

Abstract
The UL17 protein (pUL17) of herpes simplex virus 1 (HSV-1) likely associates with the surfaces of DNA-containing capsids in a heterodimer with pUL25. pUL17 is also associated with viral light particles that lack capsid proteins, suggesting its presence in the tegument of the HSV-1 virion. To help determine how pUL17 becomes incorporated into virions and its functions therein, we identified pUL17-interacting proteins by immunoprecipitation with pUL17-specific IgY at 16 h postinfection, followed by mass spectrometry. Coimmunoprecipitated proteins included cellular histone proteins H2A, H3, and H4; the intermediate filament protein vimentin; the major HSV-1 capsid protein VP5; and the HSV tegument proteins VP11/12 (pUL46) and VP13/14 (pUL47). The pUL17-VP13/14 interaction was confirmed by coimmunoprecipitation in the presence and absence of intact capsids and by affinity copurification of pUL17 and VP13/14 from lysates of cells infected with a recombinant virus encoding His-tagged pUL17. pUL17 and VP13/14-HA colocalized in the nuclear replication compartment, in the cytoplasm, and at the plasma membrane between 9 and 18 h postinfection. One possible explanation of these data is that pUL17 links the external face of the capsid to VP13/14 and associated tegument components.
Herpesvirus virions are composed of a double-stranded DNA genome encapsidated in an icosahedral shell, an amorphous proteinaceous network surrounding the capsid termed the tegument, and a glycoprotein-decorated envelope surrounding the tegument (reviewed in references 25 and 35). The predominant model of virion assembly involves primary envelopment of the nucleocapsid at the inner nuclear membrane (INM), fusion of this nascent virion envelope with the outer nuclear membrane (ONM), and subsequent attachment of tegument proteins to the de-enveloped nucleocapsid in a region of the cytoplasm derived from the Golgi apparatus and/or trans-Golgi network (25, 34). The fact that the bulk of the tegument is applied at a step after primary envelopment is consistent with the relatively sparse electron microscopic appearance of the perinuclear virion tegument as opposed to the denser tegument of the extracellular virion (3, 15). This model of virion egress suggests opportunities for a subset of tegument proteins to attach directly or indirectly to the nucleocapsid in either the nucleosol or cytosol or during budding into nuclear or cytoplasmic membranes. Supporting the idea that at least some tegumentation occurs in the nucleoplasm are the observations that pUL36, pUL37, and vhs (the UL41 gene product) are associated with intranuclear capsids (5, 30). Budding through the INM likely causes incorporation of another set of proteins into the tegument, including the peripheral membrane proteins pUL11 and pUL31, the viral kinase encoded by US3, and the nucleoplasmic proteins VP16 and VP22 (encoded by UL48 and UL49, respectively) (2, 27, 29, 31). Of these, only the pUL31 gene product is absent from extracellular virions, indicating its loss at the de-envelopment step (13, 22, 31).
Major tegument components of extracellular virions include the products of the UL46 gene (VP11/12) and UL47 gene (VP13/14) present in 400 to 600 and 1,400 to 1,880 copies per virion, respectively (16, 24, 50). Although neither protein is essential for viral replication, both play augmenting roles in VP16-dependent viral transcription (50, 51). Perhaps related to this observation, yeast two-hybrid studies detected interactions of VP16 with either VP13/14 or VP11/12 (12, 46). The VP11/12-VP16 interaction was further confirmed in a pulldown assay (46). Other potential VP13/VP14 interaction partners suggested by yeast two-hybrid assays but yet to be confirmed include the ATPase subunit of the viral terminase, the small subunit of viral ribonucleotide reductase, regulatory protein alpha22 (IE68), and the virion structural components pUL14, pUL17, pUL21, pUL49 (VP22), and pUS11 (12).
Herpes simplex virus 1 (HSV-1) VP13/14 shuttles from the nucleus to the cytoplasm during the course of infection (9), a feature shared by the VP13/14 homolog of bovine herpesvirus (44, 45). Localization in both the nucleus and cytoplasmic compartments makes it challenging to determine when and where VP13/14 becomes associated with virions. As evidence supporting a cytoplasmic site of addition to the tegument, cells infected with a pseudorabies virus mutant lacking the homolog of UL47 contain increased numbers of cytoplasmic capsids lacking electron density attributable to the tegument (19).
The role of VP11/12 in virion assembly is even less clear because viral mutants lacking this protein are unimpaired in virion egress through the nucleus or cytoplasm in all systems studied (4, 10, 19). VP11/12 can associate with membranes, and a VP11/12-glutathione S-transferase (GST) fusion protein can associate with capsids purified from infected cell nuclei when added in vitro (26). These data are consistent with functions of VP11/12 in bridging the membrane and capsid in the virion structure and perhaps during virion budding.
Although the precise contribution of HSV-1 UL17 protein (pUL17) to viral replication remains unclear, its role as a structural component of capsids is well documented. Originally classified as a DNA cleavage and packaging protein due to the exclusive production of concatameric DNA and capsids lacking DNA in cells infected with a UL17 deletion virus, pUL17 was later found to be necessary for proper capsid distribution within the intranuclear replication compartment (32, 39). pUL17 interaction with capsids was further supported by biochemical and electron microscopy studies showing association with the external surfaces of capsids (14, 40, 48). The herpes simplex virus capsid shell contains 12 pentons and 150 hexons composed of the major capsid protein VP5 (encoded by UL19), 375 triplexes made up of two molecules of VP23 (encoded by UL18) and one molecule of VP19C (encoded by UL38) and 900 copies of VP26 (UL35), which localize atop each of the 6 VP5 molecules comprising each hexon (reviewed in references 1 and 17). Recent cryoelectron microscopy studies have identified a heterodimer of pUL25/pUL17, termed the C capsid-specific component (CCSC), that is located atop triplexes, which bridge pentons to adjacent hexons in DNA-containing (type C) capsids (43). These observations are consistent with other data indicating that pUL17 localizes on the capsid surface, enhances the association of pUL25 with capsids, and coimmunoprecipitates with pUL25 most efficiently in the presence of intact triplexes (33, 40, 48). In addition to capsid association, pUL17 is a component of viral light particles which contain tegument- and membrane-associated proteins but lack capsids (32, 36, 41). Thus, pUL17 can become incorporated into tegument-like structures in the absence of capsids.
To clarify specific capsid and tegument protein interactions with pUL17, we performed coimmunoprecipitations with a pUL17-specific antibody and identified interactions with the major capsid protein VP5, as well as the tegument proteins VP11/12 and VP13/14. In light of these and previous data, we suggest two possibly related structural roles for pUL17 in virion assembly: to ensure that the capsid is competent structurally to package viral DNA and to serve as sites of attachment for a subset of virion tegument proteins, including VP13/14 and associated proteins.
MATERIALS AND METHODS
Cell lines and viruses.
Vero and Hep2 cells were obtained from the American Type Culture Collection and were propagated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% newborn calf serum (NBCS) and antibiotics as described previously (49).
