

The crystal structure of the dimethyllysine derivative of the E. coli RNA polymerase α subunit C-terminal domain is reported at 2.0 Å resolution.
Keywords: RNA polymerase, Escherichia coli, α subunit, C-terminal domain
Abstract
The α subunit C-terminal domain (αCTD) of RNA polymerase (RNAP) is a key element in transcription activation in Escherichia coli, possessing determinants responsible for the interaction of RNAP with DNA and with transcription factors. Here, the crystal structure of E. coli αCTD (α subunit residues 245–329) determined to 2.0 Å resolution is reported. Crystals were obtained after reductive methylation of the recombinantly expressed domain. The crystals belonged to space group P21 and possessed both pseudo-translational symmetry and pseudo-merohedral twinning. The refined coordinate model (R factor = 0.193, R free = 0.236) has improved geometry compared with prior lower resolution determinations of the αCTD structure [Jeon et al. (1995 ▶), Science, 270, 1495–1497; Benoff et al. (2002 ▶), Science, 297, 1562–1566]. An extensive dimerization interface formed primarily by N- and C-terminal residues is also observed. The new coordinates will facilitate the improved modeling of αCTD-containing multi-component complexes visualized at lower resolution using X-ray crystallography and electron-microscopy reconstruction.
1. Introduction
Escherichia coli RNA polymerase (RNAP) is a multisubunit enzyme with core enzyme composition α2ββ′ω and holoenzyme composition α2ββ′ωσ. Each α subunit consists of two independently folded domains: an amino-terminal domain (αNTD; α residues 1–235) and a carboxyl-terminal domain (αCTD; α residues 250–329). The two domains are connected via a long flexible linker (residues 236–249; Blatter et al., 1994 ▶; Negishi et al., 1995 ▶; Jeon et al., 1997 ▶). αNTD is responsible for α-subunit dimerization and for the assembly of the other subunits into a transcriptionally active enzyme (Zhang & Darst, 1998 ▶). αCTD plays a key role in the recruitment of RNAP holoenzyme to promoter DNA (Blatter et al., 1994 ▶; Ebright & Busby, 1995 ▶). The flexible linker facilitates the independent movement and positioning of αCTD so that one or two αCTDs can interact with DNA and/or with one or more transcription factors (Busby & Ebright, 1999 ▶). αCTD binds preferentially to upstream promoter elements (UP elements) that are A/T-rich (Gourse et al., 2000 ▶).
The structure of E. coli αCTD was initially determined in solution using NMR spectroscopy, revealing a compact fold with four helices and flexible N- and C-termini (Jeon et al., 1995 ▶). The αCTD structure was subsequently defined at 3.1 Å resolution within a ternary complex consisting of the catabolite activator protein (CAP), αCTD and DNA using X-ray crystallography (Benoff et al., 2002 ▶). In this structure, CAP and αCTD respectively bind to adjacent CAP and UP-element sites on the DNA, with αCTD binding to the narrow DNA minor groove of the UP-element A-tract. αCTD and CAP also make direct contact with each other through a small specific complementary interface.
On lac promoter DNA, the interaction between CAP (bound at promoter position −61.5) and αCTD (bound at promoter position −42) places αCTD adjacent to region 4 of the RNAP σ70 subunit (bound to the promoter −35 element) and permits functional protein–protein interaction between αCTD and σ70 region 4 (Lawson et al., 2004 ▶; Chen et al., 2003 ▶; Ross et al., 2003 ▶). The expected overall spatial arrangement of CAP, αCTD, σ70 region 4 and DNA was recently confirmed in a 20 Å resolution EM reconstruction of an intact reconstituted CAP–RNAP–promoter DNA complex (Hudson et al., 2009 ▶).
Here, we report the first crystallographic structure of free uncomplexed αCTD (α residues 245–329) determined at 2.0 Å resolution.
