

SUMMARY
The membrane mucin Muc4 is aberrantly expressed in numerous epithelial carcinomas and is currently used as a cancer diagnostic and prognostic tool. Muc4 can also potentiate signal transduction by modulating differential ErbB2 phosphorylation in the absence and in the presence of the ErbB3 soluble ligand heregulin (HRG-β1). These features of Muc4 suggest that Muc4 is not merely a cancer marker, but an oncogenic factor with a unique binding/activation relationship with the receptor ErbB2. In the present study, we examined the signaling mechanisms that are associated with the Muc4-ErbB2 module by analyzing ErbB2 differential signaling in response to Muc4 expression. Our study was carried out in the A375 human melanoma and BT-474 breast cancer cell lines as our model systems. Quantitative and comparative signaling modulations were evaluated by immunoblot using phospho-specific antibodies, and densitometry analysis. Signaling complex components were identified by chemical cross-linking, fractionation by gel filtration, immunoprecipitation and immunoblotting. Activated downstream signaling pathways were analyzed by an antibody microarray screen and immunoblot analyses. Our results indicate that Muc4 modulates ErbB2 signaling potential significantly by stabilizing and directly interacting with the ErbB2-ErbB3 heterodimer. Further analyses indicate that Muc4 promotes ErbB2 autocatalysis, but it has no effect on ErbB3 phosphorylation, although the chemical cross linking data indicated that the signaling module is composed of Muc4, ErbB2 and ErbB3. Our microarray analysis indicates that Muc4 expression promotes cell migration by increasing the phosphorylation of the focal adhesion kinase (FAK) and also through an increase in the levels of β-catenin.
INTRODUCTION
Muc4 is a heterodimeric glycoprotein that is synthesized from a single polypeptide precursor and cleaved early after synthesis, resulting in a tightly but non-covalently associated complex (Sheng et al., 1990; Rossi et al., 1996) of two subunits; mucin subunit ASGP-1 (MUC4α in human) and transmembrane subunit ASGP-2 (MUC4β in human). Muc4 is classified as a cell surface mucin and is normally expressed in epithelial tissues where it protects epithelia by lubricating these surfaces and protecting them from infections and injuries (Hattrup and Gendler, 2008). Aberrant expression of Muc4 has been reported in a variety of human carcinomas (Yonezawa et al., 2008) where Muc4 is implicated in affecting a variety of cellular phenotypes that promote cancer development, including cell adhesion (Komatsu et al., 1997), cell polarity (Ramsauer et al., 2006), promotion of oncogenesis (Karg et al., 2006; Moniaux et al., 2007; Ponnusamy et al., 2008), and cell signaling through modulation of the receptor ErbB2 activity (Carraway et al., 1999; Jepson et al., 2002; Ramsauer et al., 2006; Funes et al., 2006).
The epidermal growth factor receptor ErbB2 belongs to subclass I of the receptor tyrosine kinase superfamily. Other members of this subclass are ErbB1 (EGFR), ErbB3, and ErbB4. ErbB receptor signaling regulates large numbers of important cellular functions, including proliferation, differentiation, survival, adhesion and migration (Warren and Landgraf 2006; Citri and Yarden, 2006). These functions are mediated via an array of signaling pathways involving the downstream effector molecules Ras, Raf, mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K), Akt, phospholipase-C, Rho, and signal transducer and activator of transcription (Morandell et al., 2008). In cancer, these receptors become constitutively activated as a result of autocrine ligand production, receptor overexpression, or mutations, leading to aberrant cellular behaviors that promote neoplastic transformation and oncogenesis (Hynes and Lane, 2005).
Muc4 and ErbB2 are independently and dependently implicated in human cancer pathogenesis; Muc4 aberrant expression in a variety of carcinomas highlights the significant potential of Muc4 as a clinical tool for cancer diagnosis and prognosis (Singh et al., 2007; Shibahara et al., 2004; Chaturvedi et al., 2008), and the amplification of the ErbB2 gene locus resulting in ErbB2 aberrant expression in different cancers is linked with human cancer pathogenesis (Moasser, 2007). However, accumulating data suggest that Muc4 and ErbB2 also display a unique Muc4-ErbB2 dependent relationship in human cancer pathogenesis. Muc4 has been shown to interact with ErbB2 (Carraway et al., 1999) and to play a role in regulating the polar localization of ErbB2 (Ramsauer et al., 2003). In addition, experimental induction of Muc4 in cells lacking Muc4 leads to increased phosphorylation of ErbB2 at a carboxyl terminal tyrosine residue, Tyr1248, which is implicated in cell transformation (Carraway et al., 1999; Jepson et al., 2002). Tyrosine 1139 residue is also phosphorylated in response to Muc4 induction, facilitating apical localization of ErbB2 receptor from a baso-lateral location, leading to activation of the Akt pathway in a p38 MAPK-dependent manner (Ramsauer et al., 2006). In the presence of HRG-1β, activation of both ErbB3 (Heregulin receptor) and ErbB2 are potentiated by Muc4 and lead to the enhanced activation of ERK and Akt pathways. Furthermore, Muc4 was also shown to enhance surface accumulation of ErbB2 and ErbB3 by suppressing their internalization (Funes et al., 2006). Thus, Muc4 is portrayed as an oncogenic factor through its unique association with ErbB2 by modulating and potentiating ErbB2 cell signaling, resulting in differential effects on cell proliferation and differentiation in the absence and in the presence of the ErbB3 ligand HRG-1β.
The significance of the differential effect of Muc4 on ErbB2 and ErbB3 signaling is important to explore because it suggests that Muc4 has a unique binding relationship with the receptors, and that it can further advance our understanding of ErbB receptor activation mechanisms. ErbB receptor activation mechanisms are extensively studied, and in general, ErbB receptor activity is said to be affected by the spatial and temporal expression of ligands, formation of different homo and heterodimers, and activation of their kinase domains that result in specific tyrosine phosphorylation on their intracellular tails (Linggi and Carpenter, 2006; Burgess, 2008). However, unique structural features in ErbB2 and ErbB3, the closed ligand binding domain of ErbB2 (Garett et al., 2003) and the inactive kinase domain of ErbB3 (Plowman et al., 1993), define ErbB2 and ErbB3 receptors as obligate heterodimers with a unique signaling relationship due to their inability to signal as homodimers. ErbB2 exhibits a dimerization-competent conformation (untethered) in the absence of a soluble ligand, but cannot readily form active ErbB2 homodimers (Garrett et al., 2003), and ErbB3 assumes its dimerization competent conformation upon soluble ligand binding, but its enzymaticaly inactive kinase domain interferes with the formation of ErbB3 active homodimers. ErbB2 and ErbB3 therefore rely on complementary signaling whereby ErbB3 provides ErbB2 with a heterodimer partner and ligand binding site, and ErbB2 provides the kinase activity. In contrast to their apparent disabilities, this receptor pair forms the most potent signaling module in the ErbB receptor family in terms of cell growth and transformation (Sliwkowski et al., 1994). These specific ErbB2-ErbB3 mechanisms then suggest that the Muc4 role in modulating ErbB2 signaling may occur through receptor stabilization. In the present study, we examined the signaling mechanisms that are associated with the Muc4-ErbB2 module by analyzing ErbB2 differential signaling in response to Muc4 expression. We report that Muc4 expression results in significant ligand-independent ErbB2 activity through Muc4 ability to form stable and direct complexes with the ErbB2-ErbB3 heterodimer and to promote cell migration by increasing the phosphorylation of the focal adhesion kinase. Our work highlights the role of Muc4 as a potent oncogenic-promoting factor and further enhances our understanding of ligand-independent ErbB2 activation mechanisms.
MATERIALS AND METHODS
Reagents
Reagents were purchased as follows: Human recombinant heregulin HRG-1β (EGF domain), protease inhibitor cocktail, and anti Beta-actin (clone AC-15) mouse monoclonal antibody from Sigma; Chemical cross linking reagents BS3 (Bis [sulfosuccinimidyl] suberate) and DTSSP (3,3’- Dithiobis [sulfosuccinimidylpropionate]) from Termo Scientific; Anti-erbB-2/HER-2 rabbit polyclonal IgG, and anti-phospho-erbB-2/HER-2 (Tyr1248) from Upstate; c-erbB-2 / HER-2 / neu AB-17 (clone e2-4001 +3B5) mouse monoclonal antibody from Lab Vision Neomarkers; Phospho-HER3/ErbB3 (Tyr1289) (21D3) rabbit monoclonal antibody from Cell Signaling Technology; ErbB-3 (C-17) polyclonal antibody from Santa Cruz Biotechnology; Anti-human c-erbB-2 oncoprotein polyclonal rabbit from Dako; Anti-rabbit IgG and Anti-mouse IgG coupled to peroxidase secondary antibodies from Promega; FuGENE HD transfection reagents from Roche Applied Science; BT-474 (# HTB-20™) breast cancer cell line from American Type Culture Collection; Ready-gels 4–15% Tris-HCl precast gels from Bio-Rad. Kinase inhibitor Lapatinib (GW572016) was a gift from GlaxoSmithKline. Muc4 rCpep polyclonal rabbit antibody raised against the cytoplasmic tail of Muc4β (Price-Schiavi et al., 2000) and Muc4 mouse monoclonal antibody 4F12 raised against Muc4β subunit (Rossi et al., 1996) have been previously described.
