

Abstract

A novel strategy has been developed for the efficient syntheses of diverse arrays of heterocyclic compounds. The key elements of the approach comprise a Mannich-type, multicomponent coupling reaction in which functionalized amines, aromatic aldehydes, acylating agents, and π- and organometallic nucleophiles are combined to generate intermediates that are then further transformed into diverse heterocyclic scaffolds via a variety of cyclization manifolds. Significantly, many of these scaffolds bear functionality that may be exploited by further manipulation to create diverse collections of compounds having substructures found in biologically active natural products and clinically useful drugs. The practical utility of this strategy was exemplified by its application to the first, and extraordinarily concise synthesis of the isopavine alkaloid roelactamine.
Keywords: Imine, acyliminium ion, multi-component reaction, drug-like scaffolds, natural products, Mannich reaction, Heck reaction, ring-closing metathesis, alkaloids
1. Introduction
The discovery of potent, selective, and bioavailable small molecules that modulate biological systems of interest continues to be one of the most important endeavors for contemporary organic and medicinal chemists. Despite the increasing importance of biological macromolecules in medicine, small molecules remain as important as ever. Relative to biological therapeutics, small molecule drugs usually exhibit enhanced bioavailability, especially oral, and they are easier and less costly to manufacture. Perhaps more importantly, small molecules can be easily designed to be cell permeable, a property that gives them access to intracellular targets that may be more difficult or impossible to modulate with protein or nucleic acid derived agents. The demand for novel small molecules with favorable biological properties thus continues to be high. Accordingly, developing strategies that enable their identification is a priority for both basic biological research and translational medicine.
The isolation, identification, and direct modification of suitable natural products has traditionally been a fruitful endeavor for discovering new drugs and drug leads. However, the immense challenges and potential lack of flexibility in working with such complex molecules has inspired synthetic chemists in both industry and academia to develop alternative strategies for creating compounds with desirable biological properties. Such approaches often involve generating diverse sets of compounds that can probe a broad swath of biological space, thereby improving the odds of finding compounds with desired properties. These strategies have become an important element of drug discovery, owing in part to our inability to design molecules with predictable biological properties.
The classical strategy for new lead discovery in medicinal chemistry is the scaffold-based approach, wherein derivatives of certain core structures, often so-called privileged structures,1 are prepared and screened. More recently, a number of innovative strategies have emerged for discovering new leads. One productive approach is diversity-oriented synthesis (DOS),2,3 the objective of which is the rapid synthesis of molecules with diverse skeletons and substitution patterns derived from common starting materials. Two strategies that rely more on natural products for the creation of collections of compounds are diverted total synthesis (DTS),4 wherein intermediates in the synthesis of biologically active natural products are modified, and biology oriented synthesis (BIOS),5 wherein the core structures of natural products are used as scaffolds for generating compound collections using a structural classification of natural products (SCONP) as a design principle.
The design of synthetic approaches that enable rapid and efficient generation of collections of organic compounds that are structurally and functionally diverse and that possess skeletal frameworks found in natural products and drug-like molecules is a significant challenge.6 In our view one of the more promising ways to achieve this goal is by combining multicomponent reactions (MCRs),7 which allow the rapid synthesis of differentially substituted intermediates from three or more reactants, with further transformations involving cyclizations via selective functional group pairing to fabricate polycyclic carbo- and heterocyclic scaffolds with a variety of substituents and functional groups as depicted graphically in Figure 1.8 This general strategy has been utilized by a number of groups who have sequenced various ring forming reactions with multicomponent processes, including the Ugi four-component reaction,9 the van Leusen reaction,10 and the Petasis reaction.11 The sequencing of a MCR or related methods for constructing intermediates that may then be elaborated by ring forming reactions has recently been referred to as the “Build-Couple-Pair” approach.12
Figure 1.
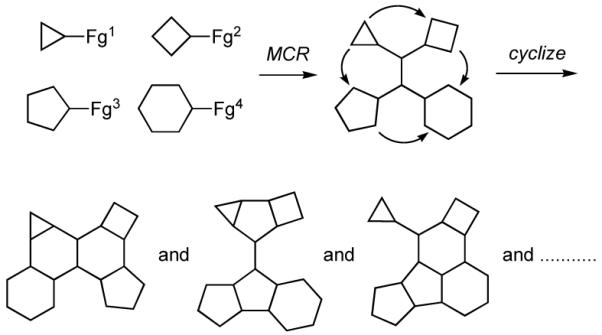
Approach to synthesis of diverse collections of polycyclic scaffolds by tandem MCR and cyclizations.
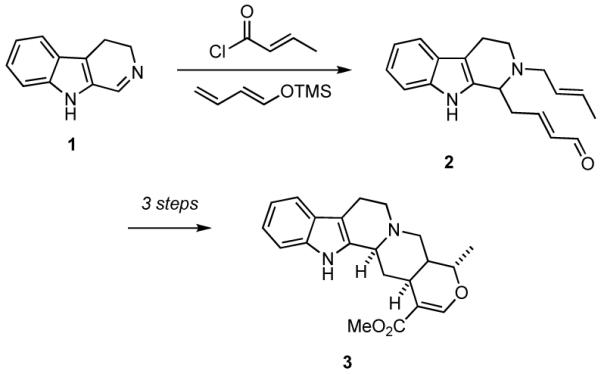
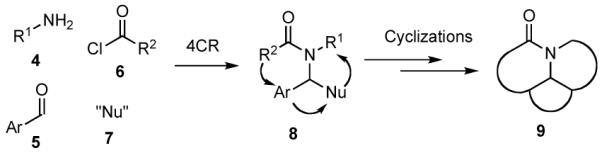
A number of years ago we discovered a multicomponent reaction that featured vinylogous Mannich reactions of electron rich dienes with N-acyl iminium ions that had been formed in situ by N-acylation of imines to give adducts that were readily transformed into complex indole alkaloids such as tetrahydroalstonine (3) (Scheme 1).13,14 Based upon this finding, we envisioned a new strategy for the rapid synthesis of diverse collections of heterocyclic compounds that entailed the combination of an alkylamine 4, an aromatic aldehyde 5, and an acylating agent 6 to generate an N-acyl iminium ion that could be subsequently trapped with a nucleophile 7 to give a highly functionalized amide or carbamate 8 (Scheme 2).15 A variant of this route to 8 would involve addition of a nucleophile to the imine to provide an intermediate amine that could then be N-acylated.16 Each of the four components could be endowed with added functionality that would then enable further transformation of 8 via a variety of ring forming reactions to generate various polycyclic scaffolds 9 bearing functionality for further diversification and for the preparation of focused libraries. We now report some of the details of our studies that have been directed toward developing the strategy outlined in Scheme 2 for the rapid synthesis of a variety of heterocyclic scaffolds.17
Scheme 1.
Scheme 2.
2. Results and Discussion
2.1 Preliminary Considerations
Before presenting specific applications of the strategy outlined in Scheme 2, a few comments are appropriate. Unlike the venerable Ugi and some other multicomponent reactions, the components in our process must be combined, at least in part, in a sequential fashion because of the inherent reactivity of the different inputs. The first stage of the sequence involves imine formation, and depending upon whether one wants to perform a three or a four component coupling, this intermediate may or may not be isolated in crude form. For example, the aldehyde and amine may be condensed in a suitable solvent in the presence of activated molecular sieves to give a solution of the imine. This mixture may then be used directly in the next step, or the molecular sieves may be removed by filtration and the resulting solution used in the next step. Alternatively, the solvent may be removed to provide the crude imine, which could either be stored or redissolved to provide a stock solution for convenient use in different reactions. When imines of allylamine are desired, a practical and convenient method for their preparation involves the reaction of the aldehyde with commercially available N,N-bis(trimethylsilyl)allylamine in the presence of catalytic amounts of trimethylsilyl triflate (TMSOTf). These solutions of N-allylimines were then used directly.
The tactics employed for completing the sequence to give 8 were dictated somewhat by the nature of the nucleophilic partner. When weak π-nucleophiles such as enol ethers and ketene acetals were used, it was necessary to treat the imine with the π-nucleophile and the acylating agent, usually in the presence of catalytic amounts of TMSOTf. With reactive nucleophiles such as allylmetals, the nucleophile could optionally be added directly to the solution of the imine and the resulting metal amide trapped by reaction with the acylating agent. Although the majority of the reactions presented herein were conducted as one-pot processes, some were performed as two-pot sequences in which the crude imines were isolated. It should be noted that overall yields in the component assembly step to give compounds related to 8 can vary depending upon how the imine-forming step is conducted. It should be noted, however, that we did not always optimize conditions for the formation of the intermediate imines or for their subsequent conversion into adducts 8, because our overall goal was to illustrate the scope and potential of the general plan outlined in Scheme 2.
Having established a protocol for the facile synthesis of intermediates 8, a number of post-condensation reactions, including Heck reactions, ring closing metathesis (RCM), Dieckmann cyclizations, and Diels-Alder and dipolar cycloadditions, together with selected combinations of these reactions, were examined to generate heterocyclic scaffolds. The results are presented according to the types of reaction(s) that were implemented to generate the final scaffolds.
2.2 Scaffolds via Heck cyclization
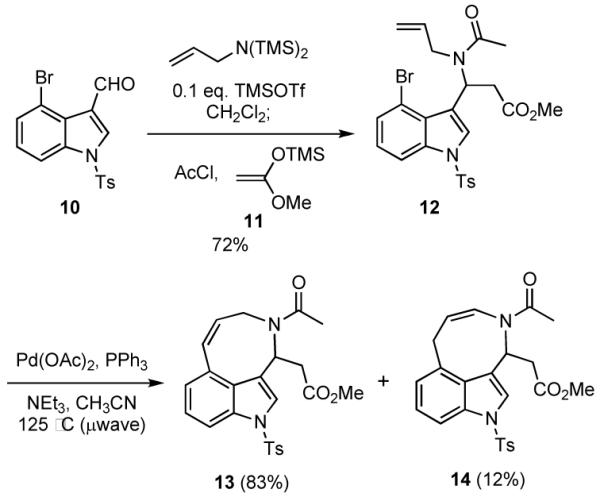
An example of using a Heck reaction in tandem with the multicomponent process in Scheme 2 is illustrated in Scheme 3. The indole 1018 was first allowed to react with commercially available N,N-bis(trimethylsilyl)allyl amine in the presence of TMSOTf, whereupon acetyl chloride and the silyl ketene acetal 1119 were added to provide 12, with TMSOTf catalyzing both imine formation and the Mannich reaction. The resulting amide 12 underwent an intramolecular Heck reaction with microwave heating to afford tricycle 13, the structure of which was established by x-ray crystallography,20 in 60% overall yield together with a small amount of the isomeric olefin 14.
Scheme 3.
2.3 Scaffolds via RCM
The reaction of unsaturated organometallic reagents as nucleophiles with imines derived from allylamine or other unsaturated amines gives rise to intermediates that may be easily elaborated by RCM reactions. For example, addition of vinylmagnesium bromide to the iminium ion generated upon reaction of the imine 16 and benzyl chloroformate gave carbamate 17, which underwent RCM to provide the 2-aryl dihydropyrrole 18 (Scheme 4). Similar yields were observed with vinylzinc chloride and vinylzinc bromide, which were generated in situ by reaction of the Grignard reagent with the corresponding zinc halide and would be expected to have better functional group tolerance. In order to obtain optimal yields and to ensure complete formation of the intermediate N-acylated imine or the corresponding α-chlorocarbamate, the solution of imine and chloroformate was generally heated prior to addition of the nucleophile. Such heating does not appear necessary when acid chlorides are employed as acylating agents. The aryl bromide function of 18 might be exploited in a variety of palladium-catalyzed cross-coupling reactions to generate focused libraries, and compound collections with alternative substitution patterns would be accessible with benzaldehydes other than 15.
Scheme 4.
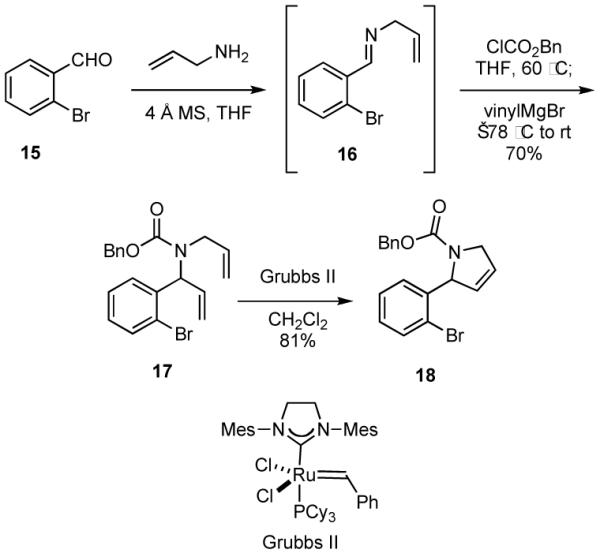
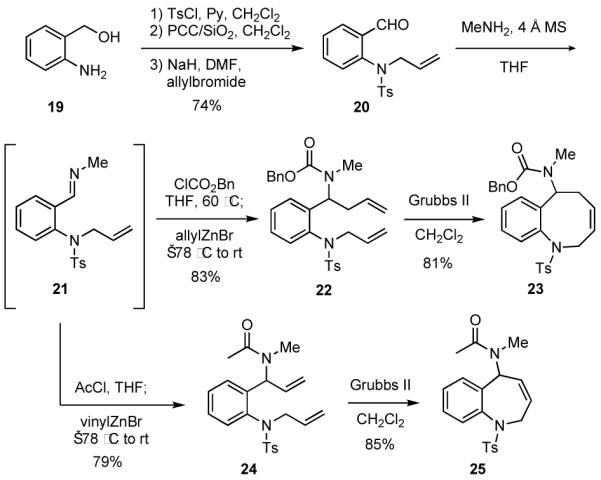
2-Aminobenzaldehydes may also be utilized as starting materials, although preliminary work suggests that better results are obtained when the amine is fully substituted. For example, the aminobenzaldehyde derivative 20 was easily prepared in 74% overall yield from commercially available 19 (Scheme 5). Condensation of 20 with methylamine gave the intermediate imine 21 that was treated in situ with carbobenzyloxy chloride and allylzinc bromide to give carbamate 22, which underwent facile RCM in the presence of Grubbs II catalyst to provide the benzazocine 23. In a parallel sequence of reactions, imine 21 was treated with acetyl chloride and vinylzinc bromide to give 24, which cyclized via a RCM in the presence of Grubbs II catalyst to give the benzazepine 25. Benzazepine rings are found in a variety of biologically active and clinically useful compounds such as Mozavaptan, which is a sodium level regulator that is marketed in Japan, and other benzazepines have been reported to act as cholesterol ester transfer protein (CETP) inhibitors.21
Scheme 5.
2-Aryltetrahydropyridines such as 27 may also be readily accessed by the reaction of imines such as 16 with benzyl chloroformate and then methyl 2-carboxylzinc bromide.22 Subsequent RCM of 26 then gave 27, which was contaminated with about 10% of an inseparable impurity that appears to be an isomeric cycloalkene.
We have thus demonstrated the underlying utility of the general strategy outlined in Scheme 2 for the rapid preparation of simple heterocycles. It then remained to apply this approach to the synthesis of more complex heterocyclic scaffolds. Toward this goal, we turned our attention to elaborating more highly functionalized adducts 8 via sequential ring forming reactions.
2.4 Scaffolds via Sequential RCM/Heck Reactions
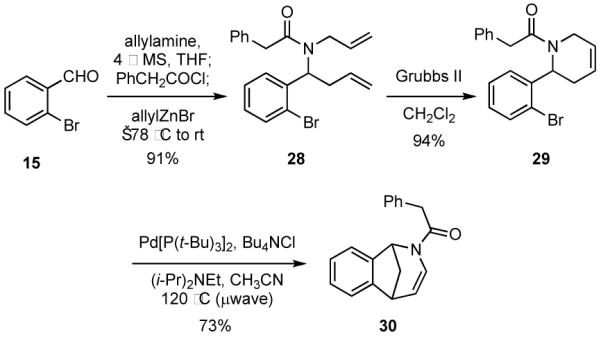
Bridged azabicycles having the framework found in 30 are known to have activity in an acute pain model for mice on the same order of magnitude as codeine.23 Compound 30 was rapidly assembled from 28, which was formed via a one-pot multicomponent reaction starting with 15 (Scheme 7). The RCM reaction of 28 proceeded smoothly in the presence of Grubbs II catalyst to give 29 in 86% overall yield from 15. Some screening of conditions was then required in order to optimize the subsequent Heck reaction to give 30. It was necessary to use microwave heating, to employ a hindered base in the reaction, and to conduct the reaction in the presence of chloride ion.
Scheme 7.
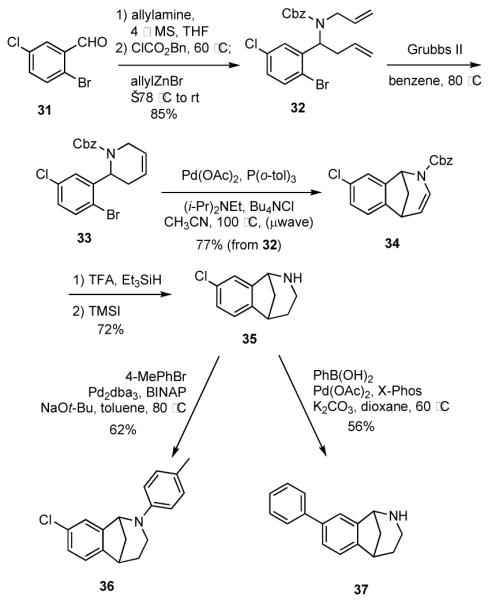
Because derivatives of azabicycles such as 30 might have useful activities, we briefly explored tactics that might be employed to create a small library of compounds related to 30. In the event, the known aldehyde 3124 was converted into the carbamate 32 (Scheme 8). Cyclization of 32 via RCM in the presence of Grubbs II catalyst gave 33. We discovered that the Heck reaction could be telescoped with the RCM step by simply adding 2-mercaptonicotinic acid and activated charcoal to remove the residual Ru catalyst.25 The solvent was exchanged with acetonitrile and Pd(OAc)2, P(o-tol)3, Hünig’s base and Bu4NCl were added, and the mixture was heated in a microwave oven to deliver 34. The olefin was reduced via ionic reduction,26 and the carbamate group was removed with TMS-I27 to give the key intermediate 35. In preliminary and unoptimized experiments, we discovered that 35 may be processed via Buchwald-Hartwig28 and Suzuki29 cross-coupling reactions to give 36 and 37, respectively.
