

Abstract
The p21-activated kinases (PAKs), immediate downstream effectors of the small G-proteins of the Rac/cdc42 family, are critical mediators of signaling pathways regulating cellular behaviors and as such, have been implicated in pathological conditions including cancer. Recent studies have validated the requirement for PAKs in promoting tumorigenesis in breast carcinoma and neurofibromatosis. Thus, there has been considerable interest in the development of inhibitors to the PAKs, as biological markers and leads for the development of therapeutics. While initial approaches were based on screening for competitive organic inhibitors, more recent efforts have focused on the identification of allosteric inhibitors, organometallic ATP-competitive inhibitors and the use of PAK1/inhibitor crystal structures for inhibitor optimization. This has led to the identification of highly selective and potent inhibitors, which will serve as a basis for further development of inhibitors for therapeutic applications.
Keywords: p21-activated kinases, organometallic, kinase inhibitors, breast cancer, neurofibromatosis
1. General background
The Rac and Cdc42 proteins are members of the Rho family of small G-proteins and are regulators of several signaling pathways, including those impinging on cellular proliferation, cytoskeleton reorganization, gene expression, and endocytosis [1, 2]. As immediate downstream effectors of Rac/Cdc42, the p21-activated kinases (PAKs) are activated by the binding of the active forms of Rac/Cdc42 (the GTP-bound forms) and as such, participate in a wide range of signaling pathways that modulate cellular behaviors. Given the major roles small G-proteins play in tumorigenesis, it is not surprising that the PAKs have also been implicated in this process. The aim of this review is to survey the efforts reported to validate the PAKs as therapeutic targets for cancer and to develop small-molecule inhibitors of the PAKs. Extensive reviews on the biology of the PAKs and the roles PAKs play in cancer have been recently published and the reader is referred to these comprehensive reviews for further details [3–5].
2. The PAK protein family and structure
The PAKs form a protein family of serine/theronine kinases, divided into 2 groups, I and II. The group I PAKs consist of three members - PAK1, PAK2 and PAK3 that share 93–95% sequence identity within the kinase domain (Figure 1A) [6, 7]. Crystal structures of the PAK1 kinase domain have been determined, in both the inhibited and active conformations. In the inactive form, PAK1 is thought to exist as a homodimer, in a “head to tail” orientation (Figure 1B) [8, 9]. In this configuration, the N-terminal regulatory domain inhibits the kinase domain in trans [10–12]. This is mediated by overlapping functional regions within the N-terminal regulatory domain. Specifically, an “inhibitory switch” domain that associates with the large lobe of the kinase domain and a “kinase inhibitory” domain directly blocks the catalytic cleft. Upon binding of Rac or Cdc42-GTP to its N-terminal tail, the PAK1 dimer is predicted to dissociate and the “kinase inhibitory” domain is removed from the catalytic cleft [8]. This allows for an active conformation that can now auto-phosphorylate threonine 423 within the activation loop and additional residues that prevent the kinase from shifting back into an inactive state (Figure 1B) [13].
Figure 1. Schematic representation of domain organization and activation following Rac/Cdc42 binding for group I PAKs.
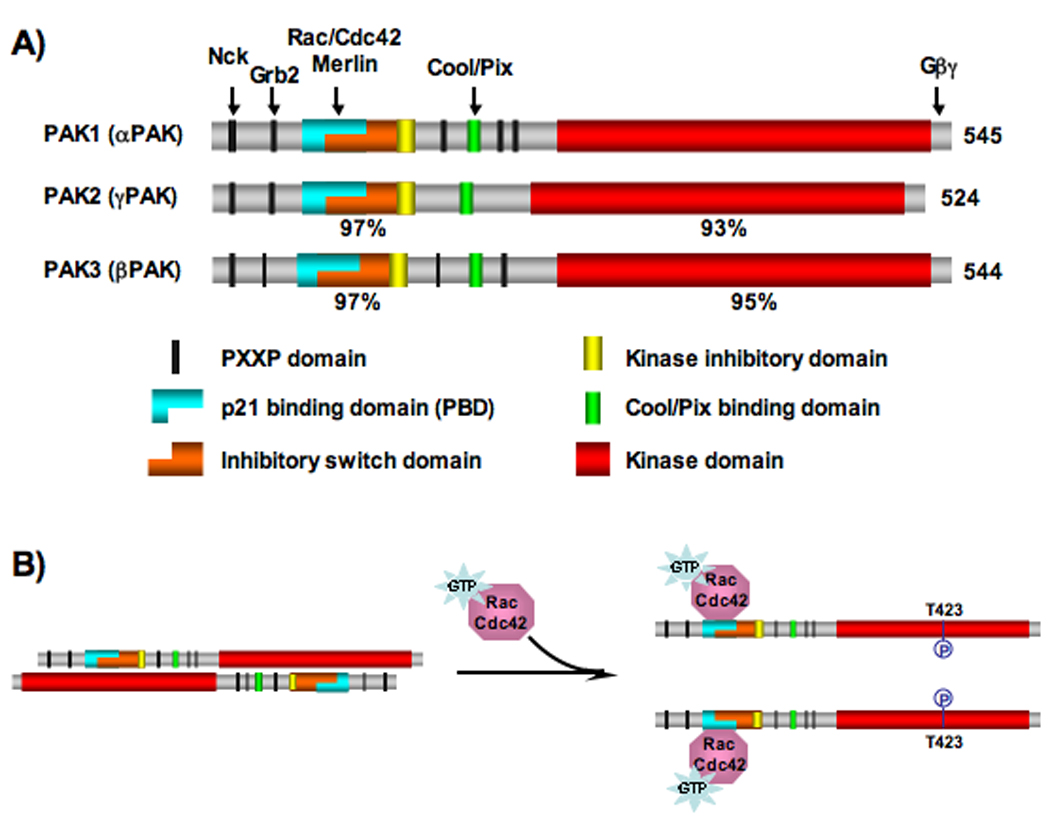
A) Domain organization of group I PAKs. Arrows indicate regions of interaction with key PAK binding partners/regulators listed above. The similarities of the regulatory and kinase domains in PAK2 and PAK3 to the corresponding domains in PAK1 are indicated under the respective domains. The size and location of each domain along the proteins reflect actual scale.
B) Conversion of PAK1 from inactive form to active form by Rac/Cdc42-GTP binding. Autophosphorylation at T423, the most critical step during PAK1 activation, is indicated.
In contrast, Group II PAKs, comprised of PAK4, PAK5 and PAK6, do not possess an auto-inhibitory domain and are not activated by Rac/Cdc42-GTP binding [14]. Given differences in the mode of regulation, overall structure and active sites between group I and group II PAKs, it is conceptually possible to develop inhibitors that would differentiate between the two groups [15]. However, for the purpose of this review we will focus our discussion on the development of group I PAK inhibitors.
