are shown, and numbers of adherent Mφ exposed to BJx109 were significantly reduced compared to PR8 in 3 independent experiments (P < 0.01; one-way ANOVA). (D) Release of infectious virus from influenza virus-infected PEC Mφ and LA-4 cells. Monolayers of cells were infected with 106 PFU of BJx109 or PR8, and samples of supernatant were removed at 2 h (to detect residual virus inoculum) and 24 h (to detect newly synthesized virions released from infected cells) postinfection. The samples were assayed for infectious virus by plaque assay on MDCK cells, and the fold increase in infectious virus was calculated by dividing the viral titer obtained at 24 h by that obtained at 2 h. The data represent the mean (plus 1 SD) from 3 independent experiments.
We next determined the consequences of exposing Mφ or epithelial cells to BJx109 or PR8 virus. First, we determined the number of adherent PEC Mφ, BAL Mφ, or murine LA-4 epithelial cells that remained adherent 24 h after exposure to 106 PFU of BJx109, PR8, or medium alone (mock infection). As seen in Fig. 1C, few adherent LA-4 epithelial cells could be detected 24 h after exposure to either BJx109 or PR8 virus. Exposure of Mφ to BJx109 virus led to a marked reduction in cell adherence (<30%); however, exposure of Mφ to an equivalent dose of PR8 had little effect on the proportion of adherent cells (>80%). Consistent with these findings, visual examination of monolayers of primary Mφ 24 h postinfection demonstrated that >80% of cells exposed to BJx109 exhibited evidence of cytopathic effect (CPE) (characterized by rounding of adherent cells, cytoplasmic granularity, cells floating in the supernatant, and cell destruction) while <10% CPE was observed in Mφ monolayers exposed to an equivalent dose of PR8 (data not shown). LA-4 epithelial cells exposed to either BJx109 or PR8 exhibited >90% CPE at 24 h postinfection.
We next compared the abilities of BJx109 and PR8 to replicate productively in primary Mφ or in airway epithelial cells. For these experiments, cell monolayers were exposed to equivalent doses of either BJx109 or PR8 and washed to remove unbound virus, and the amount of infectious virus present in the cell supernatants was determined at 2 h (to determine residual levels of infectious virus) and 24 h (to determine levels released following virus replication) postinfection. Exposure of LA-4 epithelial cells to BJx109 or PR8 led to productive virus replication and amplification, as noted from the increased ratio of infectious virus in supernatants at 2 and 24 h postinfection (Fig. 1D). In contrast, exposure of PEC or BAL Mφ to BJx109 or PR8 was not associated with release of infectious virus between 2 h and 24 h postinfection, consistent with previous reports that infection of murine Mφ by influenza virus led to abortive virus replication (52).
Following intranasal infection, BJx109 and PR8 differ markedly in their virulence for mice.
Infection of Mφ by influenza virus is likely to represent an important innate immune response, as viral replication is abortive and no infectious virus is released (52) (Fig. 1D). Given the marked differences in the capacities of virus strains BJx109 and PR8 to infect murine Mφ, it was of interest to compare the virulence of the 2 virus strains following intranasal inoculation of B6 mice. Intranasal inoculation of mice with 105 PFU of BJx109 was not associated with significant weight loss over a 10-day monitoring period (Fig. 2 A, top), and all mice survived the infection (Fig. 2A, bottom). Furthermore, no clinical signs of disease (lethargy, ruffled fur, or labored breathing) were noted. Intranasal inoculation with an equivalent dose of PR8 led to rapid weight loss, and all mice succumbed 4 to 5 days postinfection. At the time of death, PR8-infected mice showed hunched posture, lethargy, and labored breathing, consistent with development of viral pneumonia. We used reverse-genetics viruses to demonstrate that expression of the HA, but not the NA, of BJx109 on a PR8 “backbone” was sufficient to ameliorate the severe disease associated with PR8 infection. As seen in Fig. 2B, mice infected with 105 PFU of PR8-BJx109 NA lost weight rapidly (top) and succumbed to infection (bottom), while mice infected with PR8-BJx109 HA showed no weight loss or signs of disease.
FIG. 2.

Virulence of influenza viruses for C57BL/6 mice. Groups of 5 mice were infected with 105 PFU of BJx109 or PR8 (A) or reverse-genetics virus PR8-BJx109 HA or PR8-BJx109 NA (B) via the intranasal route. Control mice received an equivalent volume of PBS. (Top) Mice were weighed daily, and the results are expressed as the mean percent weight change of each group ± standard error of the mean (SEM) compared to the weight immediately prior to infection. Animals displaying evidence of pneumonia and/or having lost >25% of their original body weights were euthanized. (Bottom) Survival of mice following intranasal infection with influenza viruses. The data shown are from one experiment and are representative of two or more independent experiments. The P values for survival proportions were obtained using the Mantel-Cox log rank test. BJx109 versus PR8, P < 0.01, and PR8-BJx109 HA versus PR8-BJx109 NA, P < 0.01, in 2 independent experiments.
A range of additional virus strains were found to infect Mφ efficiently (A/New York/55/2004 [H3N2], A/Brisbane/9/2007 [H3N2], A/New Caledonia/20/1999 [H1N1], and A/Solomon Islands/3/2006 [H1N1] at 87% ± 3%, 95% ± 1%, 83% ± 2%, and 85% ± 3% infected cells, under conditions identical to those described for Fig. 1A and B), and infection of mice with 105 PFU of each virus strain was not associated with weight loss or disease over a 10-day monitoring period (data not shown). Together, these findings suggest that internalization and nonproductive infection of airway Mφ could be critical factors limiting the severity of disease induced by BJx109.
Selective depletion of Mφ from airways and lung parenchyma following treatment with CL-LIP.
To deplete Mφ from the airways and the lung, naïve mice were treated with 100 μl of CL-LIP via the intranasal route, and the efficacy of this procedure was assessed after 2 days via flow cytometric analysis of BAL fluid or lung tissue, respectively. Control mice received an equivalent volume of SL-LIP or PBS alone. Based on the studies of Kirby et al., AM were identified as CD11c+ MHC-IIint cells and pulDC as CD11c+ MHC-IIhigh (31), and representative dot plots in Fig. 3 A show selective depletion of airway Mφ in BAL fluid of CL-LIP-treated mice. Compared to SL-LIP-treated controls, CL-LIP treatment 2 days prior to analysis led to a 60 to 80% reduction in the numbers of airway Mφ in BAL fluid or lung tissue (Fig. 3B) from naive animals 48 h after treatment. While CL-LIP treatment was effective in depleting airway Mφ, it did not deplete CD11c+ MHC-IIhigh pulDC in the airways (Fig. 3C). The numbers of pulDC in BAL fluid of CL-LIP-treated mice were actually enhanced compared to PBS- or SL-LIP-treated controls, while the numbers of lung pulDC were similar to those from control animals (Fig. 3C). Intranasal treatment of naïve mice with CL-LIP did not affect the numbers of circulating monocytes/macrophages (F4/80+ or CD11chigh MHC class IIint), DC (CD11chigh MHC class IIhigh), neutrophils (Ly6Ghigh), or B220+ lymphocytes when examined 2 days after treatment (data not shown), confirming local depletion of airway Mφ using this treatment regime.