Scheme 8.
Indole 10 was also used as a starting material in a related MCR followed by sequential RCM and Heck cyclizations to generate the novel bridged indole derivative 39, which bears some resemblance to the ring system found in the ergoline alkaloids (Scheme 9). It is noteworthy that the conversion of 10 into 38 could be telescoped with a simple solvent exchange for the second step. Best results were achieved when the allylmagnesium bromide was added to the imine prior to the acylation step. Because a metal salt of an amine is the intermediate, there is considerable flexibility in selecting the acylating agent. The Heck cyclization of 38 to furnish 39 proceeded without event.
Scheme 9.
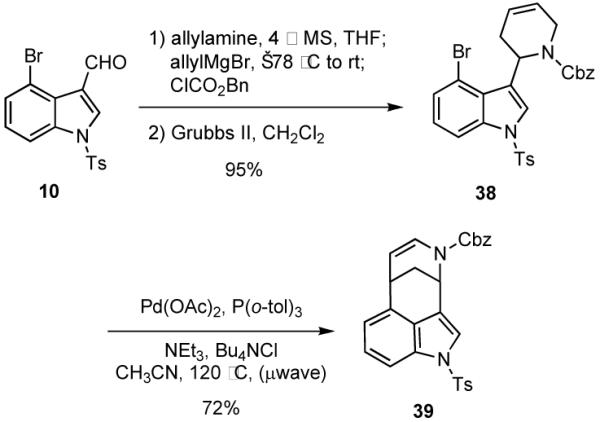
In order to broaden the scope of this approach to bridged heterocyclic ring systems, we explored the possibility of accessing different ring sizes by varying the nature of the nucleophile. For example, use of homoallylzinc chloride30 in the MCR with aldehyde 15, allylamine and benzyl chloroformate gave carbamate 40 (Scheme 10). Sequential cyclizations via a RCM and a Heck reaction generated an easily separable mixture (3.4:1) of isomers 42 and 43, whose structures were assigned by 2D NMR (COSY and HSQC). The major isomer 42 is a sub-structure of the isopavine alkaloids.
Scheme 10.
2.5 Scaffolds via Sequential RCM/Dieckmann Reactions
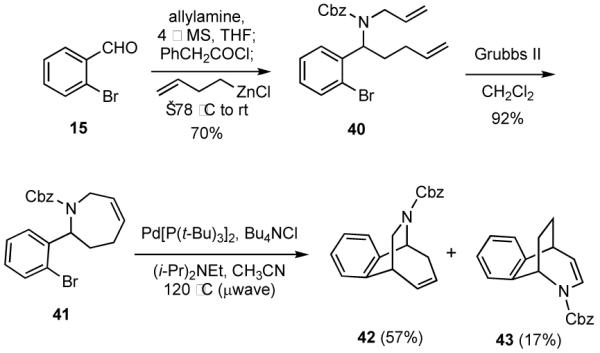
The aromatic aldehydes used thus far have contained a bromo group that was paired with alkenes in Heck cyclizations. Other functionality can be incorporated into the starting aldehyde to enable other cyclization manifolds. For example, an ester group in the starting benzaldehyde might be used in Dieckmann cyclizations. Accordingly, 4431 was employed as a starting input in a MCR using allylamine, acetyl chloride, and allylzinc bromide to give 45 (Scheme 11). Allyltributylstannane could also be used in this MCR giving 45 in 77% yield, but in a preliminary experiment use of allyl(trimethyl)silane in the presence of a catalytic amount of TMSOTf gave 45 in only 17% yield. RCM of 45 followed by Dieckmann cyclization then delivered 47 in about 70% overall yield from 45.
Scheme 11.
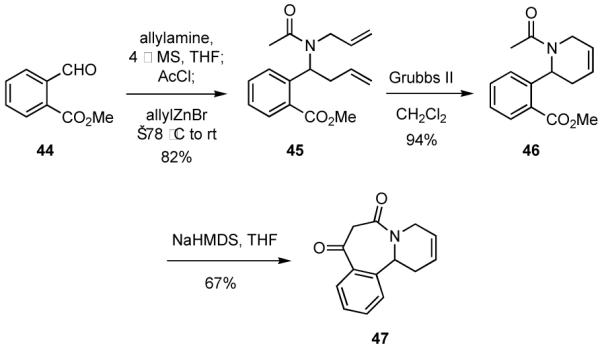
An ester group required for a Dieckmann condensation may also be introduced if a silyl ketene acetal is used as the nucleophile in the MCR. In order to exemplify this process 2-vinylbenzaldehyde (48)32 and silylketene acetal 49 were employed as two components in a MCR to generate 50 (Scheme 12). Cyclization of 50 via a Dieckmann reaction followed by RCM provided 52 in about 70% overall yield from 50. If the order of these cyclizations was reversed the overall yield of 52 was significantly lower. Moreover, the presence of the geminal groups in 50 obviates enolization α to the ester moiety and elimination of the amide side chain.
Scheme 12.
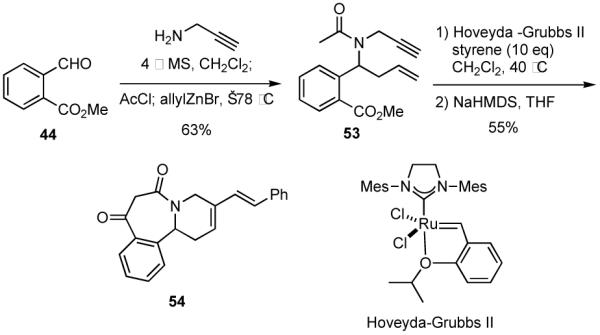
Incorporation of a carbon-carbon triple bond into MCR adducts can also enable the incorporation of a fifth component via an enyne RCM/CM sequence. For example, propargylamine was utilized as the amine component in a MCR with 44, acetyl chloride and allylzinc bromide to generate amide 53 in moderate yield from aldehyde 44 (Scheme 13). In this instance, consistently higher yields were obtained when the intermediate imine was isolated in crude form. When enyne 53 was treated with excess styrene in the presence of Hoveyda-Grubbs II catalyst, a RCM/CM process ensued to give an intermediate that was cyclized via a Dieckmann cyclization to give diene 54 in good overall yield.33 Other terminal olefins, including methyl acrylate, methyl vinyl ketone, allyltrimethylsilane, and allyl alcohol, also underwent RCM/CM,34 albeit in lower yields, but no effort was made to optimize those sequences.
Scheme 13.
2.6 Scaffolds via [3+2] Cycloadditions
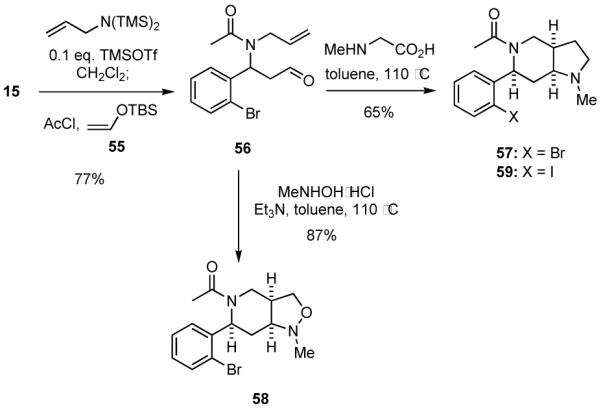
Dipolar cycloaddition reactions comprise a versatile class of reactions for generating heterocyclic ring systems.35 This fact together with the prevalence of bioactive heterocycles containing the pyrrolidine substructure inspired us to sequence our MCR process with different [3+2] reactions. Two intriguing examples are presented in Scheme 14. The aldehyde 56, which served as a common intermediate in the two sequences, was isolated in 77% yield starting from 2-bromobenzaldehyde (15) using the silyl enol ether 5536 as the nucleophile. Condensation of 56 with sarcosine in refluxing toluene led to the formation of 57 in 65% yield. The relative stereochemistry of 57 was established by comparisons of its NMR spectra with that of 59, which was synthesized from 2-iodobenzaldehyde by the same procedure used to prepare 57 and the structure of which was determined by x-ray analysis of its hydrochloride salt.20 When the aldehyde 56 was condensed with N-methylhydroxylamine, the intermediate nitrone underwent cyclization to give 58 in 87% yield. The structure of 58 was assigned by x-ray analysis of its hydrochloride salt.20 Significantly, the heterocyclic scaffolds present in 57 and 58 are found in compounds that are chemokine CCR5 receptor antagonists37 and inhibitors of dipeptidyl peptidase IV (DPP-IV).38 Of course, 56 and related compounds may be transformed by other dipolar cycloaddition reactions, and such efforts are in progress.
Scheme 14.
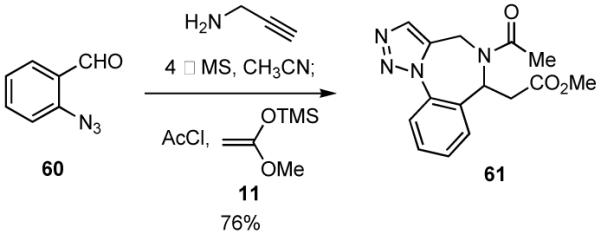
Another interesting sequence involving an intramolecular [3+2] cycloaddition is shown in Scheme 15. In this particular case the MCR adduct derived from the azido benzaldehyde 6039 underwent spontaneous intramolecular cycloaddition to give triazole 61. Preliminary attempts to cyclize 61 via a Dieckmann reaction were unsuccessful. Nevertheless, it is noteworthy that the skeletal framework in 61 is found in related compounds that have been reported to be weak inhibitors of the benzodiazepine receptor.40
Scheme 15.
2.7 Scaffolds via Diels-Alder Cycloadditions
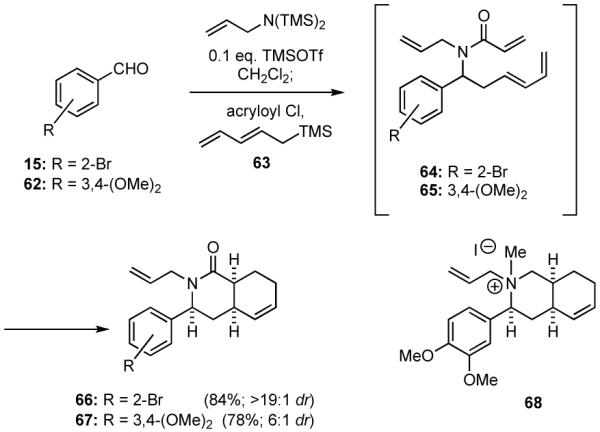
We have used intramolecular Diels-Alder reactions as key steps in the synthesis of a number of alkaloid natural products.13,41 Based upon this work, it occurred to us that such constructions might be used to access heterocyclic scaffolds comprising subunits of the berbane, yohimbine and heteroyohimbine alkaloids. In order to explore this exciting possibility, it would simply be necessary to incorporate a diene into the multicomponent process. Accordingly, when the pentadienylsilane 6342 was employed as the nucleophile in the multicomponent process involving the allylimine derived from 15 and acryloyl chloride in the presence of 10 mol % TMSOTf, the adduct 64 thus formed underwent spontaneous Diels-Alder cyclization to generate the hydroisoquinoline 66 in good yield as a single detectable diastereomer (Scheme 16). The scope of this process was expanded to include variably functionalized benzaldehydes, such as 3,4-dimethoxybenzaldehyde (62) that led to the formation of 67 in good yield and diastereoselectivity. The structure of 67 was assigned based upon the x-ray analysis of the methiodide salt of 68. Correlations of the NMR spectra of 67 and 66 allowed assignment of the relative stereochemistry of 66.
Scheme 16.
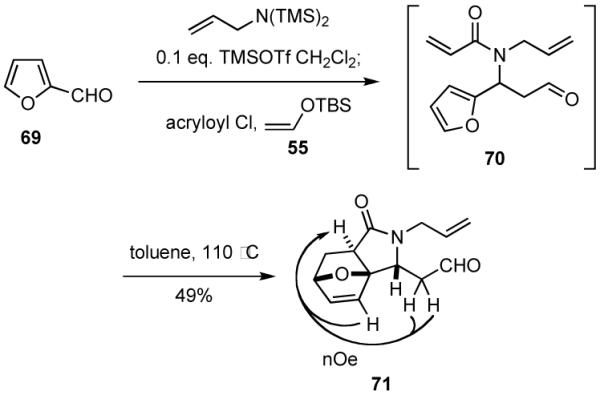
Another example of the sequencing of a Diels-Alder reaction with the MCR is illustrated in Scheme 17, wherein furfural (69) is utilized as the aldehyde component. The initially formed adduct 70 was not stable, as the Diels-Alder reaction proceeded slowly at room temperature. Consequently, 70 was simply isolated in crude form and heated under reflux in toluene to give a mixture of isomers from which the major product 71 was isolated by chromatography in 49% yield. The structure of 71 was assigned from analysis of 2D NMR and nOe spectroscopy. The stereochemical mode of this cycloaddition is consistent with literature reports for similar intramolecular Diels-Alder reactions of furans.43
Scheme 17.
2.8 Scaffolds via Friedel-Crafts Reaction
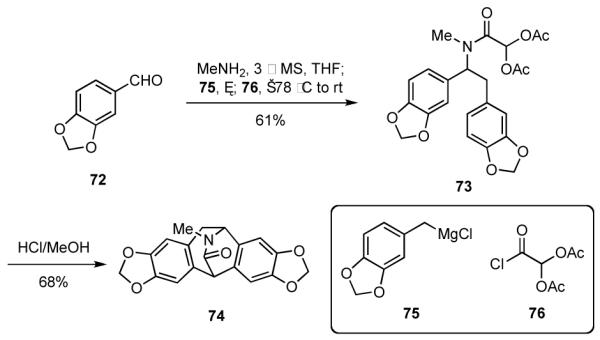
Friedel-Crafts reactions may also be used to advantage to transform adducts generated by the multicomponent process in Scheme 2. In order to illustrate how this might be accomplished, we turned our attention again to the synthesis of compounds having the isopavine skeleton, which incorporates an azabicyclo[3.2.2]nonane substructure (cf Scheme 10). Both natural and unnatural members of the isopavine family have been shown to possess interesting biological properties for the treatment of disorders like Alzheimer’s and Parkinson’s disease, Huntington’s chorea, and Down’s syndrome.44 Indeed, an approach to the isopavine alkaloid roelactamine (74)45 was developed that represents the first synthesis of this alkaloid and constitutes the most concise approach to alkaloids of this class.46
The synthesis of roelactamine commenced with the sequential reaction of piperonal (72) with methylamine, the Grignard reagent 75, and diacetoxyacetyl chloride (76), which was easily prepared in gram quantities from glyoxylic acid,47 to give amide 73 (Scheme 18). When 73 was simply treated with HCl and MeOH, roelactamine (74) was formed and isolated in good overall yield from 72. The synthetic (±)-roelactamine thus obtained gave spectral characteristics (1H and 13C NMR, IR, and mass spectral data) consistent with those reported for the natural product. Although dichloroacetyl chloride could also be used in the first step, the corresponding dichloro analog of 73 did not undergo efficient cyclization to give 74.
Scheme 18.
3. Conclusion
The representative examples presented herein clearly establish the merits of this new strategy for the facile synthesis of diverse collections of nitrogen heterocycles. The approach features an initial Mannich-type multicomponent reaction to combine functionalized amines, aromatic aldehydes, acylating agents, and π- and organometallic nucleophiles to create useful templates. These key intermediates are then further transformed into a diverse array of heterocyclic frameworks via a variety of cyclization manifolds, including Heck, RCM, Dieckmann, and Friedel-Crafts reactions as well as dipolar and Diels-Alder cycloadditions; numerous other cyclizations can be readily envisioned. Significantly, these scaffolds bear orthogonal functionality that may be selectively exploited for further manipulation and diversification to create novel sets of compounds having substructures found in biologically active natural products and clinically useful drugs. Hence, this chemistry would appear to have considerable potential for generating novel drug leads and tools for studying biological pathways and for translational medical research. Inasmuch as the underlying inspiration for this approach to creating compound libraries emerged from our work in alkaloid synthesis, it is perhaps not surprising that these basic principles were applied to an extraordinarily concise synthesis of the isopavine alkaloid roelactamine. Further applications of the concepts outlined in Scheme 2 for both the synthesis of natural products and diverse collections of scaffolds that are endowed with reactive sites or handles for the preparation of focused libraries are in progress. The results of these investigations will be reported in due course.
4. Experimental
4.1 General
Unless otherwise noted, solvents and reagents were reagent-grade and used without further purification. Dichloromethane (CH2Cl2), diisopropylamine (i-Pr2NH) and triethylamine (Et3N) were freshly distilled over CaH2. Tetrahydrofuran (THF), ether (Et2O), acetonitrile (CH3CN), toluene, and dimethylformamide (DMF) were dried according to the procedure described by Grubbs.48 1,4-Dioxane was distilled twice from sodium and stored in a Schlenk flask over molecular sieves and under nitrogen. TMSOTf and styrene were distilled from CaH2 under reduced pressure. Acetyl chloride was freshly distilled from P2O5 immediately prior to use, and acryloyl chloride was distilled immediately prior to use. Allyl bromide was dried with MgSO4 and distilled. Zinc metal was activated by washing with 1 M HCl. The 4 Å molecular sieves were activated under high vacuum at ∼300 °C for 7 h. All reactions were run under a positive pressure of N2 or argon gas in glassware that had been oven or flame dried. Vinylzinc halide solution (in THF) was made by adding a commercial solution of vinylmagnesium bromide in THF to ZnX2 (1.1 eq.). ZnCl2 was dried by fusing under vacuum. Reaction temperatures are reported as the temperatures of the bath surrounding the vessel, or programmed temperatures for microwave reactions. Microwave reactions were performed using a CEM Discover Labmate microwave synthesizer, which varies the power (generally 150 W max) automatically to reach and maintain the set temperature.
