

Abstract
Disubstituted α-hydroxy acids have been synthesized by metal-catalyzed silylene transfer to α-keto esters. A range of substituents are tolerated in the transformation with the exception of branched groups at the vinylic position. The α-hydroxy acid products can be converted into γ-lactones using a variety of lactonization conditions.
1. Introduction
α-Hydroxy acids are common structural motifs in nature.1, 2 Several recent publications have featured natural products that incorporate this moiety into their synthesis, including thapsigarin, viridiofungin, zaragozic acid, and fukinolic acid (Scheme 1).3-6 Given the prevalence of α-hydroxy acids in small molecules, new synthetic methods for their synthesis are desirable.7-13 We recently reported the development of a method to obtain α-hydroxy acids from simple α-keto esters utilizing metal-catalyzed silylene transfer.14 In this paper we report the expanded scope and utility of this methodology.
Scheme 1.

α-Hydroxy acid containing natural products.
2. Results
The silylene-mediated conversion of α-hydroxy acids is general for a range of substrates (Table 1). The highest yields were obtained with linear α-keto esters (R1 = Et, Me) and substrates featuring aromatic groups (Table 1, entry 5). The synthetic utility of the reaction could be enhanced by increasing the complexity of substituents at R1. Incorporation of a protected oxygen atom is tolerated by the reaction conditions and provides an additional handle that can be elaborated in later synthetic steps (Scheme 2).14
Table 1.
Silylene transfer to α-keto esters with substitution at R1
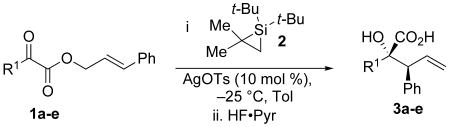 | ||||
|---|---|---|---|---|
| Entry | R1 | Product | % Yield | d.r. |
| 1 | Me | 3a | 70 | ≥97:3 |
| 2 | Et | 3b | 84 | ≥97:3 |
| 3 | i-Pr | 3c | 54 | ≥97:3 |
| 4 | t-Bu | 3d | 47 | ≥97:3 |
| 5 | Ph | 3e | 71 | ≥97:3 |
Conditions: α-keto ester (1.0 equiv), silacyclopropane 2 (1.5 equiv), AgOTs (0.10 equiv), toluene, −25 °C, 16 h. Then HF·Pyr (4.0 equiv), isolated yields.
Scheme 2.

Synthesis of α-hydroxy acids with branching at R1.
The mechanism for the conversion of α-keto esters to α-hydroxy acids involves sequential pericyclic reactions. Initial generation of a silacarbonyl ylide intermediate (7) is followed by a 6π electrocyclization and tandem Ireland– Claisen rearrangement (Scheme 3), affording α-hydroxy acid products.14-20
Scheme 3.

Postulated mechanism for the formation of α-hydroxy acids.
Functionalization at R2 (C4 of the Ireland–Claisen transition state, Scheme 3) provided direct transfer of stereochemical information to the two newly formed chiral centers in the Ireland–Claisen product (9). Substituents at this position have a preference for being pseudo-equatorial in the transition state to avoid steric congestion.19 Initial investigations employed a methyl group at R2 (11) as a single enantiomer; complete transfer of chirality was observed (Scheme 4).14
Scheme 4.

Enantiopure α-keto esters bearing a chiral substituent on the allylic ester at R2 (C4).
Extending the substrate scope to include protected hydroxyl groups at R2 did not impede the transformation. Reactions of compounds 13 and 15 both led to enantiopure acids in good yields upon exposure to the reaction conditions (Scheme 5).
Scheme 5.

Synthesis of enantiopure α-hydroxy acids.
Employing substitutions at both R1 and R2 led to the formation of diastereomeric mixtures of α-hydroxy acid products (Scheme 6). The formation of diastereomers is likely due to competing interactions in the Ireland–Claisen transition state. Aligning the benzyloxy group of 17 anti to the incipient carbon–carbon bond in the transition state creates a syn-pentane like interaction between the methyl groups at positions C1 and C6 (17a).21, 22 This interaction negates any favorable interactions gained by placing the ethyl group at C4 equatorial, leading to the observed mixture of diastereomers (17b).
Scheme 6.

Competing chiral substituents at C1 and C4 lead to the formation of diastereomers.
Unfortunately, increasing the steric bulk at the C4 position (Et to t-Bu, 19) does not improve the diastereomeric ratio of products formed. Submitting a diastereomeric mixture of α-keto esters to silylene transfer conditions (19a and 19b) led to the formation of three products (20a, 20b, and 20c): two diastereomers of the mismatched substrate and a single diastereomer from the matched substrate, respectively (Scheme 8).22 This result indicates that exocyclic stereocenters in the Ireland-Claisen transition state can influence the product distribution as effectively as stereocenters on the cyclic transition state.
Scheme 8.