2. Materials and methods
2.1. Expression and purification of αCTD
Recombinant αCTD from E. coli strain K12 was expressed from pEBT7-αCTD plasmid in E. coli BL21 (DE3) (Novagen), which encodes Met followed by α residues 245–329 (Gaal et al., 1996 ▶). Protein expression and purification were performed as described previously by Gaal et al. (1996 ▶). Briefly, protein was expressed in BL21 (DE3) cells induced with 1 mM IPTG for 4 h at 310 K. The cells were collected by centrifugation at 5000g for 10 min and frozen at 193 K. All subsequent steps were performed at 277 K. Frozen cells were resuspended in 20 ml lysis buffer (50 mM Tris–HCl pH 8.5, 233 mM NaCl, 2 mM EDTA, 0.1 mM DTT, 1 mM β-mercaptoethanol, 5% glycerol, 23 µg ml−1 PMSF, 0.5 µg ml−1 leupeptin, 1 µg ml−1 pepstatin A) and disrupted by sonication. Cell debris was removed by centrifugation at 17 000g for 30 min. A stock solution of 10%(w/v) polyethyleneimine (Sigma) was added to the supernatant to a final concentration of 0.2%; precipitated material was removed by centrifugation at 17 000g for 30 min at 277 K. The supernatant was fractionated using ammonium sulfate, with αCTD remaining in the supernatant at 35% saturation and precipitating at 100% saturation. The material was resuspended in buffer A (50 mM Tris–HCl pH 8.5, 1 mM EDTA, 0.4 mM DTT), desalted using an Econo-Pac 10DG column (Bio-Rad) and diluted to a volume of 140 ml with buffer A before application onto a HiPrep 16/10 Q XL column (GE Healthcare). αCTD was eluted with a linear gradient from 0 to 650 mM NaCl and was concentrated to a volume of ∼6 ml by ultrafiltration (Amicon Ultra-15 with Ultracel 3k membrane, Millipore). The protein was then applied onto a HiLoad 26/60 Superdex 75 gel-filtration column (GE Healthcare) equilibrated with 50 mM Tris–HCl pH 8.0, 0.1 mM EDTA, 0.4 mM DTT, 0.2 M NaCl. αCTD was eluted in a single peak, concentrated as above to ∼20–22 mg ml−1, exchanged into buffer WS [20 mM Tris–HCl pH 8.0, 0.1 mM EDTA, 1 mM DTT, 0.2 M NaCl, 0.02%(w/v) NaN3] and stored in aliquots at 277 K. Yields were typically 18–20 mg per litre of cell culture.
2.2. Cloning, expression and purification of truncated αCTD
The coding sequence for α residues 249–322 was amplified by PCR from pEBT7-αCTD with forward primer 5′-GTATCCATGGCTTTCGATCCGATCCTGCTGCG-3′ and reverse primer 5′-GACTGGATCCTTATGGCCAGTTTTCCAGGCG-3′. Amplification was performed using PfuUltra polymerase for 25 cycles with 30 pmol of each primer. The amplified DNA was digested with NcoI and BamHI restriction sites engineered into the 5′ and 3′ ends of the amplified sequence, respectively, and ligated to the NcoI and BamHI sites of the pET-28a(+) bacterial expression vector (Novagen). Expression, purification and storage were as described for αCTD (245–329). Compared with αCTD, truncated αCTD elutes earlier in the HiPrep 16/10 Q XL linear gradient. Truncated αCTD was stored in aliquots at 277 K at ∼20 mg ml−1. Yields were typically 45–50 mg per litre of cell culture.
2.3. Reductive methylation
Reductive methylation closely followed the protocol of Rayment (1997 ▶). αCTD was exchanged into 50 mM HEPES pH 7.5, 250 mM NaCl using an Econo-Pac 10DG chromatography column (Bio-Rad). 6 ml protein sample (0.5 mg ml−1) was placed in a 15 ml Falcon tube. 120 µl ABC solution (1 M borane–dimethylamine complex; Sigma–Aldrich product No. 180238) and 240 µl 1 M formaldehyde [from 37%(w/v) stock; Sigma–Aldrich product No. 33220] were added to the sample and the reaction was gently agitated for 2 h at 277 K. This last step was then repeated and the reaction was gently agitated for another 2 h period. A final 60 µl of ABC solution was added and the reaction was incubated overnight at 277 K. The reaction was quenched by the addition of 6 mg solid glycine powder. The sample was incubated for an additional 2 h at 277 K. The resulting material was desalted and concentrated by ultrafiltration in buffer WS′ [20 mM Tris–HCl pH 8.0, 0.1 mM EDTA, 1 mM DTT, 0.05 M NaCl, 0.02%(w/v) NaN3].