3. Brief outline of PAK biology
To date, more than 40 substrates have been reported for Group I PAKs, which implicate these kniases in a wide range of cellular activities including cell mobility, cell proliferation and apoptosis [3].
PAK, as part of a GIT-PIX-PAK-Nck complex located at focal adhesions, controls adhesion-induced Rac1 activation and cell spreading by regulating Rac1-β-Pix interaction [16, 17]. Furthermore, PAK also modulates cytoskeleton dynamics and cell mobility at the leading edge through phosphorylation of multiple substrates including myosin light-chain kinase (MLCK), paxillin, filamin A, cortactin, the LIM-kinases (LIMKs), Arpc1b, and stathmin [4].
During mitosis, PAK1 is recruited to the centrosomes where it interacts with a GIT1-PIX complex similar to the complex it forms at focal adhesions. Upon activation by GIT1-PIX, PAK1 phosphorylates Aurora-A and Plk1, both important regulators of mitotic events[18, 19]. In addition to driving cell cycle progression, PAK also promotes cell proliferation through phosphorylation of c-Raf (Ser338) and MEK (Ser298), two components of the MAPK pathway [20, 21].
PAK protects cells from apoptosis via multiple mechanisms. In response to survival signals, PAK phosphorylates the pro-apoptotic proteins Bad and BimL thus preventing them from interacting with anti-apoptotic protein Bcl2 [22–25]. Furthermore, PAK1 also inhibits apoptosis by phosphorylating and inactivating cell survival forkhead transcription factor, FKHR [26].
4. Validation of PAKs as therapeutic targets for cancer
Group I PAKs have long been implicated in tumorigenesis [27]. In particular, PAK1 has been reported to be widely overexpressed and/or hyperactivated in various types of benign and malignant cancers [3]. The roles of PAK1 in tumor pathogenesis and the potential therapeutic benefits of PAK inhibition are characterized in most detail in breast cancer and two types of mostly benign cancer syndrome, neurofibromatosis type 1 and 2 (NF1 and NF2).
PAK1 is upregulated in 50% of primary breast cancers [28]. Expression of a constitutively active PAK1 mutant (CA-PAK1) increases cell motility, anchorage-independent growth, and invasiveness in MCF-7 breast cancer cells and leads to development of metastatic mammary tumors and other types of breast lesions in a transgenic mouse model [29, 30]. Conversely, expression of dominant-negative PAK1 mutants (DN-PAK1s) suppresses cellular motility and invasiveness in MDA-MB-435 and MCF-7 breast cancer cells and inhibits pre-malignant progression in a 3-D cultural model for human breast cancer progression [30–33]. In addition, high PAK1 expression levels and nuclear localization have been correlated with tamoxifen resistance in ERα-positive breast cancer, which has been mechanistically linked with the ability of PAK1 to phosphorylate ERα on serine 305 [34–36]. The direct involvement of PAK1 in tumorigenesis in breast cancer and its potential role in mediating tamoxifen resistance are indications of the therapeutic potentials of PAK1 inhibition in treating malignant breast cancer, especially in the context of the resistance associated with tamoxifen-treated ERα-positive breast cancers.
NF1 and NF2 are dominantly inherited cancer disorders that develop mostly benign nerve sheath tumors of the peripheral nerves [37]. NF1 is quite common with a birth incidence of 1 in 3,000 and is caused by mutations of the NF1 tumor suppressor gene. NF2, on the other hand, is relatively less common (affecting 1 in 30,000 of the population) and has been attributed to the loss-of-heterozygosity (LOH) of the NF2 gene. NF1 encodes a Ras GTPase activating protein (GAP) named Neurofibromin. Deletion/inactivation of Neurofibromin leads to increased levels of activated GTP-bound Ras, which activates multiple oncogenic signaling cascades, including the MAPK and PI3K pathways. PAKs, as direct downstream effectors of Rac/Cdc42, are activated by signals from the PI3K pathway [38]. Activated PAKs phosphorylate c-Raf (S338) and MEK1 (S298), which are required for the full activation of the Ras-MAPK pathway [39–41]. The protein product of the NF2 gene is called Merlin, which is closely related to the cytoskeleton-linker ERM proteins (Ezrin, Radixin and Moesin). Merlin directly binds to PAKs and inhibits their activation by Rac/Cdc42 [42–45]. Therefore, despite NF1 and NF2 being pathologically and molecularly distinct diseases, they both involve mitogenic signaling pathways that are downstream of Ras and impinge upon the regulation of PAK activities [46, 47].
The importance of PAK1 in NF1 has long been suggested by pioneering studies in which dominant-negative PAK1 mutants were shown to efficiently block Ras transformation in both rat Schwann cells and a malignant peripheral nerve sheath tumor (MPNST or neurofibrosarcoma) cell line from an NF1 patient by interfering with the activation of the MAPK cascade [48]. More recently, PAK1 has been shown to play a critical role in regulating NF1 tumor environment. Several elegant studies have established that in addition to Nf1 deficiency in Schwann cells, Nf1 heterozygosity in the tumor microenvironment, particularly in bone marrow-derived mast cells (BMMCs), are required to induce neurofibroma progression in mouse models [49–51]. Importantly, Rac-GTP levels and PAK1 kinase activity are increased in Nf1+/− BMMCs after Stem cell factor (SCF) stimulation [46]. Using genetic approaches involving the crossing of Pak1−/− mice with Nf1+/− mice, McDaniel et al. demonstrated that loss of Pak1 reversed MAPK-mediated hyperproliferation and p38-regulated increased migration of Nf1 haploinsufficient BMMCs in culture and corrected dermal accumulation of Nf1+/− mast cells in vivo to levels found in wild-type mice [52]. These data established PAK1 as a key mediator of pro-tumorigenic signaling pathways in NF1 and validated PAK1 as a potential therapeutic target in this disease.
In NF2 patients, loss of Merlin is associated with elevated levels of Rac-GTP accompanied by abnormal PAK1 activation [47, 53–55]. PAK1 and PAK2 phosphorylate Merlin at serine 518, which inactivates Merlin by inducing a conformational change [56, 57]. Merlin, in a negative feedback loop, prevents PAK1 and PAK2 activation by Rac through competing with Rac for PAK binding [42–45]. Expression of a dominant-active (DA) Rac1 or PAK1 mutants overrides the inhibitory effect of Merlin on Ras-induced ERK activation and soft agar colony formation, suggesting that Merlin functions through the Rac-PAK axis to suppress Ras-ERK signaling and tumor growth [58]. To evaluate whether inhibition of PAK could be used to treat NF2 tumors, we recently employed a RNAi-based strategy to knockdown PAK1-3 expression in NIH3T3 cells expressing a dominant-negative mutant Merlin (NIH3T3/Nf2BBA) and found that depletion of all three group I PAKs severely inhibited the ability of NIH3T3/Nf2BBA cells to form tumors in a mouse xenograft model [47]. Furthermore, while attempting to knockdown PAK1 in RT4 rat schwannoma cells, we find that RT4 cells are highly addicted to PAK1 and that inhibition of PAK1 expression in these cells leads to cellular senescence [47]. More recently, Flaiz et al. applied a specific PAK inhibitor, IPA-3 (see below for further detail), to human primary schwannoma cells and found that it blocks PAK2 phosphorylation at Ser192/197, which antagonizes PAK-Pix interactions and reduces cell spreading and cell adhesion [59].
