

Abstract
Breast cancer studies implant human cancer cells under the renal capsule, subcutaneously, or orthotopically and often use estrogen supplementation and immune suppressants (etoposide) in xenograft mouse models. However, cell behavior is significantly impacted by signals from the local microenvironment. Therefore, we investigated how the combinatorial effect of the location of injection and procedural differences affected xenograft characteristics. Patient-derived breast cancer cells were injected into mouse abdominal or thoracic mammary glands ± estrogen and/or etoposide pretreatment. Abdominal xenografts had increased tumor incidence and volume, and decreased latency (P<0.001) compared to thoracic tumors. No statistically significant difference in tumor volume was found in abdominal xenografts treated ± estrogen or etoposide; however, etoposide suppressed tumor volume in thoracic xenografts (P<0.02). The combination of estrogen and etoposide significantly decreased tumor incidence in both sites. In addition, mice treated ± estradiol were injected orthotopically or subcutaneously with well-characterized breast cancer cell lines (MCF7, ZR75-1, MDA MB-231, or MCF10Ca1h). Orthotopic injection increased tumor volume; growth varied with estrogen supplementation. Location also altered methylation status of several breast cancer-related gene promoters. Lastly, vascularization of orthotopic tumors was significantly enhanced compared to subcutaneous tumors. These data suggest that optimal xenograft success occurs with orthotopic abdominal injections and illustrate molecular details of the compelling influence of the local microenvironment on in vivo models.
Keywords: cancer, mammary, mouse models, vascularization, DNA methylation
Introduction
Breast cancer remains a leading cause of cancer related death of women in the U.S. (Jemal et al., 2008) in spite of improvements in early detection and diagnosis, illustrating the importance of developing optimal in vivo models. Debate exists over the ability of mouse models of breast cancer to reflect the progression, metastatic properties and spectrum of tumor morphologies observed in patients (Cardiff, 2001; Kim et al., 2004; Richmond and Su, 2008; Wagner, 2004). While it is naive to presume that one model would emulate the varying forms and features of this non-uniform disease, the knowledge gained from individual models is indisputable. Thus, the continued improvement of models that can support the growth of human cells in a dynamic, living environment remains an important avenue of research.
Difficulties with successful engraftment of human cells in mice are a consequence associated with fundamental differences between the species. In addition to their obvious anatomical variations, there is a dramatic contrast in morphological features such as stromal composition (Fleming et al., 2008; Parmar and Cunha, 2004). In the mouse, the epithelium is embedded within a large amount of fat interspersed with a small amount of connective tissue. In contrast, human breast epithelium is more closely associated with the intra- and inter-lobular stroma, minimizing contact with adipose (Fleming et al., 2008). Therefore, injecting primary breast cancer cells directly into a more fatty mouse tissue presents a challenge for those cells to grow and retain their original pathological features.
Recently, several studies have recapitulated elements of human breast stroma in the mouse mammary gland. Kupperwasser et al. has shown that incorporation of human fibroblasts into the mouse mammary gland is necessary for the successful outgrowth of normal human breast epithelial cells (Kuperwasser et al., 2004). In addition, Hu et al. demonstrated that the development of a ductal carcinoma in situ (DCIS) xenograft model requires the presence of human myoepithelial cells (Hu et al., 2008). Collectively, these results demonstrate the supportive role human stromal cells play to enhance xenograft models, and further our understanding of the progression of breast cancer.
These studies highlight that, despite their limitations, xenograft mouse models provide a living, dynamic system and remain invaluable tools in cancer research. However, they have been performed by an array of procedures, often varying substantially between laboratories. For example, breast cancer cells and/or tissue fragments have been implanted into locations including under the renal capsule, into the omental fat pad, subcutaneously, or orthotopically into one of the five pairs of mouse mammary glands (Miller et al., 1981; Outzen and Custer, 1975; Phillips et al., 1989; Sakakibara et al., 1996; Sheffield and Welsch, 1988; Vorontsova et al., 1989). In addition, treatment with exogenous estrogens and immune suppressants varies between studies, even if they have similar endpoints of interest.
Although orthotopic injection into the abdominal or thoracic gland is a common technique for breast cancer xenograft studies, relatively little is known about the differences between these two glands in the mature mouse. Chemically-induced tumorigenesis in adult mice has been documented to preferentially develop in the thoracic gland, compared to other glands (Prehn et al., 1954; Pullinger, 1952; Vaage, 1984); however, little is known about its mechanism. The thoracic and abdominal mammary glands demonstrate equal amounts of estrogen and progesterone receptors (Haslam and Shyamala, 1979a; Haslam and Shyamala, 1979b). Nevertheless, the thoracic gland is more sensitive to hormonal stimulation in rodents, suggesting a role in the asymmetrical distribution of mammary tumor incidence (Bolander, 1990; McGrath et al., 1972; Russo and Russo, 1988). Whether these observations shown with chemically-induced tumors in the thoracic and abdominal gland correlates with xenograft success remains to be determined.
Herein, we analyzed the combinatorial effects of the location of injection and procedural variations, and identified significant differences in xenograft tumorigenicity as well as resultant tumor characteristics including gene methylation and vascularization. While previous studies reported upon tumor incidence and histology of xenografts, we are the first to identify potential mechanisms that lead to the difference in tumorigenicity and tumor characteristics. Furthermore, to the best of our knowledge, we are the first to report significant differences in the tumorigenicity between the thoracic and abdominal glands when injecting patient-derived breast cancer cells. Together these results have implications for the mechanistic understanding of xenograft success and how xenografts relate to their corresponding human counterpart.
Materials and Methods
Patient samples and generation of xenografts
This study was performed in accordance with the National Cancer Institute Review Board guidelines, protocol 04-C-0199, with written informed consent from all subjects. Estrogen receptor (ER) negative, progesterone receptor (PR) negative pleural effusion cells were collected from two breast cancer patients in order to isolate enough material to perform the experiments. Following collection, cells were pelleted by centrifugation at 835 × g for 15 min at 4°C, washed in Hank’s buffered saline solution, frozen viably in DMSO Freeze media (Invitrogen; Gaithersburg, MD) and stored in liquid nitrogen until used.
Xenografts were established by injection of 1×106 pleural effusion cells into the abdominal mammary gland of eight-week-old female NOD/SCID mice. Once palpable but no greater than 1 cm3, xenografts were removed, minced into < 1 mm pieces, and dissociated (F12 media containing 100 units/ml Collagenase type 3 (Worthington Biochemical Corp, Lakewood, NJ), 0.8 units/ml Dispase (Invitrogen), and 100 units/ml penicillin-streptomycin) at 37°C under rotating conditions for 90 to 120 min. Single cells were generated by an additional incubation in 0.05% trypsin-EDTA for 5 min at 37°C, and then sorted to eliminate the mouse cell population as described below.
Cell staining and sorting via flow cytometry
Cells were washed and resuspended in a PBS solution containing 0.1% fetal bovine serum (Invitrogen) and 100 units/ml penicillin-streptomycin (FACS buffer). Antibodies were then added according to the manufacturer’s recommendations and incubated for 25 min at 4°C. Following staining, cells were washed and then resuspended in FACS buffer. To eliminate the mouse cell population, anti-mouse CD45-FITC (Clone 3-F11 rat monoclonal, BD Biosciences; San Jose, CA), and anti-mouse H-2Kd-FITC (SF1-1.1 mouse monoclonal, BD Biosciences) positive cells were excluded during sorting. Seven-aminoactinomycin D (7AAD, BD Biosciences) was added to the final solution to distinguish live/dead cells. Cell sorting was performed on a BD FACSAria sorter using FACSDiva v6.0 software.
Xenograft cell injection into mice
All animal experiments were conducted in accordance with accepted standards of humane animal care and approved by the Animal Care and Use Committee at the NIH. Eight-week-old female NOD/SCID mice (NCI colony, Animal Production Area (APA)) were anesthetized using i.p. injections of ketamine (750 mg/kg body weight, Phoenix Pharmaceutical Inc.; St. Joseph, MO)/xylazine (50 mg/kg body weight, Lloyd, Inc.; Shenandoah, IA). Six days prior to cell injection, mice were treated ± subcutaneous 17β-estradiol pellet (0.72 mg, 90-day release; Innovative Research of America, Sarasota, FL) and ± the bone marrow suppressant VP-16 (etoposide, a commonly used immune suppressant, 0.6 mg; Calbiochem, Gibbstown, NJ) administered i.p. (Al-Hajj et al., 2003; Meyer et al., 2009; Visonneau et al., 1998). Estradiol pellets were administered, far removed from the site of injection, and each mouse received a xenograft injection at one site. For thoracic xenograft injections, the estradiol pellets were placed on the lower dorsal region of the mouse, close to the hind limb, and for abdominal xenograft injections the pellets were placed in the mid-scapular region of the mouse. Each mouse received a xenograft injection at a single site and no mouse was treated or injected twice.
Sorted cells were suspended in a 4:1 F12 (Invitrogen):Matrigel (BD Biosciencs) mixture. Thirty microliters, containing 500 cells, were injected into either the right abdominal or thoracic mammary gland. For orthotopic injections, mice were surgically incised and cells injected directly into the mammary gland. Transdermal injections were performed by injection through the skin into the gland, without incision. Weekly tumor growth was measured and volume was calculated ((Length*0.5)*(Width*0.5)*(Height*0.5)*(4/3)*3.14). All tumors were excised at the same time, when the largest tumor volume neared 1 cm3. A small piece of each tumor was frozen for DNA extraction and the remainder was fixed in formalin and embedded in paraffin. Hematoxylin and eosin (H&E) stained sections of tumors were compared.
Cell culture
The MCF7, ZR75-1, and MDA MB-231 cell lines were obtained from American Type Culture Collection (Manassas, VA), and maintained according to the repository’s instructions. MCF10Ca1h (kind gift of F.R. Miller, Wayne State University, through L.M. Wakefield, CCR, NCI) were maintained in DMEM/F12 (Invitrogen) supplemented with 5% heat-inactivated horse serum (Gemini BioProducts; W. Sacramento, CA) and 100 units/ml penicillin-streptomycin. Cells were passaged via trypsinization.