2.4. Crystallization and data collection
Crystallization screening for αCTD and truncated αCTD was performed using hanging-drop vapor diffusion at 289 K with protein concentrations in the range 5–60 mg ml−1 and employing five commercial screens (Crystal Screen, Crystal Screen II, PEG/Ion Screen I and II and Natrix; Hampton Research). Additional crystallization screening of truncated αCTD was performed at the Hauptman–Woodward Institute Center for High Throughput Structural Biology (Luft et al., 2003 ▶) with sample concentrations of 10 and 20 mg ml−1.
Crystallization screening of methylated αCTD (αCTDM) was carried out in MRC crystallization plates (Molecular Dimensions Ltd) using sitting-drop vapor diffusion with the set of commercial screens listed above. Initial incubation was for four weeks at 289 K; subsequently, plates were transferred to 277 K. Crystals obtained with 0.1 M Na HEPES pH 7.5, 1.4 M sodium citrate tribasic dihydrate (Crystal Screen reagent No. 38, Hampton Research) were soaked in crystallization reservoir solution augmented with 20% glycerol for 1 min prior to mounting on LithoLoops (Molecular Dimensions Inc.) and flash-cooling in liquid nitrogen. Crystals were screened for diffraction quality on Brookhaven National Laboratory National Synchrotron Light Source beamlines X6A and X25. The diffraction data used in the structure determination were collected from a single crystal at 100 K using an ADSC Q270 CCD detector on beamline X6A, with 0.5° rotation per image.
2.5. Structure determination, refinement and analysis
Diffraction intensities were initially integrated and scaled assuming primitive orthorhombic lattice symmetry using DENZO and SCALEPACK (Otwinowski & Minor, 1997 ▶). They were subsequently reprocessed assuming primitive monoclinic lattice symmetry using MOSFLM and SCALA (Leslie, 1992 ▶). The search model for molecular replacement trials with Phaser (McCoy et al., 2007 ▶) was αCTD from the CAP–αCTD–DNA ternary complex (PDB entry 1lb2, chain B; Benoff et al., 2002 ▶). phenix.xtriage was used for investigation of Patterson peaks and twinning tests (Adams et al., 2010 ▶).
Crystallographic refinement was performed with phenix.refine (Adams et al., 2010 ▶). Simulated annealing, individual atomic coordinate and individual atom isotropic displacement parameter refinement strategies were performed with noncrystallographic symmetry (NCS) restraints. Manual model adjustment to improve the fit to likelihood-weighted electron-density maps was carried out using Coot (Emsley & Cowtan, 2004 ▶). In the final refinement cycles NCS restraints were only applied to the peptide backbone. Water molecules were added where supported by both chemistry and geometry with likelihood-weighted difference |F obs| − |F calc| electron density >3σ and likelihood-weighted 2|F obs| − |F calc| density >1.8σ. Four metal-ion positions were assigned as Na+ based on evaluation of coordination distances (Hsin et al., 2008 ▶) and comparison of refined B-factor values with neighboring atoms. The structure has been deposited in the PDB with reference code 3k4g.
The quality and stereochemistry of the model were evaluated using Coot validation tools and MolProbity (Chen et al., 2010 ▶). Structural images were generated using UCSF Chimera (Pettersen et al., 2004 ▶). Accessible surface-area calculations were performed using PISA (Krissinel & Henrick, 2007 ▶). Secondary-structure identification was performed using Stride (Heinig & Frishman, 2004 ▶). Sequence-based prediction of preferred X—Pro peptide-bond isomerization states was performed using the PBOND server (Exarchos et al., 2009 ▶).
3. Results
3.1. αCTD crystallization
An extensive set of crystallization trials was carried out for both αCTD (α residues 245–329) and the truncated construct (α residues 249–322). Even when protein concentrations as high as 60 mg ml−1 were employed these materials failed to crystallize under any of the trial conditions.
Since the amino-acid composition of αCTD includes 7% lysine (six of 85 residues), reductive methylation was identified as a possible alternative approach to obtaining a crystal structure of the free uncomplexed domain. Reductive methylation is a simple and efficient method to alter the protein surface properties of a target sample and thereby influence its behavior during crystallization screening (Rayment, 1997 ▶; Kim et al., 2008 ▶). Methylation of the lysine side-chain amino group reduces its interaction with solvent without altering its intrinsic charge. There are now many examples of proteins that have only crystallized after reductive methylation (Rayment et al., 1993 ▶; Schubot & Waugh, 2004 ▶; Walter et al., 2006 ▶; Au et al., 2008 ▶; Kim et al., 2008 ▶).