Cell Culture and Muc4 Induction/Transfection
Construction of the A375 stable clone expressing Muc4-rep3 under tetracycline regulation (tet-off) has been previously described (Komatsu et al., 1997). The Muc4-rep3 clone contains the ASGP-2 (transmembrane) subunit of Muc4 and 3 tandem repeats from the ASGP-1 (mucin) subunit of Muc4. The stable clone was maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 1% v/v penicillin/streptomycin, 0.4 mg/mL G418, 0.15 mg/mL hygromycin, and 2 µg/mL tetracyclin. To induce Muc4 expression, cells were grown to 60% confluence, rinsed three times with PBS, and then cultured for 48 h without tetracycline. The parent A375 cells were similarly maintained but without G418, hygromycin or tetracycline. The BT-474 cell line was maintained in RPMI-1640 medium supplemented with 10% fetal calf serum and 1% v/v penicillin/streptomycin. Transient transfections of Muc4-rep1 construct (Komatsu et al., 1997) were carried out for 48 h using FuGene HD reagent as recommended by the manufacturer. Cells were serum-starved overnight (0.1% fetal calf serum) prior to HRG-1β treatment. All cells were incubated at 37°C with 5% CO2.
Analysis of Receptor Phosphorylation
The Muc4 effect on receptor phosphorylation was carried out in serum starved cells (24 h) and under different conditions, including 50 nM HRG-1β (saturating concentration) treatment for various times at 37°C and 0.2 µM kinase inhibitor (Lapatinib-GW572016) treatment for 6 h. Cells cultured in 100 mm dishes were rinsed twice with ice-cold PBS after the different treatments and lysed in 250 µL RIPA lysis buffer (50 mM MOPS, pH 7.0, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1 mM MgCl2, 1 mM Na3VO4, 1% Triton X-100, 1 mM dithiothreitol, and 1X protease inhibitors cocktail). Cells were scraped, collected, sonicated, and the lysate was cleared by centrifugation at 14,000 rpm for 30 min. at 4°C and then added to SDS-PAGE sample buffer. Lysates were resolved by 8% SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose filters. Membranes were blocked, washed, and incubated with antibodies as recommended by the phospho-antibody manufacturers. Membrane immunoblots of non phospho-antibodies were blocked in 5% non-fat dry milk in TBS-Tween (0.05 %) overnight, incubated with primary antibody for 2–4 h, washed three times in TBS-T, incubated in secondary antibody in 5% non-fat dry milk in TBS-T for 1 h, and washed three times in TBS-T. When more than one signal was required from the same membrane, the phospho-antibody signal was always acquired first. Blotted proteins were visualized by western lighting chemiluminescence reagent (PerkinElmer) and visualized on Blue Lite Autorad Film (BioExpress).
Quantitative and Statistical Analysis of receptor phosphorylation
Quantitative changes in receptor signaling in response to Muc4 expression were analyzed in triplicate from phospho-immuonoblot samples. Using densitometry software (ImageJ NIH) the signal intensities were quantitated along with the Beta actin control signals, which provided the baseline for signal normalization. The data were expressed as the mean ± standard deviation for a series of at least 3 experiments. Student’s t tests were used to compare mean values as appropriate. P values < 0.05 were considered to represent significant differences.
Immunoprecipitation
For complex formation between Muc4 and the ErbB receptors, cells were lysed in 500 uL IP buffer (50 mM Tris/HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% Na-Deoxycholate, 0.1% SDS, 1.5 mM EGTA, 1.5 mM MgCl2, 2 mM Na-Orthovanadate, and 1X protease inhibitors cocktail). Extracts were clarified by centrifugation, and 1 mg of extract was added to 30 µL protein G/A-agarose beads with bound antibodies. Samples were rotated overnight at 4°C and washed five times with cold IP buffer without the inhibitors. Immunoadsorbed proteins were eluted with 30 µL SDS-PAGE sample buffer and boiled for 5 min. The immunoprecipitated proteins were analyzed by immunoblotting with the appropriate antibodies as described above.
Chemical cross linking and Muc4 complexes analysis
To examine the Muc4 complexes with the ErbB receptors, cellular crosslinking analyses were carried out using a non-cleavable (BS3) and a reversible (DTSSP) chemical crosslinkers following the manufacturer instructions. Briefly, A375 cells (60–70% confluence) were rinsed three times with PBS and then incubated with 1 mM BS3 dissolved in 20 mM sodium phosphate, 0.15 M NaCl buffer pH 7.5 for 2 h. on ice. The reaction was quenched by adding 1 M Tris-HCl, pH 7.5 to a final concentration of 20–50 mM Tris, and incubated at room temperature for 15 min. For the reversible chemical cross-linking, 1 mM DTSSP dissolved in water was added to cells for 2 h. on ice, and the reaction was stopped by adding 1 M Tris pH 7.5 to a final concentration of 10–20 mM and incubated at room temperature for 15 min. Crosslinked cells were lysed in RIPA buffer, scraped, collected, sonicated, and cleared by centrifugation at 14,000 rpm for 30 min. at 4°C. To examine the composition of the relevant complexes, the supernatant was directly loaded by FPLC (1 mL/min) onto a high resolution Superose 6 analytical gel filtration column equilibrated at 4°C in modified RIPA buffer (without Triton X-100, dithiothreitol, protease inhibitors cocktail). The column run was programmed for flow consistency and uniform elution profile. Eluted proteins were collected in 1 mL fractions, and protein fractions containing Muc4 were then TCA precipitated and loaded on 4–15% gradient gels (Bio-Rad) for Western blot analysis. DTSSP duplicate samples were loaded side by side with either native SDS-PAGE sample buffer or with 5% β-mercaptoethanol containing SDS-PAGE sample buffer in order to observe differences between the crosslinked and reversed crosslinked complexes.
Antibody microarray analysis (KAM-1.1) with Kinexus™ Bioinformatics Corporation
The array was used to analyze further the signaling pathways that are stimulated downstream of the Muc4-ErbB2-ErbB3 complex. This array surveys differential expression and activity of over 650 signaling proteins in duplicate using two samples on the same microarray slide. The array output includes qualitative and semi-quantitative analyses of protein kinases and other signaling proteins. The two samples analyzed were A375 cells expressing Muc4 for 48 h. and a control sample. Sample lysates were prepared following the Kinexus instructions and shipped on dry ice. The validation analyses of the microarray results were carried out by Western blot analyses. The full data set is accessible at http://www.kinexus.ca/kinet array barcode K01070228, kinexus control ID 11807 and treated ID 11808.
RESULTS
Muc4 modulates ErbB2 signaling potential significantly
Earlier studies showed that Muc4 expression in the absence of soluble ligand treatment results in an elevated immunoblot ErbB2 phosphorylation signal in different systems, and that Muc4 co-immunoprecipitates with ErbB2 from detergent lysates (Jepson et al., 2002; Ramsauer et al., 2006; Ponnusamy et al., 2008). These studies support the hypothesis that Muc4 acts as a novel intramembrane ligand for the receptor ErbB2. Here we show that expression of Muc4 (Fig. 1A) increases formation of a stable complex with the phosphorylated ErbB2 receptor (Fig. 1A,B). These studies support the hypothesis that Muc4 acts as a novel intra-membrane ligand for the receptor ErbB2. We quantitatively assessed this role of Muc4 effect on ErbB2 phosphorylation signal magnitude and stability in the human melanoma cell line, A375. Quantitative assessment of the Muc4 expression effect on the ErbB2 phosphorylation signal magnitude (Fig. 1B) shows that Muc4 augments the signal magnitude significantly (p=0.0002), without significantly changing the total ErbB2 receptor levels (σ=44.94 ±1.22). Similar observations were also evident in the breast cancer cell line BT-474 transiently transfected with Muc4 (Fig. 4 compare lanes 1 and 2).
FIGURE 1.