A novel cell line, CV1-17, specifically engineered to support the replication of UL17-null viruses, was created using the Flp-In system (Invitrogen) as previously described (21). Briefly, the UL17 open reading frame (ORF) was generated by PCR from HSV-1(F) bacterial artificial chromosome (BAC) DNA (38), cloned into the FLP recombination target (FRT)-containing expression vector pcDNA5/FRT, and verified by sequencing the entire UL17 open reading frame. The pcDNA5/FRT-UL17 plasmid was cotransfected with pOG44, a Flp recombinase-encoding plasmid, into CV-1 cells previously selected for an integrated FRT locus to support recombination of the transfected DNA, as indicated previously (21). Site-specific recombination resulted in a change of cell line phenotype from zeocin resistance to hygromycin (Hyg) B resistance. Hygromycin-resistant colonies resulting from the transfection were amplified and grown into cellular monolayers. Expression of UL17 in these cells was determined by immunoblotting them with anti-pUL17 antibody (see Fig. S1 in the supplemental material) and plaque formation by the UL17-null virus. One such cell line was designated CV1-17 and was used for further studies. CV1-17 cells were maintained in DMEM supplemented with 10% newborn bovine serum and 200 μg/ml Hyg B.
The genotypes of viruses used in this study are indicated in Table 1. The wild-type HSV-1(F) strain was used in these studies and has been described previously (11). HSV-1(F) was propagated on Vero (African green monkey kidney) cells. A UL18 deletion virus derived from genetic manipulation of an HSV-1(F) BAC has also been described previously (33). The UL18-null virus was propagated on cells containing HSV-1 DNA from UL16 to UL19, as described previously (8).
TABLE 1.
Genotypes of viruses used in this study
| Virus | Genotype | Reference |
|---|---|---|
| HSV-1(F) | Wild type | 11 |
| Delta 17 | 983 bp of UL17 replaced by kanamycin resistance gene aphA1 | This paper |
| Delta 18 | Entire UL18 open reading frame replaced by aphA1 | 33 |
| 17-His | Encoded 6×His tag inserted at 3′ end of UL17 | This paper |
| VP13/14-HA | Encoded influenza virus HA tag in frame with the 3′ end of UL47 | This Paper |
A new UL17-null virus was constructed by integration and subsequent selection of a kanamycin resistance (Kanr) cassette into the UL17 ORF as follows. A gene encoding Kanr was PCR amplified with the following primers containing sequences homologous to the UL17 ORF and Kanr cassette: del17F, number 11 (CAAACTTCCAGGTCGAAATCCAG ACTCGGGCTCATGCCACCGGCGACTGTACGTGTAGGCTGGAGCTGCTTC), and del17R, number 12 (TTCCGTAGTGGTGGCGCAGGACCACGGAGATAGAACGACGGCTCCACAGCCAGTCATTCCGGGGATCCGTCGAC) (Kanr homology is underlined). The amplicons were transformed into EL250 cells containing a chloramphenicol-resistant (Cmr) wild-type HSV-1(F) BAC (38). Cmr/Kanr recombinants were selected on agar plates. Extracted BAC DNA was transfected with SuperFect (Qiagen) into CV1-17 cells, and after 3 rounds of viral plaque purification, CV1-17 cells grown in 890-cm2 roller bottles were infected with 0.01 PFU per cell for propagation of viral stocks. The stock used in these studies produced 2 × 108 PFU/ml on complementing cells and less than 100 PFU/ml on Vero cells. The final recombinant lacked the final 983 bp of UL17.
A recombinant HSV-1(F) BAC carrying a UL17 ORF in frame with 6 histidine codons (His) at the 3′ end was created by PCR using the following primers: ORF17 3′HisF, number 28 (GCCGCTGTCCTTAGGTTTTGTCGCAAGGTGTCGTCCGGGAACGGCCGTTCTCGCCACCACCACCACCACCACTAGCGTGTAGGCTGGAGCTGCTTC ) and ORF17 3′HisR, number 29 (CAACGGCGCGGGGAGGAGTGGATGGGCGAGGTGGCCGGGGGAAGGCGCCCGATTCCGGGGATCCGTCGAC) (the six His codons are italicized, the stop codon is indicated in boldface, and homology to the Kanr cassette is underlined). PCR amplicons that contained the 6×His tag immediately before the pUL17 stop codon and the Kanr cassette 3′ of the TAG flanked by FRT sites were electroporated into HSV-1(F) BAC-containing EL250 cells. Cmr/Kanr clones were identified on selective media. Overnight cultures of Cmr/Kanr colonies were grown in the presence of 10% arabinose to induce expression of the FRT recombinase, driving recombination at the FRT sites and resulting in the loss of Kanr. Purified BAC DNAs from Cmr/Kans clones were transfected by the CaCl2/Na2HPO4 method into individual wells containing Vero cells, and after 3 rounds of plaque purification, viral stocks were grown on Vero cells inoculated at 0.01 PFU per cell. The recombinant virus produced from Vero cells reached infectious titers greater than 3 × 109 PFU/ml (i.e., similar to those of wild-type virus).
An additional HSV recombinant virus, encoding an influenza virus hemagglutinin (HA) tag in frame with the 3′ end of UL47 (VP13/14-HA), was created by en passant mutagenesis, a previously described markerless BAC mutagenesis technique (42). Primers EP 47HAF, number 62 (GTGTCGGGGAGGCGCGCGACCGGGCTGGGAGGCCCGCCACGCCCATCCCATACGACGTCCCAGACTACGCTTAAAGGATGACGACGATAAGTAGGG) and EP 47HAR, number 63 (TATGCCGCGTCCAGGGCCATCGGGGCGCTTTTTATCGGGAGGAGCTTAAGCGTAGTCTGGGACGTCGTATGGGTA[r]TGGGCGTGGCGGGCCCAACCAATTAACCAATTCTGAT) (the HA tag is italicized, the stop codon is in boldface, and the pEP Kan cassette homology is underlined) were used to generate the VP13/14-HA recombinant BAC. Recombinant BAC DNA was purified and transfected into Vero cells with SuperFect (Qiagen) for virus propagation. Larger viral stocks propagated in Vero cells from this initial stock grew to titers greater than 2 × 109 PFU/ml.
Immunoprecipitation assays.
A previously described anti-pUL17 chicken IgY antibody (48) was used to coimmunoprecipitate proteins interacting with pUL17. Approximately 8.0 ×108 rabbit skin cells (RSC) were infected with 5 PFU of HSV-1(F) or UL17-null virus per cell. Cells were collected at 16 h postinfection, pelleted by centrifugation, and lysed in NP-40 lysis buffer (1.1% NP-40, 100 mM Tris, pH 7.7, 300 mM NaCl, 1 mM EDTA, pH 8.0, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1 μg/ml aprotinin, 1 μg/ml pepstatin, 1 μg/ml leupeptin, Sigma Protease Inhibitor cocktail [P8340], 1 mM Na3VO4) on ice. The lysates were clarified by a 10-min, 4,000-rpm centrifugation in an Eppendorf 5810R centrifuge with an A-4-62 rotor at 4°C 4 times to ensure removal of all cellular debris. In a preclearing step, Gamma Bind G beads, which contain a recombinant form of streptococcal protein G covalently bound to Sepharose 4B (GE Healthcare Bio-Sciences AB), were washed with phosphate-buffered saline (PBS), brought to a 50% slurry in PBS, and reacted with the solubilized cellular proteins for 1 h at 4°C and then were pelleted for 10 min at 8,000 rpm in an F34-6-38 rotor. The precleared lysates were transferred to new tubes and subsequently reacted for 1.5 h at 4°C with rabbit anti-IgY IgG plus Gamma Bind G beads with constant rotation to further remove any nonspecific binding. After removal of the bead-rabbit IgG mixture by centrifugation at 8,000 rpm in an F34-6-38 rotor at 4°C and transfer to new tubes, the supernatants were reacted with chicken anti-UL17 IgY for 2 h at 4°C, followed by reaction with rabbit anti-IgY IgG for 2 h at 4°C, and finally with Gamma Bind G beads overnight. The beads with bound proteins were pelleted and washed 4 times with ice-cold NP-40 lysis buffer, and protein was eluted from the beads in 150 μl 2× SDS-PAGE buffer (100 mM Tris-HCl, pH 6.8, 4.0% SDS, 0.2% bromophenol blue, 20% glycerol, 200 mM fresh dithiothreitol [DTT]). Samples were separated on both 8% and 15% SDS-polyacrylamide gels and stained with Coomassie blue.