Crystals of dimethyllysine αCTD (αCTDM) were obtained in Hampton Research Crystal Screen reagent No. 38 (0.1 M Na HEPES pH 7.5, 1.4 M sodium citrate). Single crystals grew after six weeks at 277 K to approximate dimensions of 120 × 120 × 80 µm. It has been reported that methylated proteins usually display reduced solubility (Schubot & Waugh, 2004 ▶). The concentration of αCTDM required for crystallization after methylation was as low as 7 mg ml−1, with an optimal value of around 17 mg ml−1.
3.2. Structure solution
The set of molecular-replacement trials leading to successful determination of the αCTDM structure is summarized in Table 1 ▶. During the initial processing of the diffraction data primitive orthorhombic lattice symmetry was assumed, with a most probable asymmetric unit content of four αCTDM protomers, but initial molecular-replacement trials encompassing all possible primitive orthorhombic space groups were unsuccessful (Table 1 ▶, trial 1). After analysis of the intensity data revealed the presence of a strong off-origin Patterson peak at u = 0, v = 0, w = ½ with 58% of the origin peak height, indicating pseudo-translational symmetry, a plausible molecular-replacement solution was identified for a primitive orthorhombic lattice with a halved unit-cell c repeat (Table 1 ▶, trial 2). However, refinement of the reduced-cell solution and the corresponding full-cell solution (created by duplication according to the pseudo-translation vector) stalled at unacceptably high R and R free values.
Table 1. αCTDM structure-solution trials.
| Trial | Space group | Unit-cell parameters (Å, °) | MR solution LLG (Phaser) | αCTDM protomers per asymmetric unit | Refinement statistics† |
|---|---|---|---|---|---|
| 1 | (P222) | a = 51.3, b = 67.6, c = 116.6 | NA | 4 | NA |
| 2 | P22121 | a = 51.3, b = 67.6, c = 58.3 | 106 | 2 | Rwork = 0.47, Rfree = 0.51 |
| 3 | P21 | a = 51.3, b = 67.6, c = 116.6, β = 90.1 | 4754 | 8 | Rwork = 0.193, Rfree = 0.236 |
R = 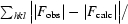
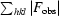 , where F
obs and F
calc are the observed and calculated structure-factor amplitudes, respectively. R
work is for reflections from the working set; R
free was calculated with 4% of the reflections chosen at random and omitted from refinement.
, where F
obs and F
calc are the observed and calculated structure-factor amplitudes, respectively. R
work is for reflections from the working set; R
free was calculated with 4% of the reflections chosen at random and omitted from refinement.
Pseudo-merohedral twinning can occur in monoclinic crystal systems with special unit-cell geometries, including β ≃ 90° (Hamdane et al., 2009 ▶), and in these cases the diffraction patterns of the individual twin domains overlap and the symmetry appears to be that of a higher symmetry space group. To investigate the possibility of pseudo-merohedral twinning, the intensity data were reprocessed as primitive monoclinic, yielding the unit-cell parameters shown in Table 1 ▶, trial 3. The second moment of intensities of acentric reflections, 〈I 2〉/〈I〉2, which is expected to be 2.0 for untwinned data and 1.5 for twinned data (Yeates, 1997 ▶), was 2.35. However, this test can behave anomalously in the presence of anisotropy, pseudo-centering or pseudo-translational symmetry (Padilla & Yeates, 2003 ▶; Brooks et al., 2008 ▶). The L-test, based on deviations in local intensities (Padilla & Yeates, 2003 ▶), yielded results that were consistent with pseudo-merohedral twinning: |L| = 0.43 (expected value of 0.500 for untwinned and 0.375 for a perfect twin) and L 2 = 0.26 (0.333 for untwinned and 0.200 for a perfect twin). The twin law found by phenix.xtriage was h, −k, −l, with an estimated twin fraction of 0.42.
The structure was ultimately solved by molecular replacement in space group P21 using the positioned pair of αCTD protomers obtained in Table 1 ▶, trial 2 as the search model, yielding an initial model with eight independent αCTDM protomers in the crystal asymmetric unit (Table 1 ▶, trial 3).