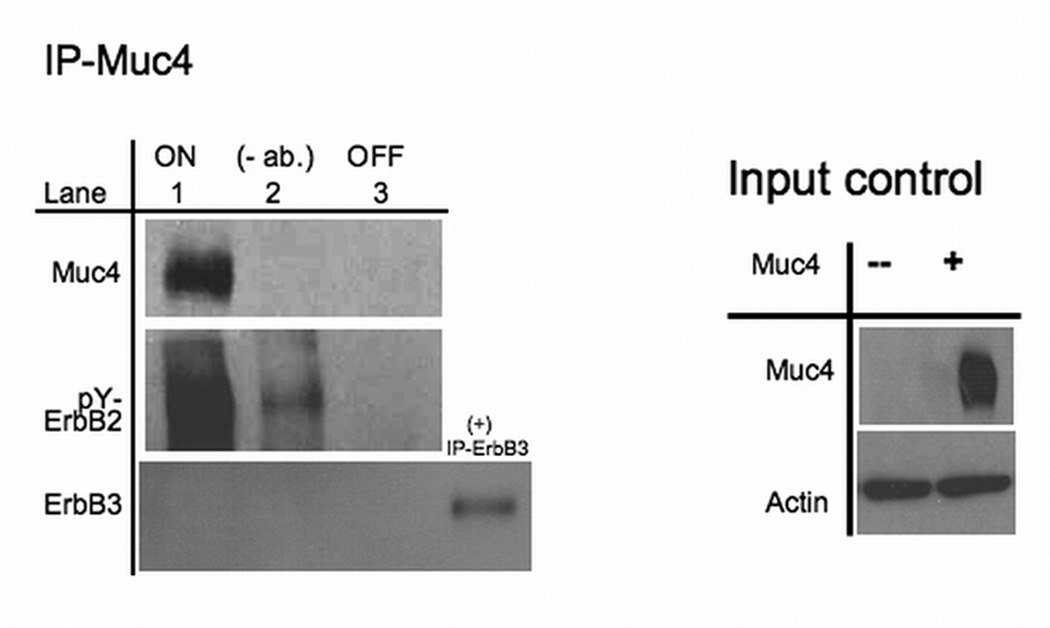
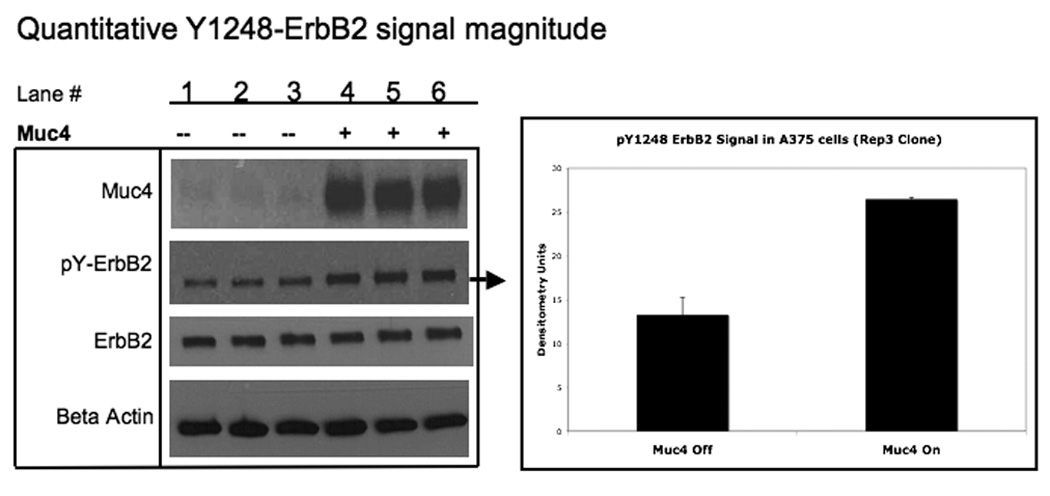
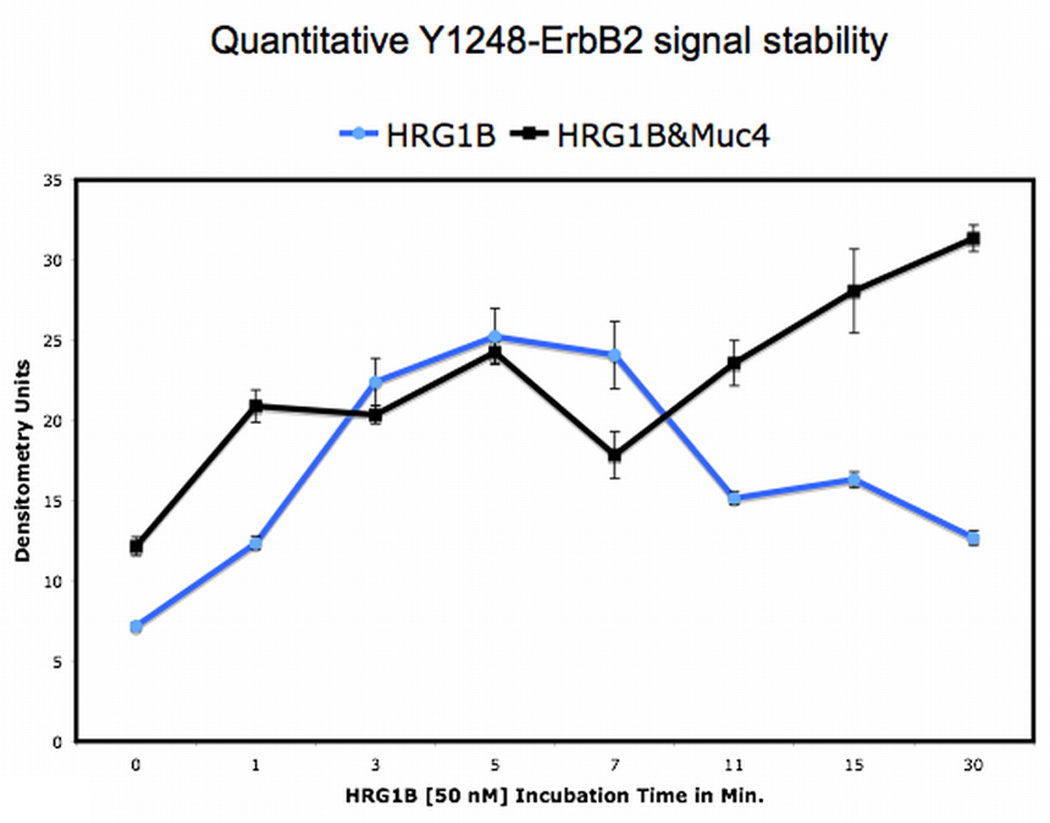
Muc4 modulates ErbB2 signaling potential . A. Muc4 forms a stable complex with phosphorylated ErbB2. Proteins from RIPA extracts of A375 Muc4-transfected cells with or without Muc4 (48 h) were subjected to immunoprecipitation with anti-rCpep, a Muc4 pAb targeting the cytoplasmic portion of Muc4, or with the pre-immune serum (−). Immunoprecipitates were immunoblotted with anti-Muc4, anti pY1248-ErbB2, and anti-ErbB3 antibodies. The control immunoprecipitation lane (+) in the ErbB3 immunoblot was carried out with the anti-ErbB3 (C-17) polyclonal antibody. The input control is an immunoblot of the lysates with anti-Muc4 and anti β-actin antibodies. B. Muc4 expression augments ErbB2 phosphorylation signal magnitude significantly (p=0.0002) without changing ErbB2 receptor levels. A375 Muc4-transfected (Rep3 clone) cells with or without Muc4 (48 h) and starved (0.1% FBS) for 24 h were immunoblotted in triplicates with anti-Muc4, anti ErbB2-Y1248, anti-ErbB2, and anti β-actin antibodies. Quantitative analysis of the ErbB2-Y1248 signal intensity is also shown. The signal intensity was calculated using the ImageJ NIH densitometry software. The ErbB2-Y1248 signal was measured, normalized with the β-actin signal, and expressed as the mean ± standard deviation for a series of at least 3 experiments. Student’s t tests were used to compare mean values as appropriate. P values < 0.05 were considered to represent significant differences. C. Muc4 expression stabilizes ErbB2 phosphorylation signal under heregulin ligand (HRG-1β) treatment. A375 Muc4-transfected (Rep 3 clone) cells with or without Muc4 (48 h) and starved (0.1% FBS) for 24 h, were incubated at 37°C with HRG-1β [50 nM] for the indicated times. Quantitative analysis of ErbB2-Y1248 immunoblot band density is shown. Using densitometry software (ImageJ NIH), the signal was measured, normalized with β-actin signal, and then expressed as the mean ± standard deviation for a series of at least three experiments. Circles represent Muc4-Off cells treated with ligand, and squares, Muc4-On cells treated with ligand.
FIGURE 4.