It should be noted that in addition to the dominant-negative, RNAi and genetic approaches discussed above, an inhibitory peptide (PAK1 83–149) that prevents PAK auto-phosphorylation has also been successfully used to inhibit PAK kinase activity in vitro [60]. Despite the usefulness of these different PAK inhibitory approaches in dissecting PAK signaling and in validating its therapeutic potential under experimental settings, they present challenges for clinical applications. Therefore, considerable efforts have also been devoted to develop small molecule inhibitors that are specific for group I PAKs.
5. Development of PAK inhibitors - early generations
As findings implicating PAKs as central players in cell signaling and human diseases such as cancer have mounted, interest in developing PAK inhibitors as biological probes and as leads for therapeutic development has increased. Initial attempts to screen for PAK1 inhibitors focused on adenosine-5’-triphosphate (ATP) competitive compounds. The catalytic domain of PAK1 adopts a typical kinase fold containing N- and C- terminal lobes connected by a hinge region forming a deep pocket for ATP binding and substrate catalysis [8, 13]. Due to the high similarity between the ATP binding pocket of kinases, it is not surprising that compounds already established as broad range kinase inhibitors, such as the natural product staurosporine and its derivative K252a, show potent, although poorly selective, inhibition of PAK1 (Figure 2A) [61–63]. Staurosporine and related derivatives are potent ATP competitive inhibitors due to their ability to mimic the highly conserved interactions between the enzyme and ATP. Specifically, the indolocarbazole moiety of staurosporine occupies the hydrophobic pocket of the enzyme that binds the adenine base of ATP and mimics the canonical hydrogen bonding pattern of ATP, while the carbohydrate moiety interacts with the ribose-binding region. This also holds true with PAK1, as an X-ray crystal structure of PAK1 in a complex with the staurosporine analogue, 3-hydroxystaurosporine (deposited in Protein Data Bank under the PDB ID 2HY8), reveals that, as expected, the indolocarbazole moiety of the compound makes two hydrogen bonds with the first and third residue of the hinge region, Glu345 and Leu347, and makes additional hydrophobic interactions with several hydrophobic residues of the binding pocket of PAK1 (Figure 3).
Figure 2. Moleculer structures of small molecule PAK inhibitors.
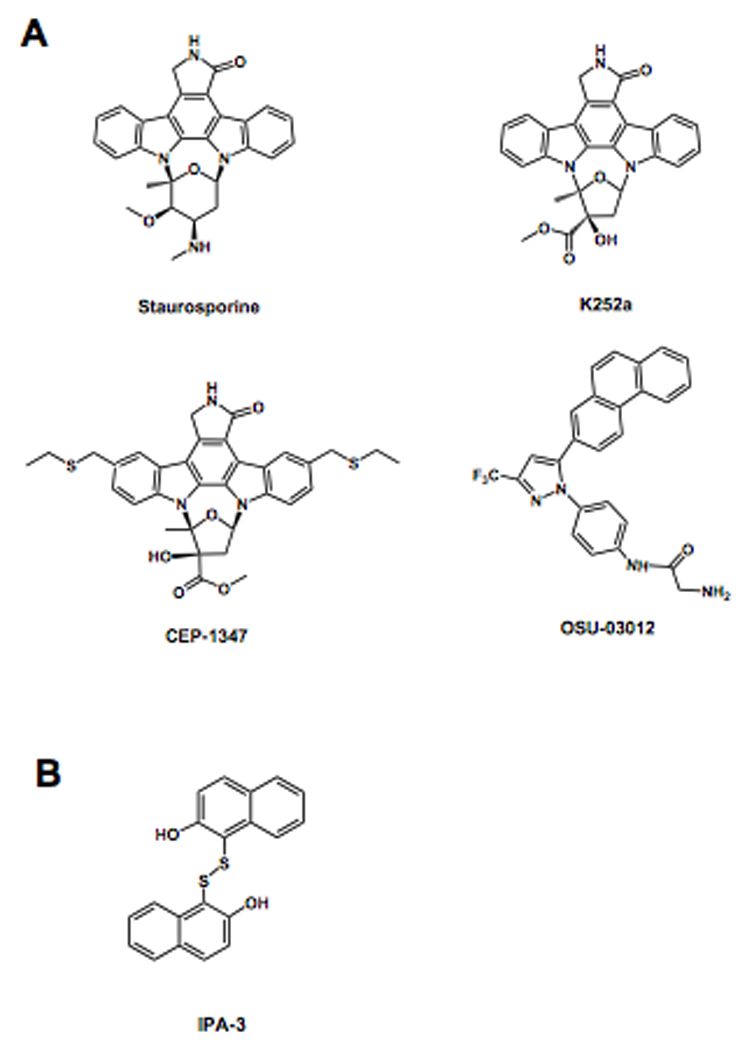
A) ATP competitive inhibitors
B) Allosteric inhibitor
Figure 3. Crystal structure of PAK1 in complex with 3-hydroxystaurosporine.