Nuclear magnetic resonance spectra were acquired at 300 K in CDCl3 unless otherwise noted. Chemical shifts are reported in parts per million (ppm, δ), downfield from tetramethylsilane (TMS, δ = 0.00 ppm) and are referenced to the residual solvent: CDCl3, δ = 7.26 ppm (1H) and 77.16 ppm (13C); CD3OD, δ = 3.31 ppm (1H) and 49.0 ppm (13C); d6-DMSO, δ = 2.50 ppm (1H) and 39.5 ppm (13C); d6-acetone, δ = 2.05 ppm (1H) and 29.8 ppm (13C). The abbreviations s, d, t, q, p, hex, hep, m and comp stand for the resonance multiplicities singlet, doublet, triplet, quartet, pentuplet, hextuplet, heptuplet, multiplet, and complex (overlapping multiplets), respectively; br = broad; app = apparent. In some cases where NMR suggested the presence of rotamers, NMR data were collected at high temperature to coalesce the signals. Otherwise, the existence of rotamers is noted in the spectral data. Infrared (IR) spectra were recorded as films on sodium chloride plates and reported as wavenumbers (cm-1).
4.2 Experimental Procedures
Methyl 3-(N-allylacetamido)-3-(4-bromo-1-tosyl-1H-indol-3-yl)propanoate (12)
TMSOTf (6.5 μL, 8 mg, 0.036 mmol) was added to a solution of 4-bromoindole-3-carboxaldehyde 1018 (140 mg, 0.37 mmol) and bis(trimethylsilyl)allylamine (0.10 mL, 82 mg, 0.40 mmol) in CH2Cl2 (1 mL), and the reaction was stirred for 3 h at room temperature. Silyl ketene acetal 1119 (135 mg, 0.95 mmol) and freshly distilled acetyl chloride (35 μL, 39 mg, 0.49 mmol) were then added, and the reaction was stirred for 4 h at room temperature. The reaction was partitioned between CH2Cl2 (5 mL) and saturated aqueous NaHCO3 (5 mL). The organic phase was removed, and the aqueous layer was extracted with CH2Cl2 (2 × 5 mL). The combined organic layers were dried (MgSO4), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography, eluting with 50% EtOAc/hexanes to give 142 mg (72%) of amide 12 as a white solid, mp 134.5-136 °C. 1H NMR (500 MHz, DMSO, 130 °C) δ 7.95 (d, J = 7.8 Hz, 1 H), 7.95 (s, 1 H), 7.77 (d, J = 8.2 Hz, 2 H), 7.45 (d, J = 7.8 Hz, 1 H), 7.36 (d, J = 8.2 Hz, 2 H), 7.23 (t, J = 8.2 Hz, 1 H), 6.41 (s, 1 H), 5.31 (dddd, J = 15.8, 10.2, 5.3, 5.3 Hz, 1 H), 4.72 (d, J = 17.1 Hz, 1 H), 4.65 (d, J = 10.2 Hz, 1H), 3.86 (dd, J = 17.3, 5.3 Hz 1 H), 3.70 (dd, J = 17.3, 5.3 Hz, 1 H), 3.54 (s, 3 H), 3.18 (dd, J = 15.3, 9.3 Hz, 1 H), 3.00 (dd, J = 15.3, 5.9 Hz, 1 H), 2.34 (s, 3 H), 2.03 (s, 3 H); 13C NMR (125 MHz, DMSO, 130 °C) δ 169.8, 169.3, 145.1, 135.5, 134.2 133.7, 129.5, 127.6, 127.4, 125.9, 125.3, 119.7, 114.7, 112.7, 112.1, 50.5, 47.2, 46.0, 36.4, 21.4, 20.2; IR (neat) 1736, 1644, 1411, 1374, 1258, 1174, 675, 574 cm-1; mass spectrum (CI) m/z 533.0754 [C24H2679BrN2O5S (M+1) requires 533.0746].
Methyl 2-((Z)-9-acetyl-2,8,9,10-tetrahydro-2-tosylazocino[3,4,5-cd]indol-10-yl)acetate (13) and methyl 2-((Z)-9-acetyl-2,6,9,10-tetrahydro-2-tosylazocino[3,4,5-cd]indol-10-yl)acetate (14)
Pd(OAc)2 (2 mg, 0.001 mmol), PPh3 (6 mg, 0.025 mmol), and Et3N (0.06 mL, 43 mg, 0.43 mmol) were added to a solution of amide 12 (0.057 mg, 0.11 mmol) in CH3CN (4.2 mL) and the reaction was heated to 125 °C in a microwave reactor for 30 min. The reaction was cooled to room temperature and concentrated under reduced pressure. The residue was purified by flash chromatography, eluting with 50% EtOAc/hexanes to give 6 mg (12%) of enamine 14 as a yellowish gum and 40 mg (83%) of amide 13 as a brownish solid. Recrystallization of 13 via slow evaporation from benzene provided large colorless prisms, mp = 170-172 °C. 13 (major): 1H NMR (500 MHz, DMSO, 130 °C) δ 7.88 (d, J = 8.3 Hz, 1 H), 7.78 (s, 1 H), 7.77 (d, J = 8.3 Hz, 2 H), 7.37 (d, J = 8.3 Hz, 2 H), 7.34 (t, J = 8.3 Hz, 1 H), 7.12 (d, J = 7.5 Hz, 1 H), 6.92 (d, J = 10.9 Hz, 1 H), 5.99 (ddd, J = 10.5, 10.5, 6.9 Hz, 1 H), 5.84 (m, 1 H), 4.34 (br, 1 H), 3.82 (br, 1 H), 3.37 (s, 3 H), 2.96 (dd, J = 15.0, 9.8 Hz, 1 H), 2.88 (dd, J = 15.0, 4.7 Hz, 1 H), 2.34 (s, 3H), 2.14 (s, 3H); 13C NMR (125 MHz, DMSO, 130 °C)δ 169.4, 168.1, 144.8, 135.2, 134.1, 133.5, 129.4, 128.6, 127.1, 126.6, 126.5, 125.9, 124.3, 124.1, 118.2, 112.5, 50.5, 50.1, 41.3, 40.6, 20.9, 20.1; IR (neat) 1736, 1638, 1419, 1371, 1307, 1259, 1176, 1149, 1091, 731 cm-1; mass spectrum (CI) m/z 453.1487 [C24H25N2O5S (M+1) requires 453.1484]. 14 (minor): 1H NMR (500 MHz, DMSO, 130 °C) δ 7.74 (d, J = 8.3 Hz, 2 H), 7.73 (d, J = 8.3 Hz, 1 H), 7.65 (s, 1 H), 7.34 (d, J = 8.3 Hz, 2 H), 7.21 (t, J = 7.4 Hz, 1 H), 7.05 (d, J = 7.4 Hz, 1 H), 6.45 (d, J = 9.1 Hz, 1 H), 5.94 (t, J = 7.1 Hz, 1 H), 5.34-5.29 (m, 1 H), 3.65 (dd, J = 7.5, 3.7 Hz, 2 H), 3.53 (s, 3 H), 3.31 (dd, J = 15.7, 6.4 Hz, 1 H), 3.10 (dd, J = 15.7, 7.7 Hz, 1 H), 2.33 (s, 3H), 2.00 (s, 3H); 13C NMR (125 MHz, DMSO, 130 °C)δ 169.8, 168.8, 144.6, 134.8, 134.2, 132.1, 129.3, 128.2, 127.2, 125.8, 125.2, 124.3, 123.2, 119.8, 115.1, 111.0, 50.4, 49.9, 38.6, 31.2, 22.2, 20.1; IR (neat) 2950, 1737, 1642, 1425, 1368, 1290, 1175, 1091, 912, 743 cm-1; mass spectrum (CI) m/z 453.1482 [C24H25N2O5S (M+1) requires 453.1484].
Allyl-[1-(2-bromophenyl)allyl]carbamic acid benzyl ester (17)
A mixture of allylamine (59 mg, 0.77 mL, 10.2 mmol) and 2-bromobenzaldehyde (1.85 g, 10.0 mmol) in THF (25 mL) containing 4 Å molecular sieves (3.0 g) was stirred for 12 h at room temperature. The sieves were removed by filtration through Celite, and the filtrate was concentrated under reduced pressure to give 2.24 g (ca 100%) of crude imine 16. A portion of 16 (56 mg, 0.25 mmol) was dissolved in THF (1 mL), benzyl chloroformate (45 mg, 38 μL, 0.26 mmol) was added, and the mixture was heated at 60 °C for 1 h. The reaction was then cooled to -78 °C, and a solution of vinylmagnesium bromide in THF (1.04 M, 288 μL, 0.30 mmol) was added. The cooling bath was removed, and the reaction was stirred at room temperature for 1.5 h. Saturated aqueous NH4Cl (∼0.5 mL) was added, and the mixture was partitioned between water (20 mL) and Et2O (20 mL), and the layers were separated. The organic layer was washed with NaHCO3 (20 mL) and brine (20 mL), dried (MgSO4), filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography, eluting with 0-25% EtOAc/hexanes to give 68 mg (70%) of 17 as a colorless oil. 1H NMR (400 MHz, CDCl3) (rotamers) δ 7.60-7.05 (comp, 9 H), 6.15-5.98 (comp, 2 H), 5.63-5.45 (comp, 1 H), 5.45-5.05 (comp, 4 H), 4.81 (d, J = 10.0 Hz, 1 H), 4.73 (d, J = 17.0 Hz, 1 H), 3.93 (dd, J = 15.9, 5.1 Hz, 1 H), 3.63 (br dd, 1 H); 13C NMR (100 MHz, CDCl3) (rotamers) δ 156.2, 141.7, 138.5, 138.3, 136.8, 134.9, 134.3, 133.3, 130.3, 129.5, 129.2, 128.5, 128.3, 128.1, 127.96, 127.93, 127.4, 126.0, 117.2, 116.1, 115.7, 73.6, 67.4, 67.3, 61.7, 47.4; IR (neat) 1702, 1441, 1404, 1277, 1251, 1140, 922, 754, 697 cm-1; mass spectrum (ESI+) m/z 386.0750 [C20H21NO279Br (M+1) requires 386.0760].
2-(2-Bromophenyl)-2,5-dihydropyrrole-1-carboxylic acid benzyl ester (18)
A solution of 17 (31 mg, 0.079 mmol) in CH2Cl2 (4 mL) containing Grubbs II catalyst (3.4 mg, 0.004 mmol) was stirred at room temperature for 3 h. The reaction was concentrated and the crude product was purified by flash chromatography, eluting with 0-25% EtOAc/hexanes to give 23 mg of dihydropyrrole 18 (81%) as a colorless oil. 1H NMR (400 MHz, CDCl3) (rotamers) δ 7.51 (d app t, J = 8.0, 1.0 Hz, 1 H), 7.40-7.06 (comp, 7 H), 6.92 (m, 1 H), 5.99 (m, 1 H), 5.89 (m, 1 H), 5.84 (m, 1 H), 5.19 (d, J = 12.3 Hz, 0.4 H), 5.09 (d, J = 12.3 Hz, 0.4 H), 5.06 (d, J = 12.7 Hz, 0.6 H), 4.97 (d, J = 12.7 Hz, 0.6 H), 4.41 (m, 2 H); 13C NMR (100 MHz, CDCl3) rotamers) δ 154.6, 154.4, 141.1, 140.3, 136.9, 136.6, 133.1, 132.8, 130.0, 128.9, 128.8, 128.6, 128.3, 128.2, 128.1, 128.0, 127.7, 127.5, 127.4, 127.1, 125.00, 124.96, 122.1, 68.2, 67.5, 67.1, 66.8, 54.6, 54.3; IR (neat) 3321, 1709, 1412, 1356, 1114, 1025, 753, 697 cm-1; mass spectrum (CI+) m/z 358.0445 [C18H17NO279Br (M+1) requires 358.0443].
N-Allyl-N-(2-formylphenyl)-4-methyl-benzenesulfonamide (20)
A solution of N-(2-formylphenyl)-4-methylbenzenesulfonamide49 (853 mg, 3.10 mmol) in dry DMF (4 mL) was added to a stirred suspension of 95% sodium hydride (89 mg, 3.5 mmol) in dry DMF (8 mL) at 0 °C. The mixture was removed from the ice bath and stirred for 15 min at room temperature, whereupon it was again cooled to 0 °C. Allyl bromide (755 mg, 0.54 mL, 6.2 mmol) was added dropwise by syringe, and the reaction was stirred for 1 h at 0 °C and then 20 h at room temperature. A solution of 1 M aqueous NaOH (20 mL) and EtOAc (80 mL) were added, and the layers were separated. The organic layer was washed with 1 M NaOH (2 × 50 mL), water (3 × 50 mL), brine (50 mL), dried (MgSO4), filtered, concentrated to give 974 mg (100%) of 20 as an off-white solid. The 20 thus obtained gave spectral data that were consistent with those reported.50
1-{2-[Allyl(toluene-4-sulfonyl)amino]phenyl}but-3-enyl)methyl carbamic acid benzyl ester (22)
A mixture of methylamine in THF (2 M, 5.0 mL, 10.0 mmol) and aldehyde 20 (315 mg, 1.00 mmol) containing 3 Å molecular sieves (600 mg) was stirred for 16 h at room temperature. The sieves were removed by filtration through Celite, and the filtrate was concentrated under reduced pressure to give 328 mg (ca 100%) of crude 21. A portion of 21 (82 mg, 0.25 mmol) was dissolved in THF (1 mL), benzyl chloroformate (45 mg, 38 μL, 0.26 mmol) was added, and the mixture was heated at 60 °C for 1 h. The reaction was then cooled to -78 °C, and a solution of allylzinc bromide51 in THF (0.90 M, 333 μL, 0.30 mmol) was added. The cooling bath was removed, and the reaction was stirred at room temperature for 1.5 h. Saturated aqueous NH4Cl (∼0.5 mL) was added, and the mixture was partitioned between water (30 mL) and Et2O (30 mL), the layers were separated, and the organic layer was washed with NaHCO3 (20 mL) and brine (20 mL), dried (MgSO4), filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography, eluting with 0-30% EtOAc/hexanes to give 105 mg (83%) of 22 as a colorless oil. 1H NMR (500 MHz, d6-DMSO, 120 °C) (rotamers) δ 7.63-7.25 (comp, 12 H), 7.22 (app td, J = 7.8, 1.7 Hz, 1 H), 6.76 (br s, 1 H), 5.83 (br s, 1 H), 5.68-5.54 (comp, 2 H), 5.19 (d, J = 12.4 Hz, 1 H), 5.14-4.83 (comp, 5 H), 4.07 (br m, 1 H), 3.94 (br m, 1 H), 2.75 (m, 1 H), 2.64 (br s), 2.63 (s, 3 H), 2.43 (s), 2.41 (br s, total 3 H); 13C NMR (125 MHz, d6-DMSO, 120 °C) (rotamers) δ 154.8, 142.9, 134.4, 131.8, 128.8, 127.5, 127.4, 127.3, 126.9, 116.0, 65.7, 20.2; IR (neat) 2928, 1695, 1450, 1401, 1349, 1165, 664, 576 cm-1; mass spectrum (CI+) m/z 505.2157 [C29H33N2O4S (M+1) requires 505.2161].
Methyl-[1-(toluene-4-sulfonyl)-1,2,5,6-tetrahydro-benzo[b]azocin-6-yl] carbamic acid benzyl ester (23)
A solution of 22 (52 mg, 0.10 mmol) in CH2Cl2 (5 mL) containing Grubbs II catalyst (8.5 mg, 0.01 mmol) was stirred at room temperature for 6 h. DMSO (44 mg, 40 μL, 0.56 mmol) was added52 and stirring continued for 4 h. The solvents were removed under reduced pressure, and the product was purified by flash chromatography, eluting with 0-50% EtOAc/pentane to give 40 mg of 23 (81%) as a colorless oil. 1H NMR (500 MHz, d6-DMSO, 120°C) (rotamers) δ 7.71 (d, J = 8.0 Hz, 2 H), 7.46 (d, J = 8.0 Hz, 2 H), 7.40-7.28 (comp, 7 H), 7.23-7.14 (comp, 2 H), 6.71 (d, J = 7.8 Hz, 1 H), 5.73 (m, 1 H), 5.63 (br s, 1 H), 5.34 (d app t, J = 11.0, 3.4 Hz, 1 H), 5.19 (s, 2 H), 2.82 (s, 3 H), 2.45 (s, 3 H); 13C NMR (125 MHz) δ 143.1, 136.3, 129.2, 128.3, 127.6, 127.0, 126.8, 126.7, 125.4, 66.0, 58.1, 20.2; IR (neat) 2936, 1696, 1491, 1445, 1401, 1344, 1325, 1163, 1096, 674, 577, 548 cm-1; mass spectrum (CI-) m/z 477.1846 [C27H29N2O4S (M) requires 477.1848].
N-(1-{2-[Allyl-(toluene-4-sulfonyl)amino]phenyl}-allyl)-N-methylacetamide (24)
Imine 21 (49 mg, 0.15 mmol) was dissolved in THF (0.5 mL), acetyl chloride (12.8 mg, 11.6 μL, 0.16 mmol) was added, and the mixture was stirred for 1 h. The reaction was then cooled to -78 °C, and a solution of vinylzinc bromide in THF solution (0.60 M, 310 μL, 0.19 mmol) was added. The cooling bath was removed, and the reaction was stirred at room temperature for 1.5 h. Saturated aqueous NH4Cl (∼0.5 mL) was added, and the mixture was partitioned between water (20 mL) and Et2O (20 mL), the layers were separated, and the organic layer was washed with NaHCO3 (15 mL) and brine (15 mL), dried (MgSO4), filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography, eluting with 0-60% EtOAc/pentane to give 47 mg (79%) of 24 as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.51 (d, J = 8.2 Hz, 1 H), 7.40 (dd, J = 7.8, 1.8 Hz, 1 H), 7.37-7.29 (comp, 3 H), 7.18 (app t d, J = 7.8, 1.6 Hz, 1 H), 6.52-6.45 (comp, 2 H), 6.05 (ddd, J = 17.3, 10.6, 3.3 Hz, 1 H), 5.49 (m, 1 H), 5.42 (dm, J = 10.6 Hz, 1 H), 5.25 (ddd, J = 17.3, 2.3, 1.0 Hz, 1 H), 4.98 (d, J = 9.6 Hz, 1 H), 4.90 (dd, J = 16.8, 1.0 Hz, 1 H), 4.44 (ddm, J = 13.5, 5.9 Hz, 1 H), 3.75 (dd, J = 13.5, 8.4 Hz, 1 H), 2.47 (s, 3 H), 2.47 (s, 3 H), 2.45 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 172.1, 144.3, 139.7, 138.9, 134.9, 133.9, 131.37, 129.6, 128.63, 128.57, 128.51, 128.0, 120.9, 116.2, 58.5, 54.9, 28.7 (2 overlapping peaks), 21.7; IR (neat) 3750, 1652, 1344, 1163, 665, 576 cm-1; mass spectrum (CI+) m/z 399.1747 [C22H27N2O3S (M+1) requires 399.1742].