Two different antibodies were used to immunoprecipitate VP13/14. One antibody (used in all except one set of experiments [see Fig. 5]) was obtained from the laboratory of David Meredith (47). Because this was available in only limiting amounts, another VP13/14 antibody was generated in our laboratory by immunization of rabbits three times, approximately 2 weeks apart, with a fusion protein purified from the BL21(DE3+) strain of Escherichia coli (Stratagene) and containing the gene encoding glutathione-S-transferase fused to the last 234 codons of HSV-1(F) UL47. The validity of the fusion protein was confirmed by sequencing the fused gene and by reactivity with the previous anti-VP13/14 antiserum. The new rabbit antiserum recognized a protein band of electrophoretic mobility indistinguishable from VP13/14 recognized by the previous antiserum in immunoblots of HSV-1(F)-infected cell lysates and of a slightly slower electrophoretic mobility in lysates of cells infected with a recombinant virus encoding an HA-tagged VP13/14 (data not shown).
FIG. 5.
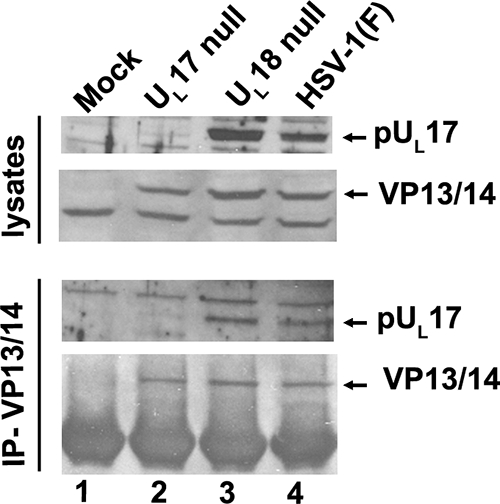
Coimmunoprecipitation of pUL17 and VP13/14 in the presence and absence of capsids. Cells were infected with 5.0 PFU per cell of HSV-1(F), UL17-null virus, or UL18-null virus. The UL18-null virus lacks intact triplexes necessary for capsid formation. At 16 h after infection, a sample of lysates was removed for immunoblotting (upper rows), and the rest of the lysate was subjected to immunoprecipitation with the VP13/14-specific antibody (lower rows). Immunoprecipitated material was electrophoretically separated and subjected to immunoblotting with antibodies to pUL17 (3rd row) or VP13/14 (4th row).
To immunoprecipitate VP13/14, CV1 cells (8.8 × 106) were infected with HSV-1(F), UL17-null, or UL18-null virus or were mock infected. Eighteen hours after infection, the cells were washed with cold PBS and lysed in cold RIPA buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate, 1 mM EDTA, and 1× protease inhibitor cocktail (Roche) for 30 min on ice. The lysates were clarified by spinning them at 14,000 rpm for 10 min at 4°C, and anti-VP13/VP14 rabbit antisera diluted 1:100 were added. Gamma Bind G protein Sepharose beads were added and rotated with the lysates at 4°C overnight. The beads were washed four times with excess RIPA buffer, and immune complexes were eluted and solubilized in SDS-containing buffer. The immunoprecipitated materials or RIPA buffer-solubilized clarified lysates were separated on SDS-12% polyacrylamide gels and transferred to nitrocellulose membranes for immunoblotting.
MALDI-MS.
Proteins of interest were cut from the polyacrylamide gel and digested with trypsin, and the masses of peptides were determined by matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS). The top 7 scoring peptide matches for each band using Mascot software are reported in Table 2.
TABLE 2.
Mass spectrometry of pUL17-interacting proteins
| Predicted protein | No. of matching peptides | SDS-PAGE mass (Da) | Predicted MW | Accession no. | Protein scorea | Total ion scoreb | Best ion scorec | Total CId (%) |
|---|---|---|---|---|---|---|---|---|
| Histone H4 (mouse) | 21 | 15,000 | 12,480 | gi|27692935 | 484 | 334 | 86 | 100 |
| Histone H2A (human H2Aj) | 10 | 17,500 | 13,927.80 | gi|7264004 | 240 | 187 | 70 | 100 |
| Histone H3 (human) | 5 | 19,000 | 15,498.50 | gi|56204740 | 265 | 119 | 92 | 100 |
| Vimentin (human) | 14 | 62,000 | 49,623.10 | gi|57471646 | 539 | 376 | 115 | 100 |
| Virion protein UL47 | 39 | 85,000 | 73,771.30 | gi|9629428 | 355 | 141 | 52 | 100 |
| Virion protein UL46 | 33 | 95,000 | 78,195.70 | gi|73826 | 431 | 263 | 85 | 100 |
| Major capsid protein HSV1 | 52 | 160,000 | 148,989.30 | gi|9629399 | 714 | 464 | 101 | 100 |
Protein score represents the combined unique ion scores of all observed mass spectra of a protein using Mascot software.
Total ion score represents the combined ion scores of all significant peptides.
Best ion score represents the score of the highest matching peptide fragment.
CI, confidence interval.
Immunofluorescence assay (IFA).
Hep2 cells grown on glass coverslips in 6-well dishes to approximately 80 to 100% confluence were infected by incubation with 5.0 PFU/cell in 199V medium (DMEM with 1% NBCS) with rocking for 1 h and then returned to stationary incubation. At various times after infection, the cells were fixed in 3% paraformaldehyde in PBS for 15 min. Cells infected with VP13/14-HA virus were fixed with ice-cold methanol for 10 min at 4°C. Autofluorescence was quenched with 50 mM ammonium chloride for 15 min; the cells were washed with PBS and permeabilized in 0.1% Triton X-100 for 2 min, followed by washing in PBS. PBS supplemented with 1% bovine serum albumin (BSA), 5% donkey serum, and 5% human serum was used to block nonspecific antibody binding for 20 to 30 min at room temperature. Primary antibodies were diluted in this blocking solution as follows: anti-pUL17, between 1:1,000 and 1:500; anti-ICP4, 1:1,000 (from Goodwin Institute, number 1114); anti-ICP8, 1:1,000 (a kind gift from Bill Ruyechan); and anti-HA (Santa Cruz), 1:100. After the 30- to 60-min incubation with primary antibody, coverslips were washed at least 3 times with PBST (PBS supplemented with 0.2% Tween 20 [Sigma]), and secondary antibodies were diluted 1:1,000 in blocking solution and reacted for 30 to 60 min. Coverslips were washed at least 4 times in PBST and then briefly in distilled water (dH2O), mounted on glass slides with Vectashield mounting medium (Vector Laboratories) containing a UV protectant, and sealed with nail polish.
Ni-NTA protein purification.