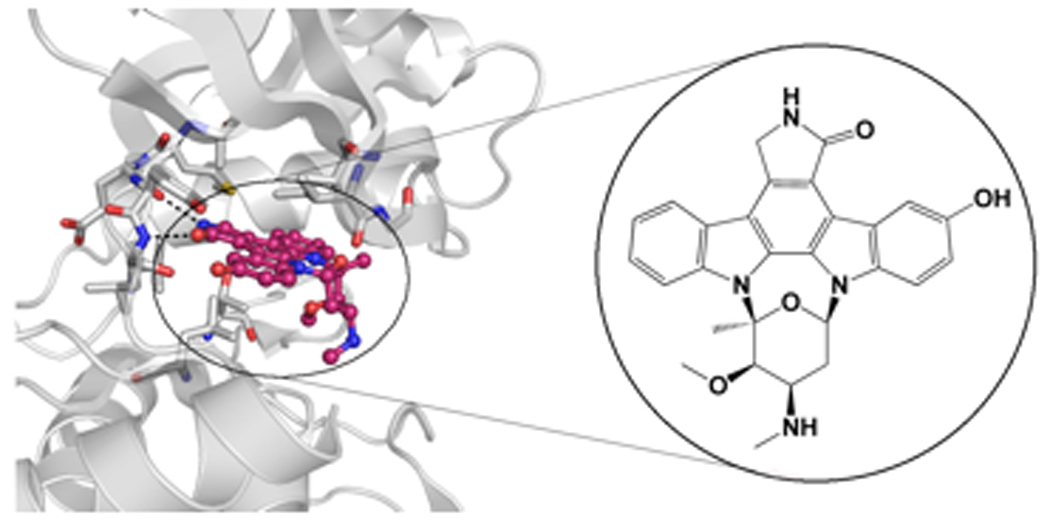
Left, a close up of the interactions between 3- hydroxystaursporine and PAK1 are shown. PAK1 residues that make hydrogen bonds with the inhibitor are labeled. Carbon atoms of 3-hydroxystaurosporine are colored pink and nitrogen and oxygen atoms are colored blue and red, respectively. Right, the molecular structure of 3-hydroxystaurosporine is shown. (Structure is deposited in Protein Data Bank under PDB ID 2HY8)
To achieve higher potency and improve selectivity for PAK1, further derivatization of the common indolocarbazole scaffold was needed. Attempts to identify such improved PAK1 inhibitors led to the identification of CEP-1347 (Figure 2A) [64]. Nheu at al. showed that CEP-1347, a synthetic derivative of the ATP antagonist K252a, directly inhibits PAK1 activity in vitro as well as PAK dependent growth of Ras-transformed cells. However, although CEP-1347 displayed greater selectivity for PAK1 inhibition over several other kinases such as PKC (protein kinase C), PKA (protein kinase A), MLCK (myosin light chain kinase) and PI3K (phosphatidylinositol-3 kinase) [65], it was later shown to be about 100-fold more selective for MLK3 (Mixed Lineage Kinase 3) [66]. Moreover CEP-1347 was shown to have relatively poor potency for PAK1 with an IC50 value of above 1 µM, and therefore not useful for animal and clinical studies.
6. Next generation PAK inhibitors
As screening technologies evolved, so have attempts to identify specific PAK inhibitors. The compound, OSU-03012, a derivative of the cyclooxygenase inhibitor, celecoxib, was developed as a PDK1 inhibitor with an IC50 value in the low micromolar range [67], but later unexpectedly shown to inhibit PAK phosphorylation at lower concentrations than PDK1-dependent AKT phosphorylation in several cell lines (Figure 2A) [68]. Subsequent studies demonstrated that OSU-03012 directly inhibits PAK1 activity in an ATP competitive fashion with an IC50 value of around 1 µM. A modeling of the OSU-03012 compound into the PAK1 ATP binding site reveals a binding mode similar to the staurosporine analogue, 3-hydroxystaurosporine, although it also reveals several non conventional protein interactions [68]. A crystal structure of the OSU-03012 compound bound to a group I PAK would directly evaluate this molecular model and reveal the precise binding mode for this inhibitor to facilitate the structure-based optimization towards the development of improved group I PAK inhibitors.
We recently reported on the combination of organoruthenium chemistry, small molecule screening and structure based design to develop a potent and selective PAK1 inhibitor [69]. Screening of a small library of inert organoruthenium compounds led to the identification of a tetrahedral ruthenium compound, DW12, as a low micromolar PAK1 inhibitor (Figure 4A). The structure of DW12 bound to PAK1 was then determined and used as an initial lead scaffold for further inhibitor optimization. A particularly striking feature of the PAK1/DW12 complex was the relative openness of the ATP binding site (Figure 4B), which led to the synthesis of bulkier octahedral ruthenium compounds to fill the space. A secondary screen of a small library of such compounds led to the identification of compound FL172 that showed significantly improved potency for PAK1 with an IC50 value of about 100nM. In addition, screening of FL172 against a panel of 264 kinases revealed that only 15 of these kinases (less than 6%) were inhibited with an IC50 similar to or better than PAK1. FL172 also showed poor inhibitory potency against the group II PAKs, thus also showing isoform selectivity. An X-ray crystal structure of PAK1 in complex with FL172 confirmed that the octahedral complex indeed fills the available space of the PAK1 active site much more efficiently than the lead compound DW12 (Figure 4B) and suggests additional potential modifications for further inhibitor improvement. Importantly, FL172 exhibited dose dependent PAK1 inhibitor activity when tested in mammalian cells. To our knowledge, FL172 represents the most potent and selective PAK1 inhibitor developed to date.
Figure 4. Organometallic PAK1 inhibitors developed through structure-based design.
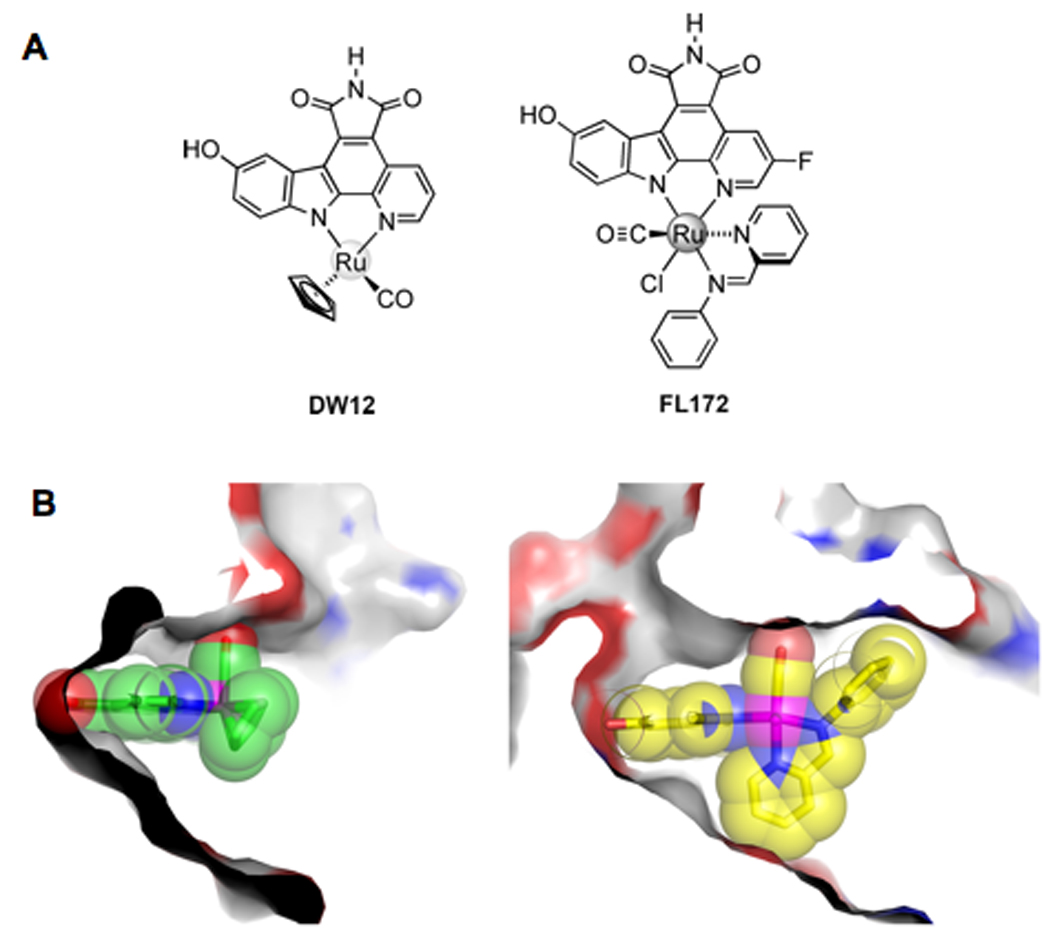
A) Molecular structures of the ruthenium-based PAK1 inhibitors. DW12 was used as a lead structure for the development of the hexavalent FL172 PAK1 inhibitor.