N-Methyl-N-[1-(toluene-4-sulfonyl)-2,5-dihydro-1H-benzo[b]azepin-5-yl]acetamide (25)
A solution of 24 (47 mg, 0.12 mmol) in CH2Cl2 (6 mL) containing Grubbs II catalyst (15.3 mg, 0.018 mmol) was stirred at room temperature for 24 h. The reaction was quenched by adding DMSO (55 mg, 50 μL, 0.70 mmol)52 and stirring for 4 h. The crude product was purified by flash chromatography, eluting with 0-100% EtOAc/pentane to give 37 mg of 25 (85%) as a brown oil. 1H NMR (400 MHz, CDCl3) (rotamers) δ 7.67 (d, J = 8.2 Hz, 2 H), 7.39-7.22 (comp, 4.3 H), 7.15 (dd, J = 7.8, 1.2 Hz, 0.7 H), 6.94 (d, J = 7.6 Hz, 1 H), 6.13-5.37 (comp, 3 H), 4.96 (d app t, J = 19.3, 3.1 Hz, 0.7 H), 4.68 (br d, J = 17.6 Hz, 0.3 H), 4.01 (br d, J = 18.2, 0.3 H), 3.84 (dm, J = 19.3 Hz, 0.7 H), 2.95 (s, 2.1 H), 2.94 (s, 0.9 H), 2.43 (s, 0.9 H), 2.43 (s, 2.1 H), 2.16 (s, 0.9 H), 1.84 (s, 2.1 H); 13C NMR (100 MHz, CDCl3) (rotamers) δ 171.8, 170.6, 143.9, 141.2, 138.6, 137.4, 130.3, 129.9, 129.3, 128.7, 128.6, 128.2, 127.7 (br), 127.3, 125.6, 59.0 (br), 48.7, 48.4, 21.7, 21.6, 21.5 (br); IR (neat) 2922, 1649, 1484, 1396, 1342, 1159, 1099, 714, 578, 542 cm-1; mass spectrum (CI+) m/z 371.1426 [C20H23N2O3S (M+1) requires 371.1429].
4-(Allylbenzyloxycarbonylamino)-4-(2-bromophenyl)-2-methylenebutyric acid methyl ester (26)
One drop of solution of methyl-2-(bromomethyl) acrylate (268 mg, 180 μL, 1.50 mmol) in THF (1 mL) was added to a mixture of acid-washed and dried zinc filings (137 mg, 2.10 mmol) and THF (0.25 mL), and the mixture was stirred vigorously for 10 min. The remaining solution of the bromide was then added dropwise over 10 min. In a separate flask, a solution of imine 16 (112 mg, 0.50 mmol) and benzyl chloroformate (90 mg, 75 μL, 0.53 mmol) in THF (2 mL) was heated at 60 °C for 1 h. The reaction was then cooled to -78 °C, and a portion of the solution of methyl-2-carboxyallylzinc bromide (ca 0.75 M based on iodometric titration) (800 μL, 0.60 mmol) was added. The cooling bath was removed, and the reaction was stirred at room temperature for 1.5 h. Saturated aqueous NH4Cl (∼0.5 mL) was added, and the mixture was partitioned between water (30 mL) and Et2O (30 mL). The layers were separated, and the organic layer was washed with NaHCO3 (20 mL) and brine (20 mL), dried (MgSO4), filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography, eluting with 0-30% EtOAc/hexanes to give 145 mg (63%) of carbamate 26 (63%) as a colorless oil. 1H NMR (500 MHz, d6-DMSO, 90 °C) δ 7.60 (dd, J = 7.8, 1.5 Hz, 1 H), 7.58 (dd, J = 7.8, 1.2 Hz, 1 H), 7.38 (ddd, J = 7.7, 7.7, 1.2 Hz, 1 H), 7.36-7.27 (comp, 4 H), 7.23 (ddd, J = 7.7, 7.7, 1.7 Hz, 1 H), 6.05 (d, J = 1.2 Hz, 1 H), 5.64 (dd, J = 9.5, 5.6 Hz, 1 H), 5.57 (d, J = 1.2 Hz, 1 H), 5.41 (dddd, J = 16.6, 10.7, 5.9, 5.9 Hz, 1 H), 5.12 (d, J = 12.7 Hz, 1 H), 5.01 (d, J = 12.7 Hz, 1 H), 4.77 (d, J = 0.7 Hz, 1 H), 4.74 (dm, J = 8.8 Hz, 1 H), 3.76 (ddm, J = 16.1, 5.6 Hz, 1 H), 3.63 (dm, J = 4.6 Hz, 1 H), 3.61 (s, 3 H), 3.04 (dd, J = 14.0, 10.0 Hz, 1 H), 2.90 (ddd, J = 14.0, 5.6, 1.0 Hz, 1 H); 13C NMR (125 MHz, d6-DMSO, 90 °C MHz) δ 165.8, 154.8, 137.6, 136.43, 136.38, 134.1, 132.5, 129.23, 129.19, 127.7, 127.2, 127.12, 127.09, 126.8, 124.7, 115.1, 66.0, 56.8, 51.0, 45.5, 33.7; IR (neat) 2950, 1698, 1437, 1406, 1228, 1134, 993, 757, 697 cm-1; mass spectrum (CI+) m/z 458.0967 [C23H25NO479Br (M+1) requires 458.0967].
N-Allyl-N-[1-(2-bromophenyl)-but-3-enyl]-2-phenylacetamide (28)
A mixture of allylamine (57 mg, 75 μL, 1.0 mmol) and 2-bromobenzaldehyde (185 mg, 1.00 mmol) in dry THF (2 mL) containing crushed 4 Å molecular sieves (300 mg) was stirred at room temperature for 7 h. Phenylacetyl chloride (170 mg, 145 μL, 1.1 mmo1) was added, and the mixture was stirred for another 1.5 h. The mixture was then cooled to -78 °C, and a solution of allylzinc bromide (∼1.4 mmol) in dry THF (1 mL) was added. The bath was removed, and the mixture was stirred for 2.5 h at room temperature. Saturated aqueous NH4Cl solution (1 mL) was added, and the mixture was partitioned between water (20 ml) and Et2O (20 mL). The layers were separated, and the aqueous layer was extracted with Et2O (10 mL). The combined organics were washed with NaHCO3 (20 ml), brine (20 mL), dried (MgSO4), filtered, and concentrated under reduced pressure to give a colorless oil. The crude product was purified by flash chromatography, eluting with 0-30% EtOAc/hexanes to give 325 mg (91%) of amide 28 as a colorless oil. 1H NMR (400 MHz, CDCl3) (rotamers) δ 7.63-7.54 (m, 1 H), 7.44-7.36 (m, 1 H), 7.34-7.20 (comp, 6 H), 7.19-7.10 (m, 1 H), 5.96 (app t, J = 7.8 Hz, 0.8 H), 5.75 (dddd, J = 17.2, 10.2, 6.9, 6.9 Hz, 0.8 H), 5.56 (m, 2 × 0.2 H), 5.37 (m, 0.8 H), 5.29 (app t, J = 7.6 Hz), 5.13-4.93 (comp, 3.8 H), 4.81 (d, J = 10.0 Hz, 0.2 H), 4.74 (d, J = 17.6 Hz, 0.2 H), 4.06 (d, J = 15.5 Hz, 0.2 H), 4.03 (d, J = 15.3 Hz, 0.2 H), 3.89-3.59 (comp, 4 H), 3.45-3.36 (comp, 0.2 H), 2.88-2.65 (m, 2 H); 13C NMR (100 MHz, CDCl3) (rotamers) δ 171.3, 171.1, 137.7, 137.3, 135.4, 135.3, 134.6, 134.5, 133.7, 133.5, 130.1, 129.7, 129.5, 129.4, 129.3, 129.2, 128.6, 127.5, 127.2, 127.1, 126.8, 126.6, 126.0, 118.6, 117.7, 116.7, 115.9, 60.2, 56.7, 46.9, 45.4, 41.3, 41.0, 36.7, 35.9; IR (neat) 3064, 2925, 1644, 1438, 1404, 1176, 1027, 992, 918, 754, 725, 697 cm-1; mass spectrum (CI+) m/z 384.0960 [C21H23NO79Br (M+1) requires 384.0963].
1-[2-(2-Bromo-phenyl)-3,6-dihydro-2H-pyridin-1-yl]-2-phenylacetamide (29)
A solution of 28 (1.40 g, 3.65 mmol) in CH2Cl2 (180 mL) containing Grubbs II catalyst (78 mg, 0.091 mmol) was stirred at room temperature for 24 h. The crude product was purified by flash chromatography, eluting with 0-25% EtOAc/hexanes to give 1.22 g (94%) of 29 as a pale brown oil. 1H NMR (500 MHz, d6-DMSO, 120 °C) (rotamers) δ 7.91 (br s 1 H), 7.63 (d, J = 8.4 Hz, 2 H), 7.49-7.00 (comp, 9 H), 5.87 (br s, 1 H), 5.77 (br m, 1 H), 5.14 (br s, 2 H), 4.31 (dd, 1 H), 2.78 (br d, 1 H), 2.45 (br d), 2.26 (br s, 2 H); 13C NMR (126 MHz, d6-DMSO, 120 °C) δ 168.2; 136.3, 135.2, 128.3, 127.5, 127.4, 126.9, 126.0, 125.6, 123.5, 123.4, 120.6, 118.4, 118.1, 112.7, 108.8, 39.8, 31.6, 28.4; IR (neat) 2924, 1646, 1406, 1026, 720 cm-1; mass spectrum (CI+) m/z 356.0651 [C19H19NO79Br (M+1) requires 356.0650].
1-(9-Azatricyclo[6.3.1.02,7]dodeca-2(7),3,5,10-tetraen-9-yl)-2-phenylacetamide (30)
A microwave vial (CEM Corp., 10 mL) containing tetrabutylammonium chloride (56 mg, 0.20 mmol), [Pd(P(t-Bu)3]2 (5.1 mg, 0.010 mmol), and amide 29 (71 mg, 0.20 mmol was sealed and flushed with argon. CH3CN (4.5 mL) and (i-Pr)2NEt (52 mg, 70 μL, 0.40 mmol) were added, and the reaction was heated via microwave for 1 h at 120 °C. The catalyst was removed by filtration through a plug of silica gel, which was washed with 50% EtOAc/hexanes. The filtrate and washings were concentrated under reduced pressure, and the residue was purified by flash chromatography, eluting with 0-35% EtOAc/hexanes to give 40 mg (72%) of azabicycle 30 as a pale yellow oil, which slowly solidified upon storing in the freezer, mp = 117-119 °C. 1H NMR (500 MHz, CDCl3) δ 7.46-7.03 (m, 9 H), rotamer 1 (major): 6.24 (dd, J = 7.9, 1.3 Hz, 0.8 H), 6.03 (d, J = 3.4 Hz, 0.8 H), 5.30 (ddd, J = 7.9, 6.5, 1.7 Hz, 0.8 H), 3.73 (d, J = 15.6 Hz, 0.8 H), 3.68 (d, 15.6 Hz, 0.8 H), 3.38 (dd, J = 6.5, 4.1 Hz, 0.8 H), 2.38 (dddd, J = 10.7, 4.1, 4.1, 1.7 Hz, 0.8 H), 2.03 (d, J = 10.7 Hz, 0.8 H); rotamer 2 (minor): 6.85 (dd, J = 8.0, 1.3 Hz, 0.2 H), 6.71 (d, J = 7.3 Hz, 0.2 H), 5.49 (ddd, J = 8.0, 6.6, 1.7 Hz, 0.2 H), 4.07 (d, J = 15.4 Hz, 0.2 H), 4.00 (d, 15.4 Hz, 0.2 H), 3.35 (dd, J = 6.6, 4.0 Hz, 0.2 H), 2.30 (dddd, J = 10.6, 4.0, 4.0, 1.7 Hz, 0.2 H), 2.09 (d, J = 10.6 Hz, 0.2 H); 13C NMR (100 MHz, CDCl3) δ (rotamer 1) 167.5, 148.8, 139.3, 134.5, 128.8, 128.6, 128.2, 126.89, 126.85, 124.3, 121.7, 120.5, 112.1, 55.6, 40.2, 38.6, 38.1; IR (neat) 2925, 1657, 1619, 1397, 1362, 1237, 1035, 756, 725, 696, 620 cm-1; mass spectrum (CI+) m/z 276.1390 [C19H18NO (M+1) requires 276.1388].
Allyl-[1-(2-bromo-5-chlorophenyl)-but-3-enyl]-carbamic acid benzyl ester (32)
A solution of allyl-(2-bromo-5-chlorobenzylidene)amine (2.20 g, 8.51 mmol), which was prepared from allylamine and 31 according to the procedure given for the preparation of 16, in THF (17 mL) and benzyl chloroformate (1.34 mL, 9.39 mmol) was heated at 60 °C for 1 h. The reaction was then cooled to -78 °C, and a solution of allylzinc bromide (ca. 13.1 mmol) in THF (10 mL) was added.51 The cooling bath was removed, and the reaction was stirred at room temperature for 1.5 h. Saturated aqueous NH4Cl (∼4 mL) was added, and the mixture was partitioned between water (100 mL) and Et2O (100 mL), and the layers were separated. The organic layer was washed with NaHCO3 (100 mL) and brine (50 mL), dried (MgSO4), filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography, eluting with 0-20% EtOAc/hexanes to give 3.31 (89%) of 32 as a pale yellow oil. 1H NMR (400 MHz, CDCl3) (rotamers) δ 7.46 (d, J = 8.4 Hz, 1 H), 7.43-7.28 (comp, 6 H), 7.12 (dd, J = 8.4, 2.4 Hz, 1 H), 5.75 (br s, 1 H), 5.60 (br m, 1 H), 5.44 (app t, J = 12.8 Hz, 1 H), 5.21 (d, J = 14.0 Hz, 1 H), 5.20 (d, J = 14.0 Hz, 1 H), 5.10 (d, J = 17.6 Hz, 1 H), 5.04 (d, J = 10.4 Hz, 1 H), 4.89 (d, J = 10.4 Hz, 1 H), 4.87 (d, J = 17.6 Hz, 1 H), 3.75 (dd, J = 16.0, 5.6 Hz, 1 H), 3.64 (br m, 1 H), 2.77 (br m, 1 H), 2.67 (br m, 1 H); 13C NMR (100 MHz, CDCl3) (rotamers) δ 155.9, 140.3, 136.7 (br), 134.5, 134.4, 134.0, 133.4, 129.6 (br), 129.3, 128.5, 128.2 (br), 128.0, 123.8, 118.1, 116.4, 67.4, 58.5, 47.0, 36.3; IR (neat) 3076, 2938, 1698, 1453, 1406, 1236, 1142, 1096, 1025, 992, 918, 814, 766, cm-1; mass spectrum (CI+) m/z 434.0524 [C21H22NO235Cl79Br (M+1) requires 434.0522].
5-Chloro-9-aza-tricyclo[6.3.1.02,7]dodeca-2,4,6,10-tetraene-9-carboxylic acid benzyl ester (34)
A solution of 32 (1.29 g, 2.97 mmol) in benzene (30 mL) containing Grubbs II catalyst (63 mg, 0.074 mmol) was heated at 80 °C for 15 h. 2-Mercaptonicotinic acid (230 mg, 1.48 mmol) was added to aid with catalyst removal, and stirring at room temperature was continued for 1 h.25 The mixture was then washed twice with aqueous NaHCO3 (20 mL) and brine (20 mL) and then stirred for 0.5 h over activated charcoal (3 g). The mixture was filtered and concentrated to ca. 10 mL. This solution was diluted to make a ∼0.04 M stock solution of 33. A 10-mL portion of this solution (ca. 0.37 mmol) was added to a 40-mL CEM microwave vial and sparged with argon for 15 min. Tetrabutylammonium chloride (139 mg, 0.50 mmol), Pd(OAc)2 (11 mg, 0.049 mmol), P(o-tol)3 (30 mg, 0.10 mmol), and (i-Pr)2NEt (129 mg, 174 μL, 1.0 mmol) were added. The vial was capped, flushed with argon, and heated via microwave at 100 °C for 1 h. The mixture was filtered through a plug of SiO2, which was washed with 50% EtOAc/hexanes. The combined filtrate and washings were concentrated, and the residue was purified by flash chromatography, eluting with 0-25% EtOAc/hexanes to yield 93 mg (77% from 32) of 34 as a colorless oil. 1H NMR (400 MHz, CDCl3) (rotamers) δ 7.49-7.30, 7.20-7.07 (comp, 8 H), 6.50 (d, J = 7.9 Hz, 0.42 H), 6.40 (d, J = 7.9 Hz, 0.58 H), 5.58 (d, J = 4.1 Hz, 0.58 H), 5.45 (d, J = 4.1 Hz, 0.42 H), 5.38-5.06 (comp, 4 H), 3.33 (dd, J = 10.7, 5.1 Hz, 1 H), 2.32 (m, 1 H), 2.10 (d, J = 10.7 Hz, 0.42 H), 2.09 (d, J = 10.7 Hz, 0.58 H); 13C NMR (100 MHz, CDCl3) (rotamers) δ 152.1, 152.0, 147.24, 147.15, 140.7, 140.5, 136.1, 135.9, 132.3, 132.1, 128.8, 128.59, 128.59, 128.56, 128.4, 128.3, 128.2, 124.4, 124.0, 122.1, 121.9, 121.75, 121.72, 110.7, 109.9, 67.90, 67.85, 57.8, 57.4, 37.99, 37.94, 37.8, 37.5; IR (neat) 1702, 1399, 1336, 1250, 1112, 694 cm-1; mass spectrum (CI+) m/z 326.0949 [C19H17NO235Cl (M+1) requires 326.0948].