FIG. 3.

Depletion of lung macrophages via treatment with clodronate-loaded liposomes. Groups of 5 naïve mice were treated with 100 μl of CL-LIP via the intranasal route. Control mice received an equivalent volume of PBS or SL-LIP. At 48 h posttreatment, the mice were killed, and the cell types present in BAL fluid and lungs were determined. (A) Representative dot plots of BAL cells from mice treated with PBS, SL-LIP, or CL-LIP. AM were identified as CD11c+ MHC class IIint and pulDC as CD11c+ MHC class IIhigh by flow cytometry. Total numbers of AM (B) and pulDC (C) in the BAL fluid and lung were determined by flow cytometry. *, CL-LIP-treated mice were significantly different from SL-LIP-treated controls (P < 0.05; one-way ANOVA).
Depletion of airway Mφ prior to and during influenza virus infection.
Intranasal infection with influenza viruses is known to induce recruitment of leukocytes, including Mφ, to the airways (11, 25). To maintain depletion of airway Mφ throughout the course of infection, we treated mice at day −2 relative to infection and every 3 days thereafter (i.e., days −2, +1, +4, and +7). Preliminary experiments established that treatment of mice with CL-LIP at days −2, +1, +4, and +7 relative to intranasal inoculation with PBS at day 0 (i.e., mock infection) did not result in any signs of overt illness (weight loss, hunched posture, or labored breathing) compared to untreated mice or mice treated identically with SL-LIP (data not shown). Mice treated with CL-LIP at days −2, +1, and +4 relative to infection with 105 PFU of BJx109 lost weight rapidly, and all mice succumbed to disease by day 7 postinfection (Fig. 4 A). In contrast, control animals infected with BJx109 and treated with SL-LIP or PBS showed no significant weight loss or disease. A similar phenomenon was observed when mice were infected with 105 PFU of Phil/82-X, a reassortant virus bearing the HA and NA of A/Philippines/2/82 and the internal components of PR8. In vitro, Phil/82-X infected murine Mφ efficiently (83% ± 6% infected Mφ, under conditions identical to those described for Fig. 1A) and did not induce weight loss or disease in mice following intranasal inoculation, although CL-LIP treatment led to severe disease and death (Fig. 4B). Mice treated with PBS, SL-LIP, or CL-LIP and infected with PR8 all lost weight rapidly and were euthanized 4 to 5 days postinfection (Fig. 4C). PR8 is a highly mouse-adapted strain, and numerous viral genes have been implicated in modulating disease severity (68); however, our findings with BJx109 and Phil82-X, both of which contain internal components derived from PR8, demonstrate that the HA/NA glycoproteins are of critical importance in enhancing the ability of the PR8 reassortant viruses to infect airway Mφ and in ameliorating disease severity in mice.
FIG. 4.

Effect of macrophage depletion on the course of disease following intranasal infection with influenza viruses. Groups of 5 B6 mice were depleted of macrophages via i.n. treatment with CL-LIP 48 h prior to infection with 105 PFU of BJx109 (A), Phil/82-X (B), or PR8 (C) and every 3 days thereafter. Control groups received an equivalent volume of SL-LIP or PBS alone. (Top) The mice were weighed daily, and the results are expressed as the mean percent weight change of each group (±SEM) compared to the original body weight. (Bottom) Animals that had lost ≥25% of their original body weight and/or presented with evidence of pneumonia were killed. The P values for survival proportions were obtained from 2 independent experiments using the Mantel-Cox log rank test. BJx109, CL-LIP versus SL-LIP, P < 0.01; Phil/82-X, CL-LIP versus SL-LIP, P < 0.01; and PR8, CL-LIP versus SL-LIP, P > 0.05.
At the time these studies were being conducted, Pribul et al. demonstrated that intranasal treatment of mice with 30% CL-LIP diluted in PBS led to effective depletion of airway Mφ with minimal associated lung inflammation (47). Our data confirmed these findings, as treatment of mice at day −2 with 30% CL-LIP reduced the percentage of airway Mφ by approximately 70% relative to SL-LIP (SL-LIP = 31.8 ± 4.2; 100% CL-LIP = 2.0 ± 0.5; 30% CL-LIP = 4.9 ± 1.9; all data are percent CD11C+ MHC-IIint cells out of total BAL cells at day 0). Furthermore, treatment of mice with 30% CL-LIP at days −2, +1, and +4 relative to infection with 105 PFU of BJx109 induced weight loss and mortality similar to that observed following treatment with 100% CL-LIP (data not shown). Thus, treatment with both 100% and 30% CL-LIP was included in all subsequent analysis for BJx109-infected mice.
Effect of Mφ depletion on virus replication in the airways during BJx109 infection of mice.
We next investigated factors contributing to the severity of clinical disease observed in Mφ-depleted mice following infection with BJx109. All subsequent analyses focused on day 7 postinfection, the time at which CL-LIP-treated mice succumbed to disease. First, we determined the viral loads in the respiratory tracts of BJx109-infected animals. By day 7 postinfection, infectious virus had been cleared from the airways of BJx109-infected mice treated with either PBS or PBS-CL (Fig. 5 A); however, 100- to 1,000-fold more virus was recovered in homogenates prepared from nasal tissues or lungs of mice treated with 30% or 100% CL-LIP. Immunohistochemical localization demonstrated abundant viral antigen in epithelial cells lining the bronchioles and large airways and throughout the alveoli of mice treated with 100% CL-LIP (Fig. 5B, ii) and 30% CL-LIP (Fig. 5B, iii), but not in PBS-treated (data not shown) or SL-LIP-treated (Fig. 5B, i) controls. Moreover, the distribution of viral antigen in the lungs of CL-LIP-treated mice infected with BJx109 was very similar to that observed in the lungs of mice infected with a similar dose of PR8 (Fig. 5B, iv). Thus, following depletion of airway Mφ via CL-LIP treatment, BJx109 is “redirected” to infect epithelial cells of the respiratory tract, thereby allowing productive virus replication and amplification in the airways. By removing airway Mφ, virus strain BJx109 induces clinical disease and pathogenesis similar to those of the mouse-adapted PR8 strain.
FIG. 5.

Depletion of airway macrophages during BJx109 infection is associated with enhanced virus replication in the airways. Groups of 5 B6 mice were depleted of macrophages via i.n. treatment with 100% CL-LIP or 30% CL-LIP 48 h prior to infection with 105 PFU of BJx109 and every 3 days thereafter. Control groups received an equivalent volume of SL-LIP or PBS alone. The mice were killed and analyzed at day 7 postinfection. (A) Lungs and nasal tissues were homogenized, and virus titers were determined by plaque assay on MDCK cells. The bars represent mean viral titers plus 1 SD. The detection limit for the plaque assays is indicated as a dotted line. *, virus titers from 100% CL-LIP- and 30% CL-LIP-treated mice that were significantly higher than those from SL-LIP-treated control animals (P < 0.01; one-way ANOVA). (B) Distribution of viral antigen in the lungs of macrophage-depleted or control mice. Representative images are shown at ×10 magnification. Cells positive for viral antigen are stained brown, and the arrows indicate representative areas of intense antigen staining.