Vero cells were infected with 5 PFU/cell UL17-His virus. Cells were collected, pelleted, and washed once with PBS. The cells were then lysed in 50 mM NaH2PO4-300 mM NaCl-10 mM imidazole-1% NP-40, pH 8.0, by sonication for 15 s on ice. Insoluble material was removed by centrifugation for 10 min at 4,000 rpm in an Eppendorf 5810R centrifuge with an A-4-62 rotor at 4°C. A 50% slurry of nickel-nitrilotriacetic acid (Ni-NTA) resin in PBS was added to the solubilized proteins and mixed by rotation at 4°C for 90 min. pUL17-His-bound Ni-NTA resin was pelleted and washed twice in 50 mM NaH2PO4-300 mM NaCl-20 mM imidazole (pH 8.0). Protein was eluted with 50 mM NaH2PO4-300 mM NaCl-250 mM imidazole (pH 8.0). The eluted proteins were electrophoretically separated on SDS-polyacrylamide gels, transferred electrically to nitrocellulose, and probed with various antibodies as described below.
Immunoblotting.
Vero cells were collected and pelleted at 4,000 rpm for 10 min. The cell pellets were resuspended and lysed in 2× SDS-PAGE sample buffer. Associated proteins were electrophoretically separated on SDS-polyacrylamide gels and transferred to nitrocellulose at 25 to 30 A for 5 to 6 h at 4°C. The membranes were blocked in 5% dried milk in PBST, rinsed with PBST, and probed with antibody diluted in PBST supplemented with 1.0% BSA. Primary antibodies were diluted as follows: anti-UL17, 1:10,000 or 1:5000 (see Fig. 5); anti-histone H3, 1:1,000; anti-vimentin, 1:1,000; anti-VP13/14, 1:1,000 or 1:5,000 (see Fig. 5). Excess primary antibody was removed with 3 10-min washes in excess PBST. Secondary horseradish peroxidase (HRP)-conjugated antibodies were diluted 1:5,000 in PBST with 5% milk and rocked for 1 h at room temperature. Excess antibody was again removed by washes in excess PBST 3 times for 10 min each time. Bound immunoglobulin was revealed by chemiluminescence using Pierce ECL detection agent exposure to X-ray film.
RESULTS
Construction of a new UL17-expressing cell line and viral deletion mutant.
Since UL17 is essential for viral replication, propagation of viral null mutants lacking this gene requires a complementing cell line engineered to express UL17. Because previously described G5 cells harbored a 16.2-kb fragment of the HSV genome, most of which flanked UL17 (8), recombination between homologous HSV sequences in the virus and cell line often resulted in genetic reversion of the UL17 deletion to wild-type sequences (data not shown).
To overcome this difficulty, a new cell line, CV1-17, containing only UL17 was created through the use of the Flp-In system (Invitrogen) as outlined in Materials and Methods. During the course of these efforts, the UL17 ORF from HSV-1(F) was subcloned and sequenced. This sequence analysis revealed differences between the UL17 ORFs in HSV-1(F) and the published HSV-1(17) sequences (23). Specifically, a +1 insertion mutation in HSV-1(F) UL17 was predicted to cause a frameshift relative to HSV-1(17) at codon 11 (Fig. 1A). The open reading frames then realign at codon 21 by a compensatory −1 deletion in UL17 of HSV-1(F). These mutations are predicted to produce 10 distinct amino acids at the N termini of the two otherwise homologous genes. Other silent mutations were observed (data not shown), as well as a single-amino-acid change at codon 352 (GenBank accession number FJ711161) (Fig. 1B). We note that the observed amino acid changes in pUL17 are also present in the recently published genomic sequences of HSV-1 strains F and H129 (37).
FIG. 1.

UL17 sequences in different HSV-1 strains. (A) An insertion between codons 10 and 11 (down arrow) causes a frameshift in the HSV-1(F) sequence compared to the published HSV-1(17) sequence. A deletion between codons 21 and 22 (up arrow) in HSV-1(F) brings the two UL17 genes back into frame. (B) A point mutation (arrow) changes a single amino acid in HSV-1(F) UL17 compared to the HSV-1(17) UL17 sequence.
After obtaining a pUL17-specific cell line, a novel HSV-1(F) BAC was generated in which a central portion of the UL17 ORF was replaced with a kanamycin resistance cassette (detailed in Materials and Methods). This deletion of 912 bp (bp 212 to 1123, with 1 representing the A in the start codon) retained 989 bp of the 3′ end of the UL17 coding sequence to preserve the UL16 promoter region. Transfection of this BAC into supporting CV1-17 cells resulted in a UL17 deletion virus (designated delta 17) with significantly reduced rates of reversion (data not shown).
Identification of UL17-interacting proteins.
To clarify pUL17's putative roles in virion assembly as both a tegument and capsid protein, we used coimmunoprecipitation to identify pUL17-interacting proteins. Cells were infected with HSV-1(F) or the novel deletion mutant lacking UL17. The proteins in either lysate were reacted separately with pUL17-specific IgY antibody, and immune complexes were purified by reaction with a rabbit anti-IgY bridging antibody that could subsequently be captured with Gamma Bind G Sepharose beads. Immunoprecipitated proteins were eluted and electrophoretically separated on both 8% and 15.0% denaturing polyacrylamide gels to resolve both high- and low-molecular-weight proteins, respectively. The results are shown in Fig. 2A and B.
FIG. 2.

Coimunoprecipitation of proteins with pUL17-specific antibody. Rabbit skin cells were infected with HSV-1(F) or the UL17 deletion virus, and pUL17 was immunoprecipitated. Immunoprecipitated proteins were electrophoretically separated on a 15% (A) or 8% (B and C) SDS-polyacrylamide gel and stained with Coomassie brilliant blue or were subjected to immunoblotting with pUL17-specific antibody as indicated. Proteins unique to or highly enriched in immunoaffinity purifications from HSV-1(F)-infected cell lysates are indicated with arrows, and their identities are given in the text. The intensely luminescent band below the pUL17 band contains the IgY heavy chain, which reacts with the secondary conjugate.
Four bands in the 8% polyacrylamide gel (indicated in Fig. 2B and corresponding to apparent Mrs of 62,000, 85,000, 95,000, and 160,000) and three bands from the 15% polyacrylamide gel (Fig. 2A) (corresponding to apparent Mrs of 15,000, 17,500, and 19,000) were exclusive to or greatly enhanced in lanes containing immunoprecipitations from lysates of HSV-1(F), as opposed to immunoprecipitations from lysates of cells infected with the UL17 deletion virus. These bands were cut from the gel and subjected to trypsin digestion. The masses of the resulting peptides were determined by MALDI-TOF mass spectrometry and were compared to available databases of vertebrate proteins.
As shown in Table 2, bands from the 8% gel with apparent Mrs of 85,000 and 95,000 contained tryptic peptides with masses consistent with the UL46 and UL47 gene products of HSV-1 (predicted molecular weights [MW], 78,239 and 73,812, respectively). In addition to these species, eight other peptide matches from the 85,000-apparent Mr band matched glycoprotein E peptides. This was not surprising, as gE/gI complexes have been shown to bind the Fc domain of IgG immunoglobulins (18). Because this complex efficiently binds rabbit immunoglobulins, we suspect that the rabbit anti-chicken IgG antibody, used to bridge the IgY and Sepharose beads in this experiment, was responsible for immunoprecipitation of the gE/gI complex. Tryptic peptides from the VP11/12-containing band also included one match to a zinc finger protein homolog and seven other peptides attributable to spindle-like microcephaly-associated proteins (ASMPs).