Injection of breast cancer cell lines into mice
Eight-week-old female athymic NCr-nu/nu mice (NCI colony, APA) were anesthetized and pretreated as described above. A 25% Matrigel solution containing 2.0×104 MCF7, 5.0×104 ZR-75-1, 1.0×105 MCF10Ca1h, or 1.0×105 MDA MB-231 cells were injected directly into the surgically-exposed abdominal mammary gland (orthotopic) or injected subcutaneously in the dorsal flank region. Tumor growth was monitored and evaluated as described above.
DNA methylation PCR array
Tumor fragments were pooled from two abdominal or two thoracic tumor xenografts derived from the ER−/PR−, HER2+ primary breast cancer cells (Fig. 1), as well as from five orthotopic or five subcutaneous MDA MD-231 tumors xenografts used for array analyses. Genomic DNA was isolated using the Qiagen QIAamp DNA kit according to the manufacturer’s instructions (Valencia, CA). The differentially methylated fractions of DNA were prepared using the Methyl-Profiler Enzyme kit, and DNA digests were analyzed using the Human Breast Cancer Methyl-Profiler DNA Methylation PCR array kits according to the manufacturer’s instructions (SABiosciences; Frederick, MD). The complete list of genes is shown in Supplemental Tables 1 and 2.
Figure 1. Effects of engraftment location, estrogen, and immune suppressant on tumorigenicity of breast cancer cells.
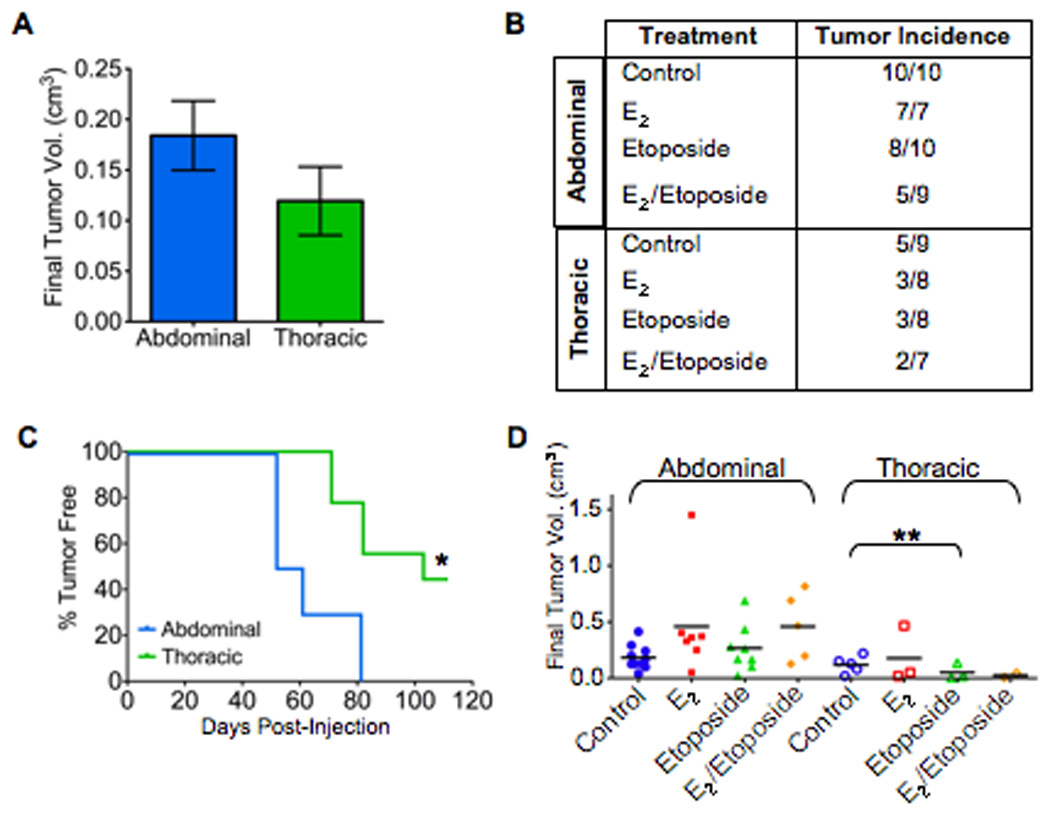
NOD/SCID mice were treated ± 17β-estradiol (E2) and etoposide as indicated. Six days later, mice were surgically incised and injected with 500 cells from a breast cancer pleural effusion in 25% Matrigel. A: final tumor volume. Data represent mean ± SE. B: tumor incidence. C: latency of tumor formation following injection (*P<0.001). Data from one representative experiment, n=5 mice per treatment group. D: final individual tumor volume; bar represents mean (**P=0.02).
Immunohistochemistry, immuofluorescence and confocal microscopy
All reagents were obtained from Sigma (St. Louis, MO) unless otherwise indicated. Five-micron-thick sections of formalin fixed, paraffin embedded tissue were prepared from all tumors. Histosections were de-paraffinized, re-hydrated, and then subjected to antigen retrieval using the DAKO citrate retrieval buffer (DAKO; Carpinteria, CA) as instructed. Endogenous peroxidase activity was blocked by a 10 min incubation in 3% hydrogen peroxide followed by PBS washes. Immunostaining was carried out using the Vectastain ABC kit (Vector Laboratories; Burlingame, CA) according to the manufacturer's instruction. The polyclonal Ki67 antibody was obtained from Novus Biologicals (catalogue number NB110-90593; Littleton, CO). Color was developed with diaminobenzidine peroxidase substrate kit (Vector), and counterstained with hematoxylin. Histosections were analyzed by manually counting cell nuclei positive for Ki67 staining; a minimum of three fields (40×) for each xenograft tumor was counted and the mean count per field calculated.
For immuofluorescence, freshly isolated tumor fragments were fixed in 4% paraformaldehyde overnight at 4°C, embedded in Tissue-Tek OCT compound, and stored at −80°C until used. Five to ten-micron-thick sections were prepared from all tumors. Histosections were blocked in 1× PBS containing 10% goat serum and 2% BSA (blocking buffer) for 30 min at room temperature, and then incubated with CD31 antibody (pre-diluted rabbit polyclonal, ab28365; Abcam, Cambridge MA) in 2% BSA and 2% goat serum for one hr at room temperature. Each slide was subsequently washed with 1× PBS and then incubated with anti-rabbit Alexa Fluor 488 (Invitrogen) secondary antibody at a 1:1000 dilution in blocking buffer for 30 min at room temperature. Coverslips were mounted using Prolong Gold antifade reagent with DAPI (Invitrogen). Imaging was performed using the Carl Zeiss LSM510 confocal imaging system (Carl Zeiss MicroImaging, Thornwood, NY). For immunohistochemistry and immuofluorescence experiments, a serial section of each sample that received all staining steps, but no primary antibody, was used as a negative control.
For detection of apoptosis, five-micron-thick sections of formalin fixed, paraffin embedded tissue were prepared from all tumors. Histosections were de-paraffinized, re-hydrated, and treated according to the Apoptag Plus Fluorescein In Situ Apoptosis Detection kit (Chemicon International, Billerica, MA). Histosections were imaged as described for immuofluorescence and the mean fluorescence intensity was calculated using Adobe Photoshop (Adobe Systems Inc., Beaverton, OR). Briefly, a minimum of three separate images from each tumor histosection were imaged and analyzed using the histogram function through the green channel, which gave the mean green intensity of the selected section. Photoshop assigns intensity values between 0 and 255 to each pixel in the selected area and then averages these intensities. The average mean intensity was calculated for each tumor, followed by a calculation to determine the mean of each treatment group.
For detection of tumor-associated blood vessels, mice were i.v. injected with 100 µl of saline containing 50 mg/ml of FITC-labeled dextran (MW 2×106; Sigma Aldrich, St. Louis, MO) and then euthanized. Tumors were excised, immediately fixed in 4% paraformaldehyde overnight at 4°C, and embedded in Tissue-Tek OCT compound. Forty-micron thick slices were cryo-sectioned from multiple regions of the periphery and center of each tumor. Five fields were visualized for each and imaged using the Carl Zeiss LSM510 confocal imaging system (Carl Zeiss MicroImaging, Thornwood, NY) at 63× magnification.
Identification of the matrix-associated vascular channels representative of vascular mimicry in xenografts was performed (Maniotis et al., 1999) with Periodic acid-Schiff (PAS); black and white photography with a green filter further highlighted the PAS-positive patterns.
Western blot analysis
Tumors were mechanically homogenized in lysis buffer (50 mM Tris-HCl pH 7.4, 1.0% NP-40, 0.25% Na-Deoxycholate, 150 mM NaCl, 1mM EDTA, 1 mM PMSF, 1mM Na3VO4) containing 1× Halt™ Protease and Phosphatase inhibitor solution (Thermo Scientific, Rockford, IL). Lysates were centrifuged at 12,000 × g for 15 min at 4°C, and total protein content of the cytosolic fraction was determined using a protein assay (Bio-Rad Laboratories, Hercules, CA). Equal amounts of protein (75 µg) were separated by electrophoresis through a 10% SDS-PAGE gel under reducing conditions. Membranes were blocked in 5% nonfat dried milk for 1 h at room temperature, incubated with primary antibody overnight at 4°C, washed, and incubated with the appropriate secondary antibody conjugated to horseradish peroxidase (Amersham, Piscataway, NJ). After washing, peroxidase activity was detected using the enhanced chemiluminescence detection system (ECL Plus; Amersham) according to the manufacturer’s recommendations.
Statistical analyses
When appropriate, data were evaluated for significance via two tailed t-tests or two-way ANOVA with Bonferroni multiple comparisons post hoc analysis, using GraphPad InStat Software version 3.0b and GraphPad Prism version 5.0b (San Diego, CA). Data was considered significant at P<0.05.