Severe clinical disease following BJx109 infection of Mφ-depleted mice is associated with excessive inflammation of the airways.
Excessive or dysregulated inflammation of the airways has been associated with severe viral pneumonia during influenza virus infections of humans (12), macaques (32), and mice (44, 60, 64). Consistent with this, the pulmonary pathology observed in lung sections from BJx109-infected mice treated with either 100% (Fig. 6 A, ii) or 30% (Fig. 6A, iii) CL-LIP was markedly more severe than that of SL-LIP-treated (Fig. 6A, i) or PBS-treated (data not shown) controls and more similar to the inflammation observed in PR8-infected mice at day 5 postinfection (Fig. 6A, iv). When scored in a blinded manner by 3 independent readers, both peribronchiolar inflammation and alveolitis were markedly enhanced in the lungs from virus-infected mice treated with 100% or 30% CL-LIP compared to PBS- or SL-LIP-treated controls (Fig. 6B).
FIG. 6.

Depletion of airway macrophages during BJx109 infection is associated with enhanced pulmonary inflammation. Groups of 5 B6 mice were depleted of airway macrophages via i.n. treatment with 100% or 30% CL-LIP 48 h prior to infection with 105 PFU of BJx109 and every 3 days thereafter. Control groups received an equivalent volume of SL-LIP or PBS alone, and all mice were killed and analyzed at day 7 postinfection. (A) Representative images of inflammation in lung sections following H&E staining. Mice infected with 105 PFU of PR8 and analyzed at day 5 postinfection are included for comparison. The images are shown at ×10 magnification. (B) Histopathological scores for lung sections from BJx109-infected mice. Lung sections were scored blind for alveolitis and peribronchiolar inflammation from 0 to 5. The data shown represent scores from individual mice (as indicated by circles) and median values (as indicated by bars) obtained from 1 of 3 independent readers. Samples were compared for statistical significance using the Kruskal-Wallis test (nonparametric). For each reader, significant differences were observed in immunopathology scores between CL-LIP-treated mice (100% or 30%) and SL-LIP-treated mice (P < 0.05). (C) Inflammatory cells present in BAL fluid of BJx109-infected mice. BAL cells recovered from the airspaces of the lung were examined by flow cytometry for total numbers of CD45+ inflammatory cells, as well as for leukocyte subsets, including neutrophils (Gr-1high), NK cells (NK1.1+ TCR-β−), CD4+ cells (CD4+), CD8+ cells (CD8+), and pulDC (CD11c+ MHC class IIhigh). A minimum of 50,000 living cells (PI−) were collected and analyzed for each mouse. *, cell numbers from 100% CL-LIP- and 30% CL-LIP-treated mice that were significantly higher than those from SL-LIP-treated control animals (P < 0.05; one-way ANOVA).
We next characterized inflammatory cells and soluble mediators present in BAL fluids from Mφ-depleted mice to gain insight into factors contributing to the severe pulmonary immunopathology observed in these animals. Total BAL cell numbers were markedly increased in BJx109-infected mice treated with 100% or 30% CL-LIP, with numbers of neutrophils, NK cells, pulDC, B220+ cells, CD4+ T cells, and CD8+ T cells all significantly enhanced in Mφ-depleted mice compared to SL-LIP-treated controls (Fig. 6C). In particular, neutrophil and CD8+ T-cell numbers were 3- to 5-fold higher in BAL fluid from Mφ-depleted mice. Of note, airway Mφ (CD11c+ MHC-IIint) were reduced by 60 to 75% in CL-LIP-treated mice (20.2% ± 6.9%, 12.2% ± 6.7%, 1.5% ± 0.4% and 1.3% ± 0.3% of total BAL cells for mice treated with PBS, SL-LIP, 30% CL-LIP, or 100% CL-LIP, respectively), indicating effective Mφ depletion at day 7 postinfection, despite the large numbers of inflammatory cells recruited to the airways of CL-LIP-treated mice.
It has been postulated that excessive or dysregulated production of inflammatory mediators may contribute to lung pathology during influenza virus infection (9, 73). At day 7 postinfection, the severe pulmonary inflammation observed in CL-LIP-treated mice infected with BJx109 was associated with a significant enhancement in local (airway) production of MCP-1 (BAL fluid, 20.2 ± 8.1 and 32.5 ± 6.1 pg/ml, and serum, 10.9 ± 2.4 and 52.0 ± 30.3 pg/ml for SL-LIP and 100% CL-LIP, respectively; P < 0.05; one-way ANOVA). In general, levels of IL-6 were enhanced in BAL fluid from Mφ-depleted mice; however, levels in the CL-LIP-treated groups were highly variable, and differences were not significant (BAL fluid, 30.2 ± 26.7 and 251.4 ± 243.7 pg/ml for SL-LIP and 100% CL-LIP, respectively; P > 0.05; one-way ANOVA). Levels of TNF-α, IFN-γ, IL-10, and IL-12 were not significantly elevated in the BAL fluid of Mφ-depleted mice compared to SL-LIP-treated controls (data not shown).
Airway Mφ have been implicated in susceptibility to secondary bacterial infections following influenza virus infections (28, 34, 58). To determine if the severe disease observed in CL-LIP-treated mice infected with BJx109 was related to secondary bacterial pneumonia, BAL supernatants recovered at day 7 postinfection were plated onto nutrient agar and horse blood agar, and after incubation at 37°C for 3 days, the number of CFU was determined. CFU counts were low (0 to 500 CFU/ml of BAL fluid for all groups) and not statistically different between BJx109-infected mice treated with PBS, SL-LIP, or CL-LIP, thereby excluding secondary bacterial infection as a major factor associated with disease.
BJx109 infection of Mφ-depleted mice is associated with vascular leakage and pulmonary edema.
Viral or inflammatory lung injury can lead to acute lung injury, characterized by alveolar barrier dysfunction and subsequent accumulation of protein-rich fluid in the alveolar compartment (42, 72). Consistent with this, total protein levels in cell-free BAL fluid were approximately 4-fold higher in Mφ-depleted mice than in SL-LIP-treated controls (Fig. 7 A). Furthermore, the lungs of Mφ-depleted mice were visibly enlarged and showed a marked enhancement in the wet/dry ratio (Fig. 7B), a measure of extravascular water. Together, these data indicate that the severe clinical disease observed following Mφ depletion of BJx109-infected mice was associated with vascular leakage and lung edema, indicative of acute respiratory distress syndrome (ARDS)-like respiratory failure (18, 72).
FIG. 7.