3.3. Crystal structure
A refined structural model for the twinned monoclinic αCTDM crystal was obtained with good agreement with the observed diffraction data (R factor = 0.193, R free = 0.236; the R free test reflections selected automatically by phenix.refine obey the highest possible symmetry of the twinned lattice, in this case 222). The 2.0 Å resolution structure provides a complete description of non-H atom positions for residues 246–329 for each of the eight αCTDM protomers in the crystal asymmetric unit. NCS restraints were initially applied to all atoms in each protomer; in the final refinement rounds, eightfold NCS was only applied to peptide backbone atoms. Final data-processing and refinement statistics are provided in Table 2 ▶. The main-chain conformation is identical for each protomer over the entire modeled residue range (Fig. 1 ▶ a); the root-mean-square deviation in Cα-atom positions after superposition of chains B–H on chain A ranges from 0.10 to 0.17 Å. Side-chain conformations are also strongly conserved between protomers, with the exception of a few surface-exposed residues. As has been reported previously (Jeon et al., 1995 ▶, 1997 ▶; Gaal et al., 1996 ▶), the domain fold (Fig. 1 ▶ b) includes four α-helices and two short 310-helical turns near the αCTDM N-terminus (Fig. 1 ▶ b, blue cylinders).
Table 2. αCTDM data collection and refinement.
| X-ray source | NSLS X6A |
| Wavelength (Å) | 1.0 |
| Data-collection temperature (K) | 100 |
| Resolution range (Å) | 47.02–2.05 (2.16–2.05) |
| Space group | P21 |
| Unit-cell parameters (Å) | a = 51.34, b = 67.61, c = 116.55, α = 90.0, β = 90.12, γ = 90.0 |
| Matthews coefficient (Å3 Da−1) | 2.63 |
| Solvent content (%) | 53.24 |
| No. of measured reflections | 187784 (26693) |
| No. of unique reflections | 50235 (7266) |
| Completeness (%) | 99.9 (99.8) |
| Redundancy | 3.7 (3.7) |
| Mean I/σ(I) | 12.9 (2.1) |
| Rmerge (%) | 8.2 (54.4) |
| Refinement resolution range (Å) | 47.02–2.05 (2.10–2.05) |
| Rwork† | 0.193 (0.261) |
| Rfree† | 0.237 (0.249) |
| Reflections, working | 50220 (3378) |
| Reflections, free | 2000 (142) |
| αCTD protomers per asymmetric unit | 8 |
| Non-H atoms | 5590 |
| Water molecules | 322 |
| Average B factor (Å2) | 31.4 |
| R.m.s.d. bond lengths‡ (Å) | 0.003 |
| R.m.s.d. bond angles‡ (°) | 0.78 |
| Twinning fraction; operator | 0.437; h, −k, −l |
Definitions provided in Table 1 ▶.
Root-mean-square deviations of bond lengths and bond angles from ideal geometry.
Figure 1.

(a) The eight chains of the αCTDM crystal asymmetric unit are shown superimposed, with main chains represented as ribbons (gray, loop or turn; blue, 310-helix; red, α-helix). Dimethyllysine (mK) residues are also displayed for each chain and are labelled with residue numbers. Regions of αCTD that interact with CAP, DNA and RNAP σ70 are outlined. (b) The αCTD primary sequence is shown with αCTDM secondary-structure elements. Lysine positions are indicated with cyan shading. Positions for which branched side-chain rotamer conformations differ between αCTDM (this work) and the lower resolution CAP–αCTD–DNA complex crystal structure (Benoff et al., 2002 ▶) are indicated by gray shading.
Owing to the near-atomic resolution of the crystal diffraction data, the refined αCTDM model has improved stereochemistry when compared with prior αCTD structure determinations, as shown in Table 3 ▶. In particular, all peptide bonds of the αCTDM model possess favored Ramachandran ϕ/ψ combinations, a result that is typically only attainable at near-atomic resolution or better (Chen et al., 2010 ▶). In addition, the improved resolution permits more reliable side-chain conformation assignments of the branched residues Thr, Val and Leu (Shapovalov & Dunbrack, 2007 ▶). A total of 19 branched side chains have revised conformations in the 2.0 Å structure of αCTDM when compared with the structure determined previously at 3.1 Å resolution (Benoff et al., 2002 ▶). The affected residues are indicated by gray ovals in Fig. 1 ▶(b).