Results
Primary breast cancer cell tumorigenicity is differentially altered by location, estrogen, and immune suppressants
Position-specific growth factor requirements vary between the thoracic and abdominal glands during mammary development (Garcia-Gasca and Spyropoulos, 2000; Hatsell and Cowin, 2006; Mailleux et al., 2002). Whether these embryological divergences result in differences in xenograft success in the adult gland remains to be determined. Therefore, we directly compared incidence, latency, and size of tumors grown in the abdominal or thoracic glands to evaluate potential site-specific differences since both are used routinely as injection sites. Two independent experiments were carried out using primary breast cancer cells obtained from two pleural effusions from separate ER−/PR− metastatic breast cancer patients. In these experiments, the abdominal region was surgically-exposed and 500 cells were injected directly into the orthotopic site (Supplementary Fig. S1).
Increased cell engraftment and growth was observed in the abdominal compared to the thoracic glands for both patient samples tested. Although average final tumor volume was greater in abdominal glands (0.18 cm3 versus 0.12 cm3, Fig. 1A), most noteworthy was the dramatic difference in tumor incidence and latency observed between the glands. While all mice receiving abdominal gland injections developed tumors, only 56% developed tumors when injected into the thoracic glands (10/10 versus 5/9, Fig. 1B). Additionally, a significant decrease in tumor latency was observed in abdominal glands compared to thoracic glands (P<0.001, Fig. 1C). Even more dramatic results were obtained when using the highly immune-compromised NOD/SCID interleukin-2 receptor gamma chain null mice. No mice developed tumors with thoracic compared with abdominal injection (Supplemental Fig. S2). The latency period was similar to that seen in NOD/SCID mice (68 vs. 60 days).
The use of etoposide to further suppress the mouse immune system has been historically used in breast cancer xenograft models (Visonneau et al., 1998), and was recently highlighted in the influential paper by Al-Hajj et al. describing the selection of a tumorigenic subpopulation of cancer cell from the bulk xenograft population. Etoposide administered as a single injection prior to xenograft cell injection has been subsequently used in other xenograft studies (Al-Hajj et al., 2003; Grimshaw et al., 2008; Meyer et al., 2009) and is a common practice to enhance cell engraftment. In addition, estrogen supplementation was used for both ER+ and ER− xenografts (Al-Hajj et al., 2003; Meyer et al., 2009), and was reported to be necessary for xenograft tumor formation using ER− human breast cancer cell lines (Gupta et al., 2007). Therefore, to evaluate the impact of estrogen and immune suppressants on cell engraftment and growth, subcutaneous slow-release pellets of 17β-estradiol (E2) and/or a single injection of etoposide were administered six days prior to cell implantation.
A non-significant increase in tumor size was observed in both glands with E2 treatment compared to controls (Fig. 1D). Within the abdominal gland group, pre-treatment with etoposide alone or in combination with E2 also had no significant impact on tumor size (Fig. 1D). In contrast, etoposide treatment in the thoracic gland demonstrated a suppressive effect, as indicated by decreased tumor incidence and size (P=0.02, Fig. 1B, D). When etoposide was used in combination with E2 in the thoracic glands, tumor growth decreased even further; however, low tumor incidence in this group did not permit statistical analysis. These observations suggest that E2 was unable to rescue cell growth from etoposide’s suppressive effect in the thoracic gland. Treatment with E2 or etoposide had no significant effect on tumor latency for any treatment.
The tumor volume is a reflection of the number of cells actively undergoing both proliferation and apoptosis. Therefore, to further investigate the xenograft characteristics, we analyzed their proliferative and apoptotic indices. A large variation in the proliferative and apoptotic indexes was observed in all treatment groups (Supplemental Fig. 3). At the termination of the study, no statistically significant difference in apoptosis was observed between treatment groups (Supplemental Fig. 3A). Additionally, no significant difference was observed between the control groups in the abdominal and thoracic glands when Ki67 was used as a cellular marker of proliferation (Supplemental Fig. 3B). E2, etoposide, or the combination also had no significant effect on the proliferative index of tumors growing in the abdominal gland. However, tumors grown in the thoracic gland had significantly lower amount of proliferation in the presence E2 alone, which corresponds with their lower tumor incidence and final tumor volume (P<0.05). It should be noted that these results were obtained at the termination of the experiment, when tumors needed to be excised due to the large size of some abdominal tumors. This data gives no information about the proliferation and apoptosis occurring during the initial stages of tumor growth, but only a snapshot of the status of the tumors at the end of the study.
Overall, supplementation with E2 had variable effects on the tumorigenicity of ER−/PR− pleural effusion cells; however, the observed suppressive effects of etoposide in the thoracic gland suggest that this pre-treatment may be detrimental in some xenograft models. Furthermore, these results suggest the abdominal gland may be comparatively more supportive to xenograft growth and differentially affected by pre-treatment with E2 and etoposide.
Tumorigenicity is unaltered by degree of procedural invasiveness
While invasive surgical procedures may stimulate an inflammation and wound healing response, one is more confident by injecting breast cancer cells directly into the exposed mammary gland. In contrast, transdermal injection eliminates surgery, potentially minimizing any type of inflammatory response, but is difficult to perform with accuracy. To evaluate the effects of invasive surgical procedures on tumorigenicity, we directly compared orthotopic and transdermal methods of injection. In this experiment, abdominal glands were utilized for both injection methods in order to minimize variation and exploit the observed increased tumor incidence (Fig. 1B). The previously described ER−/PR−, HER2− primary breast cancer cells were injected. Tumor incidence was similar for both procedures (5/5 versus 4/5, Fig. 2A), as was latency, with tumors detectable 49 days after injection (Fig. 2B). Tumor volume increased proportionally between both groups over 110 days, with similar final tumor volume for both injection methods (Fig. 2C). In summary, the more invasive surgical procedure and any associated immune response did not alter the overall tumorigenicity of the primary cells.
Figure 2. Method of cell delivery does not affect tumorigenicity.
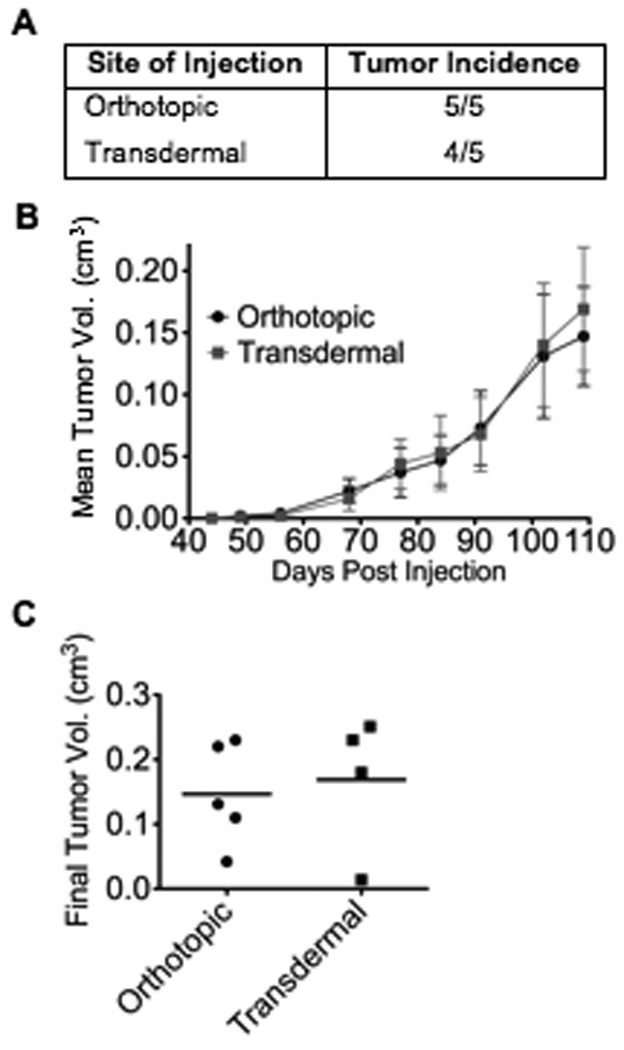
NOD/SCID mice were injected into the abdominal mammary gland with 500 cells from a breast cancer pleural effusion in 25% Matrigel. For orthotopic injections, mice were surgically incised and cells were injected directly into the gland. Transdermal injections were performed by injection through the skin into gland, without incision. A: tumor incidence. B: latency of tumor formation (n=5 mice per treatment group). Data represents mean ± SE. C: final individual tumor volume of individual mice; bar represents mean.
Location affects tumorigenicity of ER+/PR+ human breast cancer cell lines
In addition to direct mammary gland injection of breast cancer cells, another common method of xenotransplantation is a subcutaneous injection usually in a remote, dorsal location. To delineate any potential growth advantage, we compared subcutaneous and orthotopic sites of injection using ER+/PR+ MCF7 and ZR75-1 human breast cancer cell lines, chosen to avoid confounding effects of uncharacterized, primary breast cancer cells. Orthotopic injections were performed in the abdominal gland while subcutaneous injections were administered in the dorsal-flank region. E2 pellets were inserted mid-scapula, far removed from either injection sites.
Tumor incidence was higher when MCF7 cells were injected orthotopically compared to subcutaneously (8/10 versus 5/10, Fig. 3A). Tumor latency was similar for both injection sites, although a numeric increase in final tumor volume was observed in the orthotopic injection group (Fig. 3B). Using ZR75-1 cells, palpable tumors also developed concurrently in both injection sites (Fig. 3C); however, a significant increase in final tumor volume was observed in the orthotopic injection group (P<0.01, Fig. 3C). Tumor incidence was similar for both groups (8/10 versus 8/10, Fig. 3A). Overall, tumorigenicity of the ER+/PR+ cell lines was impacted by the divergent locations. Specifically, tumor incidence, latency, and volume using the orthotopic method were consistently equal to or greater than that observed using subcutaneous injection, suggesting a growth advantage in the orthotopic site.
Figure 3. Effects of injection site on the tumorigenicity of ER+/PR+ human breast cancer cell lines.