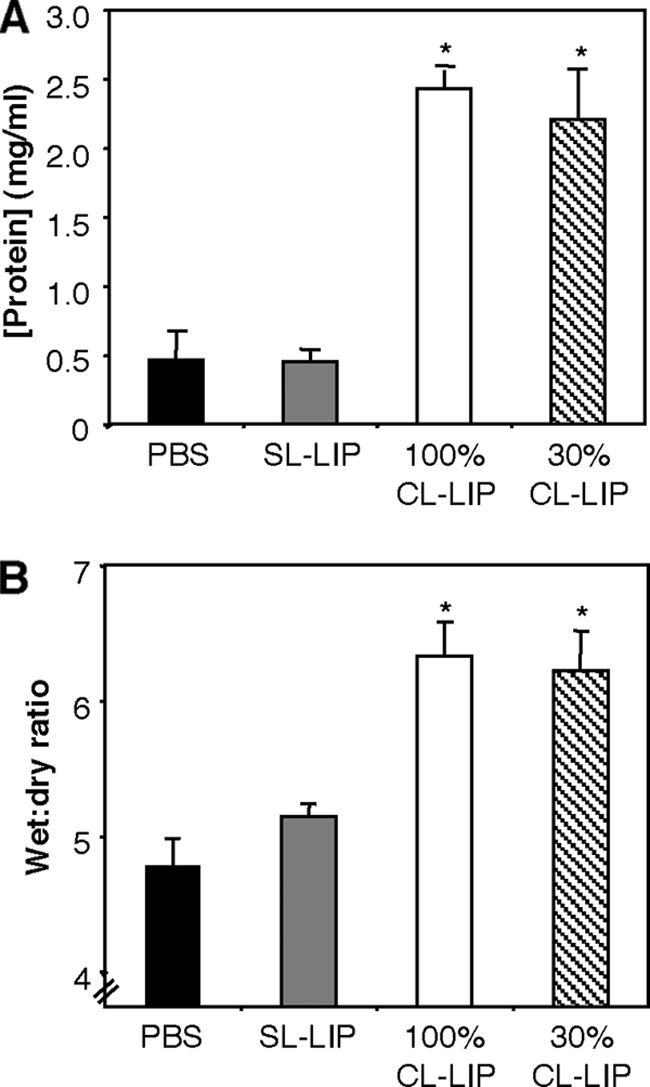
Vascular leakage and pulmonary edema in macrophage-depleted mice infected with BJx109. Groups of 5 B6 mice were depleted of macrophages via i.n. treatment with 100% or 30% CL-LIP 48 h prior to infection with 105 PFU of BJx109 and every 3 days thereafter. Control groups received an equivalent volume of SL-LIP or PBS alone, and all mice were killed and analyzed at day 7 postinfection. (A) Total protein concentrations in cell-free BAL supernatants. *, 100% CL-LIP- and 30% CL-LIP-treated mice were significantly different from SL-LIP control mice (P < 0.01; one-way ANOVA). (B) Lung wet-to-dry ratios as an assessment of pulmonary edema. The data are representative of at least 2 independent experiments. *, 100% CL-LIP- and 30% CL-LIP-treated mice were significantly different from SL-LIP-treated control mice (P < 0.01; one-way ANOVA).
Systemic responses in Mφ-depleted mice infected with BJx109.
Systemic manifestations, such as myocarditis, leukopenia, and thymic atrophy, have been associated with severe clinical disease during influenza virus infections (13, 17, 35, 54). First, infectious virus was not consistently recovered from the hearts and brains of CL-LIP-treated mice, and we did not detect evidence of edema (wet/dry ratio) in hearts recovered from Mφ-depleted mice, suggesting that myocarditis was not a major factor contributing to severe disease. We did, however, record marked reductions in the numbers of total leukocytes in the blood of CL-LIP-treated mice at day 7 postinfection (Fig. 8 A). While neutrophil numbers were not reduced, circulating B220+ cells and T-cell receptor β-positive (TCR-β+) T cells were depleted, including both CD4+ and CD8+ T-cell compartments.
FIG. 8.

Lymphopenia and thymic atrophy in macrophage-depleted mice infected with BJx109. Groups of 5 B6 mice were depleted of macrophages via i.n. treatment with 30% or 100% CL-LIP 48 h prior to infection with 105 PFU of BJx109 and every 3 days thereafter. Control groups received an equivalent volume of SL-LIP or PBS alone. All mice were killed and analyzed at day 7 postinfection. (A) Viable-cell counts were performed on whole blood to determine total leukocyte numbers, and flow cytometry was used to determine total numbers of T cells (TCR-β+), CD4+ T cells (CD4+ TCR-β+), and CD8+ T cells (CD8+ TCRβ+). *, cell numbers from 100% CL-LIP- and 30% CL-LIP-treated mice that were significantly lower than those from SL-LIP-treated control animals (P < 0.05; one-way ANOVA). (B) Viable-cell counts were performed on single-cell suspensions prepared from mouse thymi (total cells), and flow cytometry was used to determine the number of double-negative (CD4− CD8−) (DN), DP (CD4+ CD8+), and single-positive (CD4+ or CD8+) (SP) thymocytes present. *, cell numbers from macrophage-depleted mice (100% CL-LIP and 30% CL-LIP) that were significantly higher or lower than those from SL-LIP-treated controls (P < 0.05; one-way ANOVA).
In addition, the thymi of CL-LIP-treated mice infected with BJx109 showed a significant reduction in total cellularity, and flow cytometry analysis indicated a specific reduction in the number of double-positive (DP) thymocytes recovered from Mφ-depleted mice (Fig. 8B). It should be noted that in preliminary experiments we established that CL-LIP treatment of naïve animals was not associated with a reduction in total cells or DP cells in the thymi of naïve animals (data not shown).
DISCUSSION
In this study, we investigated factors involved in modulating the severity of disease in a mouse model of influenza virus infection. More specifically, we demonstrated that airway Mφ play a critical role in ameliorating disease severity following intranasal infection of mice with virus strain BJx109. In vitro studies demonstrated that BJx109 was able to infect murine Mφ, including airway Mφ, with high efficiency. Furthermore, in vivo depletion of airway Mφ during BJx109 infection of mice led to viral pneumonia and death. In contrast, virus strain PR8 was inefficient in its ability to infect murine Mφ in vitro, and the severe disease noted in PR8-infected mice was not further exacerbated when airway Mφ were depleted. Mφ depletion during BJx109 infection was associated with enhanced virus replication, pulmonary inflammation, lung edema, and vascular leakage, indicating that airway Mφ control infection and limit the development of lung injury and ARDS during influenza virus infection of mice.
In both humans and mice, bronchial and alveolar epithelial cells represent the major targets for influenza virus infection and subsequent amplification (5). Infection is initiated following attachment of the viral HA to sialic acid moieties on cell surface glycoproteins and/or glycolipids, although the specific host cell molecules that mediate this process are yet to be defined. While H3N2 and H1N1 subtype viruses can differ markedly in the types and/or linkages bound by their respective HA glycoproteins (53, 56), BJx109 and PR8 infected murine airway epithelial cells equally well in vitro (Fig. 1A), and similar levels of newly synthesized virions were released from virus-infected cells by 24 h postinfection (Fig. 1D).