Table 3. Comparison of αCTD models.
| Structure | αCTD | CAP–αCTD–DNA | αCTDM |
|---|---|---|---|
| Reference | Jeon et al. (1995 ▶) | Benoff et al. (2002 ▶) | This work |
| αCTD model residue range | 249–329 | 250–321 | 246–329 |
| Method | NMR | X-ray | X-ray |
| PDB code (chain ID) | 1coo (A) | 1lb2 (B) | 3k4g (A) |
| Cα r.m.s.d.† (Å) | 2.93 | 0.82 | — |
| Ramachandran favored‡ (%) | 78.5 | 75.7 | 100.0 |
| Ramachandran outliers‡ (%) | 1.3 | 1.4 | 0.0 |
| Poor side-chain rotamers‡ (%) | 33.8 | 14.1 | 0.9 |
Root-mean-square deviations of common Cα atoms from αCTDM chain A.
Statistics calculated using MolProbity (Chen et al., 2010 ▶).
3.4. Dimethyllysine residue environments
There are 48 lysine residues in the αCTDM crystal asymmetric unit, six in each αCTDM protomer (Fig. 1 ▶ a, displayed side chains; Fig. 1 ▶ b, cyan ovals). For 39 of the 48 lysine residues electron density is observed for the full side chain, including density consistent with dimethylation of the lysine Nζ amino N atom. The side chains of the remaining nine lysine residues appear to be disordered, with no electron density present beyond the Cβ atom. Observation of dimethylation at every ordered lysine position is a favorable indication that the reductive methylation reaction essentially proceeded to completion.
The majority of ordered dimethyllysine (mK) side chains (31 of 39) participate in either intradomain contacts or interprotomer crystal contacts (Fig. 2 ▶). Residues mK271 and mK304 form intradomain hydrogen-bonded salt bridges with Asp and Glu partners (Figs. 2 ▶ a and 2 ▶ b). The same salt-bridge pairings are present in unmethylated native αCTD (Benoff et al., 2002 ▶). mK298 forms interprotomer salt bridges with Asp/Glu partners (Fig. 2 ▶ c). The mK246 side-chain methylenes participate in interprotomer van der Waals contacts (Fig. 2 ▶ d); mK246 also participates in the crystallographic dimer interface described below.
Figure 2.

Representative dimethyllysine environments in the αCTDM crystal structure. The refined αCTDM model is shown with likelihood-weighted 2|F obs| − |F calc| electron density contoured at 1.5σ. (a, b) Intradomain hydrogen-bonded salt bridges. (a) mK271 partners with Asp258 (N—O distance range 2.8–3.2 Å). (b) mK304 partners with Glu286 (N—O distance range 2.6–3.3 Å). (c, d) Interprotomer crystal contacts. (c) mK298 forms interprotomer salt bridges with the Glu248 and Asp250 side chains. (d) mK246 participates in interprotomer van der Waals contacts with the His276 and Tyr277 side chains as well as in solvent-mediated interprotomer hydrogen bonding. The interactions depicted in (a), (b) and (d) are common to all eight αCTDM protomer copies in the crystal asymmetric unit. The interactions depicted in (c) are common to seven of the eight αCTDM protomer copies.
3.5. αCTDM dimer
The eight αCTDM protomers in the crystal asymmetric unit are arranged within the lattice as two tetramer units with approximate D2 point symmetry (Fig. 3 ▶ a); each tetramer unit consists of a pair of dimers (Fig. 3 ▶ b). The dimer interface is extensive and buries 1335 Å2 or 22% of the total accessible surface area of each protomer; in contrast, the dimer–dimer interface within each tetramer buries 435 Å2 or 7% of the total surface area of each protomer. More than half of the dimer-interface residues are located within the C-terminus (312 and 316–329), with additional residues in the N-terminus (246–254), in the α1–α2 loop (276 and 277) and in helix α2 (280). In the dimer, the pair of C-termini associate with each other, forming a short antiparallel β-sheet with a central Na+ metal-ion-binding site coordinated by two main-chain carbonyl O atoms (residues 320 and 322) in each protomer (Fig. 3 ▶ c).
Figure 3.

αCTDM crystal packing. (a) The content of one complete P21 unit cell (two crystal asymmetric units with eight αCTDM protomers per asymmetric unit) is shown with the unit cell outlined in green. For reference, one protomer is identified with rainbow coloring (blue, N-terminus; red, C-terminus). The pale red arrow indicates the approximate translational symmetry that gives rise to the Patterson map u = 0, v = 0, w = ½ pseudo-translation vector. (b) αCTDM crystallographic dimer; a sodium ion positioned at the dimer interface is shown in pink. (c) Metal-ion-binding site formed by αCTDM C-termini at the center of the crystal dimer. O atoms are colored red; N atoms are colored blue; Trp321–Pro322 cis-peptide bonds are colored cyan.