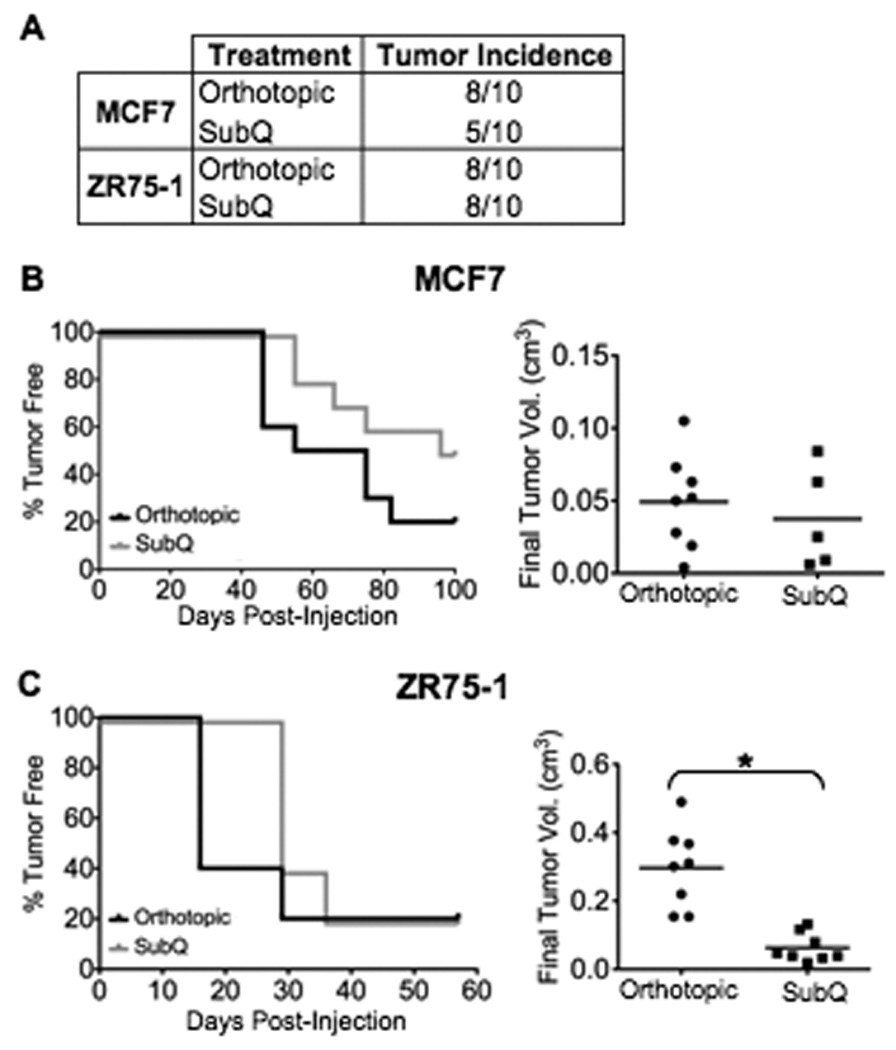
Athymic mice were treated ± 17β-estradiol six days prior to cell implantation. Mice were either surgically incised and cells injected directly into the abdominal mammary gland (Orthotopic) or injected subcutaneously (SubQ) in the dorsal flank region with either 2.0×104 MCF7 or 5.0×104 ZR75-1 cells in 25% Matrigel. A: Tumor incidence. B&C: Graphs are one representative experiment showing latency of tumor formation (n=5 mice per treatment group) for the (B) MCF7 and (C) ZR75-1 cell lines. Histograms represent final tumor volume of individual mice from all experiments; bar represents mean tumor volume (*P<0.01).
Location affects tumorigenicity of ER−/PR− human breast cancer cell lines
To further examine the effects of location and estrogen supplementation in a breast xenograft model, the ER−/PR− MDA MB-231 and MCF10Ca1h human breast cancer cell lines were also subcutaneously and orthotopically injected. Tumor latency did not vary substantially between procedural methods for either cell line (Supplemental Fig. S4). While orthotopic final tumor volume was significantly greater compared to subcutaneous final tumor volume for both cells (Fig. 4), tumor incidence and the affect of estrogen varied depending on the ER−/PR− cell line used. Using MDA MB-231 cells, subcutaneously grown xenografts had substantially reduced tumor incidence compared to orthotopic xenografts (Fig. 4A; 7/14 vs. 13/15). However, estrogen treatment appeared to enhance subcutaneous xenograft success and raise tumor incidence equal to that of the orthotopically grown tumors. Estrogen had no significant effect on orthotopically grown tumors.
Figure 4. Effects of estrogen and injection site on the tumorigenicity of ER−/PR− human breast cancer cell lines.
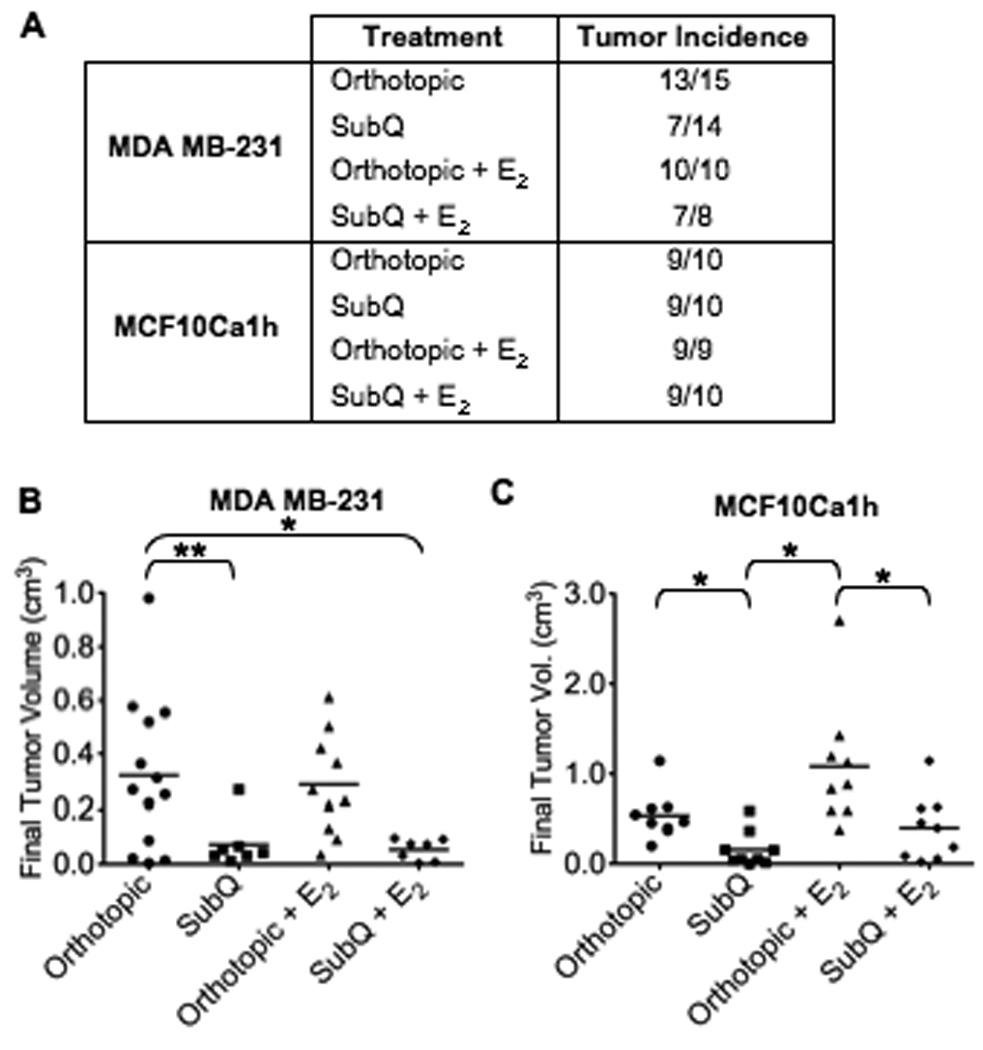
Either 1.0×104 MDA MB231 or 1.0×105 MCF10Ca1h cells in 25% Matrigel were injected into athymic mice as described in the legend to Figure 3. A: Tumor incidence. B&C: Histograms represent final individual tumor volume from all experiments for the (B) MDA MB-231 and (C) MCF10Ca1h cell lines; bar represents mean tumor volume (*P<0.01, **P<0.02).
Similar results were observed using MCF10Ca1h cells with a significantly larger final tumor volume in orthotopic compared to subcutaneous injections (P<0.01, Fig. 4C). In contrast to MDA MB-231 cells, estrogen-supplemented MCF10Ca1h groups had an increase in mean tumor size compared to their corresponding controls. Notably, final tumor volume of MCF10Ca1h tumors grown in the orthotopic site without estrogen supplementation was not statistically different from the final tumor volume of the subcutaneously grown tumors with estrogen supplementation, suggesting different regulatory effects of the location.
The proliferative and apoptotic indices were also analyzed in MDA MB-231 xenograft tumors. At the termination of the study, no statistically significant difference in apoptosis was observed between treatment groups (Fig. 5A). The orthotopically grown tumors, with or without estrogen treatment, showed significant differences in proliferative indices compared to subcutaneously grown tumors when Ki67 was used as a cellular marker of proliferation (Fig. 5B; P<0.05). The majority of the cell proliferation occurred at the periphery of the tumors, in all treatment groups, with minimal amounts of Ki67 cells found within the core of the tumors (Fig. 5C). Additionally, there was no significant difference in the proliferative indices of the tumors grown subcutaneously with estrogen compared to orthotopic tumors without estrogen treatment. This raises the possibility that the estrogen-treated subcutaneously grown tumors may have increased in size, equal to those of the orthotopically grown tumors if more time was permitted; however, the study had to be terminated for humane reasons due to the large size of several orthotopically grown tumors.
Figure 5. Proliferative and apoptotic indices of MDA MB231 xenograft tumors.