5-Chloro-9-aza-tricyclo[6.3.1.02,7]dodeca-2,4,6-triene-9-carboxylic acid benzyl ester
Triethylsilane (186 mg, 0.25 mL, 1.6 mmol) and trifluoroacetic acid (308 mg, 0.2 mL, 2.7 mmol) were added to a solution of 34 (84 mg, 0.26 mmol) in CH2Cl2 (5 mL) at 0 °C. The reaction was stirred for 2 h at 0 °C and then at room temperature for 10 h. Saturated aqueous NaHCO3 (5 mL) was added, and the layers were separated. The aqueous layer was extract with CH2Cl2 (2 × 5 mL), and the combined organic layers were dried (MgSO4), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography, eluting with 0-50% EtOAc/hexanes to yield 78 mg (91%) of product as a colorless oil. 1H NMR (400 MHz, CDCl3) (rotamers) δ 7.48-7.10 (comp, 8 H), 5.49 (br s, 0.55 H), 5.38 (br s, 0.45 H), 5.15 (m, 2 H), 3.83 (br m, 1 H), 3.25 (br m, 1 H), 2.43 (br m, 1 H), 2.19 (br m, 1 H), 1.98 (br m, 1 H), 1.87 (d, J = 11.0 Hz, 1 H), 1.57 (br m, 1 H); 13C NMR (100 MHz, CDCl3) (rotamers) δ 154.9, 154.7 (br), 144.7, 142.9 (br), 142.7 (br), 136.8 (br), 136.7, 132.6, 128.5, 127.9, 124.2, 123.8 (br), 67.1, 57.4 (br), 57.2, 43.6, 39.3, 38.5, 30.1; IR (neat) 2943, 1693, 1415, 1305, 1263, 1200, 1097, 1055, 697 cm-1; mass spectrum (CI+) m/z 328.1106 [C19H19NO235Cl (M+1) requires 328.1104].
5-Chloro-9-aza-tricyclo[6.3.1.02,7]dodeca-2,4,6-triene (35)
TMSI (258 mg, 183 μL, 1.29 mmol) was added dropwise to a stirred solution of the carbamate from the previous experiment (211 mg, 0.644 mmol) in dry CH2Cl2 (6.4 mL) at 0 °C. The reaction was stirred for 1 h, whereupon methanolic HCl (ca. 7 mmol in 2.5 mL MeOH) was added dropwise. The reaction was concentrated and then partitioned between 1 M HCl (15 mL) and Et2O (15 mL). The layers were separated, and the aqueous phase was washed with Et2O (2 × 10 mL). The aqueous layer was then basified to pH 10, saturated with NaCl, and extracted with EtOAc (2 × 10 mL). The organic phase was dried (Na2SO4), filtered, and concentrated to give 106 mg (85%) of amine 35 as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.23-7.16 (comp, 2 H), 7.12 (d, J = 7.6 Hz, 1 H), 4.19 (d, J = 4.7 Hz, 1 H), 2.72 (dd, J = 12.1, 5.9 Hz, 1 H), 2.28 (ddd, J = 12.4, 12.4, 4.7 Hz, 1 H), 2.18 (m, 1 H), 1.95 (dddd, J = 12.4, 12.4, 5.9, 2.1 Hz, 1 H), 1.91 (app br d, J = 10.6 Hz, 1 H); 13C NMR (100 MHz, CDCl3) δ 145.0, 144.7, 132.3, 127.5, 123.5, 123.3, 58.8, 45.3, 39.8, 39.1, 31.0; IR (neat) 2939, 2854, 1464, 1394, 1304, 1077, 877, 823 cm-1; mass spectrum (CI+) m/z 194.0740 [C11H13N35Cl (M+1) requires 194.0737].
5-Chloro-9-p-tolyl-9-aza-tricyclo[6.3.1.02,7]dodeca-2,4,6-triene (36)
A solution of 35 (24 mg, 0.12 mmol), sodium tert-butoxide (14 mg, 0.15 mmol), Pd2dba3 (5.7 mg, 0.006 mmol), and rac-BINAP (5.8 mg, 0.009 mmol) in anhydrous toluene (0.3 mL) was stirred at 80 °C for 15 h. The solvent was evaporated under reduced pressure, and the residue was purified by flash chromatography using a 10 mL glass pipet packed with SiO2 (3 mL), eluting with 0-25% EtOAc/hexanes to yield 22 mg (62%) of 36 as a pale yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.17 (d, J = 1.8 Hz, 1 H), 7.16 (br s, 1 H), 7.05 (d, J = 8.4 Hz, 2 H), 6.93 (m, 1 H), 6.78 (dm, J = 8.4 Hz, 2 H), 4.96 (d, J = 4.6 Hz, 1 H), 3.24 (m, 1 H), 3.10 (dd, J = 5.6, 11.4 Hz, 1 H), 2.27 (s, 3 H), 2.27 (m, 1 H), 2.21 (dd, J = 4.6, 11.4 Hz, 1 H), 2.11 (d, J = 10.8 Hz, 1 H), 2.11 (m, 1 H), 1.71 (m, 1 H); 13C NMR (100 MHz, CDCl3) δ 148.2, 144.6, 142.1, 132.2, 129.8, 128.7, 127.9, 123.6, 123.5, 116.8; IR (neat) 2937, 2854, 1614, 1511, 1462, 1379, 1297, 1192, 1079, 871 cm-1; mass spectrum (CI+) m/z 284.1208 [C18H19N35Cl (M+1) requires 284.1206].
5-Phenyl-9-aza-tricyclo[6.3.1.02,7]dodeca-2,4,6-triene (37)
A solution of 35 (19.4 mg, 0.10 mmol), PhB(OH)2 (24 mg, 0.20 mmol), Cs2CO3 (98 mg, 0.30 mmol) and PEPPSI catalyst (3.4 mg, 0.005 mmol) in 1,4-dioxane (0.5 mL) was heated at 90 °C for 1.5 h. The crude mixture was filtered through Celite, the filtrated was concentrated under reduced pressure, and the residue was dissolved in THF (2 mL). Water (0.5 mL) and 30% aqueous NaOH solution (2 drops) were added, and the mixture was stirred at room temperature for 6 h. Et2O (10 mL) was added, and the layers were separated. The aqueous layer was extracted with Et2O (2 × 5 mL), and the combined organic layers were washed with brine (15 mL), dried (Na2SO4), filtered, and concentrated. The residue was purified by flash chromatography, eluting with 10% MeOH/CH2Cl2 saturated with NH3 to yield 13.2 mg (56%) of 37 as a pale orange oil. 1H NMR (400 MHz, CDCl3) δ 7.63-7.58 (comp, 2 H), 7.49-7.40 (comp, 4 H), 7.33 (tt, J = 7.4 Hz, 1 H), 7.26 (dm, J = 7.2 Hz, 1 H), 4.27 (d, J = 4.1 Hz, 1 H), 3.22 (br m, 1 H), 2.73 (dd, J = 12.1, 5.9 Hz, 1 H), 2.37 (td, J = 12.3, 4.7 Hz, 1 H), 2.24 (m, 1 H), 1.99 (tdd, 12.7, 6.0, 2.3 Hz, 1 H), 1.95 (d, J = 10.6 Hz, 1 H), 1.55 (m, 1 H); mass spectrum (CI+) m/z 236.1434 [C17H18N (M+1) requires 236.1436].
Benzyl 6-(4-bromo-1-tosyl-1H-indol-3-yl)-5,6-dihydropyridine-1(2H)-carboxylate (38)
A mixture of allylamine (0.1 mL, 1.34 mmol) and aldehyde 10 (100 mg, 0.26 mmol) in THF (2 mL) containing crushed 4 Å molecular sieves (80 mg) was stirred at room temperature for 16 h. The sieves were removed by filtration, and the solvent was removed under reduced pressure. The crude imine was redissolved in THF (1.5 mL) and cooled to -78 °C, whereupon allylmagnesium bromide (1.12 M in Et2O, 0.26 mL, 0.29 mmol) was added slowly. The cooling bath was removed, and the reaction was stirred for 1 h at room temperature, whereupon it was recooled to -78 °C and benzyl chloroformate (97%, 56 μL, 0.38 mmol) added dropwise. The cooling bath was removed, and the mixture was stirred for 1 h. Saturated NaHCO3 (2 mL) was added, and the mixture was partitioned between water (20 mL) and Et2O (20 mL). The layers were separated, and the organic layer was washed with 1 M HCl (20 mL) and brine (20 mL). The organic phase was dried (MgSO4), filtered, and concentrated. The crude carbamate was then dissolved in CH2Cl2 (25 mL) containing 6 mol % of Grubbs II catalyst, and the solution was stirred for 17 h. The solvent was removed by flash chromatography, eluting with 5-20% EtOAc/hexanes to give 140 mg (95% from 10) of 38 as a pale yellow oil. 1H NMR (400 MHz, CDCl3) (rotamers) δ 7.91 (br m 1 H), 7.64 (app d, J = 6.8 Hz, 2 H), 7.44 (s, 1 H), 7.41-7.02 (m, 9 H), 6.33 (d, J = 6.4 Hz, 1 H), 5.87 (br s, 1 H), 5.77 (br m, 1 H), 5.15 (br s, 2 H), 4.31 (dd, J = 18.8, 3.2 Hz, 1 H), 3.75 (d, J = 18.8 Hz, 1 H), 2.79 (br d, J = 14.4 Hz, 1 H), 2.45 (br d, J = 14.4 Hz), 2.29 (br s, 3 H); 13C NMR (100 MHz, CDCl3) (rotamers) δ 155.7, 145.4, 136.4, 134.6, 130.0, 128.5 (br), 128.3, 128.0 (br), 126.9 (br), 125.6, 124.9, 124.2 (br), 113.0, 67.3, 45.0, 42.3, 29.8, 21.7; IR (neat) 1702, 1412, 1373, 1172, 1096, 1005 cm-1; mass spectrum (CI+) m/z 565.0795 [C28H26N2O4S79Br (M+1) requires 565.0797].
Indole 39
A microwave vial (CEM corp., 10 mL) containing tetrabutylammonium chloride (36 mg, 0.13 mmol), palladium(II) acetate (0.9 mg, 0.004 mmol), tri(o-tolyl)phosphine (2.4 mg, 0.008 mmol), and 38 (74 mg, 0.13 mmol) was sealed and flushed with argon, whereupon CH3CN (3.3 mL) and NEt3 (39 mg, 55 μL, 0.39 mmol) were added. The reaction was heated via microwave at 120 °C for 0.5 h. The catalyst was removed by filtering through a plug of silica gel that was washed with 50% EtOAc/hexanes. The combined filtrate and washings were concentrated, and the residue was purified by flash chromatography, eluting with 0-100% EtOAc/hexanes to give 46 mg (72%) of 39 as a colorless oil. 1H NMR (400 MHz, CDCl3) (rotamers) δ 7.79 (d, J = 8.4, 1 H), 7.66 (app dt, J = 10.6, 9.4 Hz, 2 H), 7.55-7.13 (m, 10 H), 6.98 (d, J = 7.2 Hz, 1 H), 6.79 (d, J = 8.4, 0.45 H), 6.70 (d, J = 8.2, 0.55 H), 5.54 (br s, 0.55 H), 5.45 (br s, 0.45 H), 5.37 (d, J = 11.9 Hz, 0.45 H), 5.30-5.10 (m, 2 H), 5.05 (ddd, J = 8.2, 6.5, 1.8 Hz, 0.55 H), 3.54 (m, 1 H), 2.33 (s, 3 H), 2.19 (ddd, J = 12.5, 5.3, 3.1 Hz, 1 H), 2.08 (ddd, J = 12.7, 4.3, 2.4 Hz, 0.55 H), 2.01 (ddd, 12.5, 4.3, 2.4 Hz, 0.45 H); 13C NMR (100 MHz, CDCl3) (rotamers) δ 152.9, 152.6, 145.0, 144.9, 136.6, 136.5, 136.13, 136.07, 135.8, 135.6, 133.7, 133.6, 130.03, 130.00, 129.0, 128.9, 128.8, 128.7, 128.4, 128.2, 128.1, 127.9, 127.1, 126.9, 125.6, 125.5, 123.7, 123.4, 121.3, 120.8, 120.7, 120.6, 118.8, 118.7, 111.6, 111.5, 110.3, 109.6, 68.2, 67.9, 43.9, 43.7, 30.4, 30.2, 30.0, 21.7; IR (neat) 1706, 1640, 1402, 1340, 1176, 1110, 1017, 951, 746, 702, 662 cm-1; mass spectrum (CI+) m/z 485.1532 [C28H25N2O4S (M+1) requires 485.1535].
Allyl-[1-(2-bromophenyl)-pent-4-enyl]-carbamic acid benzyl ester (40)
A solution of benzyl chloroformate (92 mg, 77 μL, 0.54 mmol) and imine 16 (112 mg, 0.50 mmol) in dry THF (2 mL) was heated at 60 °C for 1 h. The mixture was then cooled to -78 °C, whereupon a solution of butenylzinc chloride (0.8M, 0.75 mL, 0.60 mmol)30 was added slowly. The bath was removed, and the reaction stirred at room temperature for 2 h. Saturated aqueous NH4Cl (1 mL) was added, and the mixture was partitioned between water (20 ml) and Et2O (20 mL). The layers were separated, and the aqueous layer was extracted with Et2O (10 mL). The combined organic layers were washed with NaHCO3 solution (20 ml) and brine (20 mL), dried (MgSO4), filtered, and concentrated. The crude product was purified by flash chromatography, eluting with 0-30% EtOAc/hexanes to give 146 mg (70%) of diene 40 as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.55 (d, J = 8.0 Hz), 7.48-7.27 (m, 6 H), 7.14 (app t d, J = 7.8, 1.8 Hz, 1 H), 5.80 (br s, 1 H), 5.56 (br m, 1 H), 5.42 (br m, 1 H), 5.20 (br s, 2 H), 4.96 (br m, 2 H), 4.84 (br m, 2 H), 3.61 (br s, 2 H), 2.08 (br m, 4 H); 13C NMR (100 MHz, CDCl3) δ (rotamer 1) 156.2, 138.5, 137.7, 134.8, 133.6, 129.3, 128.5, 128.0 (br), 127.3, 126.4, 116.2, 115.2, 67.4, 58.7 (br), 46.7, 31.5, 30.7; IR (neat) 2937, 1698, 1406, 1248, 1137, 914, 753, 697 cm-1; mass spectrum (ESI+) m/z 414.1063 [C22H25NO279Br (M+1) requires 414.1071].
2-(2-Bromophenyl)-2,3,4,7-tetrahydroazepine-1-carboxylic acid benzyl ester (41)
A solution of 40 (100 mg, 0.24 mmol) in CH2Cl2 (12 mL) containing Grubbs II catalyst (20 mg, 0.024 mmol) was stirred at room temperature for 24 h. The solvent was evaporated under reduced pressure, and the crude product was purified by flash chromatography, eluting with 0-25% EtOAc/hexanes to give 86 mg (92%) of 41 as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.53 (m, 1 H), 7.38-7.16 (comp, 6 H), 7.10 (m, 1 H), 6.97 (m, 1 H), 5.51 (dd, J = 12.3, 3.8 Hz, 0.4 H), 5.39 (dd, J = 12.3, 3.8 Hz, 0.6 H), 5.15 (d, J = 12.3 Hz, 0.4 H), 5.11 (d, J = 12.7 Hz, 0.4 H), 5.05 (d, J = 13.0 Hz, 0.6 H), 5.00 (d, J = 12.7 Hz, 0.6 H), 4.70 (dd, J = 16.7, 7.2 Hz), 4.50 (dd, J = 16.8, 6.2 Hz, 0.4 H), 4.18 (m, 1 H), 2.55-2.20 (comp, 3 H), 2.08 (m, 1 H); 13C NMR (100 MHz, CDCl3) δ 156.2, 143.4, 142.7, 137.0, 136.6, 133.5, 133.2, 131.8, 131.5, 128.6, 128.5, 128.3, 128.0, 127.8, 127.7, 127.6, 127.1, 127.0, 125.7 (br), 122.9, 122.4, 67.3, 67.1, 61.6, 41.7, 31.4, 31.1, 29.9, 29.4; IR (neat) 2937, 1702, 1411, 1242, 1208, 1112, 748 cm-1; mass spectrum (CI+) m/z 386.0753 [C20H21NO279Br (M+1) requires 386.0756].
12-Azatricyclo[6.3.2.02,7]trideca-2(7),3,5,9-tetraene-12-carboxylic acid benzyl ester (42) and 12-Azatricyclo[6.3.2.02,7]trideca-2(7),3,5,9-tetraene-12-carboxylic acid benzyl ester (43)
A microwave vial (CEM Corp., 10 mL) containing tetrabutylammonium chloride (76 mg, 0.27 mmol), [Pd(P(t-Bu)3]2 (6.9 mg, 0.014 mmol), and 41 (104 mg, 0.27 mmol) was sealed and flushed with argon, whereupon CH3CN (4.5 mL) and (i-Pr)2NEt (70 mg, 95 μL, 0.54 mmol) were added. The reaction was heated via microwave for 1 h at 120 °C. The catalyst was removed by filtering through a plug of silica gel, which was washed with 50% EtOAc/hexanes (20 mL). The combined filtrate and washings were concentrated, and the residue was purified by flash chromatography, eluting with 0-25% EtOAc/hexanes to give 43 (14 mg, 17%) and 42 (47 mg, 57%) as colorless oils. 42: 1H NMR (400 MHz, CDCl3) (rotamers) δ 7.43-7.14 (comp, 8 H), 7.10 (m, 1 H), 6.07 (m, 1 H), 5.41-5.10 (comp, 4 H), 4.11 (dd, J = 11.2, 2.0 Hz, 0.4 H), 4.06 (dd, J = 11.0, 2.0 Hz, 0.6 H), 3.43 (m, 1 H), 3.38 (dm, J = 8.4 Hz, 0.4 H), 3.31 (dm, J = 8.4 Hz, 0.6 H), 2.88 (dm, J = 18.6 Hz, 0.6 H), 2.78 (dm, J = 18.4 Hz, 0.4 H), 2.28 (m, 1 H); 13C NMR (100 MHz) (rotamers) δ 155.9, 155.6,, 142.8, 142.6, 137.6, 137.4, 137.01, 136.97, 130.2, 130.0, 129.0, 128.7, 128.60, 128.56, 128.1, 128.1, 128.0, 127.9, 127.7, 127.59, 127.57, 127.1, 126.9, 126.8, 125.9, 125.7, 124.3, 124.1, 67.3, 67.1, 55.0, 54.8, 52.0, 51.2, 38.05, 38.02, 36.8, 36.2; IR (neat) 3025, 2881, 1697, 1411, 1331, 1219, 1115, 1094, 753, 697 cm-1; mass spectrum (CI+) m/z 306.1489 [C20H20NO2 (M+1) requires 306.1494]. 43: 1H NMR (400 MHz, CDCl3) δ 7.44-7.14 (comp, 8 H), 7.09 (m, 1 H), 6.51 (d, J = 9.2 Hz, 0.3 H), 6.43 (d, J = 9.2 Hz, 0.7 H), 5.61 (d, J = 6.1 Hz, 0.7 H), 5.45 (d, J = 6.3 Hz, 0.3 H), 5.28-5.04 (comp, 3 H), 3.09 (m, 1 H), 2.45 (m, 1 H), 2.09 (m, 1 H), 1.85 (m, 1 H); 13C NMR (100 MHz, CDCl3) δ 153.3, 145.3, 136.2, 135.1, 128.8. 128.6, 128.3, 128.2, 128.1, 127.4, 127.0, 126.9, 126.8, 125.4, 124.7, 124.5, 124.3, 112.1, 111.3, 68.0, 53.9, 53.7, 36.8, 36.7, 30.8, 30.6, 27.6; IR (neat) 1702, 1388, 1321, 1272, 1221, 1006, 754, 697 cm-1; mass spectrum (CI+) m/z 306.1495 [C20H20NO2 (M+1) requires 306.1494].