The three bands from the 15% gel containing proteins with apparent Mrs of 15,000, 17,500, and 19,000 most abundantly contained peptides consistent with cellular histone proteins H4 (predicted MW, 11,236), H2A (predicted MW, 13,927), and H3 (predicted MW, 15,498), respectively (Fig. 2A and Table 2). These masses matched those of tryptic peptides from a number of animal histones due to the high conservation of these proteins (data not shown).
A predominant band of 62,000 apparent Mr was detected in both the 8% and 15% SDS-polyacrylamide gels. This band was much more prominent in the HSV-1(F) immunoprecipitation reactions and contained tryptic peptides with masses consistent with those from the human intermediate-filament protein vimentin and vimentin homologs, which are components of intermediate filaments in several animal species. The other two most highly ranked matches included unnamed human proteins, both with homology to the N-terminal head region of intermediate-filament proteins (data not shown).
The slowest-migrating band in this experiment produced tryptic peptides consistent with the major capsid protein VP5 from a variety of herpesviruses, with human herpesviruses 1 and 2 scoring the highest and second highest, respectively. This was consistent with pUL17 association with capsids which contain abundant amounts of VP5 (see Discussion).
A band consistent with pUL17 alone was not apparent in the immunoprecipitation, an observation that is likely attributable to the fact that pUL17 and VP13/14 comigrate on denaturing polyacrylamide gels (32). To determine whether pUL17 was immunoprecipitated, we transferred the electrophoretically separated proteins immunoprecipitated with anti-pUL17 antibody to nitrocellulose and probed the proteins with anti-pUL17 antibody. Bound immunoglobulin was revealed by reaction with horseradish peroxidase-conjugated anti-chicken antibody, followed by chemiluminescence as detailed in Materials and Methods. As shown in Fig. 2C, pUL17 was readily immunoprecipitated by its cognate antibody, inasmuch as it was present in the HSV-1(F) sample.
To confirm the putative pUL17 interaction with VP13/14, lysates of cells infected with HSV-1(F) and the UL17 deletion virus were reacted with polyclonal antibody directed against VP13/14 (a kind gift from the laboratory of David Meredith), and the presence of pUL17 in immunoprecipitated material was assessed by immunoblotting. As shown in Fig. 3C, approximately equal amounts of VP13/14 were immunoprecipitated from cells infected with either the UL17 deletion or wild-type virus. More important for the purposes of this report, however, pUL17 was readily coimmunoprecipitated with the VP13/14-specific antibody (Fig. 3A). These data therefore confirmed the interaction between pUL17 and VP13/14.
FIG. 3.

Coimmunoprecipitation of proteins with anti-VP13/14 antibody. Cells were mock infected or were infected with HSV-1(F) or the UL17 deletion virus, and lysates were reacted with VP13/VP14-specific antibody. Immune complexes were purified, electrophoretically separated, and subjected to immunoblotting with antibodies against pUL17 (A), VP16 (B), or VP13/14 (C). Bound immunoglobulins were revealed by reaction with appropriately conjugated anti-immunogobulins followed by chemiluminescent exposure to X-ray film. The arrow in panel A indicates a band containing pUL17. The arrow in panel C refers to the band containing VP13/14. The heavy band below this is the heavy chain of rabbit IgG, which reacts with the conjugated secondary antibody. The lines to the left of each panel refer to the positions of size standards. From top to bottom, they are Mrs 180,000, 130,000, 100,000, 72,000, 55,000, 40,000, and 33,000.
In the expectation that VP13/14 interacted with VP16 as well, the immunoblot of immunoprecipitated proteins was probed with antibody against VP16. As shown in Fig. 3B, VP16 was coimmunoprecipitated by the VP13/14-specific antibody. Surprisingly, the interaction between VP13/14 and VP16 was greatly diminished in lysates of cells infected with the UL17-null virus. Thus, UL17 substantially augmented coimmunoprecipitation of VP13/14 and VP16.
In a second set of experiments to confirm the pUL17-VP13/14 interaction, we sought to copurify VP13/14 with pUL17 by means other than coimmunoprecipitation. Therefore, a novel virus encoding pUL17 with 6 histidine codons fused to its C terminus (pUL17-His) was constructed as described in Materials and Methods. Cells were infected with this virus and 16 h later were lysed in 1% NP-40. pUL17-His was then purified from these lysates by affinity chromatography on Ni2+-containing agarose beads. As a negative control, lysates of cells infected with HSV-1(F) were treated identically. Proteins bound to the affinity matrix were eluted sequentially 4 times in the presence of imidazole, denatured in SDS, electrophoretically separated, and transferred to nitrocellulose. As shown in Fig. 4, probing the nitrocellulose with pUL17-specific antibody revealed pUL17-His within the eluates from pUL17-His-infected cells but not from eluates derived from cells infected with HSV-1(F). pUL17 was most concentrated in the first elution, with subsequent elutions diminishing in signal as expected (Fig. 5A, lanes 1 and 3).
FIG. 4.
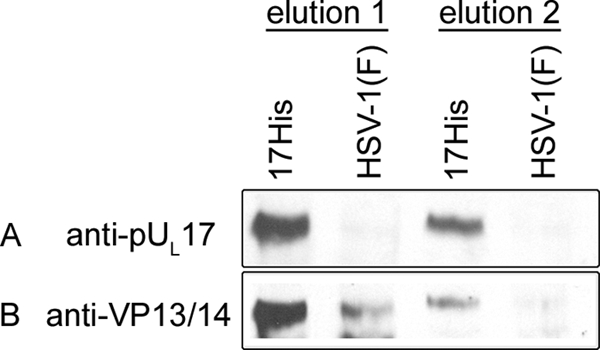
Affinity copurification of VP13/14 and pUL17. Hep2 cells were infected with either HSV-1(F) or a recombinant HSV-1 bearing a histidine tag fused to the C terminus of pUL17. Lysates of the cells were clarified, and soluble proteins were subjected to affinity chromatography on Ni2+-containing beads. After extensive washing, proteins bound to the beads were eluted in SDS, electrophoretically separated, and subjected to immunoblotting with antibodies to pUL17 (A) or VP13/14 (B).
The same immunoblot was then stripped and reprobed for the presence of VP13/14. As shown in Fig. 4B, although a background level of VP13/14 was detectable in the first elution from beads containing HSV-1(F)-infected cell proteins, analysis by NIH image software indicated that VP13/14 was increased at least 5-fold in eluates derived from cells infected with the pUL17-His virus. Moreover, VP13/14 was readily detectable in the second elution from beads bearing proteins from cells infected with UL17-His virus, but not from cells infected with HSV-1(F). These data therefore indicate that pUL17 and VP13/14 can be copurified via the His tag incorporated into the C terminus of pUL17 and further confirm the VP13/14-pUL17 interaction.
Because it is possible that the coimmunoprecipitation and affinity copurification of pUL17 and VP13/14 were consequential to immunoprecipitation or purification of intact capsids with which both proteins might associate, we asked whether the VP13/14-pUL17 interaction was dependent on fully assembled capsids. To this end, cells were infected with HSV-1(F), the UL17 deletion virus, or a virus lacking the capacity to encode VP23 (an essential component of capsid triplexes encoded by UL18). Lysates of the infected cells were then immunoprecipitated with anti-VP13/14 antibody. As shown in Fig. 5, VP13/14 was readily immunoprecipitated with its cognate antibody from lysates of cells infected with HSV-1(F), UL18-null, or UL17-null virus. Most importantly, pUL17 was coimmunoprecipitated with this antibody from lysates of cells infected with HSV-1(F) and the UL18-null virus. We conclude that the pUL17 and VP13/14 interaction is not dependent on the presence of intact capsids.