4. Discussion
Recombinant E. coli RNA polymerase α subunit C-terminal domain (α residues 245–329) is recalcitrant to crystallization in the absence of other macromolecular partners. A truncated fragment, in which the residues that are disordered in the 3.1 Å CAP–αCTD–DNA complex crystal structure of Benoff et al. (2002 ▶) were removed, also did not yield crystals. However, reductive methylation of the full-length αCTD construct yielded crystals of a dimethyllysine derivative that diffracted to 2.0 Å resolution. Methylation increased the propensity of αCTD to form crystals by reducing solubility and by increasing the availability of lysines for the formation of crystal contacts.
Methylated αCTD assembles into dimers in the monoclinic twinned crystal lattice. At present we do not know whether the observed dimer, which is formed nearly entirely by interactions between N- and C-terminal residues, represents a biologically relevant assembly. Blatter et al. (1994 ▶) reported that αCTD prepared by limited proteolysis from whole recombinant α subunit behaved as a dimer during analytical size-exclusion chromatography, but the residues involved were not identified. There is one dimethyllysine residue in the crystallographic dimer interface (mK246; Fig. 2 ▶ d) but the dimethylamino group does not directly participate in interface formation. There would be no stereochemical barrier to prevent native αCTD from forming an essentially identical interface, either as an isolated domain or in the context of intact RNAP enzyme.
Formation of the observed dimer does involve discrete conformations of the αCTD N- and C-termini, including a trans-peptide bond between Asp250 and Pro251, a cis-peptide bond between Trp321 and Pro322 (Fig. 3 ▶ c, cyan bond) and a trans-peptide bond between Pro322 and Pro323. The PBOND server (Exarchos et al., 2009 ▶) predicts a slight preference for the cis conformation (confidence level 0.6) for all three X–Pro peptide bonds based on extended sequence contexts. In the CAP–αCTD–DNA complex described by Benoff and coworkers, electron density for αCTD is only evident for residues 250–322, thus it is probable that some or all of the X–Pro peptide bonds exist as mixed cis–trans conformers in this structure. The long incubation period required for formation of αCTDM crystals (six weeks) supports the hypothesis that slow cis–trans X–Pro conversions accompany the crystallization process. Thermodynamic analysis using the PISA server (Krissinel & Henrick, 2007 ▶) indicates that once formed the dimer is likely to be stable in solution.
Structure determination of methylated αCTD has yielded a revised high-resolution model with significantly improved stereochemistry compared with prior determinations. The new αCTDM coordinates will facilitate the production of improved models of αCTD-containing transcription complexes visualized at low resolution, including subassemblies determined using X-ray crystallography and whole complexes determined using electron-microscopy reconstruction. These structures will be described in future publications by our laboratory.
Supplementary Material
PDB reference: RNA polymerase α subunit C-terminal domain, 3k4g
Acknowledgments
We thank Brian P. Hudson for comments on the manuscript, Helen M. Berman for advice and for providing laboratory space to carry out the work, and Richard H. Ebright for providing the pEBT7-αCTD plasmid and for helpful discussions. NSLS beamline X6A is funded by NIH-NIGMS and NSLS beamline X25 is supported by DOE-OBER and NIH-NCRR. This work was supported by National Institutes of Health GM21589 to CLL.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Au, K., Ren, J., Walter, T. S., Harlos, K., Nettleship, J. E., Owens, R. J., Stuart, D. I. & Esnouf, R. M. (2008). Acta Cryst. F64, 327–333. [DOI] [PMC free article] [PubMed]