B) Surface representation cut away of the PAK1 active site in complex with organoruthenium compounds. Left, PAK1 in complex with DW12 is shown illustrating the open available space in the kinase active site. Right, PAK1 in complex with FL172 is shown illustrating the shape complementarity of the kinase and the inhibitor. Carbon atoms of DW12 and FL172 are shown in green and yellow respectively; the nitrogen, oxygen and ruthenium atoms colored blue, red and purple, respectively.
(Figure adapted from Maksimoska et al., J. Am. Chem. Soc., [72]).
An alternative to ATP-competitive kinase inhibitors are allosteric inhibitors that bind outside of the ATP-binding pocket. An advantage of allosteric inhibitors is that they have the potential for achieving greater selectivity than ATP-competitive inhibitors since they target unique kinase-specific features, while a disadvantage is that they tend to have reduced potency relative to ATP-competitive inhibitors since the protein pockets that they target are typically not as deep and rich in inhibitor binding residues. Attempts to identify allosteric group I PAK inhibitors have met with some success, as recently described by Deacon et al. [70]. Deacon et al. screened a library of 33,000 compounds for inhibitors that decrease the activation of PAK1 by GTP-bound Cdc42 and then rescreened these compounds at high ATP concentrations to further restrict the hits to compounds that worked through an allosteric mechanism. This process led to the identification of the small molecule PAK1 inhibitor, 2,2’-dihydroxy-1,1’-dinaphthyldisulfide (IPA-3) (Figure 2B), which was shown to have an IC50 of about 2.5 µM for PAK1 [70]. IPA-3 was also shown to be specific to group I PAKs with limited inhibitory capacity towards the group II PAKs and a panel of diverse kinases representing the mammalian kinome. Importantly, it was found that IPA-3 was inefficient in inhibiting pre-activated PAK1, indicating that IPA-3 functions by inhibiting one or more steps in the activation process of PAK1. Indeed, follow-up studies demonstrated that IPA-3 binds covalently to the regulatory domain of PAK1, thus inhibiting the binding of Cdc42-GTP. While the exact nature of the bonds are unknown, they are sufficient to impede PAK1 activation [71].
An important aspect of IPA-3 is the presence of a disulfide bond that is likely to be reduced in cell culture and under physiological conditions. Furthermore, the binding of IPA-3 to PAK1 itself is reversed under reducing environmental conditions [71]. This likely explains the requirement for higher concentrations of IPA-3 to inhibit PAKs in cells and the requirement for a large exchangeable reservoir of the compound in the growth media [70]. Therefore, although IPA-3 can selectively target PAK1 under controlled experimental conditions, the compound is unlikely to be a useful tool for inhibiting PAK1 activity in biological systems. Nonetheless, the approach taken to identify allosteric inhibitors of the PAKs is exciting and likely to yield other interesting hits in the future.
7. Future prospects
To date, a handful of group I PAK inhibitors have been developed. The organic ATP-competitive inhibitors display IC50 values in the single digit micromolar range and do not yet have favorable selectivity profiles. These inhibitors therefore require additional structure-based development to increase their potency and selectivity prior to further preclinical development. The allosteric IPA-3 inhibitor also shows relatively poor potency for group I PAKs although a favorable selectivity profile. Unfortunately, the instability of this compound significantly limits its effectiveness in cellular systems. Nonetheless, the complex regulatory mechanism of the group I PAKs leaves open the possibility of developing other allosteric PAK inhibitors.
The most potent and selective PAK1 inhibitor developed to date is the organometalic FL172 compound that exhibits a mid nanomolar IC50 value and a favorable selectivity profile as well as the ability to inhibit PAK1 activity in cells. Although, this compound has been shown to be relatively inert in vitro, it is still unclear how such a compound will behave at an organismal level and future experiments to test this compound in appropriate mouse models will dictate whether such compounds might be amenable to further development along the drug development pathway. One clear result that came out of the studies to develop FL172 was the finding that the ATP binding site of PAK1 was large relative to other kinases. This feature could be exploited to develop potent and selective group I PAK inhibitors, either of the organic or organometallic variety.
Clearly, the identification, development and improvement of inhibitors that can distinguish between the group I and II PAKs will be instrumental to our understanding of the functions of these two groups of proteins under normal physiological conditions. Likewise, this will allow further validation and assessment of the requirement for the group I PAKs in various types of cancers. There are a significant number of reports implicating PAK overexpression and activation in various types of cancer [3]. These studies affirm the rationale behind the identification and development of highly potent and specific PAK inhibitors as potential therapeutics.