Colocalization of pUL17, VP11/12, and VP13/14 in infected cells.
Because it is possible that the detected interactions occurred after cell lysis rather than in intact cells, we next asked if VP13/14 and pUL17 colocalize in infected cells. Because the polyclonal antibodies directed against VP13/14 were not useful for indirect immunofluorescence (data not shown), we constructed a recombinant virus with an HA tag placed at the C terminus of VP13/14.
Cells were infected with the VP13/14-HA virus, fixed at various times after infection, permeabilized, and immunostained with anti-pUL17 and anti-HA antibodies. Bound immunoglobulins were revealed by reaction with anti-immunoglobulins conjugated to different fluorescent markers. The results, shown in Fig. 6, indicate that pUL17 and VP13/14 (as indicated by reaction with the anti-HA antibody) are detectable by 6 h after infection. At this time point, some pUL17 and VP13/VP14 colocalized in either the cytoplasm in a perinuclear region of cells or at low levels in the nucleus, whereas some pUL17 and VP13/14 immunoreactivity did not colocalize. By 9 h, both pUL17 and VP13/14 were predominantly located in the intranuclear replication compartment, as determined by their distribution in a pattern identical to that obtained upon immunostaining with ICP4-specific antibody (7). Some cells contained more pUL17-specific staining than VP13/14 staining, whereas others contained more VP13/14- than pUL17-specific immunostaining. Although previous reports indicated that pUL17 localized to intranuclear aggregates containing VP5 and ICP35 (14), such distribution was seen only rarely in the current study. At 12 to 15 h postinfection, small amounts of immunoreactivity attributable to both VP13/14 and pUL17 colocalized at the plasma membrane (Fig. 6M, N, O, Q, R, and S). These data indicate that at least as viewed by immunofluorescence, VP13/14 and pUL17 colocalize at later times postinfection in the replication compartment of the nucleus, in regions of the cytoplasm, and at the plasma membrane.
FIG. 6.

Immunofluorescence staining of cells infected with a virus encoding an HA-tagged VP13/14. At the indicated times after infection, Hep2 cells were fixed, permeabilized, and reacted with antibodies to pUL17 (leftmost column), HA (indicating VP13/14 localization; second column from the left), or ICP4 (rightmost column). The third column from the left is a merge of the two columns to its left. Panels U, V, and W are higher magnifications of the areas within the rectangles in panels M, N, and O, respectively. The arrows in panels M, N, O, R, and S indicate regions of the plasma membrane bearing pUL17 and VP13/14-HA-specific signals. RC, replication compartment; PM, plasma membrane.
DISCUSSION
The main goal of this study was to identify pUL17-interacting proteins to shed light on its role in tegument and capsid assembly. The studies were initiated by coimmunoprecipitation with pUL17 antibody, followed by analysis of bands predominantly or exclusively visualized in electrophoretic profiles of proteins immunoprecipitated from lysates of wild-type virus as opposed to those from a UL17 deletion virus. Although this approach might preclude detection of interesting interacting proteins if they were to (i) represent minor components of a band, (ii) comigrate with proteins immunoprecipitated nonspecifically, or (iii) comigrate with the heavy or light chains of the IgY and IgG antibodies, we were able to identify specific interactions between pUL17 and vimentin, cellular histone proteins, the capsid component VP5, and the tegument proteins VP13/14 and VP11/12. Histone proteins that coimmunoprecipitated with pUL17 included H4, H2A, and H3, which represent core proteins of the nucleosome. The exclusion of histone H2B from this group may reflect the fact that the encoding gene is transcriptionally downregulated in lytic HSV infection as a consequence of competition by HSV-1 VP16 for the transcription factor Oct1 (20). We should also note that this approach does not distinguish indirect from direct interactions.
As a subsequent step to ensure that the detected pUL17 interactions occurred in intact cells as opposed to cellular lysates, we performed colocalization experiments in permeabilized but otherwise intact infected cells. In these experiments, VP13/14 colocalized with pUL17 (Fig. 6). In contrast, an interaction between histones and pUL17 was not supported, inasmuch as pUL17 and histone H3 did not colocalize to any appreciable extent in infected-cell nuclei (data not shown). We are also skeptical of the pUL17-vimentin interaction, as vimentin is mostly cytoplasmic, whereas pUL17 is largely nuclear in infected cells, and extensive colocalization between these two proteins was not observed (not shown). Thus, the histone-pUL17 and vimentin-pUL17 interactions occur in cell lysates, but whether these proteins interact in intact cells requires further verification.
Accumulating evidence suggests that the detected interaction between pUL17 and VP5 is indirect and occurs through pUL17's interaction with triplexes. This conclusion is supported by the observations that (i) pUL17 and VP5 do not coimmunoprecipitate from lysates of cells infected with a UL18-null virus that lacks assembled triplexes (28, 33), (ii) the immunoreactivity of pUL17 and coimmunoprecipitation of pUL17 with its interaction partner pUL25 were reduced in lysates of cells infected with the UL18-null virus (33), and (iii) the deduced location of pUL17 within the CCSC located atop capsid triplexes that link capsid pentons to adjacent hexons (43). We did not expect to see triplex proteins in the pUL17-immunoprecipitated proteins detected here because the broad bands of comigrating IgY subunits precluded us from subjecting appropriate regions of the gel to mass spectrometry. Similarly, potential comigration with vimentin may have precluded detection of pUL25 (predicted Mr, 62,666) peptides because of the great abundance of vimentin peptides in the harvested gel slice.
We should note that the deduced presence of pUL17 in the CCSC was observed exclusively in DNA-containing capsids (43), whereas others using biochemical techniques have detected pUL17 on all capsid forms, including procapsids (40). Reconciling these observations will require further studies, but one possibility is that some triplexes bearing pUL17 are incorporated into the procapsid, whereas structural changes induced by DNA insertion stabilizes the conformation of the pUL17-pUL25 complex or promotes pUL17-pUL25 binding to the capsid. Further studies will be required to determine which triplex components interact directly or indirectly with pUL17.
Perhaps the most important finding of the current study is the observation that pUL17 interacts with VP13/14, thus supporting an interaction previously suggested by yeast two-hybrid assays (12). Although we detected an interaction between pUL17 and VP11/12, verification of this interaction was hampered by the lack of an appropriate antibody to VP11/12. We also noted that an interaction between VP16 and VP13/14 was precluded in cells infected with a UL17-null virus, further supporting a role for pUL17 in tegument assembly. Although there are many possible explanations for this result, one possibility is that the interaction with pUL17 helps ensure VP13/14 competency to bind VP16. Further studies to test this hypothesis are required.