Major differences were noted, however, in the course of disease (Fig. 2) and in the distribution of viral antigen in airway epithelial cells of BJx109- versus PR8-infected mice (Fig. 5B, i and iv, respectively), indicating that components of the innate immune system limit infection and/or amplification of BJx109 in the lung. BJx109 is highly sensitive to neutralization and destruction by the collectin SP-D, while PR8 is largely resistant (23, 49). SP-D is found in lung fluids and other respiratory secretions lining the airways (10, 40, 49) and binds to mannose-rich glycans expressed on the HA and NA of influenza viruses (23, 39, 49) to aggregate virions, neutralize virus infectivity, and opsonize virus for its interactions with neutrophils (21, 22). The related collectin mannose-binding lectin (MBL) may also transude from the circulation into the inflamed lung during influenza virus infections (49). The potent antiviral activity of SP-D (and related innate immune proteins) is likely to inhibit attachment and/or entry of BJx109 into bronchial and alveolar epithelial cells and may also hinder release and spread of BJx109 from virus-infected epithelial cells following productive virus replication. In contrast, PR8 appears to be largely resistant to these host defenses and so replicates extensively in epithelial cells throughout the respiratory tract.
In contrast to epithelial cells, Mφ did not support productive viral replication, as there was no increase in levels of infectious virus released from primary Mφ exposed to either BJx109 or PR8 by 24 h postinfection (Fig. 1D). Also, the receptors mediating infectious entry into Mφ are different from epithelial cell receptors, as the MMR, implicated in infection of murine Mφ by influenza viruses (49), is not expressed on epithelial cell populations. Exposure of Mφ to influenza virus has been shown to induce potent release of proinflammatory cytokines and chemokines (26, 27, 45, 55), and these may play a critical role in limiting virus infection. Such responses must be tightly regulated, as dysregulated cytokine responses have been associated with immunopathology and disease severity during infections with H5N1 (9, 73) and 1918 (32) pandemic viruses. While both Mφ and epithelial cells respond to influenza virus infection by releasing soluble mediators, the spectra of chemokines and cytokines produced differ markedly between cell types (reviewed in reference 29). Depletion of airway Mφ prior to respiratory syncytial virus (RSV) infection of mice led to profound inhibition of the early release of inflammatory cytokines in the airways but had little effect on disease severity, weight loss, or lung function (47). In the mouse model of influenza virus infection, it is likely that Mφ depletion may also inhibit early protective cytokine responses to BJx109, and the absence of such responses would contribute to the uncontrolled virus replication observed in the lungs of CL-LIP-treated animals (Fig. 5). Interestingly, local production of MCP-1 was markedly enhanced in the later stages of infection (data not shown), suggesting that dysregulated cytokine production by infected epithelial cells and/or additional leukocytes recruited to the airways may also contribute to the pulmonary inflammation, lung injury and severe disease observed at day 7 postinfection. The inability of PR8 to infect airway Mφ (Fig. 1A) suggests the virus is also likely be poor in its ability to induce early inflammatory mediators from Mφ; consequently, the pathogenesis of BJx109 in Mφ-depleted mice is rather similar to that of PR8 in untreated animals.
Intranasal infection of mice with BJx109 induced very mild disease, while infection of Mφ-depleted mice led to severe disease and death. A number of factors have been proposed to contribute to the severity of influenza disease, including uncontrolled virus replication and spread (19, 38), secondary bacterial infection (7, 20, 57), and excessive or dysregulated inflammatory responses (44, 73). In humans, severe influenza virus infections have been associated with histopathological lesions indicative of ARDS (18), a condition in which the integrity of the lung alveolar-capillary barrier may be damaged, leading to flooding of the alveoli with plasma liquid and proteins. Depletion of airway Mφ led to enhanced virus replication in the airways during BJx109 infection, and this in turn was associated with hallmark features of ARDS, such as severe pulmonary inflammation (Fig. 6), alveolar leakage (Fig. 7A), and lung edema (Fig. 7B), similar to that described in a mouse model of ARDS infection induced by H5N1 infection (72). Together, these findings demonstrate a critical role for airway Mφ in containing early virus replication and in ameliorating epithelial injury and vascular permeability in the lungs of virus-infected mice.
In animal models of influenza virus infection, we and others (30, 64, 71) have demonstrated the importance of airway Mφ in limiting virus replication and disease severity. While infection of mouse airway Mφ by RSV is also abortive (14), depletion of airway macrophages prior to RSV infection has been associated with exacerbated virus replication and airway occlusion (51) and with reduced early inflammatory cytokines and NK cells in the airways (47), suggesting that factors such as the virus strain, inoculum dose, and mouse strain can also influence the disease outcome. Despite these differences, airway macrophages play a clear role in viral clearance and resolution of inflammation associated with respiratory viruses.
In the context of influenza virus infections, mononuclear phagocytes recruited to inflamed lung tissues in a CCR2-dependent manner (26) can contribute to alveolar epithelial cell apoptosis by the release of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) in acute viral pneumonia, thereby leading to alveolar damage and vascular leakage (25). Thus, while “resting” airway Mφ are the first immune cells to sense and respond to influenza virus infection, the subsequent recruitment and activation of inflammatory Mφ must be tightly controlled during infection to promote clearance of virus-infected epithelial cells while minimizing excessive damage and associated vascular leakage into the airways. Early and excessive macrophage infiltration has also been associated with severe lung pathology and disease following infection of mice with highly pathogenic H1N1 and H5N1 viruses (44).
A recombinant virus expressing the HA and NA glycoproteins of the 1918 pandemic virus was shown to be highly virulent for mice, and depletion of airway Mφ during infection was associated with increased virus replication in the lungs (and spread to the brain), as well as significant reductions in cytokines and chemokines in the airways at day 5 postinfection (64). The exceptional virulence of the 1918 pandemic virus is likely due to the constellation of virus genes; however, the HA and NA have been implicated in contributing to its pathogenicity in mice (33, 63, 64). In contrast to PR8, the 1918 virus can infect and replicate productively to some degree in mouse Mφ (44), and consistent with these findings, Mφ depletion further exacerbated disease in mice infected with the recombinant virus expressing the HA/NA derived from the 1918 strain (64). The reduced proinflammatory mediators in the lungs of macrophage-depleted mice led the authors to propose that the HA/NA glycoproteins of the 1918 virus induced high levels of cytokines and chemokines in the lung and that airway Mφ were required to facilitate this process. Together, these findings suggest that mechanisms underlying the virulence of distinct influenza virus strains for mice can be very different.
The PR8 strain is highly adapted to growth in the mouse lung and induces interstitial pneumonia similar to that observed in human cases of viral pneumonia (15, 37). Multiple factors have been proposed to contribute to the virulence of PR8, including its high growth capacity, a function of the internal genes of PR8 that led to its use is the generation of high-yielding reassortant strains with vaccine potential (3). In addition, we propose that during adaptation to growth in the mouse lung, viruses may also have been selected for virulence by virtue of their ability to evade airway Mφ. Direct comparison of inflammatory responses to BJx109 and PR8 in vivo are complicated by the rapid growth of PR8 in the airways (50); however, our current findings that BJx109 induces a disease similar to that induced by PR8 in mice depleted of airway macrophages indicates that these cells play a critical role in containing viral replication, inflammation, and disease. Together, our data demonstrate a critical role for airway Mφ in modulating disease severity and indicate that the susceptibility of particular virus strains to the antiviral responses of airway Mφ is likely to be an important factor contributing to their virulence for mice.