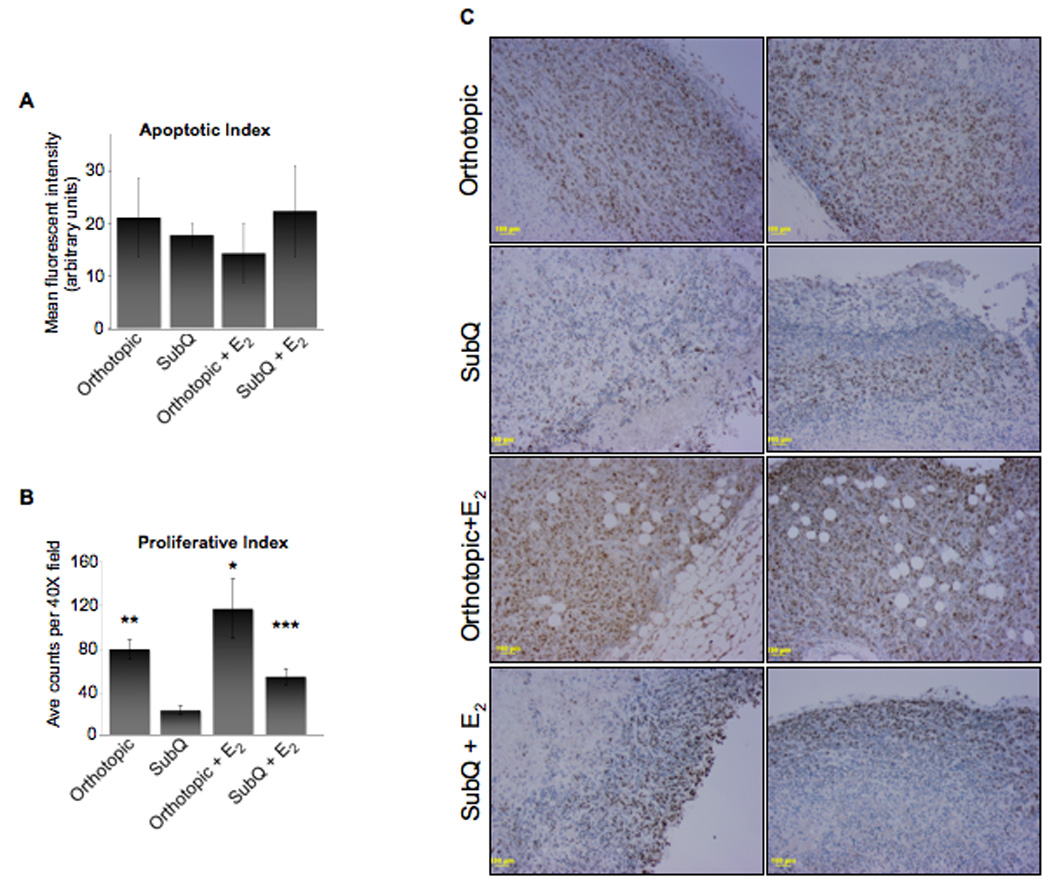
MDA MB 231 cells (1.0×104) in 25% Matrigel were injected into athymic mice as described in the legend to Figure 3. A: Histogram representing the mean fluorescence intensity of positively stained apoptotic cells. A minimum of three separate images from each tumor histosection (20×) was imaged, analyzed, and means calculated. Subsequently, the mean for each treatment was calculated, ± SD. B: Histogram represents the average number of cells positively stained for Ki67 (40×); a minimum of 3 fields was counted for each xenograft tumor. The mean for each tumor was calculated and, subsequently, the mean for each treatment was calculated, ± SD (*P<0.05 above subQ). C: Panels are two representative histosections of xenograft tumors for each treatment group, stained for Ki67. Images taken at 10×; scale bar = 100 µm.
Although tumor growth in the orthotopic versus subcutaneous site varied between cell lines, subcutaneous injections never had the advantage regardless of estrogen supplementation. Growth in the subcutaneous site was either equal to, or less than, that observed in the orthotopic site for each cell line used. These results demonstrate that, similar to results observed with the primary breast cancer cell, tumorigenicity is improved when injecting cells into the orthotopic site.
Location changes DNA methylation patterns of gene promoters
Alterations in DNA methylation patterns are associated with the development of a variety of human cancers, including breast cancer (Jones and Baylin, 2002). Therefore, the methylation status of 24 gene promoters with reported alterations in a variety of breast cancers was investigated via quantative real-time PCR arrays (Genes listed in Supplemental Tables 1 and 2). Genomic DNA was isolated from abdominal and thoracic tumor xenografts derived from the previously examined ER−/PR−, HER2+ primary breast cancer cells (Fig. 1). Analysis revealed that four gene promoters had ~10% change in the hypermethylation status between the thoracic and abdominal tumors (Table 1). The genes encoding ERα1, TRAIL-R4, and p73 had an increased percentage of promoter hypermethylation in abdominal tumors, while Cox2 exhibited an increase in promoter hypermethylation in thoracic tumors. Significantly greater differences in the methylation patterns of breast-cancer associated genes were observed between tumors grown orthotopically compared to subcutaneously using the MDA MB-231 and ZR75-1 cells (Tables 2 & 3); a greater than 10% change from hyper- to un-methylated was observed in over 14 genes, with some exhibiting as high as a 50% change. Promoters for the genes ADAM metallopeptidase domain 2, Cyclin D1, and O-6-methylguanine-DNA methyltransferase were more highly hypermethylated in subcutaneous tumors, while stratifin demonstrated higher methylation in orthotopic tumors from both cell lines tested. Collectively, the observed divergence in methylation status of all xenograft tumors may have arisen from clonal selection via microenvironmental pressures, direct influence of the microenvironment on the cells, or a combination of the two.
Table 1.
Promoter methylation status of genes in breast cancer xenograft tumors
| Abdominal | Thoracic | |||||
|---|---|---|---|---|---|---|
| Gene | Symbol | Gene ID | %HM | %UM | %HM | %UM |
| ERα | ESR1 | 2099 | 21.7 | 78.3 | 11.5 | 88.5 |
| COX2 | PTGS2 | 469616 | 50.0 | 50.0 | 59.0 | 41.0 |
| TRAIL-4R | TNFRS10D | 8793 | 92.9 | 7.1 | 82.9 | 17.1 |
| P73 | TP73 | 7161 | 90.0 | 10.0 | 81.6 | 18.4 |
HM = hypermethylated; UM = unmethylated, IM = intermediately methylation
Table 2.
Promoter methylation status of genes in MDA MB-231 xenograft tumors
| Orthotopic | Subcutaneous | ||||||
|---|---|---|---|---|---|---|---|
| Gene | Symbol | %HM | %UM | %IM | %HM | %UM | %IM |
| ADAM metallopeptidase domain 2 | ADAM23 | 42.2 | 57.8 | 0.0 | 83.8 | 16.2 | 0.0 |
| Cyclin D2 | CCND2 | 40.5 | 59.5 | 0.0 | 95.3 | 4.7 | 0.0 |
| E-cadherin | CDH1 | 48.3 | 17.9 | 33.8 | 26.2 | 12.9 | 60.9 |
| H-cadherin | CDH13 | 95.5 | 5.5 | 0.0 | 55.5 | 0.4 | 44.1 |
| Estrogen receptor alpha 1 | ESR1 | 86.8 | 13.3 | 0.0 | 96.4 | 3.6 | 0.0 |
| O-6-methlyguanine-DNA methyltransferase | MGMT | 40.2 | 59.8 | 0.0 | 91.6 | 8.4 | 0.0 |
| PYD and CARD domain containing | PYCARD | 32.6 | 7.1 | 60.3 | 14.3 | 0.8 | 85.0 |
| Ras associated domain family member 1 | RASSF1 | 84.0 | 16.0 | 0.0 | 94.5 | 5.5 | 0.0 |
| Stratifin | SFN | 31.1 | 56.1 | 12.8 | 12.3 | 56.8 | 30.9 |
| Tumor necrosis factor receptor superfamily | TNFRSF10C | 88.0 | 12.0 | 0.0 | 53.7 | 1.8 | 44.5 |
HM = hypermethylated; UM = unmethylated, IM = intermediately methylation
Table 3.
Promoter methylation status of genes in ZR75-1 xenograft tumors
| Orthotopic | Subcutaneous | ||||||
|---|---|---|---|---|---|---|---|
| Gene | Symbol | %HM | %UM | %IM | %HM | %UM | %IM |
| ADAM metallopeptidase domain 2 | ADAM23 | 50.0 | 50.0 | 0.0 | 59.1 | 40.9 | 0.0 |
| Cyclin D2 | CCND2 | 83.6 | 16.4 | 0.0 | 91.2 | 8.8 | 0.0 |
| Cyclin-dependent kinase inhibitor 2A | CDKN1C | 23.0 | 77.0 | 0.0 | 16.2 | 83.8 | 0.0 |
| O-6-methlyguanine-DNA methyltransferase | MGMT | 75.9 | 24.1 | 0.0 | 89.6 | 10.4 | 0.0 |
| Stratifin | SFN | 15.3 | 84.7 | 0.0 | 4.6 | 95.4 | 0.0 |
HM = hypermethylated; UM = unmethylated, IM = intermediately methylation
Xenografts grown orthotopically exhibit enhanced vascularization
One striking observation when excising the tumors was the distinct lack of blood vessels in the subcutaneous tumors (Fig. 6A), suggesting that the increased size of orthotopic tumors could be due to enhanced vasculature. To visualize the vessels, FITC-dextran was injected i.v. into mice with palpable tumors, the tumors excised, and blood vessels imaged (Fig. 6). Histosections from ER−/PR− MDA MB-231 and ER+/PR+ ZR75-1 derived-tumors were also immunohistochemically analyzed with the endothelial marker CD31 (Supplemental Fig. S5). Orthotopic xenografts had more blood vessels, regardless of cell type (Fig. 6A); the vessel diameter was also larger and more heavily branched (Fig. 6B, C). This suggests that either tumors from the orthotopic site are more efficient at neoangiogenesis, or there is already a well-established network of blood vessels in the mammary gland that the tumors utilize.
Figure 6. Differences in the vascularization between orthotopically and subcutaneously grown xenograft tumors.