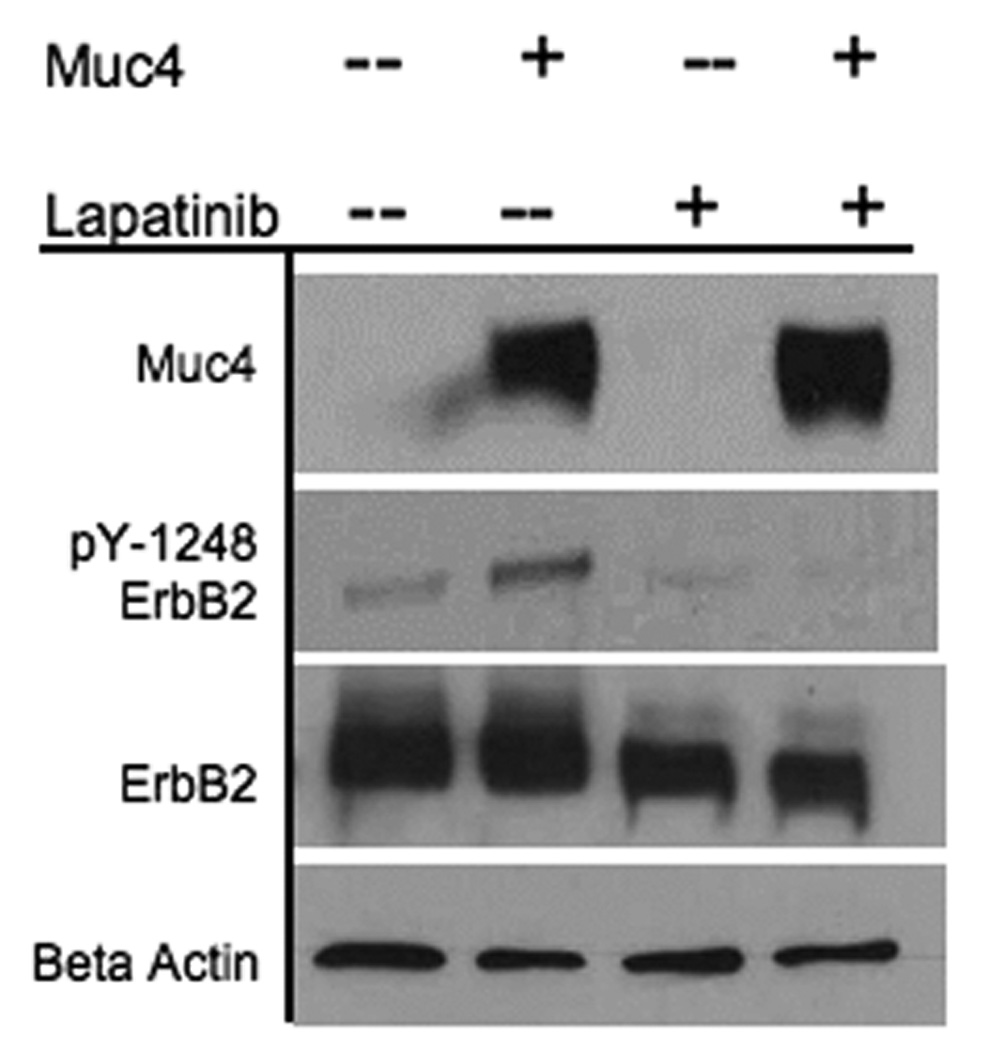
Muc4 promotes ErbB2 autocatalysis. BT-474 cells (Muc4-0ff) and BT-474 cells transfected with Muc4-Rep 3 plasmid for 48 h (Muc4-On) were serum starved (24 h) and incubated with the kinase inhibitor, Lapatinib-GW572016 [0.2 µM] for 6 h. Cells were rinsed twice with ice-cold PBS saline and lysed in RIPA buffer pH 7.2. Cleared lysates were immunoblotted with anti-Muc4, anti ErbB2-Y1248, anti-ErbB2, and anti β-actin antibodies.
The effect of Muc4 on the ErbB2 phosphorylation signal stability was assessed using a comparative analysis of the signal in A375 cells stimulated with a soluble ligand (HRG-1β) and with cells similarly stimulated and expressing Muc4. Fig.1C (circle) shows that ligand treatment resulted in a transient ErbB2 phosphorylation signal pattern that saturated fast, peaking at the 5 min. time point and was almost lost shortly afterward by the 11 min. time point. The ErbB2 phosphorylation signal analysis of Muc4-transfected cells was quantitatively compared; however, it displayed a stabilizing ErbB2 phosphorylation signal pattern (square). This signal reached a similar saturation level at the 5 min. time point, but was not lost, exhibiting an overall increase in signal intensity with time. These data indicate that both phosphorylated ErbB2 signal magnitude and signal stability were increased by Muc4 expression. Because alterations in receptor signaling down regulation are implicated in driving aberrant cellular proliferation and resulting in oncogenic phenotypes, Muc4 in these experiments is depicted as a potent cell surface oncogenic signaling modulator for the receptor ErbB2.
Muc4 modulates ErbB2 signaling potential by stabilizing and directly interacting with the ErbB2-ErbB3 heterodimer
Elevated and constitutive ErbB2 phosphorylation signals have been implicated in numerous human cancers, and the mechanism by which ErbB2 acquires this oncogenic potential is believed to occur in response to heterodimer formation of ErbB2 with other members of the family, resulting in functional oncogenic units that can drive tumor cell proliferation (Holbro et al., 2003). Although ErbB2 homodimers are also suspected to be the result of elevated phosphorylated ErbB2 signaling in cancer, there is no current structure data that supports this mechanism (Garrett et al., 2003). In our model system (A375), ErbB3 expression is robust, making it the most likely ErbB2 partner. We therefore evaluated the role of the ErbB2-ErbB3 unit in the Muc4 modulation effect using ErbB3 co-immunoprecipitation analysis. Fig. 2A shows that ErbB3 co-immunoprecipitates with both ErbB2 and Muc4, but in cells not expressing Muc4, the stable association between ErbB2 and ErbB3 is lost. This result was inconsistent with the Muc4 co-immunoprecipitation observation in Fig.1A where ErbB3 was not stably associated with Muc4; however such inconsistency is possible when a co-immunoprecipitation analysis is carried out with antibodies that are raised against different proteins within a complex. The enhanced stability of the Muc4-ErbB2-ErbB3 association also suggested that Muc4 potentiates ErbB2 phosphorylation by directly interacting with the ErbB2-ErbB3 heterodimer.
FIGURE 2.
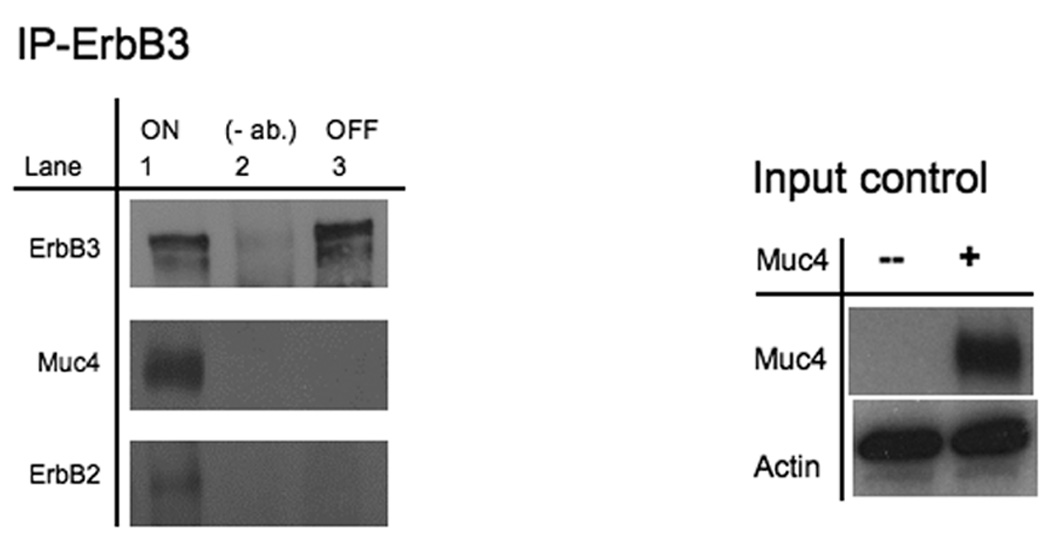
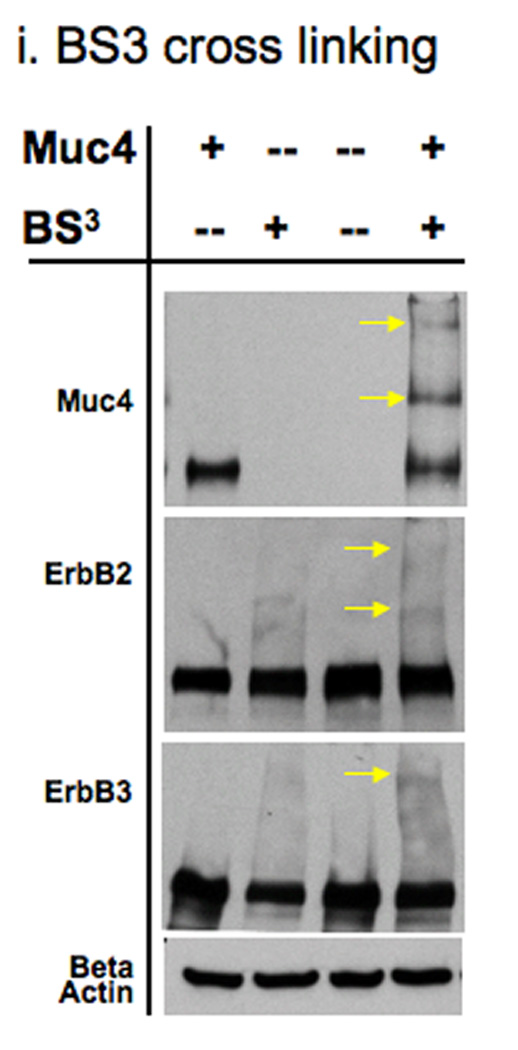
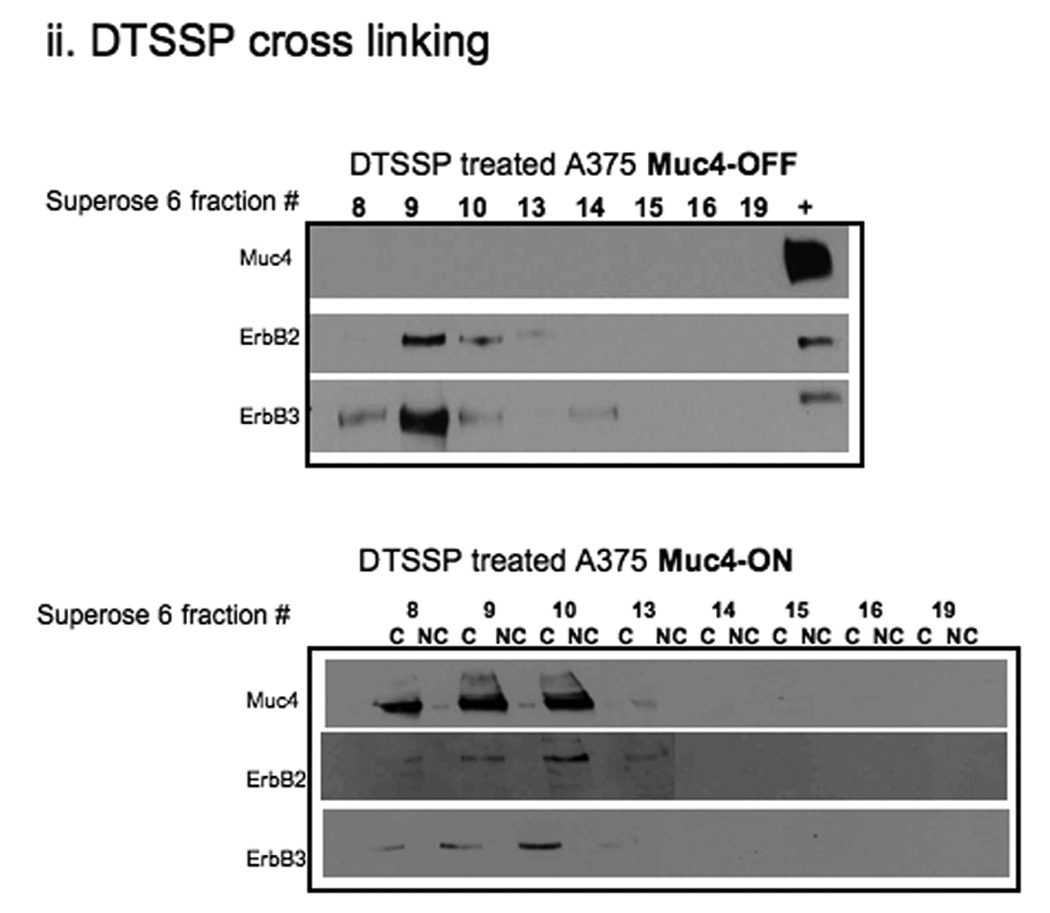
Muc4 modulates ErbB2 signaling potential by stabilizing and directly interacting with the ErbB2-ErbB3 heterodimer. A. Muc4 promotes stable ErbB2-ErbB3 interaction. Proteins from RIPA extracts of A375 cells with or without Muc4 (48 h) were subjected to immunoprecipitation with anti-ErbB3 (C-17) antibody. Immunoprecipitates were immunoblotted with anti-ErbB3, anti-Muc4, and anti-ErbB2 antibodies. The input control is an immunoblot of the lysates with anti-Muc4 and anti β-actin antibodies. B. Muc4 expression results in Muc4-ErbB2-ErbB3 complexes. Cellular chemical cross-linking immunoblots. A375 Muc4-transfected (Rep 3 clone) cells with or without Muc4 (48 h) were crossed linked with chemical cross-linking agents. i). With BS3 [1 mM] for 2 h on ice, and quenched with Tris-HCl [20–50 mM], pH 7.5 at room temperature for 15 min. Yellow arrows indicate cross-linked products. Cell lysates were separated on 4–15% gradient gels, and blotted with anti-Muc4, anti-ErbB2, anti-ErbB3, and β-actin antibodies. ii). With the reversible chemical cross linking analogue, DTSSP, at [1 mM] for 2 h, quenched with Tris-HCl [20–50 mM], pH 7.5 