It is likely that the tegument is anchored to the capsid by a series of interactions. Previous work to identify such anchoring points on the capsid focused on the pentonic vertices which bear densities of primary tegument when viewed in intact virions (52). The precise identities of the protein(s) responsible for the penton-associated densities are unknown. One candidate is the protein pUL36 (VP1/2), which has been shown to interact with HSV nucleocapsids and with pUL25 in the pseudorabies virus system, suggesting its role as a primary tegument protein (5, 6). The current study suggests another anchoring point of tegument on the capsid, specifically through the interactions between pUL17 on the capsid surface and VP13/14 in the next layer of tegument. We favor the possibility that the interaction is predominantly cytoplasmic because attempts to determine whether VP13/14 was associated with intranuclear capsids were hampered by migration of VP13/14 throughout sucrose gradients and poor immunoreactivity of intranuclear capsids using VP13/14-specific antisera in immunogold electron microscopy experiments (data not shown). These results, however, do not exclude an intranuclear interaction. In any event, a cytoplasmic interaction might reflect one between capsid-associated pUL17 and the tegument protein VP13/14, which accumulates at secondary envelopment sites in the cytoplasm.
Alternatively, the evidence is also consistent with the existence of a second population of pUL17 that associates with the tegument independently of the capsid. This evidence includes the observation that pUL17 is readily detectable in light particles that bear most tegument proteins (including VP13/14) but lack capsids (32, 41) and levels of pUL17 in the virion that are greater than can be accounted for by the amounts associated with capsids (41). Therefore, the pUL17-VP13/14 interaction detected in the current study may reflect a tegument assembly step that occurs separately from the capsid. This is consistent with the observation that pUL17 and VP13/14 still coimmunoprecipitate in the absence of intact capsids (Fig. 5).
Supplementary Material
Acknowledgments
We thank the David Meredith laboratory for the polyclonal antibody to VP13/14.
These studies were supported by grants R01GM50741 and T32 AI07618 from the National Institutes of Health.
Footnotes
Published ahead of print on 26 May 2010.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Baines, J. D., and C. Duffy. 2006. Nucleocapsid assembly and envelopment of herpes simplex virus, p. 175-204. In R. M. Sandri-Goldin (ed.), Alpha herpesviruses: pathogenesis, molecular biology and infection control. Caister Scientific Press, Norfolk, United Kingdom.
- 2.Baines, J. D., R. J. Jacob, L. Simmerman, and B. Roizman. 1995. The UL11 gene products of herpes simplex virus 1 are present in the nuclear and cytoplasmic membranes, and intranuclear dense bodies of infected cells. J. Virol. 69:825-833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baines, J. D., C. E. Hsieh, E. Wills, C. Mannella, and M. Marko. 2007. Electron tomography of nascent herpes simplex virions. J. Virol. 81:2726-2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barker, D. E., and B. Roizman. 1990. Identification of 3 genes nonessential for growth in cell culture near the right terminus of the unique sequences of long component of herpes simplex virus 1. Virology 177:684-691. [DOI] [PubMed] [Google Scholar]
- 5.Bucks, M. A., K. J. O'Regan, M. A. Murphy, J. W. Wills, and R. J. Courtney. 2007. Herpes simplex virus type 1 tegument proteins VP1/2 and UL37 are associated with intranuclear capsids. Virology 361:316-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coller, K. E., J. I. H. Lee, A. Ueda, and G. A. Smith. 2007. The capsid and tegument of the alphaherpesviruses are linked by an interaction between the UL25 and VP1/2 proteins. J. Virol. 81:11790-11797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Bruyn Kops, A., S. L. Uprichard, M. Chen, and D. M. Knipe. 1998. Comparison of the intranuclear distributions of herpes simplex virus proteins involved in various viral functions. Virology 252:162-178. [DOI] [PubMed] [Google Scholar]
- 8.Desai, P., N. A. DeLuca, J. C. Glorioso, and S. Person. 1993. Mutations in herpes simplex virus type 1 genes encoding VP5 and VP23 abrogate capsid formation and cleavage of replicated DNA. J. Virol. 67:1357-1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donnelly, M., and G. Elliott. 2001. Nuclear localization and shuttling of herpes simplex virus tegument protein VP13/14. J. Virol. 75:2566-2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dorange, F., B. K. Tischer, J. F. Vautherot, and N. Osterrieder. 2002. Characterization of Marek's disease virus serotype 1 (MDV-1) deletion mutants that lack UL46 to UL49 genes: MDV-1 UL49, encoding VP22, is indispensable for virus growth. J. Virol. 76:1959-1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ejercito, P. M., E. D. Kieff, and B. Roizman. 1968. Characterization of herpes simplex virus strains differing in their effects on social behavior of infected cells. J. Gen. Virol. 2:357-364. [DOI] [PubMed] [Google Scholar]
- 12.Fossum, E., C. C. Friedel, S. V. Rajagopala, B. R. Titz, A. Baiker, T. Schmidt, T. Kraus, T. Stellberger, C. Rutenberg, S. Suthram, S. Bandyopadhyay, D. Rose, A. von Brunn, M. Uhlmann, C. Zeretzke, Y. A. Dong, H. Boulet, M. Koegl, S. M. Bailer, U. Koszinowski, T. Ideker, P. Uetz, R. Zimmer, and J. Haas. 2009. Evolutionarily conserved herpesviral protein interaction networks. PLoS Pathog. 5:e1000570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuchs, W., B. G. Klupp, H. Granzow, N. Osterrieder, and T. C. Mettenleiter. 2002. The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host-cell nucleus and represent components of primary enveloped but not mature virions. J. Virol. 76:364-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goshima, F., D. Watanabe, H. Takakuwa, K. Wada, T. Daikoku, H. Yamada, and Y. Nishiyama. 2000. Herpes simplex virus UL17 protein is associated with B capsids and colocalizes with ICP35 and VP5 in infected cells. Arch. Virol. 145:417-426. [DOI] [PubMed] [Google Scholar]
- 15.Granzow, H., B. G. Klupp, W. Fuchs, J. Veits, N. Osterrieder, and T. C. Mettenleiter. 2001. Egress of alphaherpesviruses: comparative ultrastructural study. J. Virol. 75:3675-3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heine, J. W., R. W. Honess, E. Cassai, and B. Roizman. 1974. Proteins specified by herpes simplex virus. XII. The virion polypeptides of type 1 strains. J. Virol. 14:640-651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Homa, F. L., and J. C. Brown. 1997. Capsid assembly and DNA packaging in herpes simplex virus. Rev. Med. Virol. 7:107-122. [DOI] [PubMed] [Google Scholar]
- 18.Johnson, D. C., M. C. Frame, M. W. Ligas, A. M. Cross, and N. D. Stow. 1988. Herpes simplex virus immunoglobulin G Fc receptor activity depends on a complex of two viral glycoproteins, gE and gI. J. Virol. 62:1347-1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kopp, M., B. G. Klupp, H. Granzow, W. Fuchs, and T. C. Mettenleiter. 2002. Identification and characterization of the pseudorabies virus tegument proteins UL46 and UL47: role for UL47 in virion morphogenesis in the cytoplasm. J. Virol. 76:8820-8833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Latchman, D. S., J. F. Pratridge, and L. M. Kemp. 1989. The different competitive abilities of viral TAATGARAT elements and cellular octamer motifs mediate the induction of viral immediate-early genes and the repression of the histone H2B gene in herpes simplex virus infected cells. Nucleic Acids Res. 17:8533-8542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang, L., M. Tanaka, Y. Kawaguchi, and J. D. Baines. 2004. Cell lines that support replication of a novel herpes simplex 1 UL31 deletion mutant can properly target UL34 protein to the nuclear rim in the absence of UL31. Virology 329:68-76. [DOI] [PubMed] [Google Scholar]