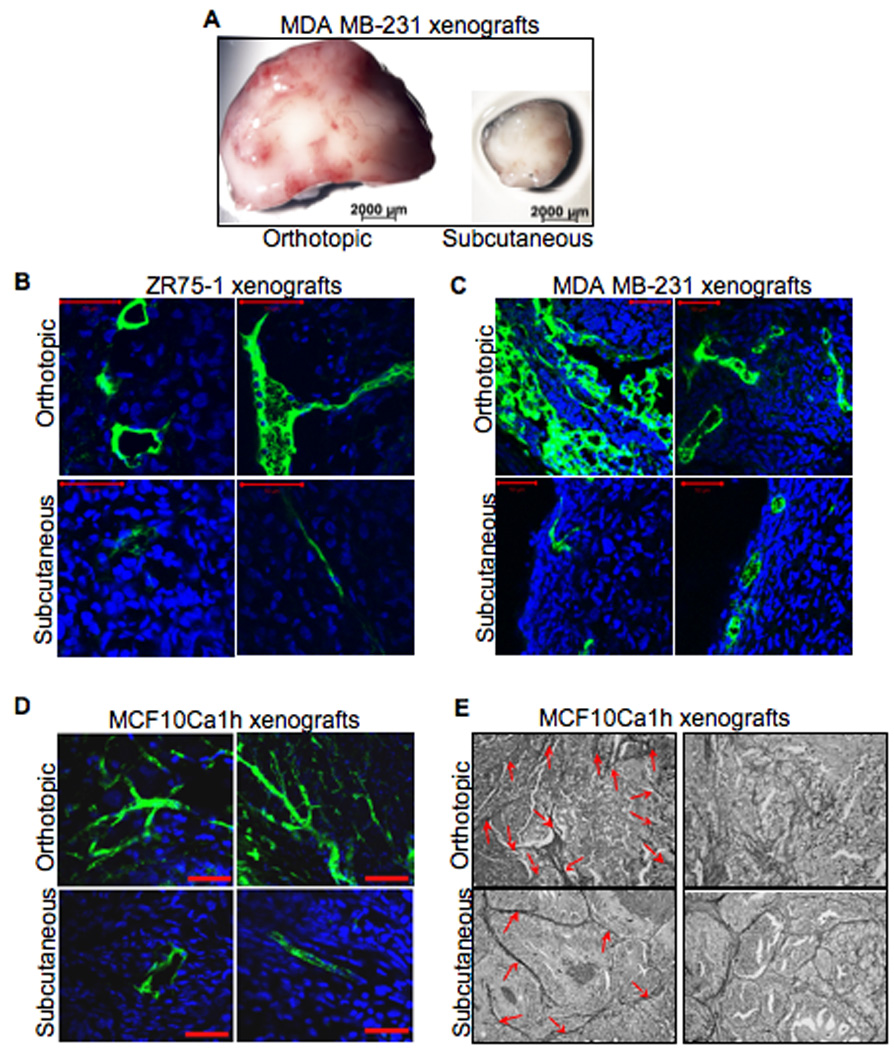
Mice were i.v. injected with FITC-dextran; the tumors were excised, immediately fixed and imaged (n=5 per treatment group). A: representative whole xenograft tumors. B – D: representative images of FITC-dextran stained blood vessels (20 – 40 µm sections) of tumor xenografts. Scale bar = 200 µm. E: representative images of Periodic acid-Schiff (PAS) stained xenograft tumor sections (20×; n=5 per treatment group). Red arrows denote the stained vascular channels.
Tumor cells are also able to supply blood and nutrients by vascular mimicry, whereby tumor cells express endothelium-associated genes and form extracellular matrix-rich vasculogenic-like networks (Maniotis et al., 1999). To visualize whether there were differences in vascular mimicry between the two sites, MCF10Ca1h tumor tissue sections were Periodic acid-Schiff (PAS) stained and imaged. Similar to the MDA MB-231 and ZR75-1 cells, MCF10Ca1h derived xenografts grown orthotopically had increased vascularization compared to subcutaneously grown tumors (Fig. 6D). Moreover, the orthotopic xenografts had increased PAS staining of the hallmark tube-like networks of vascular mimicry (Fig. 6E), suggesting yet another mechanism by which the orthotopic xenografts exhibited increased tumorigenicity.
Discussion
Recent reports have eloquently illustrated the importance of the local environment on the development and progression of cancer (Booth et al., 2008; Boulanger et al., 2007; Hu et al., 2008; Kuperwasser et al., 2004; Quintana et al., 2008); therefore, we hypothesized that experimental variations in cancer xenograft models would affect tumor characteristics. Indeed, we show that the location of xenograft injection had a profound effect on both tumorigenicity and characteristics of the resultant tumor. Although previous reports investigated injection site, strain, and estrogen and etoposide on the tumorigenicity of cancer cell lines and primary tissue in mice (Laidlaw et al., 1995; Miller et al., 1981; Osborne et al., 1985; Outzen and Custer, 1975; Phillips et al., 1989; Sakakibara et al., 1996; Sheffield and Welsch, 1988; Taghian et al., 1993; Visonneau et al., 1998; Xie et al., 1992), we are the first to observe factors such as vascularity and promoter methylation, which suggest potential mechanisms that cause these diverse tumor characteristics and lays the groundwork for future mechanistic studies. To the best of our knowledge, we are also the first to report significant differences in the tumorigenicity between the thoracic and abdominal glands in xenograft models. Furthermore, this report highlights the importance of choosing an appropriate model when initiating experiments or building upon a previous study, as well as how results relate to one another.
Seemingly diminutive procedural differences such as injection into the thoracic versus abdominal mammary gland had dramatic effects on xenograft success and behavior. The finite molecular differences between the thoracic and abdominal mammary glands have not been extensively investigated in the adult murine gland. We found, however, that xenograft tumor incidence and growth was superior in the abdominal compared to the thoracic gland. The molecular composition and robustness to alterations in the normal signaling of the adult glands may be one explanation for the observed differences in xenograft success, as alterations in gene function in the embryonic mammary gland differentially affects thoracic and abdominal gland development. For example, mouse embryos mutated for FGF10 or FGFR2b only form the abdominal mammary placodes, and fail to form all other mammary placodes (Mailleux et al., 2002). Additionally, embryos with a deletion of Gli3, a mediator of Hedgehog signaling, completely fail to develop the thoracic glands (Hatsell and Cowin, 2006). Adult female mice with disruption of the HOXC6 homeobox gene possess thoracic mammary glands that are completely devoid of epithelium; in contrast, the epithelial ducts within the abdominal glands are significantly enlarged and fail to regress in response to ovariectomy (Garcia-Gasca and Spyropoulos, 2000). Collectively, embryonic studies suggest that there is a higher susceptibility in the thoracic glands for developmental failure and a relatively higher resistance for loss of gene function in the abdominal glands. This divergence in growth factor requirement may be translated into differences within the adult glands and potentially reflected in xenograft tumorigenicity. In rats, administration of 1-methyl-1-nitrosourea for the induction of mammary carcinomas results in differences between the thoracic and abdominal glands (Thompson and Meeker, 1983). The thoracic glands develop 2.4 times more carcinomas than in the abdominal glands, suggesting fundamental differences in the glands across rodent species.
Variations in promoter methylation patterns of genes known to be differentially methylated in breast cancer were observed between thoracic and abdominal tumors derived from primary metastatic breast cancer cells. TRAIL-R4, a member of the TNF-receptor superfamily known to play an essential role in TNFR1- and Fas-mediated apoptosis (Tomkova et al., 2008), and p73, a member of the p53 family of transcription factors known to function as a tumor suppressor (Seitz et al., 2002), were both found to have an increased percentage of promoter hypermethylation in abdominal xenograft tumors. Altered cell signaling due to inhibition of the expression of these two genes via promoter hypermethylation may have contributed to the increase in tumorigenicity of the cells grown in the abdominal gland. Conversely, increased hypermethylation of the COX2 promoter in thoracic tumors may have contributed to the decreased thoracic tumor incidence, latency and final volume. COX2, a prostaglandin-endoperoxide synthase, is a key enzyme in the biosynthesis of prostaglandin, and expression in breast cancer has been associated with large tumor size, high histological grade, negative hormone receptor status, high proliferation rate, and ductal type of histology (Singh-Ranger et al., 2008). Although thoracic xenografts retained negative hormone status, consistent with other characteristics of loss of COX2 expression, thoracic tumors were smaller than the abdominal tumors, which did not exhibit COX2 hypermethylation.
Four gene promoters were found to have greater than ~10 to 50% change in methylation status between the orthotopic and subcutaneous sites in both the MDA MB-231 and ZR75-1 xenograft tumors. Stratifin, a known inhibitor of AKT mediated growth, demonstrated increased promoter hypermethylation in the orthotopic tumors compared to subcutaneous tumors (Umbricht et al., 2001). This inhibition of AKT suppression may have enhanced the tumorigenicity of the cells in the orthotopic site, as AKT activation stimulates cell proliferation and survival. Conversely, the promoters for Cyclin D2, ADAM23, and MGMT had comparatively higher hypermethylation in the thoracic tumors derived from either cell line. These genes are involved in positive regulation of G1 progression, cell-to-cell adhesion, and cellular DNA repair, respectively (Costa et al., 2004; Esteller et al., 1999; Evron et al., 2001). These selective alterations in promoter methylation may have contributed to the resultant xenograft characteristics.
These results in promoter methylation may have arisen from the clonal selection of a particular cell and/or from signals within the local microenvironment. The observed methylation changes may provide a cell growth/survival advantage within a specific niche. In the present study, E-cadherin demonstrated a 22% increase in promoter hypermethylation in the orthotopically grown MDA MD-231 tumors. Consistent with these results, the E-cadherin promoter was shown to dynamically shift its methylation status in relation to the microenvironment in cultured breast cancer cells (Graff et al., 2000). During in vitro invasion, the density of promoter methylation of E-cadherin markedly increases and E-cadherin expression decreases. Conversely, when cells are cultured as spheroids, which require homotypic cell adhesion, promoter methylation decreases dramatically and E-cadherin is re-expressed. Collectively, these data suggest that methylation of cancer-associated genes exhibit plasticity that may contribute to the dynamic, phenotypic heterogeneity, which is necessary for successful xenograft growth.