at room temperature for 15 min. Cross-linked cells were lysed in RIPA buffer without phosphatase inhibitors or dithiothreitol. Cleared lystes were loaded by FPLC (1 mL/min) onto a high resolution Superose 6 analytical gel filtration column equilibrated at 4°C in RIPA buffer without Triton X-100, inhibitors or dithiothreitol. Collected fractions were TCA-precipitated and separated on 4–15% gradient gels. Muc4-Off fractions were treated with 5% β-mercaptoethanol-containing SDS-PAGE sample buffer (top). Muc4-On fractions (bottom) were loaded side by side in duplicates with native SDS-PAGE sample buffer to represent non-cleaved (NC) samples, or with 5% β-mercaptoethanol containing SDS-PAGE sample buffer to examine the cleaved complex components (C). Samples were then immunoblotted with the indicated antibodies.
Previous reports on the Muc4 potentiation mechanism of ErbB2 phosphorylation showed that in insect cells, the Muc4-ErbB2 interaction was dependent on one of the EGF-like domains within Muc4 (Carraway et al., 1999). To further explore the role of the ErbB2-ErbB3 heterodimer in Muc4 signaling, we chemically cross-linked A375 cells with or without Muc4 using BS3, a water soluble, non-cleavable, and membrane impermeable chemical cross-linker with a spacer arm length of 11.4 Å. We predicted that the cross-linked Muc4 expressing samples would result in changes in the immunoblot patterns of Muc4, ErbB2, and ErbB3, possibly through the generation of additional, unique slower mobility bands. Fig. 2B-i is depicted with arrows that point to the additional slower mobility unique bands that were observed in Muc4, ErbB2 and ErbB3 immunoblots after this treatment. We noted that one cross-linked product of slow mobility at a similar position on the gel was observed in all immunoblots (Muc4, ErbB2, and ErbB3), indicating that Muc4 expression resulted in Muc4-ErbB2-ErbB3 complexes. To further test the direct Muc4 effect, we proceeded to characterize these large complexes by identifying their composition. We repeated the cross-linking experiment with a reversible chemical cross linker analogue, DTSSP, and included a fractionation step by gel filtration chromatography on Superose6 column prior to cleavage and immunoblot analysis of samples. An observation of a Muc4 band in unique fractions, i.e. without ErbB2 and ErbB3 bands, supports an indirect Muc4 mechanism, while an observation of Muc4 band in the same fractions with ErbB2 and ErbB3 bands will support a direct Muc4 mechanism. The eluted fractions were TCA precipitated and reduced to reverse the cross-linking prior to the immunoblot analysis. The results in Fig. 2B-ii show that in the Muc4 expressing sample, Muc4 was eluted in the same fractions (8–10 mL) that were observed for the receptors, indicating that Muc4 expression results in Muc4-ErbB2-ErbB3 complexes, and further supporting a direct role for Muc4 in the mechanism by which it potentiates ErbB2 phosphorylation signaling. This figure also shows that ErbB2 and ErbB3 were eluted together in the control cells; however, the pattern of ErbB2 and ErbB3 association was different, displaying an extended interaction across the fractions in the Muc4 expressing cells, and a more centered association in the control sample. These results were also in agreement with the ErbB3 co-immunoprecipitation (Fig. 2A) and suggested that Muc4 stabilizes the ErbB2-ErbB3 heterodimer directly, and that in the absence of Muc4, the interaction between ErbB2 and ErbB3 is more transient in nature.
Muc4 has no effect on ErbB3 phosphorylation
The Muc4 stable association with both ErbB2 and ErbB3 suggested that Muc4 could also result in differential ErbB3 phosphorylation signal. We therefore examined changes in ErbB3 phosphorylation levels in response to Muc4 expression, anticipating that the increase in phosphorylated ErbB2 will accompany similar changes in the ErbB3 phosphorylation levels. Fig. 3 shows in triplicate samples that Muc4 expression has no effect on ErbB3 phosphorylation, while the positive control lanes of samples treated with the ErbB3 soluble ligand HRG-1β do generate phosphorylated ErbB3 signal upon stimulation for 3 min. Similar results were also obtained in immunoblots with other phospho-tyrosine antibodies (not shown). These results indicate that Muc4 has no effect on ErbB3 phosphorylation, and suggest that the role of ErbB3 in Muc4 signaling is to provide added structural stability to the Muc4-ErbB2-ErbB3 complex.
FIGURE 3.