Acknowledgments
This work was supported by Project Grant 509230 from The National Health and Medical Research Council (NHMRC) of Australia. P.C.R. is an NHMRC R. D. Wright Research Fellow. The Melbourne WHO Collaborating Centre for Reference and Research on Influenza is supported by the Australian Government Department of Health and Ageing.
Footnotes
Published ahead of print on 26 May 2010.
REFERENCES
- 1.Anders, E. M., C. A. Hartley, P. C. Reading, and R. A. Ezekowitz. 1994. Complement-dependent neutralization of influenza virus by a serum mannose-binding lectin. J. Gen. Virol. 75:615-622. [DOI] [PubMed] [Google Scholar]
- 2.Ayala, A., C. S. Chung, J. L. Lomas, G. Y. Song, L. A. Doughty, S. H. Gregory, W. G. Cioffi, B. W. LeBlanc, J. Reichner, H. H. Simms, and P. S. Grutkoski. 2002. Shock-induced neutrophil mediated priming for acute lung injury in mice: divergent effects of TLR-4 and TLR-4/FasL deficiency. Am. J. Pathol. 161:2283-2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baez, M., P. Palese, and E. D. Kilbourne. 1980. Gene composition of high-yielding influenza vaccine strains obtained by recombination. J. Infect. Dis. 141:362-365. [DOI] [PubMed] [Google Scholar]
- 4.Bem, R. A., A. W. Farnand, V. Wong, A. Koski, M. E. Rosenfeld, N. van Rooijen, C. W. Frevert, T. R. Martin, and G. Matute-Bello. 2008. Depletion of resident alveolar macrophages does not prevent Fas-mediated lung injury in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 295:L314-L325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bender, B. S., and P. A. Small, Jr. 1992. Influenza: pathogenesis and host defense. Semin. Respir. Infect. 7:38-45. [PubMed] [Google Scholar]
- 6.Benoit, A., Y. Huang, J. Proctor, G. Rowden, and R. Anderson. 2006. Effects of alveolar macrophage depletion on liposomal vaccine protection against respiratory syncytial virus (RSV). Clin. Exp. Immunol. 145:147-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bullough, P. A., F. M. Hughson, J. J. Skehel, and D. C. Wiley. 1994. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 371:37-43. [DOI] [PubMed] [Google Scholar]
- 8.Carbonetti, N. H., G. V. Artamonova, N. Van Rooijen, and V. I. Ayala. 2007. Pertussis toxin targets airway macrophages to promote Bordetella pertussis infection of the respiratory tract. Infect. Immun. 75:1713-1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheung, C. Y., L. L. Poon, A. S. Lau, W. Luk, Y. L. Lau, K. F. Shortridge, S. Gordon, Y. Guan, and J. S. Peiris. 2002. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet 360:1831-1837. [DOI] [PubMed] [Google Scholar]
- 10.Crouch, E. C. 1998. Collectins and pulmonary host defense. Am. J. Respir. Cell Mol. Biol. 19:177-201. [DOI] [PubMed] [Google Scholar]
- 11.Dawson, T. C., M. A. Beck, W. A. Kuziel, F. Henderson, and N. Maeda. 2000. Contrasting effects of CCR5 and CCR2 deficiency in the pulmonary inflammatory response to influenza A virus. Am. J. Pathol. 156:1951-1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Jong, M. D., C. P. Simmons, T. T. Thanh, V. M. Hien, G. J. Smith, T. N. Chau, D. M. Hoang, N. V. Chau, T. H. Khanh, V. C. Dong, P. T. Qui, B. V. Cam, Q. Ha do, Y. Guan, J. S. Peiris, N. T. Chinh, T. T. Hien, and J. Farrar. 2006. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat. Med. 12:1203-1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engblom, E., T. O. Ekfors, O. H. Meurman, A. Toivanen, and J. Nikoskelainen. 1983. Fatal influenza A myocarditis with isolation of virus from the myocardium. Acta Med. Scand. 213:75-78. [DOI] [PubMed] [Google Scholar]
- 14.Franke-Ullmann, G., C. Pfortner, P. Walter, C. Steinmuller, M. L. Lohmann-Matthes, L. Kobzik, and J. Freihorst. 1995. Alteration of pulmonary macrophage function by respiratory syncytial virus infection in vitro. J. Immunol. 154:268-280. [PubMed] [Google Scholar]
- 15.Frankova, V. 1975. Inhalatory infection of mice with influenza A0/PR8 virus. I. The site of primary virus replication and its spread in the respiratory tract. Acta Virol. 19:29-34. [PubMed] [Google Scholar]
- 16.Fujisawa, H., S. Tsuru, M. Taniguchi, Y. Zinnaka, and K. Nomoto. 1987. Protective mechanisms against pulmonary infection with influenza virus. I. Relative contribution of polymorphonuclear leukocytes and of alveolar macrophages to protection during the early phase of intranasal infection. J. Gen. Virol. 68:425-432. [DOI] [PubMed] [Google Scholar]
- 17.Gao, P., S. Watanabe, T. Ito, H. Goto, K. Wells, M. McGregor, A. J. Cooley, and Y. Kawaoka. 1999. Biological heterogeneity, including systemic replication in mice, of H5N1 influenza A virus isolates from humans in Hong Kong. J. Virol. 73:3184-3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grose, C., and K. Chokephaibulkit. 2004. Avian influenza virus infection of children in Vietnam and Thailand. Pediatr. Infect. Dis. J. 23:793-794. [DOI] [PubMed] [Google Scholar]
- 19.Gu, J., Z. Xie, Z. Gao, J. Liu, C. Korteweg, J. Ye, L. T. Lau, J. Lu, Z. Gao, B. Zhang, M. A. McNutt, M. Lu, V. M. Anderson, E. Gong, A. C. Yu, and W. I. Lipkin. 2007. H5N1 infection of the respiratory tract and beyond: a molecular pathology study. Lancet 370:1137-1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hall, J. N., M. C. Stone, and J. C. Simpson. 1918. The epidemic of pneumonia following influenza at Camp Logan, Texas. JAMA 71:1986-1987. [Google Scholar]
- 21.Hartshorn, K. L., E. C. Crouch, M. R. White, P. Eggleton, A. I. Tauber, D. Chang, and K. Sastry. 1994. Evidence for a protective role of pulmonary surfactant protein D (SP-D) against influenza A viruses. J. Clin. Invest. 94:311-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hartshorn, K. L., K. B. Reid, M. R. White, J. C. Jensenius, S. M. Morris, A. I. Tauber, and E. Crouch. 1996. Neutrophil deactivation by influenza A viruses: mechanisms of protection after viral opsonization with collectins and hemagglutination-inhibiting antibodies. Blood 87:3450-3461. [PubMed] [Google Scholar]
- 23.Hartshorn, K. L., M. R. White, V. Shepherd, K. Reid, J. C. Jensenius, and E. C. Crouch. 1997. Mechanisms of anti-influenza activity of surfactant proteins A and D: comparison with serum collectins. Am. J. Physiol. 273:L1156-L1166. [DOI] [PubMed] [Google Scholar]