Methyl 2-(1-(N-allylacetamido)but-3-enyl)benzoate (45)
A mixture containing freshly distilled methyl formyl benzoate (44)31 (135 mg, 0.83 mmol), allyl amine (0.06 mL, 46 mg, 0.80 mmol) and activated 4 Å molecular sieves (∼500 mg) in CH2Cl2 (2.6 mL) was stirred for 12 h. Freshly distilled acetyl chloride (0.06 mL, 66 mg, 0.84 mmol) was added, and the reaction was stirred for 1 h. The reaction was then cooled to -78 °C, freshly prepared allylzinc bromide (2.1 M in THF, 0.5 mL, 1.05 mmol) was added, and the reaction was stirred for 1.5 h. The dry ice bath was removed, and the reaction was filtered through a Celite pad that was washed with CH2Cl2 (3 × 5 mL). The combined filtrate and washings were washed with saturated aqueous NH4Cl (10 mL). The aqueous layer was backwashed with CH2Cl2 (2 × 5 mL), and the combined organics were dried (MgSO4), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography, eluting with 40% EtOAc/hexanes to give 189 mg (82%) of amide 45 as a viscous colorless oil. 1H NMR (500 MHz, DMSO, 100 °C) δ 7.62 (d, J = 7.9 Hz, 1 H), 7.6 (d, J = 7.2, 1 H), 7.52 (t, J = 7.5 Hz, 1 H), 7.38 (t, J = 7.5 Hz, 1H), 6.00 (br, 1 H), 5.73 (dddd, J = 17.0, 10.2, 6.7, 6.7, 1 H), 5.51-5.43 (m, 1 H), 5.09 (d, J=17.2 Hz, 1 H), 5.00 (d, J =10.2, 1 H), 4.88 (comp, 2 H), 3.80 (s, 3 H) 3.82-3.68 (comp, 2 H), 2.79-2.69 (comp, 2 H), 2.00 (s, 3 H); 13C NMR (125 MHz, DMSO, 130 °C) δ 169.3, 167.6, 138.1, 134.7, 134.5, 132.0, 130.2, 128.4, 127.7, 126.7, 116.3, 114.9, 54.0, 51.3, 45.8, 35.3, 21.1; IR (neat) 1722, 1649, 1405, 1270, 918, 749, cm-1; mass spectrum (CI) m/z 288.1601 [C17H22NO3 (M+1) requires 288.1600].
Methyl 2-(1-acetyl-1,2,3,6-tetrahydropyridin-2-yl)benzoate (46)
Grubbs II catalyst (4 mg, 0.0053 mmol) was added to a solution of 45 (152 mg, 0.53 mmol) in CH2Cl2 (20 mL) and the reaction was heated under reflux for 3 h. The reaction was cooled the room temperature and concentrated under reduced pressure. The residue was purified by flash chromatography, eluting with 50% EtOAc/hexanes to give 129 mg (94%) of amide 46 as a tan solid, mp 104-106 °C. 1H NMR (500 MHz, DMSO, 130 °C) δ 7.70 (d, J = 7.6, 1.4 Hz, 1 H), 7.46 (t, J = 7.6 Hz) 7.33 (t, J = 7.6 Hz, 1 H), 7.25 (d, J = 7.6 Hz, 1H), 6.11 (br, 1 H), 5.85 (ddd, J = 6.2, 3.1, 3.1, 1 H), 5.81-5.77 (m, 1 H) 4.23 (dd, J = 18.4, 2.3 Hz, 1 H), 3.86 (s, 3 H), 3.82 (d, J = 18.4 Hz, 1 H), 2.74-2.68 (m, 1 H), 2.34 (dd, J = 17.5, 6.2 Hz, 1 H), 1.97 (m, 3 H); 13C NMR (125 MHz, DMSO, 130 °C) δ 168.8, 167.2, 142.8, 130.5, 129.1, 128.9, 130.2, 126.1, 126.0, 123.8, 122.5, 51.2, 48.2, 41.4, 29.1, 20.7; IR (neat) 1722, 1649, 1405, 1270, 918, 749, cm-1; mass spectrum (CI) m/z 260.1291 [C15H18NO3 (M+1) requires 260.1287].
1,12b-Dihydrobenzo[c]pyrido[1,2-a]azepine-6,8(4H,7H)-dione (47)
NaHMDS (1.85 M in THF, 0.93 mL, 1.72 mmol) was added to a solution of 46 (179 mg, 0.69 mmol) in THF (30 mL) and the reaction was stirred for 2 h at room temperature. Saturated aqueous NH4Cl (10 mL) was then added. The layers were separated, and the aqueous layer was backwashed with CH2Cl2 (2 × 5 mL). The organics were combined and dried (MgSO4), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography, eluting with 50% EtOAc/hexanes to give 106 mg (67%) of amide 47 as a tan solid, mp 151.5-153 °C. 1H NMR (400 MHz, CDCl3) δ 7.95 (dd, J = 7.7, 1.4 H, 1 H), 7.50 (td, J = 7.7, 1.4 Hz, 1 H), 7.38 (td, J = 7.7, 1.1 Hz, 1 H), 7.28 (d, J = 7.7 Hz, 1H), 6.15-6.11 (m, 1 H), 5.84 (ddd, J = 6.3, 3.1, 3.1, 1 H), 5.69 (d, J = 5.6 Hz, 1 H), 4.43 (d, J = 17.3 Hz, 1 H), 4.19-4.12 (m, 1 H), 3.79 (d, J = 17.3 Hz, 1 H), 3.55-3.47 (m, 1 H), 2.95-2.86 (m, 1 H), 2.82 (dd. J = 17.4, 5.6 Hz, 1 H); 13C NMR (100 MHz, CDCl3) δ 192.1, 169.8, 142.2, 136.5, 134.1, 131.3, 129.2, 126.0, 124.5, 122.6, 54.7, 51.6, 41.6, 26.0; IR (neat) 2906, 2853, 1681, 1650, 1409, 1248, 1137, 923, cm-1; mass spectrum (CI) m/z 228.1027 [C14H14NO2 (M+1) requires 228.1025].
Methyl 3-(N-allylacetamido)-2,2-dimethyl-3-(2-vinylphenyl)propanoate (50)
TMSOTf (23.0 μL, 28 mg, 0.13 mmol) was added to a solution of 2-vinylbenzaldehyde32 (172 mg, 1.30 mmol) and bis(trimethylsilyl)allylamine (0.33 mL, 269 mg, 1.34 mmol) in CH2Cl2 (3.2 mL), and the reaction was stirred for 30 min at room temperature. Silyl ketene acetal 49 (0.53 mL, 455 mg, 2.60 mmol) and freshly distilled acetyl chloride (0.11 mL, 121 mg, 1.55 mmol) were added, and the reaction was stirred for 19 h at room temperature. The reaction was partitioned between CH2Cl2 (5 mL) and saturated aqueous NaHCO3 (5 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (2 × 5 mL). The combined organic layers were dried (MgSO4), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography, eluting with 25% EtOAc/hexanes to give 328 mg (80%) of amide 50 as a viscous yellowish oil. 1H NMR (400 MHz, CDCl3) δ 7.47 (d, J = 7.5 Hz, 1 H), 7.38 (d, J = 7.9 Hz, 1 H), 7.26 (t, J = 7.5 Hz, 1H), 7.20 (dt, J = 7.9, 1.5 Hz, 1H), 7.04 (dd, J = 17.1, 10.9 Hz, 1 H), 6.36 (s, 1 H), 5.57 (dd, J = 17.1, 1.7 Hz, 1 H), 5.32 (dd, J = 10.9, 1.7 Hz, 1 H), 5.07-4.98 (m, 1 H), 4.80 (dd, J = 7.5, 1.4 Hz, 1 H), 4.77 (dd, J = 14.2, 1.4 Hz, 1 H), 3.78-3.79 (comp, 2 H), 3.57 (s, 3 H), 2.11 (s, 3 H), 1.45 (s, 3 H), 1.33 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 178.3, 173.1, 140.4, 136.2, 136.1, 135.4, 128.8, 128.5, 128.2, 127.8, 117.6, 117.1, 58.9, 52.8, 50.6, 48.5, 27.5, 24.9, 23.5; IR (neat) 2981, 1729, 1651, 1456, 1396, 1252, 1147, 918, 779 cm-1; mass spectrum (CI) m/z 316.1912 [C19H25NO3 (M+1) requires 316.1913].
1-Allyl-5,5-dimethyl-6-(2-vinylphenyl)piperidine-2,4-dione (51)
NaHMDS (540 mg, 2.95 mmol) was added to a solution of 50 (308 mg, 0.98 mmol) in THF (38 mL) and the reaction was stirred for 2 h at room temperature. Saturated NH4Cl (15 mL) and H2O (15 mL) were then added. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (2 × 10 mL). The combined organic layers were washed with saturated aqueous NaCl (20 mL), dried (MgSO4), filtered, and concentrated. The residue was purified by flash chromatography, eluting with 30% EtOAc/hexanes to give 240 mg (87%) of 51 as a light brownish solid. 1H NMR (400 MHz, CDCl3) δ 7.43 (dd, J = 7.5, 1.7 Hz, 1 H), 7.29-7.21 (comp, 2 H), 6.99 (dd, J = 17.1, 10.9 Hz, 1 H), 6.74 (dd, J = 7.7, 1.2 Hz, 1 H), (dddd, J = 17.1, 9.9, 8.9, 4.4 Hz, 1H), 5.64 (dd, J = 17.1, 1.0 Hz, 1 H), 5.42 (dd, J = 10.9, 1.4 Hz, 1 H), 5.28 (d, J = 9.9 Hz, 1 H), 5.23 (d, J = 17.1 Hz, 1 H), 4.80 (ddt, J = 14.7, 4.4, 1.7 Hz, 1 H), 4.74 (s, 1 H), 3.63 (d, J = 21.2 Hz, 1 H), 3.5 (d, J = 21.2 Hz, 1 H), 2.96 (dd, J = 14.7, 8.9 Hz, 1 H), 1.42 (s, 3 H), 0.80 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 207.3, 166.9, 139.2, 135.9, 135.1, 133.3, 129.7, 129.5, 128.1, 125.8, 120.6, 119.5, 64.2, 50.7, 48.5, 45.4, 26.6, 20.3; IR (neat) 2979, 1723, 1650, 1463, 1282, 931, 755 cm-1; mass spectrum (CI) m/z 284.1658 [C18H21NO2 (M+1) requires 284.1651].
1,1-Dimethyl-1,12b-dihydrobenzo[c]pyrido[1,2-a]azepine-2,4(3H,6H)-dione (52)
Grubbs II catalyst (4 mg, 0.0053 mmol) was added to a solution of 51 (76 mg, 0.27 mmol) in CH2Cl2 (10 mL), and the reaction was heated under reflux for 22 h. The reaction was then cooled to room temperature and concentrated under reduced pressure. The residue was purified by flash chromatography, eluting with 40% EtOAc/hexanes to give 55 mg (80%) of 52 as a brownish solid. 1H NMR (400 MHz, CDCl3) δ 7.29-7.24 (comp, 2 H), 7.15 (dt, J = 8.2, 4.2 Hz, 1 H) 6.97 (d, J = 7.9 Hz, 1 H), 6.60 (d, J = 12.3 Hz, 1 H), 5.95 (dt, J = 12.3, 3.4 Hz, 1 H), 5.30 (d, J = 19.2 Hz, 1 H), 4.57 (s, 1 H), 3.85 (d, J = 19.2 Hz, 1 H), 3.32 (d, J = 20.5 Hz, 1 H), 3.17 (d, J = 20.5 Hz, 1 H), 1.45 (s, 3 H), 1.30 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 209.4, 166.6, 137.4, 136.1, 132.2, 130.3, 129.9, 128.8, 127.6, 125.3, 65.2, 49.2, 47.8, 47.0, 26.2, 21.5; IR (neat) 3426, 2974, 2244, 1722, 1660, 1462, 1376, 1310, 1269,1181, 952, 734 cm-1; mass spectrum (CI) m/z 256.1328 [C16H17NO2 (M+1) requires 256.1337].
Methyl 2-(1-(N-(prop-2-ynyl)acetamido)but-3-enyl)benzoate (53)
A mixture containing freshly distilled methyl formyl benzoate (44) (146 mg, 0.89 mmol), propargyl amine (0.06 mL, 48 mg, 0.87 mmol) and activated 4 Å molecular sieves (∼ 500 mg) in CH2Cl2 (2.2 mL) was stirred for 12 h. Freshly distilled acetyl chloride (65 μL, 72 mg, 0.91 mmol) was added, and the reaction was stirred for 1 h. The reaction was then cooled to -78 °C, freshly prepared allylzinc bromide (2.2 M in THF, 0.46 mL, 1.01 mmol) was added, and the reaction was stirred for 1 h. The dry ice bath was removed, and the reaction was filtered through a Celite pad that was washed with CH2Cl2 (3 × 5 mL). The combined filtrate and washings were washed with saturated aqueous NH4Cl (10 mL). The aqueous layer was backwashed with CH2Cl2 (2 × 5 mL), and the combined organics were dried (MgSO4), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography, eluting with 25% EtOAc/hexanes to give 158 mg (63%) of amide 53 as a viscous colorless oil. 1H NMR (500 MHz, DMSO, 120 °C) δ 7.67 (d, J = 8.0 Hz, 1H), 7.65 (d, J = 9.9 Hz, 1 H), 7.55 (t, J = 7.6 Hz, 1 H), 7.39 (t, J = 7.6 Hz, 1 H), 6.02 (t, J = 7.3 Hz, 1 H), 5.77 (dddd, J = 17.1, 10.4, 6.7, 6.7 Hz, 1 H), 5.12 (d, J = 17.1 Hz, 1 H), 5.02 (d, J = 10.2 Hz, 1 H), 4.03 (d, J = 18.4 Hz, 1 H), 3.80 (s, 3 H), 3.80-3.78 (comp, 1 H), 2.82-2.79 (comp, 3 H), 2.08 (s, 3 H); 13C NMR (125 MHz, DMSO, 120 °C) δ 169.2, 167.4, 138.0, 134.3, 131.5, 130.5, 128.8, 127.6, 126.9, 116.5, 80.1, 72.5, 54.3, 51.4, 35.2, 32.4, 21.2; IR (neat) 1721, 1654, 1406, 1269, 913, 747 cm-1; mass spectrum (CI) m/z 286.1448 [C17H20NO3 (M+1) requires 286.1443].
(E)-3-Styryl-1,12b-dihydrobenzo[c]pyrido[1,2-a]azepine-6,8(4H,7H)-dione (54)
The Hoveyda-Grubbs II catalyst (0.012 g, 0.018 mmol) was added to a solution of amide 53 (0.077 mg, 0.26 mmol) and freshly distilled styrene (0.46 mL, 42 mg, 4.01 mmol) in CH2Cl2 (11 mL) and the reaction was heated under reflux for 24 h. The reaction was cooled to room temperature and DMSO (0.25 mL) was added. The reaction was stirred for 20 h and then concentrated under reduced pressure. The residue was quickly purified by flash chromatography, eluting with 50% EtOAc/Hexane to give a brownish oil, which was immediately dissolved in THF (11 mL). NaHMDS (2M in THF, 0.26 mL, 0.52 mmol) was added, and the reaction was stirred for 5 min upon which sat. aqueous NH4Cl (5 mL) was added. The organic phase was removed, and the aqueous layer was washed with Et2O (2 × 5 mL). The combined organic layers were dried (MgSO4), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography, eluting with 30% EtOAc/hexanes to give 52 mg (58%) of amide 54 as a solid, contaminated with approximately 3% of the terminal vinyl derivative. 1H NMR (400 MHz, CDCl3) δ 7.99 (dd, J = 7.9, 1.0 Hz, 1H), 7.49 (td, J = 7.5, 1.4 Hz, 1H), 7.41-7.20 (comp, 7H), 6.79 (d, J = 16.4 Hz, 1H), 6.49 (d, J = 16.4 Hz, 1H), 6.24 (m, 1H), 5.72 (d, J = 6.2 Hz, 1H), 4.46 (d, J = 17.4 Hz, 1H), 4.43 (d, J = 18.1 Hz, 1H), 3.88 (d, J = 17.4 Hz, 1H), 3.84 (d, J = 18.1 Hz, 1H), 3.11-3.06 (m, 1H), 2.98 (dd, J = 18.3, 5.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 192.0, 169.9, 142.0, 137.8, 136.6, 134.3, 133.4, 131.4, 129.6, 129.4, 128.6, 128.5, 128.1, 127.2, 126.0, 123.8, 55.0, 51.8, 41.4, 26.9; IR (neat) 2358, 1680, 1641, 1596, 1410, 1247, 957 cm-1; mass spectrum (CI) m/z 330.1492 [C22H20NO2 (M+1) requires 330.1494].