Whether estrogen plays a systemic or local stromal effect and whether this effect is necessary for successful xenograft transplantation has been debated. Our results demonstrated that pretreatment with estrogen did not statistically enhance the tumorigenicity of primary ER− human breast cancer cells and had a variable effect on tumorigenicity of ER− breast cancer cell lines.
Estrogen appeared to slightly, but not significantly, enhance the final tumor volume for the MCF10Ca1h cells at both injection sites; tumor latency and incidence were unaffected. In contrast, estrogen increased tumor incidence in the subcutaneous site for the MDA MB-231 cells, but had no effect on final tumor volume or latency. Although the proliferative index for the subcutaneously injected MDA MB-231 cells suggests that, given more time, estrogen supplementation may have increased their final tumor volume, it remains unknown if they would reach the size of the orthotopic tumors. We can only conclude from our data that there was no significant effect of estrogen on the final tumor volume. Similar to our findings, estrogen treatment of xenograft-bearing mice has no significant effect on the level of cell proliferation observed in ER−, comedo DCIS specimens (Holland et al., 1997). However, contrary to these data, Gupta et al. demonstrated in a xenograft model of parturition-induced breast carcinoma formation that ER− tumors require circulating estrogens for their formation (Gupta et al., 2007). Weakly tumorigenic ER− breast cancer cells generated via introduction of the SV40 large T antigen, hTERT, and RasV12 formed tumors when injected subcutaneously post-pregnancy. However, this cell line failed to form subcutaneous tumors when systemic estrogen formation was blocked with the use of letrozole, an inhibitor of aromatase-dependent estrogen synthesis. In addition, systemic estrogen supplementation was sufficient to induce subcutaneous tumor formation in the absence of pregnancy, while placebo-treated controls failed to develop any palpable tumors. In the present study, we did not observe any significant enhancement of tumor latency or volume with estrogen supplementation in either of the two primary breast cancer cells cell lines tested, and an increase in tumor incidence for only one cell line. Although we injected relatively low numbers of cells specifically to observe differences in tumorigenicity, both cell lines are highly tumorigenic. The cell line used by Gupta et al. was reported as weakly tumorigenic, potentially providing an even lower baseline above which estrogenic effects could be more easily measured. Furthermore, we did not observe any enhancement of tumorigenicity with estrogen supplementation using the two primary breast cancer samples. However, these cells were not injected subcutaneously as in the Gupta report, but orthotopically into the mammary fat pad. The mouse mammary stroma may have compensated for the need of estrogen supplementation, for, as we show with the cell lines, the orthotopic injection appears to be a more robust model, potentially negating the need for systemic estrogen.
It should be noted that the majority of cancers used in xenograft studies are derived from post-menopausal patients. The circulating estrogen levels of these patients are comparable to the circulating levels in a mouse (50 – 100 pg/ml vs. 10 – 60 pg/ml, respectively) (Fotherby, 1984). Thus, estrogen supplementation introduces the tumor cells to significantly higher levels than in the patient. Exogenous 17β-estradiol is generally given in the form of a 0.72 mg 60-day slow release pellet which results in circulating levels of estrogen that is comparable to women in mid-cycle (300–400 pg/ml). Such high levels of estrogen may potentially have a suppressive effect on xenograft growth. Furthermore, estrogen supplementation has deleterious effects on the mouse host, including urinary retention with cystitis (Pearse et al., 2009), renal damage, and even death (Kang et al., 2009).
In this study, the method of cell delivery did not influence tumor latency, incidence, or final volume. One might expect that an invasive open surgery would elicit a more robust immune response compared to a transdermal injection. However, this phenomenon may be specific for xenograft models given the recent evidence that chronic infiltration of tissue by some innate immune cell types contributes to epithelial cancer development (DeNardo and Coussens, 2007), and that wound healing promotes tumor growth in a syngeneic mouse model (Stuelten et al., 2008). Additionally, no significant promotion of tumor growth was observed with pre-treatment with the immune suppressant etoposide in the abdominal gland, and a significant suppressive effect on final tumor volume, as well as decrease in incidence, was observed in the thoracic gland. This data, similar to developmental studies in the embryonic mammary glands, suggest that there are fundamental differences between the glands and potentially different regulatory effects of location. The intended effect of a single-dose etoposide pre-treatment is to suppress the host’s immune system, thus allowing for higher xenograft engraftment and growth. The clearance of etoposide from the mouse system is approximately one hour (Sengupta et al., 2000) and the bone marrow suppression lasts approximately four to six days (Visonneau et al., 1998); therefore, the etoposide should only have suppressed the host’s immune system and not the growth of the xenograft cells injected six days post-etoposide treatment. This study suggests that the use of etoposide should be re-evaluated, especially given the recent study highlighting the potential side effects of the drug; 38% of female rats and 6.7% of female mice receiving a single injection of etoposide developed breast cancer (Fomina et al., 2007).
Our study underscores potential molecular mechanisms for success in xenograft model systems of breast cancer, and lays the groundwork for future detailed, mechanistic studies. Ultimately, any xenograft experiment testing a particular mechanism should be robust enough to overcome procedural differences, thus reflecting a global mechanism. However, higher success of cell engraftment and growth is an imperative first step in any cancer xenograft model.
Supplementary Material
Acknowledgments
This research was supported by the Center for Cancer Research, an Intramural Research Program of the National Cancer Institute and by Breast Cancer Research Stamp proceeds awarded through competitive peer review.
Footnotes
The authors declare no conflicts of interest.
References
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolander FF., Jr Differential characteristics of the thoracic and abdominal mammary glands from mice. Exp Cell Res. 1990;189(1):142–144. doi: 10.1016/0014-4827(90)90266-d. [DOI] [PubMed] [Google Scholar]
- Booth BW, Mack DL, Androutsellis-Theotokis A, McKay RD, Boulanger CA, Smith GH. The mammary microenvironment alters the differentiation repertoire of neural stem cells. Proc Natl Acad Sci U S A. 2008;105(39):14891–14896. doi: 10.1073/pnas.0803214105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulanger CA, Mack DL, Booth BW, Smith GH. Interaction with the mammary microenvironment redirects spermatogenic cell fate in vivo. Proc Natl Acad Sci U S A. 2007;104(10):3871–3876. doi: 10.1073/pnas.0611637104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardiff RD. Validity of mouse mammary tumour models for human breast cancer: comparative pathology. Microsc Res Tech. 2001;52(2):224–230. doi: 10.1002/1097-0029(20010115)52:2<224::AID-JEMT1007>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Costa FF, Verbisck NV, Salim AC, Ierardi DF, Pires LC, Sasahara RM, Sogayar MC, Zanata SM, Mackay A, O'Hare M, Soares F, Simpson AJ, Camargo AA. Epigenetic silencing of the adhesion molecule ADAM23 is highly frequent in breast tumors. Oncogene. 2004;23(7):1481–1488. doi: 10.1038/sj.onc.1207263. [DOI] [PubMed] [Google Scholar]
- DeNardo DG, Coussens LM. Inflammation and breast cancer. Balancing immune response: crosstalk between adaptive and innate immune cells during breast cancer progression. Breast Cancer Res. 2007;9(4):212. doi: 10.1186/bcr1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 1999;59(4):793–797. [PubMed] [Google Scholar]
- Evron E, Umbricht CB, Korz D, Raman V, Loeb DM, Niranjan B, Buluwela L, Weitzman SA, Marks J, Sukumar S. Loss of cyclin D2 expression in the majority of breast cancers is associated with promoter hypermethylation. Cancer Res. 2001;61(6):2782–2787. [PubMed] [Google Scholar]
- Fleming JM, Long EL, Ginsburg E, Gerscovich D, Meltzer PS, Vonderhaar BK. Interlobular and intralobular mammary stroma: genotype may not reflect phenotype. BMC Cell Biol. 2008;9:46. doi: 10.1186/1471-2121-9-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fomina TI, Perelmuter VM, Vtorushin SV, Zavjalova MV, Borovskaja TG, Timina EA. Carcinogenic effect of an antitumor drug etoposide in laboratory animals. Bull Exp Biol Med. 2007;144(5):725–727. doi: 10.1007/s10517-007-0416-0. [DOI] [PubMed] [Google Scholar]
- Fotherby K. Endocrinology of menstrual cycle and pregnancy. In: Making HL, editor. Biochemistry of Steroid Hormones. Oxford: Blackwell Scientific Publications, Oxford; 1984. [Google Scholar]
- Garcia-Gasca A, Spyropoulos DD. Differential mammary morphogenesis along the anteroposterior axis in Hoxc6 gene targeted mice. Dev Dyn. 2000;219(2):261–276. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1048>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Graff JR, Gabrielson E, Fujii H, Baylin SB, Herman JG. Methylation patterns of the E-cadherin 5' CpG island are unstable and reflect the dynamic, heterogeneous loss of E-cadherin expression during metastatic progression. J Biol Chem. 2000;275(4):2727–2732. doi: 10.1074/jbc.275.4.2727. [DOI] [PubMed] [Google Scholar]