Acknowledgements
Work in the authors lab was supported in part by the Children’s Tumor Foundation and the Department of Defense Neurofibromatosis Research Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- 2.Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 3.Dummler B, Ohshiro K, Kumar R, Field J. Pak protein kinases and their role in cancer. Cancer Metastasis Rev. 2009;28:51–63. doi: 10.1007/s10555-008-9168-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Molli PR, Li DQ, Murray BW, Rayala SK, Kumar R. PAK signaling in oncogenesis. Oncogene. 2009;28:2545–2555. doi: 10.1038/onc.2009.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arias-Romero LE, Chernoff J. A tale of two Paks. Biology of the cell / under the auspices of the European Cell Biology Organization. 2008;100:97–108. doi: 10.1042/BC20070109. [DOI] [PubMed] [Google Scholar]
- 6.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 7.Bishop AL, Hall A. Rho GTPases and their effector proteins. Biochem J. 2000;348(Pt 2):241–255. [PMC free article] [PubMed] [Google Scholar]
- 8.Lei M, Lu W, Meng W, Parrini MC, Eck MJ, Mayer BJ, et al. Structure of PAK1 in an autoinhibited conformation reveals a multistage activation switch. Cell. 2000;102:387–397. doi: 10.1016/s0092-8674(00)00043-x. [DOI] [PubMed] [Google Scholar]
- 9.Parrini MC, Lei M, Harrison SC, Mayer BJ. Pak1 kinase homodimers are autoinhibited in trans and dissociated upon activation by Cdc42 and Rac1. Mol Cell. 2002;9:73–83. doi: 10.1016/s1097-2765(01)00428-2. [DOI] [PubMed] [Google Scholar]
- 10.Frost JA, Khokhlatchev A, Stippec S, White MA, Cobb MH. Differential effects of PAK1-activating mutations reveal activity-dependent and -independent effects on cytoskeletal regulation. J Biol Chem. 1998;273:28191–28198. doi: 10.1074/jbc.273.43.28191. [DOI] [PubMed] [Google Scholar]
- 11.Zhao ZS, Manser E, Chen XQ, Chong C, Leung T, Lim L. A conserved negative regulatory region in alphaPAK: inhibition of PAK kinases reveals their morphological roles downstream of Cdc42 and Rac1. Mol Cell Biol. 1998;18:2153–2163. doi: 10.1128/mcb.18.4.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zenke FT, King CC, Bohl BP, Bokoch GM. Identification of a central phosphorylation site in p21-activated kinase regulating autoinhibition and kinase activity. J Biol Chem. 1999;274:32565–32573. doi: 10.1074/jbc.274.46.32565. [DOI] [PubMed] [Google Scholar]
- 13.Lei M, Robinson MA, Harrison SC. The active conformation of the PAK1 kinase domain. Structure. 2005;13:769–778. doi: 10.1016/j.str.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 14.Wells CM, Jones GE. The emerging importance of group II PAKs. Biochem J. 425:465–473. doi: 10.1042/BJ20091173. [DOI] [PubMed] [Google Scholar]
- 15.Eswaran J, Soundararajan M, Knapp S. Targeting group II PAKs in cancer and metastasis. Cancer Metastasis Rev. 2009;28:209–217. doi: 10.1007/s10555-008-9181-4. [DOI] [PubMed] [Google Scholar]
- 16.Brown MC, Cary LA, Jamieson JS, Cooper JA, Turner CE. Src and FAK kinases cooperate to phosphorylate paxillin kinase linker, stimulate its focal adhesion localization, and regulate cell spreading and protrusiveness. Mol Biol Cell. 2005;16:4316–4328. doi: 10.1091/mbc.E05-02-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cau J, Hall A. Cdc42 controls the polarity of the actin and microtubule cytoskeletons through two distinct signal transduction pathways. J Cell Sci. 2005;118:2579–2587. doi: 10.1242/jcs.02385. [DOI] [PubMed] [Google Scholar]
- 18.Zhao ZS, Lim JP, Ng YW, Lim L, Manser E. The GIT-associated kinase PAK targets to the centrosome and regulates Aurora-A. Mol Cell. 2005;20:237–249. doi: 10.1016/j.molcel.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 19.Maroto B, Ye MB, von Lohneysen K, Schnelzer A, Knaus UG. P21-activated kinase is required for mitotic progression and regulates Plk1. Oncogene. 2008;27:4900–4908. doi: 10.1038/onc.2008.131. [DOI] [PubMed] [Google Scholar]
- 20.Beeser A, Jaffer ZM, Hofmann C, Chernoff J. Role of group A p21-activated kinases in activation of extracellular-regulated kinase by growth factors. J Biol Chem. 2005;280:36609–36615. doi: 10.1074/jbc.M502306200. [DOI] [PubMed] [Google Scholar]
- 21.Tran NH, Frost JA. Phosphorylation of Raf-1 by p21-activated kinase 1 and Src regulates Raf-1 autoinhibition. J Biol Chem. 2003;278:11221–11226. doi: 10.1074/jbc.M210318200. [DOI] [PubMed] [Google Scholar]
- 22.Sastry KS, Karpova Y, Kulik G. Epidermal growth factor protects prostate cancer cells from apoptosis by inducing BAD phosphorylation via redundant signaling pathways. J Biol Chem. 2006;281:27367–27377. doi: 10.1074/jbc.M511485200. [DOI] [PubMed] [Google Scholar]
- 23.Jin S, Zhuo Y, Guo W, Field J. p21-activated Kinase 1 (Pak1)-dependent phosphorylation of Raf-1 regulates its mitochondrial localization, phosphorylation of BAD, and Bcl-2 association. J Biol Chem. 2005;280:24698–24705. doi: 10.1074/jbc.M413374200. [DOI] [PubMed] [Google Scholar]
- 24.Schurmann A, Mooney AF, Sanders LC, Sells MA, Wang HG, Reed JC, et al. p21-activated kinase 1 phosphorylates the death agonist bad and protects cells from apoptosis. Mol Cell Biol. 2000;20:453–461. doi: 10.1128/mcb.20.2.453-461.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vadlamudi RK, Bagheri-Yarmand R, Yang Z, Balasenthil S, Nguyen D, Sahin AA, et al. Dynein light chain 1, a p21-activated kinase 1-interacting substrate, promotes cancerous phenotypes. Cancer Cell. 2004;5:575–585. doi: 10.1016/j.ccr.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 26.Mazumdar A, Kumar R. Estrogen regulation of Pak1 and FKHR pathways in breast cancer cells. FEBS Lett. 2003;535:6–10. doi: 10.1016/s0014-5793(02)03846-2. [DOI] [PubMed] [Google Scholar]
- 27.Kumar R, Vadlamudi RK. Emerging functions of p21-activated kinases in human cancer cells. J Cell Physiol. 2002;193:133–144. doi: 10.1002/jcp.10167. [DOI] [PubMed] [Google Scholar]