N-Allyl-N-(1-(2-bromophenyl)-2-formylethyl)acetamide (56)
TMSOTf (15.5 μL, 19 mg, 0.086 mmol) was added to a solution of 2-bromobenzaldehyde (0.1 mL, 158 mg, 0.86 mmol) and bis(trimethylsilyl)allylamine (0.22 mL, 179 mg, 0.89 mmol) in CH2Cl2 (2.2 mL), and the reaction was stirred for 1 h at room temperature. The reaction was cooled to 0 °C and silyl enol ether 5536 (420 mg, 2.66 mmol) and freshly distilled acetyl chloride (0.07 mL, 77 mg, 0.98 mmol) were then added, and the reaction was stirred for 10 min. The ice bath was removed and the reaction was stirred for 40 h at room temperature. The reaction was concentrated under reduced pressure. The residue was purified by flash chromatography,, eluting with 50% EtOAc/hexanes to give 207 mg (77%) of amide 56 as a brownish solid, mp = 56-58 °C. 1H NMR (500 MHz, DMSO, 100 °C) δ 9.66 (s, 1 H), 7.64 (d, J = 7.8 Hz, 1H), 7.54 (d J = 7.8 Hz, 1H), 7.40 (t, J = 7.8 Hz, 1H), 7.26 (t, J = 7.8 Hz, 1H), 6.02 (br, 1H), 5.52-5.45 (m, 1H), 4.94-4.92 (comp, 2H), 3.81 (dd, J = 17.2, 5.2 Hz, 2H), 3.73 (dd, J = 17.2, 5.7 Hz), 3.24-3.19 (m, 1H), 3.10 (dd, J = 16.4, 7.6 Hz, 1H), 2.08 (s, 3H); 13C NMR (125 MHz, DMSO, 130 °C) δ 199.2, 169.3, 137.1, 134.0, 132.5, 129.2, 129.0, 127.0, 124.0, 115.3, 52.7, 46.2, 44.9, 21.0; IR (neat) 1720, 1645, 1405, 1024, 913, 749 cm-1; mass spectrum (CI) m/z 310.0446 [C14H1679BrNO2 (M+1) requires 310.0443] 312.0426.
1-(Hexahydro-6-(2-bromophenyl)-1-methyl-1H-pyrrolo[3,2-c]pyridin-5(6H)-yl)ethanone (57)
Sarcosine (84 mg, 0.95 mmol) was added to a solution of 56 (166 mg, 0.54 mmol) in toluene (1 mL). The reaction was heated under reflux for 3 h. The reaction was cooled to room temperature and then was partitioned between water (5 mL) and CH2Cl2 (5 mL). The organic layer was removed and the aqueous layer was washed with CH2Cl2 (2 × 5 mL). The combined organic layers were dried (MgSO4), filtered and concentrated under reduced pressure. The residue was purified by flash chromatography, eluting with 25% MeOH/EtOAc to give 118 mg (65%) of 57 as a brownish gum. 1H NMR (500 MHz, DMSO, 130 °C) δ 7.54 (d, J = 7.9 Hz, 1 H), 7.33-7.27 (comp, 2 H), 7.14 (t, J = 7.0 Hz, 1 H), 4.97 (dd, J = 12.8, 4.8 Hz, 1 H), 4.20 (br, 1 H), 3.14 (br, 1 H), 2.88-2.84 (m, 1 H), 2.50-2.47 (m, 1 H), 2.38 (br, 1 H), 2.30 (s, 3 H), 2.27-2.23 (comp, 2 H), 1.94-1.84 (comp, 4 H), 1.51-1.38 (comp, 2 H); 13C NMR (125 MHz, DMSO, 130 °C) δ 168.4, 143.1, 131.9, 127.5, 127.5, 125.8, 120.2, 61.0, 54.2, 37.9, 32.6, 27.3, 20.8, 19.9; IR (neat) 1648, 1413, 1243, 1023, 757 cm-1; mass spectrum (CI) m/z 337.0912 [C16H2179BrN2O (M+1) requires 337.0915] 339.0902.
1-((3aR,6S,7aS)-6-(2-Bromophenyl)-tetrahydro-1-methylisoxazolo[4,3-c]pyridin-5(1H,3H,6H)-yl)ethanone (58)
N-methyl hydroxylamine hydrochloride (70 mg, 0.84 mmol) and Et3N (0.24 mL, 174 mg, 1.72 mmol) were added to a solution of 55 (166 mg, 0.54 mmol) in toluene (10 mL). The reaction was heated to reflux for 5 h. The reaction was cooled to room temperature and then concentrated under reduced pressure. The residue was then partitioned between water (5 mL) and CH2Cl2 (5 mL). The organic phase was removed, and the aqueous layer was washed with CH2Cl2 (3 × 5 mL). The combined organic layers were dried (MgSO4), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography, eluting with EtOAc to give 158 mg (87%) of amide 58 as a pale orange gum. 1H NMR (500 MHz, DMSO, 130 °C) δ 7.55 (d, J = 7.9 Hz, 1 H), 7.34 (t, J = 7.1 Hz, 1 H), 7.28 (d, J = 7.1 Hz, 1 H), 7.16 (t, J = 7.3 Hz, 1 H), 5.04 (dd, J = 12.9, 5.0 Hz, 1 H), 4.10 (br s, 1 H), 4.04 (t, J = 8.3 Hz, 1 H), 3.46 (dd, J = 8.7, 5.0 Hz, 1H), 3.26 (br s, 1 H), 3.06-3.00 (m, 1 H), 2.95 (s, 1 H) 2.58 (s, 3 H), 2.20 (dt, J = 13.7, 5.2 Hz, 1 H) 1.86 (s, 3 H) 1.59 (q, J = 12.7 Hz, 1 H); 13C NMR (100 MHz, CDCl3, mixture of rotomers, major rotomer reported) δ 172.2, 143.1, 134.0, 130.0, 129.6, 127.0, 122.0, 69.1, 65.3, 57.1, 44.5, 43.4, 40.3, 33.8, 22.6; IR (neat) 1651, 1417, 1361, 1253, 1026, 998, 967, 917, 757, 726, 642 cm-1; mass spectrum (CI) m/z 339.0705 [C15H1979BrN2O2 (M+1) requires 339.0708] 341.0692.
14,14a-Dihydrobenzo[f]pyrido[1,2-d][1,2,3]triazolo[1,5-a][1,4]diazepine-11,13(9H,12H)-dione (61)
A mixture containing propargyl amine (0.06 mL, 48 mg, 0.87 mmol), 2-azidobenzaldehyde39 (116 mg, 0.79 mmol), activated 4 Å molecular sieves (∼500 mg) in CH3CN (2 mL) was stirred for 12 h. Freshly distilled acetyl chloride (0.07 mL, 77 mg, 0.98 mmol) and silyl ketene acetal 11 (286 mg, 2.01 mg) were added, and the reaction was stirred for 28 h. The reaction was then filtered through Celite washing with CH2Cl2 (3 × 5 mL), and the combined washings were concentrated under reduced pressure. The residue was purified by flash chromatography, eluting with EtOAc to give 181 mg (76%) of triazole 61 as a viscous colorless oil. 1H NMR (500 MHz, DMSO, 130 °C) δ 7.94 (d, J = 7.9 Hz, 1 H), 7.91 (s, 1 H), 7.63 (t, J = 7.9 Hz, 1 H), 7.58 (d, J = 7.3 Hz, 1 H), 7.51 (t, J = 7.3 Hz, 1 H), 5.84 (s, 1 H), 5.21 (d, J = 15.5 Hz, 1 H), 4.28 (d, J = 15.5 Hz, 1H), 3.43 (s, 3 H), 2.41 (dd, J = 15.3, 6.1 Hz, 1 H), 2.21 (s, 3 H), 1.80 (m, 1 H); 13C NMR (125 MHz, DMSO, 130 °C) δ 168.7, 168.1, 134.1, 132.2, 132.1, 131.0, 129.4, 128.5, 122.6, 55.1, 50.4, 38.0, 36.6, 20.8; IR (neat) 1738, 1650, 1500, 1411, 1305, 1234, 1168, 980, 767 cm-1; mass spectrum (CI) m/z 301.1304 [C15H17N4O3 (M+1) requires 301.1301].
2-Allyl-3-(2-bromophenyl)-2,3,4,4a,8,8a-hexahydroisoquinolin-1(7H)-one (66)
TMSOTf (15.5 μL, 19 mg, 0.086 mmol) was added to a solution of 2-bromobenzaldehyde (0.1 mL, 158 mg, 0.86 mmol) and bis(trimethylsilyl)allylamine (0.22 mL, 179 mg, 0.89 mmol) in CH2Cl2 (2.2 mL), and the reaction was stirred for 30 min at room temperature. Pentadienyl TMS 6342 (303 mg, 2.16 mmol) and freshly distilled acryloyl chloride (85 μL, 94 mg, 1.05 mmol) were then added and the reaction stirred for 22 h. The reaction was partitioned between CH2Cl2 (5 mL) and saturated aqueous NaHCO3 (5 mL). The organic phase was removed, and the aqueous layer was washed with CH2Cl2 (3 × 4 mL). The combined organic layers were dried (MgSO4), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography, eluting with 15% EtOAc/hexanes to give 251 mg (84%) of amide 66 as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.51 (d, J = 7.6 Hz, 1 H), 7.29 (t, J = 7.6 Hz, 1 H), 7.16 (d, J = 7.6 Hz, 1 H), 7.11 (t, J = 7.6 Hz, 1 H), 5.74-5.66 (comp, 2 H), 5.55-5.52 (m, 1 H), 5.08 (d, J = 10.0 Hz, 1 H), 4.97 (dd, J = 10.1, 5.4 Hz, 1 H), 4.87 (d, J = 17.2 Hz, 1 H), 4.66 (dd, J = 14.8, 4.2 Hz, 1 H), 2.94 (dd, J = 14.8, 7.7 Hz, 1 H), 2.7 (d, J = 10.9 Hz, 1 H), 2.56-2.55 (m, 1 H), 2.21-2.08 (comp, 4 H), 1.85-1.77 (m, 1 H), 1.64-1.56 (m, 1 H); 13C NMR (125 MHz, CDCl3) δ 174.9, 141.2, 133.8, 133.0, 129.7, 129.2, 128.9, 128.8, 128.8, 123.5, 118.9, 60.4, 47.6, 42.0, 35.5, 32.6, 25.9, 23.6; IR (neat) 2933, 1651, 1568, 1410, 1338, 1283, 1188, 1024, 925, 760 cm-1; mass spectrum (CI) m/z 346.0799 [C18H2079BrNO2 (M+1) requires 346.0807], 348.0784.
2-Allyl-2,3,4,4a,8,8a-hexahydro-3-(3,4-dimethoxyphenyl)isoquinolin-1(7H)-one (67)
TMSOTf (12 μL, 55 mg, 0.066 mmol) was added to a solution of 3,4-dimethoxybenzaldehyde (62) (111 mg, 0.67 mmol) and bis(trimethylsilyl)allylamine (0.17 mL, 139 mg, 0.69 mmol) in CH2Cl2 (1.7 mL), and the reaction was stirred for 30 min at room temperature. Freshly distilled acryloyl chloride (65 μL, 72 mg, 0.80 mmol) and pentadienylsilane 63 (214 mg, 1.53 mmol) and were then added, and the reaction was stirred for 24 h at room temperature. The reaction was partitioned between CH2Cl2 (5 mL) and saturated aqueous NaHCO3 (5 mL). The organic phase was removed, and the aqueous layer was extracted with CH2Cl2 (2 × 5 mL). The combined organic layers were dried (MgSO4), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography, eluting with 40% EtOAc/hexanes to give 172 mg (78%) of amide 67 (6:1 inseparable mixture of diastereomers) as a viscous colorless oil. 1H NMR (400 MHz, CDCl3, major diastereomer) δ 6.82 (d, J = 8.2 Hz, 1 H), 6.74 (dd, J = 8.2, 2.0 Hz, 1 H), 6.66 (d, J = 1.7 Hz, 1 H), 5.78-5.65 (comp, 2 H), 5.58-5.54 (m, 1H), 5.08 (dd, J = 10.2, 1.0 Hz, 1 H), 4.89 (dd, J = 17.1, 1.0 Hz, 1 H), 4.62 (dd, J = 15.0, 4.4 Hz, 1 H), 4.42 (dd, J = 10.9, 4.8 Hz, 1 H), 3.88 (s, 3 H), 3.86 (s, 3 H), 3.07 (dd, J = 15.0, 1.0 Hz, 1 H), 2.72-2.68 (m, 1 H), 2.58-2.52 (m, 1 H), 2.24-2.14 (comp, 3 H), 2.12-2.04 (comp, 2 H), 1.89-1.74 (comp, 2 H); 13C NMR (100 MHz, CDCl3, major diastereomer) δ 174.2, 149.5, 148.8, 134.2, 133.2, 128.7, 128.1, 119.6, 117.3, 111.3, 109.8, 61.3, 56.1, 56.1, 46.5, 41.5, 37.1, 32.3, 25.5, 23.3; IR (neat) 2934, 1634, 1516, 1442, 1410, 1262, 1138, 1027, 928; mass spectrum (CI) m/z 328.1913 [C20H26NO3 (M+1) requires 328.1913].
Epoxy isoindolone 71
TMSOTf (21 μL, 26 mg, 0.12 mmol) was added to a solution of furfural (0.1 mL, 116 mg, 1.21 mmol) and bis(trimethylsilyl)allylamine (0.31 mL, 253 mg, 1.26 mmol) in CH2Cl2 (3.0 mL) and the reaction was stirred for 30 min. Freshly distilled acetyl chloride (0.12 mL, 132 mg, 1.48 mmol) and silyl enol ether 55 (411 mg, 2.60 mmol) were added and the reaction was stirred for 6 h. The reaction was then partitioned between CH2Cl2 (5 mL) and saturated aqueous NaHCO3 (5 mL). The organic phase was removed, and the aqueous layer was washed with CH2Cl2 (3 × 4 mL). The combined organic layers were dried (MgSO4), filtered, and concentrated under reduced pressure. The residue was dissolved in toluene (35 mL) and heated under reflux for 18 h. The reaction was cooled to room temperature and concentrated under reduced pressure. The residue was purified by flash chromatography, eluting with 75-100% Et2O/hexanes followed by EtOAc to give 139.2 mg (49%) of amide 71 as a dark yellowish oil. 1H NMR (500 MHz, CDCl3) δ 9.90 (s, 1 H), 6.41 (d, J = 5.9 Hz, 1 H), 6.27 (d, J = 5.9 Hz, 1 H), 5.73 (dddd, J = 17.0, 10.3, 5.9, 5.9 Hz, 1 H), 5.25 (d, J = 17.0 Hz), 5.21 (d, J = 10.3 Hz, 1 H), 5.06 (d, J = 3.9 Hz, 1 H), 4.34 (t, J = 6.1 Hz, 1 H), 4.27 (dd, J = 15.9, 4.0 Hz, 1H), 3.61 (dd, J = 15.9, 6.5 Hz, 1 H), 2.96 (d, J = 6.1 Hz, 2 H), 2.53 (dd, J = 8.8, 3.9 Hz, 1 H), 2.20 (d, J = 11.7 Hz, 1 H), 1.60 (dd, J = 11.7, 8.8 Hz, 1 H); 13C NMR (100 MHz, CDCl3) δ 198.4, 174.0, 137.5, 132.1, 131.4, 118.1, 92.0, 79.0, 54.5, 45.9, 44.4, 43.9, 28.3; IR (neat) 2951, 2735, 1721, 1688, 1440, 1348, 1253, 1045, 946, 700 cm-1; mass spectrum (CI) m/z 234.1131 [C13H15NO3 (M+1) requires 234.1130].
N-[1,2-bis(3,4-Ethylenedioxyphenyl)ethyl]-2,2-diacetoxy-N-methylacetamide (73)
A solution of methylamine (∼1.9 M in THF, 0.5 mL, 0.95 mmol) was added to a mixture of piperonal (72) (0.92 mg, 0.61 mmol) in THF (1 mL) containing activated 3 Å molecular sieves (8-12 mesh, ∼750 mg), and the reaction was stirred for 18 h. In a separate flask, a solution of piperonyl chloride53 (728 mg, 4.27 mmol) in THF (6 mL) was added dropwise over 2 h to a suspension of magnesium turnings (170 mg) in THF (2.5 mL) at 0 °C, and the reaction was stirred for 3 h at 0 °C. A solution of the Grignard reagent (75) thus prepared (0.4 M in THF, 3.5 mL, 1.40 mmol) was added to the mixture of the imine, and the reaction was heated under reflux for 20 h. The mixture was cooled to -78 °C, and diacetoxyacetyl chloride (76)47 (358 mg, 1.83 mmol) was added. The reaction was stirred for 2.5 h at -78 °C. The cold bath was removed, and the mixture was partitioned between CH2Cl2 (5 mL) and sat. NaHCO3 (5 mL). The organic phase was removed, and the aqueous layer was washed with CH2Cl2 (3 × 4 mL). The combined organic layers were dried (MgSO4), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography, eluting with 30% EtOAc/hexanes to give 173 mg (61%) of amide 73 as a pale yellow solid, mp 141-143 °C. 1H NMR (500 MHz, DMSO, 130 °C) δ 6.95 (s, 1 H), 6.88 (s, 1 H), 6.84 (comp, 2 H), 6.77 (d, J = 1.4 Hz, 1 H), 6.74 (d, J = 7.9 Hz, 1 H), 6.70 (dd, J = 7.9, 1.5 Hz, 1 H), 5.97 (d, J = 1.5 Hz 2 H), 5.91 (s, 2 H), 5.63 (br, 1 H), 3.26 (dd, J = 14.3, 6.1 Hz, 1H), 3.10 (dd, J = 14.3, 9.5 Hz, 1 H), 2.74 (s, 3 H), 2.04 (s, 6 H); 13C NMR (125 MHz, DMSO, 130 °C) δ 167.4, 167.3, 162.8, 147.0, 146.6 146.1, 145.1, 132.1, 131.0, 121.2, 120.2, 108.4, 107.3, 107.3 107.2, 100.3, 99.9, 83.9, 57.5, 35.0, 28.4, 19.3, 19.2; IR (neat) 1767, 1668, 1490, 1441, 1246, 1198, 1038, 924 cm-1; mass spectrum (CI) m/z 458.1453 [C23H23NO9 (M+1) requires 458.1451].
(±)-Roelactamine (74)
A solution of conc. HCl/MeOH (2:1, 5 mL) was added to amide 73 and the reaction was stirred for 8 h at room temperature. The reaction was diluted with CH2Cl2 (5 mL) and carefully quenched with 2 M Na2CO3 (10 mL) and sat. NaHCO3 (5 mL). The organic phase was removed, and the aqueous layer was washed with CH2Cl2 (3 × 5 mL). The combined organic layers were dried (MgSO4), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography, eluting with 40% EtOAc/hexanes to give 58 mg (68%) of 74 as a white foam. 1H NMR (400 MHz, CDCl3,) δ 6.75 (s, 1 H), 6.73 (s, 1 H), 6.69 (s, 1 H), 6.46 (s, 1 H), 5.93 (d, J = 1.4 Hz, 1 H), 5.89 (d, J = 1.4 Hz, 1 H), 5.87 (d, J = 1.4 Hz, 1 H), 5.85 (d, J = 1.4 Hz, 1 H), 4.39 (dd, J = 4.2, 2.6 Hz, 1 H), 4.25 (s, 1 H), 3.33 (dd, J = 17.3, 4.2 Hz, 1 H), 3.10 (s, 3 H), 2.95 (dd, J = 17.3, 2.6 Hz, 1 H); 13C NMR (100 MHz, CDCl3) δ 173.6, 148.1, 148.0, 147.4, 147.0, 135.1, 130.3, 130.2, 127.0, 112.2, 110.0, 107.0, 106.3, 102.1, 101.9, 61.5, 57.4, 34.8, 33.3; IR (neat) 2895, 1661, 1504, 1482, 1228, 1038, 941, 873 cm-1; mass spectrum (CI) m/z 338.1026 [C19H16NO5 (M+1) requires 338.1028].