- 24.Hashimoto, Y., T. Moki, T. Takizawa, A. Shiratsuchi, and Y. Nakanishi. 2007. Evidence for phagocytosis of influenza virus-infected, apoptotic cells by neutrophils and macrophages in mice. J. Immunol. 178:2448-2457. [DOI] [PubMed] [Google Scholar]
- 25.Herold, S., M. Steinmueller, W. von Wulffen, L. Cakarova, R. Pinto, S. Pleschka, M. Mack, W. A. Kuziel, N. Corazza, T. Brunner, W. Seeger, and J. Lohmeyer. 2008. Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J. Exp. Med. 205:3065-3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herold, S., W. von Wulffen, M. Steinmueller, S. Pleschka, W. A. Kuziel, M. Mack, M. Srivastava, W. Seeger, U. A. Maus, and J. Lohmeyer. 2006. Alveolar epithelial cells direct monocyte transepithelial migration upon influenza virus infection: impact of chemokines and adhesion molecules. J. Immunol. 177:1817-1824. [DOI] [PubMed] [Google Scholar]
- 27.Hofmann, P., H. Sprenger, A. Kaufmann, A. Bender, C. Hasse, M. Nain, and D. Gemsa. 1997. Susceptibility of mononuclear phagocytes to influenza A virus infection and possible role in the antiviral response. J. Leukoc. Biol. 61:408-414. [DOI] [PubMed] [Google Scholar]
- 28.Jakab, G. J. 1982. Immune impairment of alveolar macrophage phagocytosis during influenza virus pneumonia. Am. Rev. Respir. Dis. 126:778-782. [DOI] [PubMed] [Google Scholar]
- 29.Julkunen, I., T. Sareneva, J. Pirhonen, T. Ronni, K. Melen, and S. Matikainen. 2001. Molecular pathogenesis of influenza A virus infection and virus-induced regulation of cytokine gene expression. Cytokine Growth Factor Rev. 12:171-180. [DOI] [PubMed] [Google Scholar]
- 30.Kim, H. M., Y. W. Lee, K. J. Lee, H. S. Kim, S. W. Cho, N. van Rooijen, Y. Guan, and S. H. Seo. 2008. Alveolar macrophages are indispensable for controlling influenza viruses in lungs of pigs. J. Virol. 82:4265-4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kirby, A. C., J. G. Raynes, and P. M. Kaye. 2006. CD11b regulates recruitment of alveolar macrophages but not pulmonary dendritic cells after pneumococcal challenge. J. Infect. Dis. 193:205-213. [DOI] [PubMed] [Google Scholar]
- 32.Kobasa, D., S. M. Jones, K. Shinya, J. C. Kash, J. Copps, H. Ebihara, Y. Hatta, J. H. Kim, P. Halfmann, M. Hatta, F. Feldmann, J. B. Alimonti, L. Fernando, Y. Li, M. G. Katze, H. Feldmann, and Y. Kawaoka. 2007. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature 445:319-323. [DOI] [PubMed] [Google Scholar]
- 33.Kobasa, D., A. Takada, K. Shinya, M. Hatta, P. Halfmann, S. Theriault, H. Suzuki, H. Nishimura, K. Mitamura, N. Sugaya, T. Usui, T. Murata, Y. Maeda, S. Watanabe, M. Suresh, T. Suzuki, Y. Suzuki, H. Feldmann, and Y. Kawaoka. 2004. Enhanced virulence of influenza A viruses with the haemagglutinin of the 1918 pandemic virus. Nature 431:703-707. [DOI] [PubMed] [Google Scholar]
- 34.Kodihalli, S., V. Sivanandan, K. V. Nagaraja, D. Shaw, and D. A. Halvorson. 1994. Effect of avian influenza virus infection on the phagocytic function of systemic phagocytes and pulmonary macrophages of turkeys. Avian Dis. 38:93-102. [PubMed] [Google Scholar]
- 35.Kotaka, M., Y. Kitaura, H. Deguchi, and K. Kawamura. 1990. Experimental influenza A virus myocarditis in mice. Light and electron microscopic, virologic, and hemodynamic study. Am. J. Pathol. 136:409-419. [PMC free article] [PubMed] [Google Scholar]
- 36.Leemans, J. C., N. P. Juffermans, S. Florquin, N. van Rooijen, M. J. Vervoordeldonk, A. Verbon, S. J. van Deventer, and T. van der Poll. 2001. Depletion of alveolar macrophages exerts protective effects in pulmonary tuberculosis in mice. J. Immunol. 166:4604-4611. [DOI] [PubMed] [Google Scholar]
- 37.Louria, D. B., H. L. Blumenfeld, J. T. Ellis, E. D. Kilbourne, and D. E. Rogers. 1959. Studies on influenza in the pandemic of 1957-1958. II. Pulmonary complications of influenza. J. Clin. Invest. 38:213-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu, X., T. M. Tumpey, T. Morken, S. R. Zaki, N. J. Cox, and J. M. Katz. 1999. A mouse model for the evaluation of pathogenesis and immunity to influenza A (H5N1) viruses isolated from humans. J. Virol. 73:5903-5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Malhotra, R., J. S. Haurum, S. Thiel, and R. B. Sim. 1994. Binding of human collectins (SP-A and MBP) to influenza virus. Biochem. J. 304:455-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCormack, F. X., and J. A. Whitsett. 2002. The pulmonary collectins, SP-A and SP-D, orchestrate innate immunity in the lung. J. Clin. Invest. 109:707-712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McGill, J., N. Van Rooijen, and K. L. Legge. 2008. Protective influenza-specific CD8 T cell responses require interactions with dendritic cells in the lungs. J. Exp. Med. 205:1635-1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morty, R. E., O. Eickelberg, and W. Seeger. 2007. Alveolar fluid clearance in acute lung injury: what have we learned from animal models and clinical studies? Intensive Care Med. 33:1229-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neumann, G., T. Watanabe, H. Ito, S. Watanabe, H. Goto, P. Gao, M. Hughes, D. R. Perez, R. Donis, E. Hoffmann, G. Hobom, and Y. Kawaoka. 1999. Generation of influenza A viruses entirely from cloned cDNAs. Proc. Natl. Acad. Sci. U. S. A. 96:9345-9350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perrone, L. A., J. K. Plowden, A. Garcia-Sastre, J. M. Katz, and T. M. Tumpey. 2008. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog. 4:e1000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peschke, T., A. Bender, M. Nain, and D. Gemsa. 1993. Role of macrophage cytokines in influenza A virus infections. Immunobiology 189:340-355. [DOI] [PubMed] [Google Scholar]
- 46.Pontow, S. E., V. Kery, and P. D. Stahl. 1992. Mannose receptor. Int. Rev. Cytol. 137B:221-244. [DOI] [PubMed] [Google Scholar]