- 28.Balasenthil S, Sahin AA, Barnes CJ, Wang RA, Pestell RG, Vadlamudi RK, et al. p21-activated kinase-1 signaling mediates cyclin D1 expression in mammary epithelial and cancer cells. J Biol Chem. 2004;279:1422–1428. doi: 10.1074/jbc.M309937200. [DOI] [PubMed] [Google Scholar]
- 29.Wang RA, Zhang H, Balasenthil S, Medina D, Kumar R. PAK1 hyperactivation is sufficient for mammary gland tumor formation. Oncogene. 2006;25:2931–2936. doi: 10.1038/sj.onc.1209309. [DOI] [PubMed] [Google Scholar]
- 30.Vadlamudi RK, Adam L, Wang RA, Mandal M, Nguyen D, Sahin A, et al. Regulatable expression of p21-activated kinase-1 promotes anchorage-independent growth and abnormal organization of mitotic spindles in human epithelial breast cancer cells. J Biol Chem. 2000;275:36238–36244. doi: 10.1074/jbc.M002138200. [DOI] [PubMed] [Google Scholar]
- 31.Adam L, Vadlamudi R, Mandal M, Chernoff J, Kumar R. Regulation of microfilament reorganization and invasiveness of breast cancer cells by kinase dead p21-activated kinase-1. J Biol Chem. 2000;275:12041–12050. doi: 10.1074/jbc.275.16.12041. [DOI] [PubMed] [Google Scholar]
- 32.Adam L, Vadlamudi R, Kondapaka SB, Chernoff J, Mendelsohn J, Kumar R. Heregulin regulates cytoskeletal reorganization and cell migration through the p21-activated kinase-1 via phosphatidylinositol-3 kinase. J Biol Chem. 1998;273:28238–28246. doi: 10.1074/jbc.273.43.28238. [DOI] [PubMed] [Google Scholar]
- 33.Li Q, Mullins SR, Sloane BF, Mattingly RR. p21-Activated kinase 1 coordinates aberrant cell survival and pericellular proteolysis in a three-dimensional culture model for premalignant progression of human breast cancer. Neoplasia. 2008;10:314–329. doi: 10.1593/neo.07970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rayala SK, Talukder AH, Balasenthil S, Tharakan R, Barnes CJ, Wang RA, et al. P21-activated kinase 1 regulation of estrogen receptor-alpha activation involves serine 305 activation linked with serine 118 phosphorylation. Cancer Res. 2006;66:1694–1701. doi: 10.1158/0008-5472.CAN-05-2922. [DOI] [PubMed] [Google Scholar]
- 35.Holm C, Rayala S, Jirstrom K, Stal O, Kumar R, Landberg G. Association between Pak1 expression and subcellular localization and tamoxifen resistance in breast cancer patients. J Natl Cancer Inst. 2006;98:671–680. doi: 10.1093/jnci/djj185. [DOI] [PubMed] [Google Scholar]
- 36.Bostner J, Ahnstrom Waltersson M, Fornander T, Skoog L, Nordenskjold B, Stal O. Amplification of CCND1 and PAK1 as predictors of recurrence and tamoxifen resistance in postmenopausal breast cancer. Oncogene. 2007;26:6997–7005. doi: 10.1038/sj.onc.1210506. [DOI] [PubMed] [Google Scholar]
- 37.Gerber PA, Antal AS, Neumann NJ, Homey B, Matuschek C, Peiper M, et al. Neurofibromatosis. Eur J Med Res. 2009;14:102–105. doi: 10.1186/2047-783X-14-3-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsakiridis T, Taha C, Grinstein S, Klip A. Insulin activates a p21-activated kinase in muscle cells via phosphatidylinositol 3-kinase. J Biol Chem. 1996;271:19664–19667. doi: 10.1074/jbc.271.33.19664. [DOI] [PubMed] [Google Scholar]
- 39.Slack-Davis JK, Eblen ST, Zecevic M, Boerner SA, Tarcsafalvi A, Diaz HB, et al. PAK1 phosphorylation of MEK1 regulates fibronectin-stimulated MAPK activation. J Cell Biol. 2003;162:281–291. doi: 10.1083/jcb.200212141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chaudhary A, King WG, Mattaliano MD, Frost JA, Diaz B, Morrison DK, et al. Phosphatidylinositol 3-kinase regulates Raf1 through Pak phosphorylation of serine 338. Curr Biol. 2000;10:551–554. doi: 10.1016/s0960-9822(00)00475-9. [DOI] [PubMed] [Google Scholar]
- 41.King AJ, Sun H, Diaz B, Barnard D, Miao W, Bagrodia S, et al. The protein kinase Pak3 positively regulates Raf-1 activity through phosphorylation of serine 338. Nature. 1998;396:180–183. doi: 10.1038/24184. [DOI] [PubMed] [Google Scholar]
- 42.Hirokawa Y, Tikoo A, Huynh J, Utermark T, Hanemann CO, Giovannini M, et al. A clue to the therapy of neurofibromatosis type 2: NF2/merlin is a PAK1 inhibitor. Cancer J. 2004;10:20–26. doi: 10.1097/00130404-200401000-00006. [DOI] [PubMed] [Google Scholar]
- 43.Kissil JL, Wilker EW, Johnson KC, Eckman MS, Yaffe MB, Jacks T. Merlin, the product of the Nf2 tumor suppressor gene, is an inhibitor of the p21-activated kinase, Pak1. Mol Cell. 2003;12:841–849. doi: 10.1016/s1097-2765(03)00382-4. [DOI] [PubMed] [Google Scholar]
- 44.Xiao GH, Gallagher R, Shetler J, Skele K, Altomare DA, Pestell RG, et al. The NF2 tumor suppressor gene product, merlin, inhibits cell proliferation and cell cycle progression by repressing cyclin D1 expression. Mol Cell Biol. 2005;25:2384–2394. doi: 10.1128/MCB.25.6.2384-2394.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilkes MC, Repellin CE, Hong M, Bracamonte M, Penheiter SG, Borg JP, et al. Erbin and the NF2 tumor suppressor Merlin cooperatively regulate cell-type-specific activation of PAK2 by TGF-beta. Dev Cell. 2009;16:433–444. doi: 10.1016/j.devcel.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ingram DA, Hiatt K, King AJ, Fisher L, Shivakumar R, Derstine C, et al. Hyperactivation of p21(ras) and the hematopoietic-specific Rho GTPase, Rac2, cooperate to alter the proliferation of neurofibromin-deficient mast cells in vivo and in vitro. J Exp Med. 2001;194:57–69. doi: 10.1084/jem.194.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yi C, Wilker EW, Yaffe MB, Stemmer-Rachamimov A, Kissil JL. Validation of the p21-activated kinases as targets for inhibition in neurofibromatosis type 2. Cancer Res. 2008;68:7932–7937. doi: 10.1158/0008-5472.CAN-08-0866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tang Y, Chen Z, Ambrose D, Liu J, Gibbs JB, Chernoff J, et al. Kinase-deficient Pak1 mutants inhibit Ras transformation of Rat-1 fibroblasts. Mol Cell Biol. 1997;17:4454–4464. doi: 10.1128/mcb.17.8.4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang FC, Ingram DA, Chen S, Hingtgen CM, Ratner N, Monk KR, et al. Neurofibromin-deficient Schwann cells secrete a potent migratory stimulus for Nf1+/− mast cells. J Clin Invest. 