- Benoff, B., Yang, H., Lawson, C. L., Parkinson, G., Liu, J., Blatter, E., Ebright, Y. W., Berman, H. M. & Ebright, R. H. (2002). Science, 297, 1562–1566. [DOI] [PubMed]
- Blatter, E. E., Ross, W., Tang, H., Gourse, R. L. & Ebright, R. H. (1994). Cell, 78, 889–896. [DOI] [PubMed]
- Brooks, C. L., Blackler, R. J., Gerstenbruch, S., Kosma, P., Müller-Loennies, S., Brade, H. & Evans, S. V. (2008). Acta Cryst. D64, 1250–1258. [DOI] [PubMed]
- Busby, S. & Ebright, R. H. (1999). J. Mol. Biol.293, 199–213. [DOI] [PubMed]
- Chen, H., Tang, H. & Ebright, R. H. (2003). Mol. Cell, 11, 1621–1633. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Ebright, R. H. & Busby, S. (1995). Curr. Opin. Genet. Dev.5, 197–203. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Exarchos, K. P., Exarchos, T. P., Papaloukas, C., Troganis, A. N. & Fotiadis, D. I. (2009). Genomics Proteomics Bioinformatics, 7, 138–142. [DOI] [PMC free article] [PubMed]
- Gaal, T., Ross, W., Blatter, E. E., Tang, H., Jia, X., Krishnan, V. V., Assa-Munt, N., Ebright, R. H. & Gourse, R. L. (1996). Genes Dev.10, 16–26. [DOI] [PubMed]
- Gourse, R. L., Ross, W. & Gaal, T. (2000). Mol. Microbiol.37, 687–695. [DOI] [PubMed]
- Hamdane, D., Lechauve, C., Marden, M. C. & Golinelli-Pimpaneau, B. (2009). Acta Cryst. D65, 388–392. [DOI] [PubMed]
- Heinig, M. & Frishman, D. (2004). Nucleic Acids Res.32, W500–W502. [DOI] [PMC free article] [PubMed]
- Hsin, K., Sheng, Y., Harding, M. M., Taylor, P. & Walkinshaw, M. D. (2008). J. Appl. Cryst.41, 963–968.
- Hudson, B., Quispe, J., Lara, S., Kim, Y., Berman, H. M., Arnold, E., Ebright, R. E. & Lawson, C. L. (2009). Proc. Natl Acad. Sci. USA, 106, 19830–19835. [DOI] [PMC free article] [PubMed]
- Jeon, Y. H., Negishi, T., Shirakawa, M., Yamazaki, T., Fujita, N., Ishihama, A. & Kyogoku, Y. (1995). Science, 270, 1495–1497. [DOI] [PubMed]
- Jeon, Y. H., Yamazaki, T., Otomo, T., Ishihama, A. & Kyogoku, Y. (1997). J. Mol. Biol.267, 953–962. [DOI] [PubMed]
- Kim, Y. et al. (2008). Nature Methods, 5, 853–854. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol.372, 774–797. [DOI] [PubMed]
- Lawson, C. L., Swigon, D., Murakami, K., Darst, S. A., Berman, H. & Ebright, R. E. (2004). Curr. Opin. Struct. Biol.14, 10–20. [DOI] [PMC free article] [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Luft, J. R., Collins, R. J., Fehrman, N. A., Lauricella, A. M., Veatch, C. K. & DeTitta, G. T. (2003). J. Struct. Biol.142, 170–179. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst.40, 658–674. [DOI] [PMC free article] [PubMed]
- Negishi, T., Fujita, N. & Ishihama, A. (1995). J. Mol. Biol.248, 723–728. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Padilla, J. E. & Yeates, T. O. (2003). Acta Cryst. D59, 1124–1130. [DOI] [PubMed]
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C. & Ferrin, T. E. (2004). J. Comput. Chem.25, 1605–1612. [DOI] [PubMed]
- Rayment, I. (1997). Methods Enzymol.276, 171–179. [PubMed]
- Rayment, I., Rypniewski, W. R., Schmidt-Base, K., Smith, R., Tomchick, D. R., Benning, M. M., Winkelmann, D. A., Wesenberg, G. & Holden, H. M. (1993). Science, 261, 50–58. [DOI] [PubMed]
- Ross, W., Schneider, D. A., Paul, B. J., Mertens, A. & Gourse, R. L. (2003). Genes Dev.17, 1293–1307. [DOI] [PMC free article] [PubMed]
- Schubot, F. D. & Waugh, D. S. (2004). Acta Cryst. D60, 1981–1986. [DOI] [PubMed]
- Shapovalov, M. V. & Dunbrack, R. L. Jr (2007). Proteins, 66, 279–303. [DOI] [PubMed]
- Walter, T. S., Meier, C., Assenberg, R., Au, K. F., Ren, J., Verma, A., Nettleship, J. E., Owens, R. J., Stuart, D. I. & Grimes, J. M. (2006). Structure, 14, 1617–1622. [DOI] [PMC free article] [PubMed]
- Yeates, T. O. (1997). Methods Enzymol.276, 344–357. [PubMed]
- Zhang, G. & Darst, S. A. (1998). Science, 281, 262–266. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: RNA polymerase α subunit C-terminal domain, 3k4g