Scheme 6.
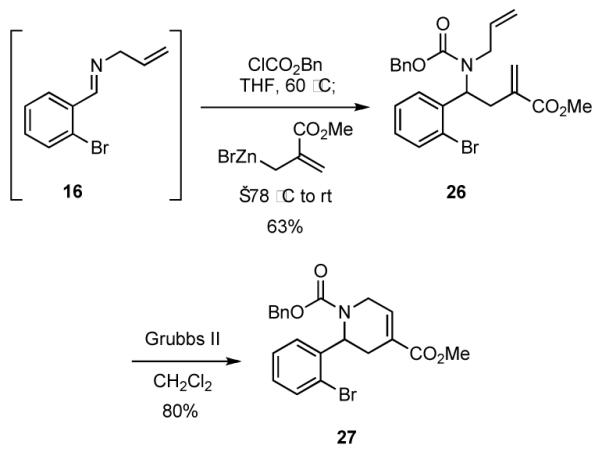
Acknowledgments
We thank the National Institutes of Health (GM 25439 and GM 86192), the Robert A. Welch Foundation (F-0652), The Texas Institute for Drug and Diagnostic Development, and Welch Foundation Grant #H-F-0032, Merck Research Laboratories, and Boehringer Ingelheim Pharmaceuticals for their generous support of this research. We are also grateful to Dr. Richard Pederson (Materia, Inc.) for providing metathesis catalysts used in these studies. We additionally thank Dr. Vincent Lynch (The University of Texas) for X-ray crystallography data and Dr. James Sahn and Dr. Jotham Coe (Pfizer Laboratories) for other assistance and helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.a) Horton DA, Bourne GT, Smythe ML. Chem. Rev. 2003;103:893. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]; b) Constantino L, Barlocco D. Curr. Med. Chem. 2006;13:65. [PubMed] [Google Scholar]
- 2.See for example: Strausberg RL, Schreiber SL. Science. 2003;300:294. doi: 10.1126/science.1083395. Burke MD, Schreiber SL. Angew. Chem. Int. Ed. 2004;43:46. doi: 10.1002/anie.200300626. Arya P, Joseph R, Gan Z, Rakic B. Chem. Biol. 2005;12:163. doi: 10.1016/j.chembiol.2005.01.011. Tan DS. Nature Chem. Biol. 2005;1:74. doi: 10.1038/nchembio0705-74. Spandl RJ, Bender A, Spring DR. Org. Biomol. Chem. 2008;6:1149. doi: 10.1039/b719372f.
- 3.For some recent applications of DOS strategies to identifying small molecules with important bioactivities, see: Antibiotic discovery: Wyatt EE, Galloway WRJD, Thomas GL, Welch M, Loiseleur O, Plowright AT, Spring DR. Chem. Commun. 2008:4962. doi: 10.1039/b812901k. Thomas GL, Spandl RJ, Glansdorp FG, Welch W, Bender A, Cockfield J, Lindsay JA, Bryant C, Brown DFJ, Loiseleur O, Rudyk H, Ladlow M, Spring DR. Angew. Chem. Int. Ed. 2008;47:2808. doi: 10.1002/anie.200705415. c) Inhibition of a protein-protein interaction in a cancerrelated pathway: Stanton BZ, Peng LF, Maloof N, Nakai K, Wang X, Duffner JL, Taveras KM, Hyman JM, Lee SW, Koehler AN, Chen JK, Fox JL, Mandinova A, Schreiber SL. Nature Chem. Bio. 2009;5:154. doi: 10.1038/nchembio.142.
- 4.For some leading references, see: Gaul C, Njardarson JT, Shan D, Dorn DC, Wu K-D, Tong WP, Huang X-Y, Moore MAS, Danishefsky SJ. J. Am. Chem. Soc. 2004;126:11326. doi: 10.1021/ja048779q. Wilson RM, Danishefsky SJ. J. Org. Chem. 2006;71:8329. doi: 10.1021/jo0610053. Wilson RM, Danishefsky SJ. J. Org. Chem. 2007;72:4293. doi: 10.1021/jo070871s.
- 5.For leading references, see: Koch MA, Schuffenhauer A, Scheck M, Wetzel S, Casaulta Ma., Odermatt A, Ertl P, Waldmann H. Proc. Nat. Acad. Sci. (USA) 2005;102:17272. doi: 10.1073/pnas.0503647102. Wetzel S, Schuffenhauer A, Roggo S, Ertl P, Waldmann H. Chimia. 2007;61:355. doi: 10.1021/ci600338x.
- 6.Shaw JT. Nat. Prod. Rep. 2009;26:11. doi: 10.1039/b814468k. [DOI] [PubMed] [Google Scholar]
- 7.For an excellent review of MCRs, see: Zhu J, Bienayme H, editors. Multicomponent Reactions. Wiley-VCH; Wienheim: 2005.
- 8.For a recent mini-review, see: Sunderhaus JD, Martin SF. Chem. Eur. J. 2009;15:1300. doi: 10.1002/chem.200802140.
- 9.For reviews, see: Dömling A. Chem. Rev. 2006;106:17. doi: 10.1021/cr0505728. Akritopoulou-Zanze I, Djuric SW. Heterocycles. 2007;73:125. See also: Ribelin TP, Judd AS, Akritopoulou-Zanze I, Henry RF, Cross JL, Whittern DN, Djuric SW. Org. Lett. 2007;9:5119. doi: 10.1021/ol7023373.
- 10.a) Gracias V, Gasiecki AF, Djuric SW. Org. Lett. 2005;7:3183. doi: 10.1021/ol050852+. [DOI] [PubMed] [Google Scholar]; b) Gracias V, Gasiecki AF, Djuric SW. Tetrahedron Lett. 2005;46:9049. doi: 10.1021/ol050852+. [DOI] [PubMed] [Google Scholar]; c) Gracias V, Darczak D, Gasiecki AF, Djuric SW. Tetrahedron Lett. 2005;46:9053. [Google Scholar]; d) Beebe X, Gracias V, Djuric SW. Tetrahedron Lett. 2006;47:3225. [Google Scholar]; e) Gracias V, Gasiecki AF, Pagano TG, Djuric SW. Tetrahedron Lett. 2006;47:8873. [Google Scholar]
- 11.Kumagai N, Muncipinto G, Schreiber SL. Angew. Chem. Int. Ed. 2006;45:3635. doi: 10.1002/anie.200600497. [DOI] [PubMed] [Google Scholar]
- 12.Nielsen TE, Schreiber SL. Angew. Chem. Int. Ed. 2008;47:48. doi: 10.1002/anie.200703073. See also: Comer E, Rohan E, Deng L, Porco JA., Jr. Org. Lett. 2007;9:2123. doi: 10.1021/ol070606t.
- 13.For some examples, see: Martin SF, Benage B, Geraci LS, Hunter JE, Mortimore M. J. Am. Chem. Soc. 1991;113:6161. Martin SF, Clark CC, Corbett JW. J. Org. Chem. 1995;60:3236. Ito M, Clark CC, Mortimore M, Goh JB, Martin SF. J. Am. Chem. Soc. 2001;123:8003. doi: 10.1021/ja010935v.
- 14.For a review of the vinylogous Mannich reaction, see: Martin SF. Acc. Chem. Res. 2002;35:895. doi: 10.1021/ar950230w. For other leading examples, see also: Martin SF, Barr KJ, Smith DW, Bur SK. J. Am. Chem. Soc. 1999;121:6990. Martin SF, Chen KX, Eary CT. Org. Lett. 1999;1:79. doi: 10.1021/ol990554a. Martin SF, Lopez OD. Tetrahedron Lett. 1999;40:8949. Martin SF, Bur SK. Tetrahedron. 1999;55:8905. Liras S, Lynch CL, Fryer AM, Vu BT, Martin SF. J. Am. Chem. Soc. 2001;123:5918. doi: 10.1021/ja010577w. Reichelt A, Bur SK, Martin SF. Tetrahedron. 2002;58:6323.
- 15.For some related MCRs, see: Black DA, Arndtsen BA. Org. Lett. 2004;6:1107. doi: 10.1021/ol036462+. Fischer C, Carriera EM. Org. Lett. 2004;6:1497. doi: 10.1021/ol049578u. Davis JL, Dhawan R, Arndtsen BA. Angew. Chem. Int. Ed. 2004;43:590. doi: 10.1002/anie.200352123. Black DA, Arndtsen BA. J. Org. Chem. 2005;70:5133. doi: 10.1021/jo0503557. Black DA, Arndtsen BA. Tetrahedron. 2005;61:11317. Wei C, Li C-J. Lett. Org. Chem. 2005;2:410. Black DA, Arndtsen BA. Org. Lett. 2006;8:1991. doi: 10.1021/ol060277p. Zhang L, Malinakova HC. J. Org. Chem. 2007;72:1484. doi: 10.1021/jo0621773.
- 16.During the course of this work, a related MCR was reported: Yin Y, Zhao G, Li G-L. Tetrahedron. 2005;61:12042.
- 17.For a preliminary account of some of this work, see: Sunderhaus JD, Dockendorff C, Martin SF. Org. Lett. 2007;9:4223. doi: 10.1021/ol7018357.
- 18.a) Kalinin AV, Chauder BA, Rakhit S, Snieckus V. Org. Lett. 2003;5:3519. doi: 10.1021/ol035398t. [DOI] [PubMed] [Google Scholar]; b) Ralbovsky JL, Scola PM, Sugino E, Burgos-Garcia C, Weinreb SM, Parvez M. Heterocycles. 1996;43:1497. [Google Scholar]
- 19.Oisaki K, Suto Y, Kanai M, Shibasaki M. J. Am. Chem. Soc. 2003;125:5644. doi: 10.1021/ja034993n. [DOI] [PubMed] [Google Scholar]
- 20.Crystallographic data, excluding structure factors, for the structures of 13, the hydrochloride salts of 57 and 58, and 68 have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication numbers CCDC 666629-666630, and 724900, respectively. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [fax: 144-(0)1223-336033 or [deposit@ccdc.cam.ac.uk].
- 21.See for example: Cao G, Escribano AM, Fernandez MC, Fields F, Gernert DL, Cioffi CL, Herr RJ, Mantlo NB, Martin de la Nava EM, Mateo-Herranz AI, Mayhugh DR, Wang X. WO 2005/037796 2005
- 22.Alami NE, Belaud C, Villieras J. Tetrahedron Lett. 1987;28:59. [Google Scholar]
- 23.Jacobsen AE, Mokotoff M. J. Med. Chem. 1970;13:7. doi: 10.1021/jm00295a002. [DOI] [PubMed] [Google Scholar]
- 24.Spring DR, Krishnan S, Blackwell HE, Schreiber SL. J. Am. Chem. Soc. 2002;124:1354. doi: 10.1021/ja017248o. [DOI] [PubMed] [Google Scholar]
- 25.Yee NK, Farina V, Houpis IN, Haddad N, Frutos RP, Gallou F, Wang X-J, Wei X, Simpson RD, Feng X, Fuchs V, Xu Y, Tan J, Zhang L, Xu J, Smith-Keenan LL, Vitous J, Ridges MD, Spinelli EM, Johnson M, Donsbach K, Nicola T, Brenner M, Winter E, Kreye P, Samstag W. J. Org. Chem. 2006;71:7133. doi: 10.1021/jo060285j. [DOI] [PubMed] [Google Scholar]
- 26.For a related example, see: Comins DL, Weglarz MA. J. Org. Chem. 1991;56:2506.
- 27.Jung ME, Lyster MA. J. Chem. Soc., Chem. Comm. 1978:315. [Google Scholar]
- 28.Larsen SB, Bang-Andersen B, Johansen TN, Jorgensen M. Tetrahedron. 2008;64:2938. [Google Scholar]
- 29.O’Brien CJ, Kantchev EAB, Valente C, Hadei N, Chass GA, Lough A, Hopkinson AC, Organ MG. Chem. Eur. J. 2006;12:4743. doi: 10.1002/chem.200600251. [DOI] [PubMed] [Google Scholar]
- 30.Negishi E, Valente LF, Kobayashi M. J. Am. Chem. Soc. 1980;102:3298. [Google Scholar]
- 31.Ye B-H, Naruta Y. Tetrahedron. 2003;59:3593. [Google Scholar]
- 32.Kerins F, O’Shea DF. J. Org. Chem. 2002;67:4968. doi: 10.1021/jo020074o. [DOI] [PubMed] [Google Scholar]
- 33.Approximately 3% of the unsubstituted vinyl derivative of 54 was observed in the 1H NMR spectrum of 54.
- 34.For a review, see: Diver ST, Giessert AJ. Chem. Rev. 2004;104:1317. doi: 10.1021/cr020009e.
- 35.For some leading reviews, see: Pandey G, Banerjee P, Gadre SR. Chem. Rev. 2006;106:4484. doi: 10.1021/cr050011g. Coldham I, Hufton R. Chem. Rev. 2005;105:2765. doi: 10.1021/cr040004c.
- 36.Kawakamie Y, Aoki T, Yamashita Y. Polymer Bulletin. 1987;18:473. [Google Scholar]
- 37.Shah SK, Chen N, Guthikonda RN, Mills SG, Malkowitz L, Springer MS, Gould SL, DeMartino JA, Carella A, Carver G, Holmes K, Schleif WA, Danzeisen R, Hazuda D, Kessler J, Lineberger J, Miller M, Emini EA, MacCoss M. Bioorg. Med. Chem. Lett. 2005;15:977. doi: 10.1016/j.bmcl.2004.12.044. [DOI] [PubMed] [Google Scholar]
- 38.Ashton W, Sisco RM, Dong H, Lyons KA, He H, Doss GA, Leiting B, Patel RA, Wu JK, Marsilio F, Thornberry NA, Weber AE. Bioorg. Med. Chem. Lett. 2005;15:2253. doi: 10.1016/j.bmcl.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 39.Capperucci A, Degl’Innocenti A, Funicello M, Mauriello G, Scafato P, Spagnolo P. J. Org. Chem. 1995;60:2254. [Google Scholar]
- 40.Bertelli L, Biagi G, Giorgi I, Livi O, Manera C, Scartoni V, Martini C, Giannaccini G, Trincavelli L, Barili PL. Farmaco. 1998;53:305. doi: 10.1016/s0014-827x(98)00025-1. [DOI] [PubMed] [Google Scholar]
- 41.For some examples, see: Martin SF, Desai SR, Phillips GW, Miller AC. J. Am. Chem. Soc. 1980;102:3294. Martin SF, Williamson SA, Gist RP, Smith KM. J. Org. Chem. 1983;48:5170. Martin SF, Rueger H, Williamson SA, Grzejszczak S. J. Am. Chem. Soc. 1987;109:6124. Martin SF, Li W. J. Org. Chem. 1991;56:642. Humphrey JM, Liao Y, Ali A, Rein T, Wong Y-L, Chen H-J, Courtney AK, Martin SF. J. Am. Chem. Soc. 2002;124:8584. doi: 10.1021/ja0202964.
- 42.Ainsworth PJ, Craig D, Reader JC, Slawin AMZ, White AJP, Williams DJ. Tetrahedron. 1996;52:695. [Google Scholar]
- 43.For a review, see: Kappe CO, Murphree SS, Padwa A. Tetrahedron. 1997;53:14179.
- 44.(a) Weber E, Keana J, Barmettler P. PCT Int. Appl. WO 9012575. 1990 [Google Scholar]; Chem. Abstr. 1991;115:106019w. [Google Scholar]; (b) Childers WE, Jr., Abou-Gharbia MA. 4,940,789 U.S. Patent. 1990; Chem. Abstr. 1990;113:19190w. [Google Scholar]
- 45.Gözler B, Önür MA, Bilir S, Hesse M. Helv. Chem. Acta. 1992;75:260. [Google Scholar]
- 46.For selected syntheses of isopavine alkaloids, see: Gozler B. In: The Alkaloids. Brossi A, editor. Vol. 31. Academic Press; New York, New York: 1987. p. 343. Gottlieb L, Meyers AI. J. Org. Chem. 1990;55:5659. Carrillo L, Badía D, Domínguez E, Vicario JL, Tellitu I. J. Org. Chem. 1997;62:6716. Shinohara T, Takeda A, Toda J, Sano T. Heterocycles. 1998;48:981. Dragoli DR, Burdett MT, Ellman JA. J. Am. Chem. Soc. 2001;123:10127. doi: 10.1021/ja016349j. Tambar UK, Enber DC, Stoltz BM. J. Am. Chem. Soc. 2006;128:11752. doi: 10.1021/ja0651815.
- 47.Rewcastle GW, Sutherland HS, Weir CA, Blackburn AG, Denny WA. Tetrahedron Lett. 2005;46:8719. [Google Scholar]
- 48.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518. [Google Scholar]
- 49.Synthesized according to: Fonseca MH, Eibler E, Zabel M, König B. Tetrahedron: Asymmetry. 2003;14:1989. We added SiO2 to the PCC oxidation reaction to facilitate removal of the chromium tars.
- 50.Mahmud H, Lovely CJ, Dias HVR. Tetrahedron. 2001;57:4095. [Google Scholar]
- 51.Negishi E, Boardman LD, Sawada H, Bagheri V, Stoll AT, Tour JM, Rand CL. J. Am. Chem. Soc. 1988;110:5383. [Google Scholar]
- 52.DMSO was added to facilitate removal of the Ru catalyst. See: Anh YM, Yang K, Georg GI. Org. Lett. 2001;3:1411. doi: 10.1021/ol010045k. We have since found that use of 2-mercaptonicotinic acid is more effective (reference 25).
- 53.Bourry A, Akué-Gédu R, Rigo B, Hénichart J-P, Sanz G, Couturier D. J. Heterocyclic Chem. 2003;40:989. [Google Scholar]