2003;112:1851–1861. doi: 10.1172/JCI19195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science. 2002;296:920–922. doi: 10.1126/science.1068452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang FC, Ingram DA, Chen S, Zhu Y, Yuan J, Li X, et al. Nf1-dependent tumors require a microenvironment containing Nf1+/−- and c-kit-dependent bone marrow. Cell. 2008;135:437–448. doi: 10.1016/j.cell.2008.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McDaniel AS, Allen JD, Park SJ, Jaffer ZM, Michels EG, Burgin SJ, et al. Pak1 regulates multiple c-Kit mediated Ras-MAPK gain-in-function phenotypes in Nf1+/− mast cells. Blood. 2008;112:4646–4654. doi: 10.1182/blood-2008-04-155085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakai Y, Zheng Y, MacCollin M, Ratner N. Temporal control of Rac in Schwann cell-axon interaction is disrupted in NF2-mutant schwannoma cells. J Neurosci. 2006;26:3390–3395. doi: 10.1523/JNEUROSCI.4865-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kaempchen K, Mielke K, Utermark T, Langmesser S, Hanemann CO. Upregulation of the Rac1/JNK signaling pathway in primary human schwannoma cells. Hum Mol Genet. 2003;12:1211–1221. doi: 10.1093/hmg/ddg146. [DOI] [PubMed] [Google Scholar]
- 55.Flaiz C, Kaempchen K, Matthies C, Hanemann CO. Actin-rich protrusions and nonlocalized GTPase activation in Merlin-deficient schwannomas. J Neuropathol Exp Neurol. 2007;66:608–616. doi: 10.1097/nen.0b013e318093e555. [DOI] [PubMed] [Google Scholar]
- 56.Xiao GH, Beeser A, Chernoff J, Testa JR. p21-activated kinase links Rac/Cdc42 signaling to merlin. J Biol Chem. 2002;277:883–886. doi: 10.1074/jbc.C100553200. [DOI] [PubMed] [Google Scholar]
- 57.Kissil JL, Johnson KC, Eckman MS, Jacks T. Merlin phosphorylation by p21-activated kinase 2 and effects of phosphorylation on merlin localization. J Biol Chem. 2002;277:10394–10399. doi: 10.1074/jbc.M200083200. [DOI] [PubMed] [Google Scholar]
- 58.Morrison H, Sperka T, Manent J, Giovannini M, Ponta H, Herrlich P. Merlin/neurofibromatosis type 2 suppresses growth by inhibiting the activation of Ras and Rac. Cancer Res. 2007;67:520–527. doi: 10.1158/0008-5472.CAN-06-1608. [DOI] [PubMed] [Google Scholar]
- 59.Flaiz C, Chernoff J, Ammoun S, Peterson JR, Hanemann CO. PAK kinase regulates Rac GTPase and is a potential target in human schwannomas. Exp Neurol. 2009;218:137–144. doi: 10.1016/j.expneurol.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhao ZS, Manser E, Chen XQ, Chong C, Leung T, Lim L. A conserved negative regulatory region in alphaPAK: inhibition of PAK kinases reveals their morphological roles downstream of Cdc42 and Rac1. Mol Cell Biol. 1998;18:2153–2163. doi: 10.1128/mcb.18.4.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ghoreschi K, Laurence A, O'Shea JJ. Selectivity and therapeutic inhibition of kinases: to be or not to be? Nat Immunol. 2009;10:356–360. doi: 10.1038/ni.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kaneko M, Saito Y, Saito H, Matsumoto T, Matsuda Y, Vaught JL, et al. Neurotrophic 3,9-bis[(alkylthio)methyl]-and-bis(alkoxymethyl)-K-252a derivatives. J Med Chem. 1997;40:1863–1869. doi: 10.1021/jm970031d. [DOI] [PubMed] [Google Scholar]
- 64.Nheu TV, He H, Hirokawa Y, Tamaki K, Florin L, Schmitz ML, et al. The K252a derivatives, inhibitors for the PAK/MLK kinase family selectively block the growth of RAS transformants. Cancer J. 2002;8:328–336. doi: 10.1097/00130404-200207000-00009. [DOI] [PubMed] [Google Scholar]
- 65.Maroney AC, Glicksman MA, Basma AN, Walton KM, Knight E, Jr, Murphy CA, et al. Motoneuron apoptosis is blocked by CEP-1347 (KT 7515), a novel inhibitor of the JNK signaling pathway. J Neurosci. 1998;18:104–111. doi: 10.1523/JNEUROSCI.18-01-00104.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maroney AC, Finn JP, Connors TJ, Durkin JT, Angeles T, Gessner G, et al. CEP-1347 (KT7515), a Semisynthetic Inhibitor of the Mixed Lineage Kinase Family. Journal of Biological Chemistry. 2001;276:25302–25308. doi: 10.1074/jbc.M011601200. [DOI] [PubMed] [Google Scholar]
- 67.Zhu J, Huang J-W, Tseng P-H, Yang Y-T, Fowble J, Shiau CW, et al. From the Cyclooxygenase-2 Inhibitor Celecoxib to a Novel Class of 3-Phosphoinositide-Dependent Protein Kinase-1 Inhibitors. Cancer Res. 2004;64:4309–4318. doi: 10.1158/0008-5472.CAN-03-4063. [DOI] [PubMed] [Google Scholar]
- 68.Porchia LM, Guerra M, Wang YC, Zhang Y, Espinosa AV, Shinohara M, et al. 2-amino-N-{4-[5-(2-phenanthrenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]-phe nyl} acetamide (OSU-03012), a celecoxib derivative, directly targets p21-activated kinase. Mol Pharmacol. 2007;72:1124–1131. doi: 10.1124/mol.107.037556. [DOI] [PubMed] [Google Scholar]
- 69.Maksimoska J, Feng L, Harms K, Yi C, Kissil J, Marmorstein R, et al. Targeting large kinase active site with rigid, bulky octahedral ruthenium complexes. Journal of the American Chemical Society. 2008;130:15764–15765. doi: 10.1021/ja805555a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Deacon SW, Beeser A, Fukui JA, Rennefahrt UE, Myers C, Chernoff J, et al. An isoform-selective, small-molecule inhibitor targets the autoregulatory mechanism of p21-activated kinase. Chemistry & biology. 2008;15:322–331. doi: 10.1016/j.chembiol.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Viaud J, Peterson JR. An allosteric kinase inhibitor binds the p21-activated kinase autoregulatory domain covalently. Molecular cancer therapeutics. 2009;8:2559–2565. doi: 10.1158/1535-7163.MCT-09-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maksimoska J, Feng L, Harms K, Yi C, Kissil J, Marmorstein R, et al. Targeting large kinase active site with rigid, bulky octahedral ruthenium complexes. J Am Chem Soc. 2008;130:15764–15765. doi: 10.1021/ja805555a. [DOI] [PMC free article] [PubMed] [Google Scholar]