- 47.Pribul, P. K., J. Harker, B. Wang, H. Wang, J. S. Tregoning, J. Schwarze, and P. J. Openshaw. 2008. Alveolar macrophages are a major determinant of early responses to viral lung infection but do not influence subsequent disease development. J. Virol. 82:4441-4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reading, P. C., J. L. Miller, and E. M. Anders. 2000. Involvement of the mannose receptor in infection of macrophages by influenza virus. J. Virol. 74:5190-5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reading, P. C., L. S. Morey, E. C. Crouch, and E. M. Anders. 1997. Collectin-mediated antiviral host defense of the lung: evidence from influenza virus infection of mice. J. Virol. 71:8204-8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reading, P. C., P. G. Whitney, D. L. Pickett, M. D. Tate, and A. G. Brooks. February 2010. Influenza viruses differ in ability to infect macrophages and to induce a local inflammatory response following intraperitoneal injection of mice. Immunol. Cell Biol. doi: 10.1038/icb.2010.11. [DOI] [PubMed]
- 51.Reed, J. L., Y. A. Brewah, T. Delaney, T. Welliver, T. Burwell, E. Benjamin, E. Kuta, A. Kozhich, L. McKinney, J. Suzich, P. A. Kiener, L. Avendano, L. Velozo, A. Humbles, R. C. Welliver, Sr., and A. J. Coyle. 2008. Macrophage impairment underlies airway occlusion in primary respiratory syncytial virus bronchiolitis. J. Infect. Dis. 198:1783-1793. [DOI] [PubMed] [Google Scholar]
- 52.Rodgers, B., and C. A. Mims. 1981. Interaction of influenza virus with mouse macrophages. Infect. Immun. 31:751-757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rogers, G. N., J. C. Paulson, R. S. Daniels, J. J. Skehel, I. A. Wilson, and D. C. Wiley. 1983. Single amino acid substitutions in influenza haemagglutinin change receptor binding specificity. Nature 304:76-78. [DOI] [PubMed] [Google Scholar]
- 54.Sakamoto, M., F. Suzuki, S. Arai, T. Takishima, and N. Ishida. 1981. Experimental myocarditis induced in mice by infection with influenza A2 virus. Microbiol. Immunol. 25:173-181. [DOI] [PubMed] [Google Scholar]
- 55.Seo, S. H., R. Webby, and R. G. Webster. 2004. No apoptotic deaths and different levels of inductions of inflammatory cytokines in alveolar macrophages infected with influenza viruses. Virology 329:270-279. [DOI] [PubMed] [Google Scholar]
- 56.Slepushkin, V. A., P. D. Staber, G. Wang, P. B. McCray, Jr., and B. L. Davidson. 2001. Infection of human airway epithelia with H1N1, H2N2, and H3N2 influenza A virus strains. Mol. Ther. 3:395-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Spooner, L. H., L. H. Scoot, and, E. H. Heathe. 1919. A bacteriologic study of the influenza epidemic at Camp Devens, Mass. JAMA 72:155-159. [Google Scholar]
- 58.Sun, K., and D. W. Metzger. 2008. Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nat. Med. 14:558-564. [DOI] [PubMed] [Google Scholar]
- 59.Sur, S., J. S. Wild, B. K. Choudhury, N. Sur, R. Alam, and D. M. Klinman. 1999. Long term prevention of allergic lung inflammation in a mouse model of asthma by CpG oligodeoxynucleotides. J. Immunol. 162:6284-6293. [PubMed] [Google Scholar]
- 60.Szretter, K. J., S. Gangappa, X. Lu, C. Smith, W. J. Shieh, S. R. Zaki, S. Sambhara, T. M. Tumpey, and J. M. Katz. 2007. Role of host cytokine responses in the pathogenesis of avian H5N1 influenza viruses in mice. J. Virol. 81:2736-2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tate, M. D., A. G. Brooks, and P. C. Reading. 2008. The role of neutrophils in the upper and lower respiratory tract during influenza virus infection of mice. Respir. Res. 9:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tate, M. D., Y. M. Deng, J. E. Jones, G. P. Anderson, A. G. Brooks, and P. C. Reading. 2009. Neutrophils ameliorate lung injury and the development of severe disease during influenza infection. J. Immunol. 183:7441-7450. [DOI] [PubMed] [Google Scholar]
- 63.Tumpey, T. M., A. Garcia-Sastre, J. K. Taubenberger, P. Palese, D. E. Swayne, and C. F. Basler. 2004. Pathogenicity and immunogenicity of influenza viruses with genes from the 1918 pandemic virus. Proc. Natl. Acad. Sci. U. S. A. 101:3166-3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tumpey, T. M., A. Garcia-Sastre, J. K. Taubenberger, P. Palese, D. E. Swayne, M. J. Pantin-Jackwood, S. Schultz-Cherry, A. Solorzano, N. Van Rooijen, J. M. Katz, and C. F. Basler. 2005. Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J. Virol. 79:14933-14944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Van Rooijen, N., and A. Sanders. 1994. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J. Immunol. Methods 174:83-93. [DOI] [PubMed] [Google Scholar]
- 66.van Rooijen, N., A. Sanders, and T. K. van den Berg. 1996. Apoptosis of macrophages induced by liposome-mediated intracellular delivery of clodronate and propamidine. J. Immunol. Methods 193:93-99. [DOI] [PubMed] [Google Scholar]
- 67.Wagner, R., M. Matrosovich, and H. D. Klenk. 2002. Functional balance between haemagglutinin and neuraminidase in influenza virus infections. Rev. Med. Virol. 12:159-166. [DOI] [PubMed] [Google Scholar]
- 68.Ward, A. C. 1997. Virulence of influenza A virus for mouse lung. Virus Genes 14:187-194. [DOI] [PubMed] [Google Scholar]
- 69.Wells, M. A., P. Albrecht, S. Daniel, and F. A. Ennis. 1978. Host defense mechanisms against influenza virus: interaction of influenza virus with murine macrophages in vitro. Infect. Immun. 22:758-762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Whitehead, G. S., L. H. Burch, K. G. Berman, C. A. Piantadosi, and D. A. Schwartz. 2006. Genetic basis of murine responses to hyperoxia-induced lung injury. Immunogenetics 58:793-804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wijburg, O. L., S. DiNatale, J. Vadolas, N. van Rooijen, and R. A. Strugnell. 1997. Alveolar macrophages regulate the induction of primary cytotoxic T-lymphocyte responses during influenza virus infection. J. Virol. 71:9450-9457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xu, T., J. Qiao, L. Zhao, G. Wang, G. He, K. Li, Y. Tian, M. Gao, J. Wang, H. Wang, and C. Dong. 2006. Acute respiratory distress syndrome induced by avian influenza A (H5N1) virus in mice. Am. J. Respir. Crit. Care Med. 174:1011-1017. [DOI] [PubMed] [Google Scholar]
- 73.Yuen, K. Y., P. K. Chan, M. Peiris, D. N. Tsang, T. L. Que, K. F. Shortridge, P. T. Cheung, W. K. To, E. T. Ho, R. Sung, and A. F. Cheng. 1998. Clinical features and rapid viral diagnosis of human disease associated with avian influenza A H5N1 virus. Lancet 351:467-471. [DOI] [PubMed] [Google Scholar]