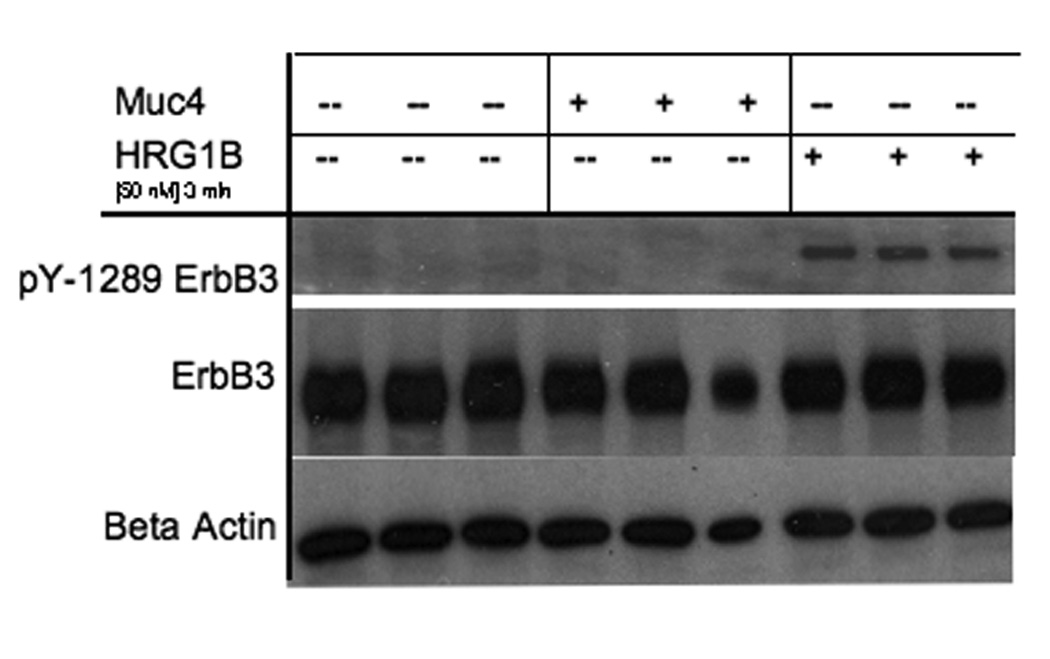
Muc4 has no effect on ErbB3 phosphorylation. A375 Muc4-transfected (Rep 3 clone) cells with or without Muc4 (48 h) and starved (0.1% FBS) for 24 h were incubated at 37°C with HRG-1β [50 nM] for 3 min. Cleared lysates were loaded in triplicates and immunoblotted with anti-ErbB3, anti-ErbB Y1289, and anti β-actin antibodies.
Muc4 promotes ErbB2 autocatalysis
The mechanism by which Muc4 potentiates ErbB2 signaling was assessed further by testing the mechanistic role of Muc4 in affecting ErbB2 kinase activity. One possibility was that Muc4 association with the ErbB2-ErbB3 dimer promotes ErbB2 autocatalytic activity, leading to an increase in ErbB2 phosphorylation signal. To test this possibility we examined the effect of an ErbB2 kinase inhibitor on Muc4 promotion of ErbB2 phosphorylation. Two different outcomes were possible with Muc4 expression and effective inhibitor treatment (Fig. 4 lane 4); ErbB2 phosphorylation will be lost, suggesting that Muc4 promotes ErbB2 autocatalysis, or the signal will be maintained, suggesting a membrane trapping role for Muc4, in which other intracellular kinases that are not effected by the inhibitor treatment phosphorylate the ErbB2 cytoplamsic tail.
The kinase inhibitor Lapatinib is a reversible dual inhibitor of EGFR and ErbB2 auto phosphorylation and activation. The A375 cell line was not used in this experiment because our analysis indicated that this cell line was not sensitive to Lapatinib treatment (data not shown). Instead, 14 the breast cancer cell line BT-474 was used for the following reasons; it is a Lapatinib sensitive cancer cell line at relatively low concentration (Penuel et al., 2002); it has no detected endogenous Muc4 expression; the Muc4 plasmid is readily transfected into it; and importantly, it exhibits similar phenotype regarding an increased ErbB2 phosphorylation level as was observed in the A375 line (see figure 4 lanes 1, 2). Our results in Fig. 4 indicate that with Muc4 expression and effective Lapatinib inhibition, the phosphorylated ErbB2 signal is lost. These data therefore indicate that Muc4 acts by promoting ErbB2 autocatalysis.
Downstream signaling analysis
The emerging model from our study indicated that in Muc4 expressing cells, large complexes containing Muc4-ErbB2-ErbB3 were forming at the cell membrane (Fig. 2), displaying a potent signaling potential through augmented and stable ErbB2 phosphorylation (Fig. 1), but lacking in ErbB3 phosphorylation (Fig. 3). To better understand the role that Muc4 plays in down stream signaling we examined the signaling pathways that are differentially stimulated by its expression using an antibody microarray screen. Previous Muc4 dependent signaling studies showed that Muc4 expression activates the MAP kinase p38 pathway (Ramsauer et al., 2003) and that MUC4 silencing in ovarian cancer cells activates the Erk pathway (Ponnusamy et al., 2008).
The antibody microarray analysis surveyed over 650 signaling components from the major established signaling pathways, and probed both phosphorylated and total protein levels. Using the A375 human melanoma cell line as a model system, signaling hits between cells expressing Muc4 for 48 h. and cells not expressing Muc4 were compared. The screen generated several hits that were at least two fold different between the Muc4 expressing cells and the control cells (Table 1). We noted that several of these hits were signaling proteins that are implicated in cell migration pathways (FAK, β-catenin, Paxillin). We validated several of these targets by western blot analysis, and our results (Fig. 5) were in agreement with the results of the screen. Some of the validated signals displayed a more significant change in signal intensity than was observed in the screen, possibly due to the stringent signal background threshold criteria settings in the array. Our results suggest that Muc4 expression promotes cell migration by increasing the phosphorylation of the focal adhesion kinase (FAK), and also through an increase in the levels of β-catenin. These results further support the involvement of the Muc4-ErbB2-ErbB3 interaction in cell migration, and are in agreement with a study in ovarian tumor cells in which MUC4 promoted FAK phosphorylation, cytoskeletal rearrangements and cell migration (Ponnusamy et al., 2008).
Table 1. Muc4 expression results in differential signaling regulation.
Summary of the antibody microarray screen analysis (Kinexus™ Bioinformatics Corporation- KAM-1.1) indicating hits with two fold increase or decrease in signal outputs. A375 Muc4-transfected (Rep 3 clone) cells induced or not to express Muc4 (48 h) were lysed in the recommended lysis buffer, and the cleared lysates were assayed for protein concentration using standard Bradford protocol. Samples were adjusted to [2 mg/mL] and shipped on dry ice. Samples were surveyed by labeling the control and treated lysate samples with the same dye, and analyzing both samples separately but on the same chip. The output included qualitative and semi-quantitative analyses of the differential expression and phosphorylation states of over 650 protein kinases and other cell signaling proteins. The full data set is accessible at http://www.kinexus.ca/kinet. The array barcode is K01070228, and the kinexus IDs are 11807 and 11808 for the control and treated samples respectively.
| Target Protein Name |
Phospho Site (Human) |
Full Target Protein Name | Swiss-prot Link |
Fold Change |
|---|---|---|---|---|
| RSK1/2 | S363/S369 | Ribosomal S6 protein-serine kinase 1/2 | Q15418 | 4.41 |
| FAK | S722 | Focal adhesion protein-tyrosine kinase | Q05397 | 3.83 |
| PKCq | S676 | Protein-serine kinase C theta | Q04759 | 3.27 |
| Csk | Pan-specific | C-terminus of Src tyrosine kinase | P41240 | 2.90 |
| SOX9 | S181 | SRY (sex determining region Y)-box 9 (campomelic dysplasia, autosomal sex- reversal) |
P48436 | 2.58 |
| FAK | S843 | Focal adhesion protein-tyrosine kinase | Q05397 | 2.41 |
| PP4/A'2 | Pan-specific | Protein-serine phosphatase 4 - regulatory subunit (PPX/A'2) |
Q8TF05 | 2.39 |
| PDK1 | Pan-specific | 3-phosphoinositide-dependent protein- serine kinase 1 |
O15530 | 2.34 |
| Rb | T821 | Retinoblastoma-associated protein 1 | P06400 | 2.24 |
| Cofilin 1 | S3 | Cofilin 1 | P23528 | 2.21 |
| STAT1 | Y701 | Signal transducer and activator of transcription 1 |
P42224 | 2.19 |
| Tau | S720 | Microtubule-associated protein tau | P10636 | 2.03 |
| Paxillin 1 | Y118 | Paxillin 1 | P49023 | 0.39 |
| Catenin b | S45 | Catenin (cadherin-associated protein) beta 1 |
P35222 | 0.46 |
| CDK10 | Pan-specific | Cyclin-dependent protein-serine kinase 10 PISSLRE |
Q15131 | 0.46 |
| CaMKK (CaMKK2) | Pan-specific | Calcium/calmodulin-dependent protein- serine kinase kinase |
Q8N5S9 | 0.47 |
| Histone H2B | S14 | Histone H2B | P33778 | 0.47 |
| PI3KR4 | Pan-specific | Phospohoinositide-3-kinase, regulatory subunit 4 |
Q99570 | 0.47 |
FIGURE 5.