- 22.Loret, S., G. Guay, and R. Lippe. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J. Virol. 82:8605-8618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGeoch, D. J., M. A. Dalrymple, A. J. Davison, A. Dolan, M. C. Frame, D. McNab, L. J. Perry, J. E. Scott, and P. Taylor. 1988. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 69:1531-1574. [DOI] [PubMed] [Google Scholar]
- 24.McLean, G., F. Rixon, N. Langeland, L. Haarr, and H. Marsden. 1990. Identification and characterization of the virion protein products of herpes simplex virus type 1 gene UL47. J. Gen. Virol. 71:2953-2960. [DOI] [PubMed] [Google Scholar]
- 25.Mettenleiter, T. C., B. G. Klupp, and H. Granzow. 2006. Herpesvirus assembly: a tale of two membranes. Curr. Opin. Microbiol. 9:423-429. [DOI] [PubMed] [Google Scholar]
- 26.Murphy, M. A., M. A. Bucks, K. J. O'Regan, and R. J. Courtney. 2008. The HSV-1 tegument protein pUL46 associates with cellular membranes and viral capsids. Virology 376:279-289. [DOI] [PubMed] [Google Scholar]
- 27.Naldinho-Souto, R., H. Browne, and T. Minson. 2006. Herpes simplex virus tegument protein VP16 is a component of primary enveloped virions. J. Virol. 80:2582-2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Newcomb, W. W., B. L. Trus, F. P. Booy, A. C. Steven, J. S. Wall, and S. C. Brown. 1993. Structure of the herpes simplex virus capsid molecular composition of the pentons and the triplexes. J. Mol. Biol. 232:499-511. [DOI] [PubMed] [Google Scholar]
- 29.Padula, M. E., M. L. Sydnor, and D. W. Wilson. 2009. Isolation and preliminary characterization of herpes simplex virus 1 primary enveloped virions from the perinuclear space. J. Virol. 83:4757-4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Read, G. S., and M. Patterson. 2007. Packaging of the virion host shutoff (Vhs) protein of herpes simplex virus: two forms of the Vhs polypeptide are associated with intranuclear B and C capsids, but only one is associated with enveloped virions. J. Virol. 81:1148-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reynolds, A. E., E. G. Wills, R. J. Roller, B. J. Ryckman, and J. D. Baines. 2002. Ultrastructural localization of the HSV-1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 76:8939-8952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salmon, B., C. Cunningham, A. J. Davison, W. J. Harris, and J. D. Baines. 1998. The herpes simplex virus 1 UL17 gene encodes virion tegument proteins that are required for cleavage and packaging of viral DNA. J. Virol. 72:3779-3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scholtes, L., and J. D. Baines. 2009. Effects of major capsid proteins, capsid assembly, and DNA cleavage/packaging on the pUL17/pUL25 complex of herpes simplex virus 1. J. Virol. 83:12725-12737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stackpole, C. W. 1969. Herpes-type virus of the frog renal adenocarcinoma. I. Virus development in tumor transplants maintained at low temperature. J. Virol. 4:75-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steven, A. C., and P. G. Spear. 1997. Herpesvirus capsid assembly and envelopment, p. 312-351. In W. Chiu, R. M. Burnett, and R. L. Garcea (ed.), Structural biology of viruses. Oxford University Press, New York, NY.
- 36.Szilágyi, J. F., and C. Cunningham. 1991. Identification and characterization of a novel non-infectious herpes simplex virus-related particle. J. Gen. Virol. 72:661-668. [DOI] [PubMed] [Google Scholar]
- 37.Szpara, M. L., L. Parsons, and L. W. Enquist. 2010. Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations. J. Virol. 84:5303-5313. doi: 10.1128/JVI.00312-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tanaka, M., H. Kagawa, Y. Yamanashi, T. Sata, and Y. Kawaguchi. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 77:1382-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taus, N. S., B. Salmon, and J. D. Baines. 1998. The herpes simplex virus 1 UL17 gene is required for localization of capsids and major and minor capsid proteins to intranuclear sites where viral DNA is cleaved and packaged. Virology 252:115-125. [DOI] [PubMed] [Google Scholar]
- 40.Thurlow, J. K., M. Murphy, N. D. Stow, and V. G. Preston. 2006. Herpes simplex virus type 1 DNA-packaging protein UL17 is required for efficient binding of UL25 to capsids. J. Virol. 80:2118-2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thurlow, J. K., F. J. Rixon, M. Murphy, P. Targett-Adams, M. Hughes, and V. G. Preston. 2005. The herpes simplex virus type 1 DNA packaging protein UL17 is a virion protein that is present in both the capsid and the tegument compartments. J. Virol. 79:150-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tischer, B. K., J. von Einem, B. Kaufer, and N. Osterrieder. 2006. Two-step RED-mediated recombination for versatile high-efficiency markerless DNA manipulation in Eschericia coli. Biotechniques 40:191-197. [DOI] [PubMed] [Google Scholar]
- 43.Trus, B. L., W. W. Newcomb, N. Cheng, G. Cardone, L. Marekov, F. L. Homa, J. C. Brown, and A. C. Steven. 2007. Allosteric signaling and a nuclear exit strategy: binding of UL25/UL17 heterodimers to DNA-filled HSV-1 capsids. Mol. Cell 26:479-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Verhagen, J., M. Donnelly, and G. Elliott. 2006. Characterization of a novel transferable CRM-1-independent nuclear export signal in a herpesvirus tegument protein that shuttles between the nucleus and cytoplasm. J. Virol. 80:10021-10035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Verhagen, J., I. Hutchinson, and G. Elliott. 2006. Nucleocytoplasmic shuttling of bovine herpesvirus 1 UL47 protein in infected cells. J. Virol. 80:1059-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vittone, V., E. Diefenbach, D. Triffett, M. W. Douglas, A. L. Cunningham, and R. J. Diefenbach. 2005. Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 79:9566-9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whittaker, G. R., M. P. Riggio, I. W. Halliburton, R. A. Killington, G. P. Allen, and D. M. Meredith. 1991. Antigenic and protein sequence homology between VP13/14, a herpes simplex virus type 1 tegument protein, and gp10, a glycoprotein of equine herpes virus 1 and 4. J. Virol. 65:2320-2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wills, E., L. Scholtes, and J. D. Baines. 2006. Herpes simplex virus 1 DNA packaging proteins encoded by UL6, UL15, UL17, UL28, and UL33 are located on the external surface of the viral capsid. J. Virol. 80:10894-10899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang, K., and J. D. Baines. 2006. The putative terminase subunit of herpes simplex virus 1 encoded by UL28 is necessary and sufficient to mediate interaction between pUL15 and pUL33. J. Virol. 80:5733-5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang, Y., and J. L. McKnight. 1993. Herpes simplex virus type 1 UL46 and UL47 deletion mutants lack VP11 and VP12 or VP13 and VP14, respectively, and exhibit altered viral thymidine kinase expression. J. Virol. 67:1482-1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang, Y., D. A. Sirko, and J. L. C. McKnight. 1991. Role of herpes simplex virus type 1 UL46 and UL47 in αTIF mediated induction: characterization of three viral deletion mutants. J. Virol. 65:829-841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou, Z. H., D. H. Chen, J. Jakana, F. J. Rixon, and W. Chiu. 1999. Visualization of tegument-capsid interactions and DNA in intact herpes simplex virus type 1 virions. J. Virol. 73:3210-3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.