- Grimshaw MJ, Cooper L, Papazisis K, Coleman JA, Bohnenkamp HR, Chiapero-Stanke L, Taylor-Papadimitriou J, Burchell JM. Mammosphere culture of metastatic breast cancer cells enriches for tumorigenic breast cancer cells. Breast Cancer Res. 2008;10(3):R52. doi: 10.1186/bcr2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta PB, Proia D, Cingoz O, Weremowicz J, Naber SP, Weinberg RA, Kuperwasser C. Systemic stromal effects of estrogen promote the growth of estrogen receptor-negative cancers. Cancer Res. 2007;67(5):2062–2071. doi: 10.1158/0008-5472.CAN-06-3895. [DOI] [PubMed] [Google Scholar]
- Haslam SZ, Shyamala G. Effect of oestradiol on progesterone receptors in normal mammary glands and its relationship with lactation. Biochem J. 1979a;182(1):127–131. doi: 10.1042/bj1820127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslam SZ, Shyamala G. Progesterone receptors in normal mammary glands of mice: characterization and relationship to development. Endocrinology. 1979b;105(3):786–795. doi: 10.1210/endo-105-3-786. [DOI] [PubMed] [Google Scholar]
- Hatsell SJ, Cowin P. Gli3-mediated repression of Hedgehog targets is required for normal mammary development. Development. 2006;133(18):3661–3670. doi: 10.1242/dev.02542. [DOI] [PubMed] [Google Scholar]
- Holland PA, Knox WF, Potten CS, Howell A, Anderson E, Baildam AD, Bundred NJ. Assessment of hormone dependence of comedo ductal carcinoma in situ of the breast. J Natl Cancer Inst. 1997;89(14):1059–1065. doi: 10.1093/jnci/89.14.1059. [DOI] [PubMed] [Google Scholar]
- Hu M, Yao J, Carroll DK, Weremowicz S, Chen H, Carrasco D, Richardson A, Violette S, Nikolskaya T, Nikolsky Y, Bauerlein EL, Hahn WC, Gelman RS, Allred C, Bissell MJ, Schnitt S, Polyak K. Regulation of in situ to invasive breast carcinoma transition. Cancer Cell. 2008;13(5):394–406. doi: 10.1016/j.ccr.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin. 2008;58(2):71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3(6):415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- Kang JS, Kang MR, Han SB, Yoon WK, Kim JH, Lee TC, Lee CW, Lee KH, Lee K, Park SK, Kim HM. Low dose estrogen supplementation reduces mortality of mice in estrogen-dependent human tumor xenograft model. Biol Pharm Bull. 2009;32(1):150–152. doi: 10.1248/bpb.32.150. [DOI] [PubMed] [Google Scholar]
- Kim JB, O'Hare MJ, Stein R. Models of breast cancer: is merging human and animal models the future? Breast Cancer Res. 2004;6(1):22–30. doi: 10.1186/bcr645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuperwasser C, Chavarria T, Wu M, Magrane G, Gray JW, Carey L, Richardson A, Weinberg RA. Reconstruction of functionally normal and malignant human breast tissues in mice. Proc Natl Acad Sci U S A. 2004;101(14):4966–4971. doi: 10.1073/pnas.0401064101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laidlaw IJ, Clarke RB, Howell A, Owen AW, Potten CS, Anderson E. The proliferation of normal human breast tissue implanted into athymic nude mice is stimulated by estrogen but not progesterone. Endocrinology. 1995;136(1):164–171. doi: 10.1210/endo.136.1.7828527. [DOI] [PubMed] [Google Scholar]
- Mailleux AA, Spencer-Dene B, Dillon C, Ndiaye D, Savona-Baron C, Itoh N, Kato S, Dickson C, Thiery JP, Bellusci S. Role of FGF10/FGFR2b signaling during mammary gland development in the mouse embryo. Development. 2002;129(1):53–60. doi: 10.1242/dev.129.1.53. [DOI] [PubMed] [Google Scholar]
- Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LM, Pe'er J, Trent JM, Meltzer PS, Hendrix MJ. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. 1999;155(3):739–752. doi: 10.1016/S0002-9440(10)65173-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath CM, Nandi S, Young L. Relationship between organization of mammary tumors and the ability of tumor cells to replicate mammary tumor virus and to recognize growth-inhibitory contact signals in vitro. J Virol. 1972;9(2):367–376. doi: 10.1128/jvi.9.2.367-376.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MJ, Fleming JM, Ali MA, Pesesky MW, Ginsburg E, Vonderhaar BK. Dynamic regulation of CD24 and the invasive, CD44posCD24neg phenotype in breast cancer cell lines. Breast Cancer Res. 2009;11(6):R82. doi: 10.1186/bcr2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller FR, Medina D, Heppner GH. Preferential growth of mammary tumors in intact mammary fatpads. Cancer Res. 1981;41(10):3863–3867. [PubMed] [Google Scholar]
- Osborne CK, Hobbs K, Clark GM. Effect of estrogens and antiestrogens on growth of human breast cancer cells in athymic nude mice. Cancer Res. 1985;45(2):584–590. [PubMed] [Google Scholar]
- Outzen HC, Custer RP. Growth of human normal and neoplastic mammary tissues in the cleared mammary fat pad of the nude mouse. J Natl Cancer Inst. 1975;55(6):1461–1466. doi: 10.1093/jnci/55.6.1461. [DOI] [PubMed] [Google Scholar]
- Parmar H, Cunha GR. Epithelial-stromal interactions in the mouse and human mammary gland in vivo. Endocr Relat Cancer. 2004;11(3):437–458. doi: 10.1677/erc.1.00659. [DOI] [PubMed] [Google Scholar]
- Pearse G, Frith J, Randall KJ, Klinowska T. Urinary retention and cystitis associated with subcutaneous estradiol pellets in female nude mice. Toxicol Pathol. 2009;37(2):227–234. doi: 10.1177/0192623308329281. [DOI] [PubMed] [Google Scholar]
- Phillips RA, Jewett MA, Gallie BL. Growth of human tumors in immune-deficient scid mice and nude mice. Curr Top Microbiol Immunol. 1989;152:259–263. doi: 10.1007/978-3-642-74974-2_31. [DOI] [PubMed] [Google Scholar]
- Prehn RT, Main JM, Schneiderman M. Factors influencing tumor distribution among the mammary glands of the mouse. J Natl Cancer Inst. 1954;14(4):895–904. [PubMed] [Google Scholar]
- Pullinger BD. A gland of predilection for mammary nodules in a strain of mice deprived of the milk agent. Br J Cancer. 1952;6(1):78–79. doi: 10.1038/bjc.1952.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008;456(7222):593–598. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond A, Su Y. Mouse xenograft models vs GEM models for human cancer therapeutics. Dis Model Mech. 2008;1(2–3):78–82. doi: 10.1242/dmm.000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo IH, Russo J. Hormone prevention of mammary carcinogenesis: a new approach in anticancer research. Anticancer Res. 1988;8(6):1247–1264. [PubMed] [Google Scholar]
- Sakakibara T, Xu Y, Bumpers HL, Chen FA, Bankert RB, Arredondo MA, Edge SB, Repasky EA. Growth and metastasis of surgical specimens of human breast carcinomas in SCID mice. Cancer J Sci Am. 1996;2(5):291–300. [PubMed] [Google Scholar]
- Seitz S, Wassmuth P, Fischer J, Nothnagel A, Jandrig B, Schlag PM, Scherneck S. Mutation analysis and mRNA expression of trail-receptors in human breast cancer. Int J Cancer. 2002;102(2):117–128. doi: 10.1002/ijc.10694. [DOI] [PubMed] [Google Scholar]
- Sengupta S, Tyagi P, Velpandian T, Gupta YK, Gupta SK. Etoposide encapsulated in positively charged liposomes: pharmacokinetic studies in mice and formulation stability studies. Pharmacol Res. 2000;42(5):459–464. doi: 10.1006/phrs.2000.0714. [DOI] [PubMed] [Google Scholar]
- Sheffield LG, Welsch CW. Transplantation of human breast epithelia to mammary-gland-free fat-pads of athymic nude mice: influence of mammotrophic hormones on growth of breast epithelia. Int J Cancer. 1988;41(5):713–719. doi: 10.1002/ijc.2910410513. [DOI] [PubMed] [Google Scholar]
- Singh-Ranger G, Salhab M, Mokbel K. The role of cyclooxygenase-2 in breast cancer: review. Breast Cancer Res Treat. 2008;109(2):189–198. doi: 10.1007/s10549-007-9641-5. [DOI] [PubMed] [Google Scholar]
- Stuelten CH, Barbul A, Busch JI, Sutton E, Katz R, Sato M, Wakefield LM, Roberts AB, Niederhuber JE. Acute wounds accelerate tumorigenesis by a T cell-dependent mechanism. Cancer Res. 2008;68(18):7278–7282. doi: 10.1158/0008-5472.CAN-08-1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taghian A, Budach W, Zietman A, Freeman J, Gioioso D, Ruka W, Suit HD. Quantitative comparison between the transplantability of human and murine tumors into the subcutaneous tissue of NCr/Sed-nu/nu nude and severe combined immunodeficient mice. Cancer Res. 1993;53(20):5012–5017. [PubMed] [Google Scholar]
- Thompson HJ, Meeker LD. Induction of mammary gland carcinomas by the subcutaneous injection of 1-methyl-1-nitrosourea. Cancer Res. 1983;43(4):1628–1629. [PubMed] [Google Scholar]
- Tomkova K, Tomka M, Zajac V. Contribution of p53, p63, and p73 to the developmental diseases and cancer. Neoplasma. 2008;55(3):177–181. [PubMed] [Google Scholar]
- Umbricht CB, Evron E, Gabrielson E, Ferguson A, Marks J, Sukumar S. Hypermethylation of 14-3-3 sigma (stratifin) is an early event in breast cancer. Oncogene. 2001;20(26):3348–3353. doi: 10.1038/sj.onc.1204438. [DOI] [PubMed] [Google Scholar]
- Vaage J. Relationship between tumor growth characteristics and preferential sites of growth. J Natl Cancer Inst. 1984;72(5):1199–1203. [PubMed] [Google Scholar]
- Visonneau S, Cesano A, Torosian MH, Miller EJ, Santoli D. Growth characteristics and metastatic properties of human breast cancer xenografts in immunodeficient mice. Am J Pathol. 1998;152(5):1299–1311. [PMC free article] [PubMed] [Google Scholar]
- Vorontsova AL, Garina GB, Kudriavets Iu I, Ponomarev IN, Korolev VI. [The cellular sensitivity of the primary tumor and its metastases in breast cancer patients to cytostatics, interferon and tumor necrosis factor] Vopr Onkol. 1989;35(11):1315–1318. [PubMed] [Google Scholar]
- Wagner KU. Models of breast cancer: quo vadis, animal modeling? Breast Cancer Res. 2004;6(1):31–38. doi: 10.1186/bcr723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Brunner N, Jensen G, Albrectsen J, Gotthardsen B, Rygaard J. Comparative studies between nude and scid mice on the growth and metastatic behavior of xenografted human tumors. Clin Exp Metastasis. 1992;10(3):201–210. doi: 10.1007/BF00132752. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.