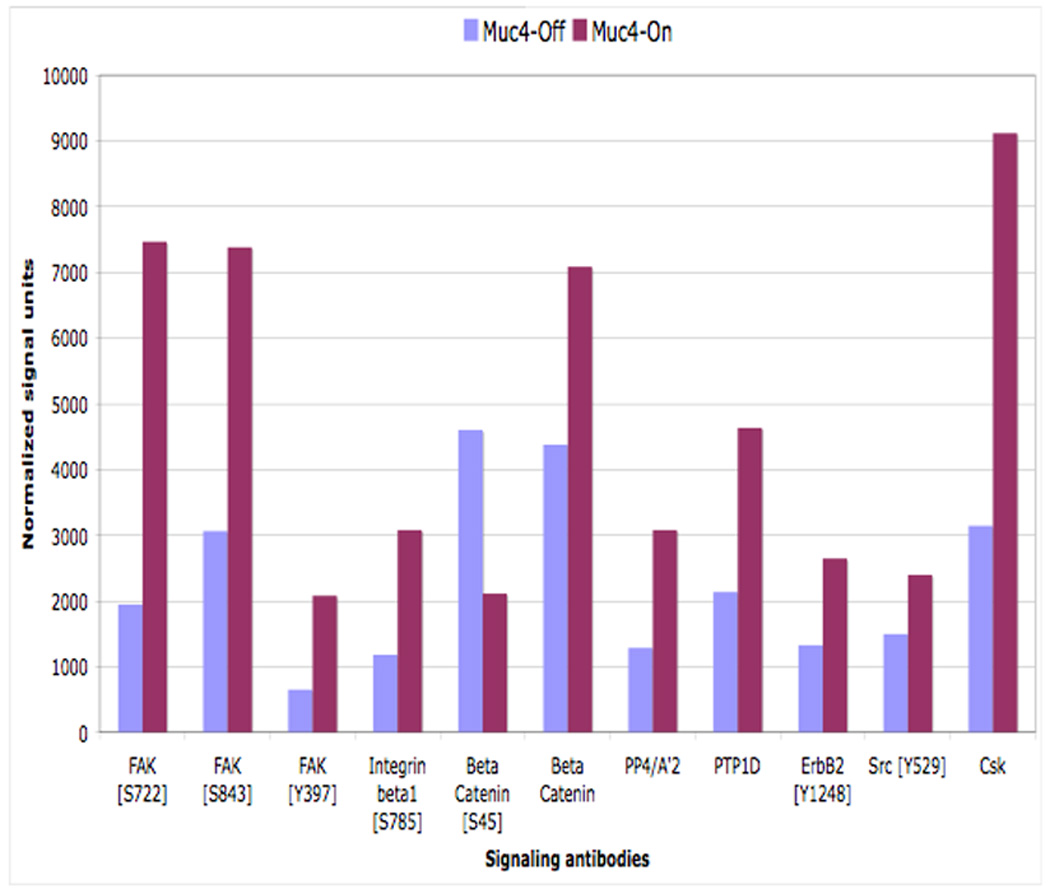
Muc4 expression promotes cell migration signaling by increasing the phosphorylation of focal adhesion kinase (FAK). Immunoblot validation analyses of the microarry screen results. Proteins from RIPA extracts of A375 Muc4-transfected cells induced or not to express Muc4 (48 h) were immunoblotted with the indicated signaling antibodies. Normalized signal units were calculated using the ImageJ NIH densitometry software, and normalized with the corresponding β-actin singal.
DISCUSSION
ErbB receptor activation mechanism is intensively studied due to the important role that these receptors play in several cellular processes and in cancer pathogenesis. These studies indicate that the ErbB receptor activation mechanism is complex, and can result in unpredicted signaling outcomes. Investigating the mechanisms of unpredicted ErbB signaling outcomes further enhance our understanding of these receptor activation mechanisms and our ability to design specific regulating agents. This study examined unpredicted ErbB2 signaling outcomes that were generated in response to the membrane mucin expression, Muc4. Our signaling analysis shows that Muc4 imparts ligand-independent ErbB2 signaling. Our mechanistic analysis indicates that Muc4 acts through direct and stable interaction with the ErbB2-ErbB3 heterodimer, resulting in ErbB2 autocatalysis. The downstream signaling analysis shows that the Muc4-ErbB2-ErbB3 complex may also stimulate cellular migration via the FAK signaling pathway. Together these results suggest a model in which Muc4 effectively stimulates cellular signaling via its direct and stable interaction with the ErbB2-ErbB3 heterodimer (Carraway et al., 2009), possibly a result of Muc4 unique structural complementary relationship with the heterodimer.
ErbB receptor activation mechanisms that are derived from in vivo studies suggest that these receptors can become activated via unique mechanisms that are not always evident in in vitro or through structural studies. Our results generated unpredicted ErbB2 signaling outcomes showing that Muc4, a non-ErbB family member, effect ErbB2 signaling quantitatively and temporally in a cellular background lacking soluble ligand stimulation. The outcome of ErbB2 ligand-independent activation has been previously observed in cellular systems (Nagy et al., 1999; Penuel et al., 2002; Zhang et al., 2006), and has been predicted as a possible outcome in molecular models of ErbB2 as a result of increased receptor local concentration on the surface of living cells through multiple ErbB2 interfaces interactions (Zhang et al., 2006; Kumagi et al., 2003). However, in our analysis, the increased ErbB2 phosphorylation in Muc4 induced cells did not accompany an increase in ErbB2 local concentration or receptor expression levels. Instead, our cross linking data suggests that the ErbB2 signaling stability is imparted by Muc4 expression through direct interaction at the membrane. Interestingly, the Muc4-ErbB2-ErbB3 complex does not similarly stimulate ErbB3 phosphorylation, suggesting that the role of ErbB3 in this complex is to provide added structural stability, and that the Muc4 effect is unique to ErbB2 but is dependent on prior ErbB2-ErbB3 association. The proposed activation mechanism is that Muc4 stimulates ErbB2 autocatalysis through stable Muc4-ErbB2-ErbB3 complex formation.
The single pass transmembrane domain of MUC4/Muc4 (UniProtKB/Swiss-Prot file # Q99102, # Q63661, #P04626) within ASGP-2 may also play a significant role in this stable association. Experimental evaluation of the effect of the transmembrane domain on ErbB receptor activity using mutational analyses and molecular models indicate that dimerization can also occur within this domain ( Tanner and Kyte, 1999; Sharpe et al., 2000; Mendrola et al., 2002; Samna Soumana et al., 2008), and that the specific alignment of transmembrane helices can facilitate ligand independent activity. In addition, these studies showed that interactions in the transmembrane domains may increase dimer stability, and highlighted the predictive power of such dimerization based on the transmembrane sequence, specifically the presence of the putative GXXXG motif in the correct orientation (Samna Soumana et al., 2007, 2008). In this motif, G is Gly and X is any residue, and a variation of this motif, SmxxxSm, in which a Gly residue can be replaced by small a residue such as Ala, Ser or Thr (Samna Soumana et al., 2008), was similarly shown to promote transmembrane association stability. All ErbB receptors as well as Muc4 contain these motifs, and the orientation of these within the transmembrane domains can be assessed to predict their role in facilitating stable membrane association (Dawson et al., 2002). Molecular modeling and or mutational analyses of the Muc4-ErbB2-ErbB3 transmembrane domain dimerization potential is necessary in order to better understand the role that this domain may play in the formation of stable Muc4-ErbB2-ErbB3 complexes that can yield ligand independent signaling.
Additional support for our model was generated in a recent study that solved the extracellular domain structure of the single EGF receptor family member in Drosophila melanogaster (dEGFR), the closest structural relative of ErbB2 (Alvarado et al., 2009). Through comparison of the extracellular structures, growth factor ligand regulation, and autoinhibition through interdomain interactions, the authors suggest that ErbB2 signaling may be regulated by ligands in the same way as dEGFR. Reports that the dEGFR ligands must be palmitoylated to drive their membrane association and increase the local ligand concentration (Miura et al., 2006) support the view that membrane association is a key feature of ligands that activate ErbB receptors that adopt an extended confirmation, i.e. ErbB2 and dEGFR. Together these studies reinforce the usage of the term intramembrane-ligand when referring to the Muc4 ASGP-2 subunit role as a ligand for the receptor ErbB2.
The large mucin subunit of Muc4, ASGP-1, which extends a few nM out into the extracellular matrix, was established as an anti-adhesive factor that disrupts cell-cell and cell-matrix interactions (Komatsu et al., 1997). Our downstream signaling analysis showed that the transmembrane subunit of Muc4, Muc4-rep3, which did not show an anti-adhesive effect due to its truncated mucin subunit, effectively stimulated ErbB2 to transduce cellular migration signaling. This observation further illustrates the dual role that Muc4 plays as a signaling potentiation factor, and that the Muc4 ASGP-2 subunit through its action with the ErbB2-ErbB3 heterodimer can influence cell migration without directly altering cell adhesion (Carraway et al., 2009).
In conclusion, our model suggests that the transmembrane subunit of Muc4, ASGP-2, can result in significant ErbB2 phosphorylation and downstream signaling through membrane association with the heterodimer ErbB2-ErbB3. In addition, the Muc4 membrane association dependent signaling is proposed to be unique to ErbB2.
ACKNOWLEDGMENTS
We thank Malik Keshwani from Dr. T.K. Harris’ laboratory at the University of Miami School of Medicine for technical help with the gel filtration fractionation. We also would like to thank GlaxoSmithKline for providing Lapatinib for our Kinase inhibition experiments.
Contract grant sponsor: National Institutes of Health; Contract grant numbers: RO1 CA52498
REFERENCES
- Alvarado D, Klein DE, Lemmon MA. Nature. 2009;461(7261):287–291. doi: 10.1038/nature08297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess AW. Growth Factors. 2008;26(5):263–274. doi: 10.1080/08977190802312844. [DOI] [PubMed] [Google Scholar]
- Carraway KL, III, Rossi EA, Komatsu M, Price-Schiavi SA, Huang D, Guy PM, Carvajal ME, Fregien N, Carraway CAC, Carraway KL. J. Biol. Chem. 1999;274(9):5263–5266. doi: 10.1074/jbc.274.9.5263. [DOI] [PubMed] [Google Scholar]
- Carraway KL, Theodoropoulos G, Kozloski GA, Carothers Carraway CA. Future Oncol. 2009;5(10):1631–1640. doi: 10.2217/fon.09.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi P, Singh AP, Chakraborty S, Chauhan SC, Bafna S, Meza JL, Singh PK, Hollingsworth MA, Mehta PP, Batra SK. Cancer Res. 2008;68(7):2065–2070. doi: 10.1158/0008-5472.CAN-07-6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri A, Yarden Y. Nat Rev Mol Cell Biol. 2006;7(7):505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- Dawson JP, Weinger JS, Engelman DM. J Mol Biol. 2002;316(3):799–805. doi: 10.1006/jmbi.2001.5353. [DOI] [PubMed] [Google Scholar]
- Funes M, Miller JK, Lai C, Carraway KL, 3rd, Sweeney C. J Biol Chem. 2006;281(28):19310–19319. doi: 10.1074/jbc.M603225200. [DOI] [PubMed] [Google Scholar]
- Garrett TP, McKern NM, Lou M, Elleman TC, Adams TE, Lovrecz GO, Kofler M, Jorissen RN, Nice EC, Burgess AW, Ward CW. Mol Cell. 2003;11(2):495–505. doi: 10.1016/s1097-2765(03)00048-0. [DOI] [PubMed] [Google Scholar]
- Hattrup CL, Gendler SJ. Annu Rev Physiol. 2008;70:431–457. doi: 10.1146/annurev.physiol.70.113006.100659. [DOI] [PubMed] [Google Scholar]
- Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF, 3rd, Hynes NE. Proc Natl Acad Sci USA. 2003;100(15):8933–8938. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes NE, Lane HA. Nat Rev Cancer. 2005;5(5):341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- Jepson S, Komatsu M, Haq B, Arango ME, Huang D, Carraway CA, Carraway KL. Oncogene. 2002;21(49):7524–7532. doi: 10.1038/sj.onc.1205970. [DOI] [PubMed] [Google Scholar]
- Karg A, Dinc ZA, Basok O, Ucvet A. Pathol Res Pract. 2006;202(8):577–583. doi: 10.1016/j.prp.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Carraway CAC, Freigien N, Carraway KL. J Biol Chem. 1997;272(52):33245–33254. doi: 10.1074/jbc.272.52.33245. [DOI] [PubMed] [Google Scholar]
- Kumagai T, Katsumata M, Hasegawa A, Furuuchi K, Funakoshi T, Kawase I, Greene MI. Proc Natl Acad Sci USA. 2003;100(16):9220–9225. doi: 10.1073/pnas.1633546100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linggi B, Carpenter G. Trends Cell Biol. 2006;16(12):649–656. doi: 10.1016/j.tcb.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Mendrola JM, Berger MB, King MC, Lemmon MA. J Biol Chem. 2002;277(7):4704–4712. doi: 10.1074/jbc.M108681200. [DOI] [PubMed] [Google Scholar]
- Miura GI, Buglino J, Alvarado D, Lemmon MA, Resh MD, Treisman JE. Dev Cell. 2006;10(2):167–176. doi: 10.1016/j.devcel.2005.11.017. [DOI] [PubMed] [Google Scholar]
- Moasser MM. Oncogene. 2007;26(45):6469–6487. doi: 10.1038/sj.onc.1210477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moniaux N, Chaturvedi P, Varshney GC, Meza JL, Rodriguez-Sierra JF, Aubert JP, Batra SK. Br J Cancer. 2007;97(3):345–357. doi: 10.1038/sj.bjc.6603868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morandell S, Stasyk T, Skvortsov S, Ascher S, Huber LA. Proteomics. 2008;8(21):4383–4401. doi: 10.1002/pmic.200800204. [DOI] [PubMed] [Google Scholar]
- Nagy P, Jenei A, Kirsch AK, Szollosi J, Damjanovich S, Jovin TM. J Cell Sci. 1999;112(Pt 11):1733–1741. doi: 10.1242/jcs.112.11.1733. [DOI] [PubMed] [Google Scholar]
- Penuel E, Akita RW, Sliwkowski MX. J Biol Chem. 2002;277(32):28468–28473. doi: 10.1074/jbc.M202510200. [DOI] [PubMed] [Google Scholar]
- Plowman GD, Culouscou JM, Whitney GS, Green JM, Carlton GW, Foy L, Neubauer MG, Shoyab M. Proc Natl Acad Sci USA. 1993;90(5):1746–1750. doi: 10.1073/pnas.90.5.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnusamy MP, Singh AP, Jain M, Chakraborty S, Moniaux N, Batra SK. Br J Cancer. 2008;99(3):520–526. doi: 10.1038/sj.bjc.6604517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price-Schiavi SA, Zhu X, Aquinin R, Carraway KL. J Biol Chem. 2000;275(23):17800–17807. doi: 10.1074/jbc.275.23.17800. [DOI] [PubMed] [Google Scholar]
- Ramsauer VP, Carraway CAC, Salas PJI, Carraway KL. J Biol Chem. 2003;278(32):30142–30147. doi: 10.1074/jbc.M303220200. [DOI] [PubMed] [Google Scholar]
- Ramsauer VP, Pino V, Farooq A, Carothers Carraway CA, Salas PJ, Carraway KL. Mol Biol Cell. 2006;17(7):2931–2941. doi: 10.1091/mbc.E05-09-0895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi EA, McNeer RR, Price-Schiavi SA, Van den Brande JMH, Komatsu M, Thompson JF, Carraway CAC, Fregien NL, Carraway KL. J Biol Chem. 1996;271(52):33476–33485. doi: 10.1074/jbc.271.52.33476. [DOI] [PubMed] [Google Scholar]
- Samna Soumana O, Garnier N, Genest M. Eur Biophys J. 2007;36(8):1071–1082. doi: 10.1007/s00249-007-0195-6. [DOI] [PubMed] [Google Scholar]
- Samna Soumana O, Garnier N, Genest M. Eur Biophys J. 2008;37(6):851–864. doi: 10.1007/s00249-008-0293-0. [DOI] [PubMed] [Google Scholar]
- Sharpe S, Barber KR, Grant CW. Biochemistry. 2000;39(21):6572–6580. doi: 10.1021/bi000038o. [DOI] [PubMed] [Google Scholar]
- Sheng ZQ, Hull SR, Carraway KL. J Biol Chem. 1990;265(15):8505–8510. [PubMed] [Google Scholar]
- Shibahara H, Tamada S, Higashi M, Goto M, Batra SK, Hollingsworth MA, Imai K, Yonezawa S. Hepatology. 2004;39(1):220–229. doi: 10.1002/hep.20031. [DOI] [PubMed] [Google Scholar]
- Singh AP, Chaturvedi P, Batra SK. Cancer Res. 2007;67(2):433–436. doi: 10.1158/0008-5472.CAN-06-3114. [DOI] [PubMed] [Google Scholar]
- Sliwkowski MX, Schaefer G, Akita RW, Lofgren JA, Fitzpatrick VD, Nuijens A, Fendly BM, Cerione RA, Vandlen RL, Carraway KL., 3rd J Biol Chem. 1994;269(20):14661–14665. [PubMed] [Google Scholar]
- Tanner KG, Kyte J. J Biol Chem. 1999;274(50):35985–35990. doi: 10.1074/jbc.274.50.35985. [DOI] [PubMed] [Google Scholar]
- Warren CM, Landgraf R. Cell Signal. 2006;18(7):923–933. doi: 10.1016/j.cellsig.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Yonezawa S, Goto M, Yamada N, Higashi M, Nomoto M. Proteomics. 2008;8(16):3329–3341. doi: 10.1002/pmic.200800040. [DOI] [PubMed] [Google Scholar]
- Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. Cell. 2006;125(6):1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]