Incorporation of a t-butyl group at R2 does not change diastereoselectivity
Substitutions at R3 are generally well tolerated (Table 2).14 Substrates that feature branching at that position, however, do not give rearrangement products (Schemes 9, 10, and 11). α-Keto esters that did not give α-hydroxy acid products include terminally disubstituted allylic α-keto ester 25 and substrates that would require a dearomatization event (21). Branching at R3 may not be tolerated because of the proximal silyl group in the Ireland–Claisen transition state. Of all the positions available for substitution on the various substrates, R3 is the closest to the silyl functionality.
Table 2.
Silylene transfer to α-keto esters with substitution at R3
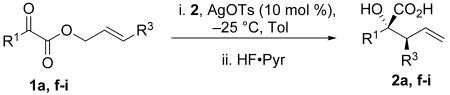 | |||||
|---|---|---|---|---|---|
| Entry | R1 | R3 | Product | % Yield | d.r. |
| 1 | Me | Ph | 2a | 70 | ≥97:3 |
| 2 | Ph | Me | 2f | 62 | ≥97:3 |
| 3 | Ph | n-Bu | 2g | 72 | ≥97:3 |
| 4 | Ph | CH2OTBDMS | 2h | 71 | ≥97:3 |
| 5 | Et | (CH2)2OBn | 2i | 75 | ≥97:3 |
Conditions: α-keto ester (1.0 equiv), silacyclopropane 2 (1.5 equiv), AgOTs (0.10 equiv), toluene, −25 °C, 16 h then, HF·Pyr (4.0 equiv), isolated yields.
Scheme 9.

Substrates requiring dearomatization are not tolerated.
Scheme 10.

α-Keto esters with bulky substituents at R3 are not tolerated.
Scheme 11.

Terminally disubstituted alkenes are not tolerated.
Ultimately, the silylene transfer products feature a homoallylic carboxylic acid that can be elaborated into γ-lactones by several different lactonization conditions. Traditional iodolactonization methods provided moderate to good yield of the desired lactone as a mixture of diastereomers (Table 3, entries 1-5).23-26 Use of N-bromosuccinimide led to slightly improved diastereomeric ratios and decreased yield. Surprisingly, asymmetric dihydroxylation conditions yielded no product, but standard dihydroxylation conditions provided the requisite product 28c as a single diastereomer.27 It is conceivable that the catalyst-ligand complex in the asymmetric dihydroxylation is unable to accommodate the sterically congested α-hydroxy acid substrates.
Table 3.
Lactonization conditions and results for the conversion of 27 to 28
| Entry | R1 | R2 | R3 | X | Conditions | Product | % Yield | d.r. |
|---|---|---|---|---|---|---|---|---|
| 1 | Ph | H | H | I | NaHCO3, I2, CH3CN, 0 °C | 28a | 91 | 60:40 |
| 2 | Me | n-Bu | Me | I | I2, CH3CN | 28b | 48 | 80:20 |
| 3 | Me | n-Bu | Me | I | KI, I2, THF, NaHCO3 | 28b | 34 | 80:20 |
| 4 | Et | n-Bu | H | OH | H2O, t-BuOH, AD mix | 28c | - | - |
| 5 | Et | n-Bu | H | I | NaHCO3, I2, CH3CN, 0 °C | 28d | 37 | 80:20 |
| 6 | Et | n-Bu | H | Br | NBS, THF, 0 °C | 28e | 76 | 83:17 |
| 7 | Et | n-Bu | H | I | NIS, THF 0 °C | 28d | 92 | 60:40 |
| 8 | Et | n-Bu | H | I | Ti(Oi-Pr)4, NIS, CH2Cl2 −20 °C | 28d | 34 | 56:44 |
| 9 | Et | n-Bu | H | OH | OsO4, NMO, CH2Cl2 | 28c | 73 | 100:0 |
Access to a variety of lactones with differing functionality provided an opportunity to synthesize a range of compounds. Several elaborations have led to differentiated products, including diol 29, a potential precursor to the secondary marine metabolite (−)-delesserine (Scheme 13). Utilization of a slightly modified α-keto ester in the silylene transfer reaction could afford the natural product in a few additional steps.28 Elaboration of the diol 29 to olefin 30 via reductive elimination provides a potential handle for ring-closing metathesis reactions (Scheme 13).29
Scheme 13.

Elaboration of lactone products.
3. Experimentals
3.1 General remarks
Melting points were obtained using a Büchi 510 melting point apparatus and are reported uncorrected. Analytical thin later chromatography was performed on EMD Silical Gel 60 F254 precoated plates. Liquid chromatography utilized force flow (flash chromatography) of the indicated solvent system on Silacycle Sila-P silica gel (SiO2) 60Å pore size, 40-63 μm mesh. Infrared spectroscopy was performed on an Mattson Instruments FTIR, Galaxy System. High-resolution mass spectra were acquired on a Walters LCT Premier and were obtained by peak matching. Microanalyses were performed by Atlantic Microlabs, Atlanta, GA. 1H NMR and 13C NMR spectra were recorded at 25 °C at 400 and 100, and 500 and 125 MHz, respectively, using Bruker DRX 400 or DRX 500 spectrometers as indicated. These data are reported as follows: chemical shift in ppm from internal tetramethylsilane on the δ scale, multiciplity (br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constants (Hz), and integration. Silacyclopropanes were stored and manipulated in an Innovative Technologies nitrogen-atmosphere dry box. All reactions were performed under an atmosphere of nitrogen or argon in glassware that had been flame-dried under vacuum prior to use. Solvents were distilled or filtered before use. Compounds 1–5, 11, 12, 27a, 27b, and 27c, were synthesized according to previously published reports.
3.2 (S,E)-1-(tert-Butyldimethylsilyloxy)-5-(4-methoxybenzyloxy)pent-3-en-2-yl 2-oxopropanoate (13)
To a solution of (S,E)-1-(tert-butyldimethylsilyloxy)-5-(4-methoxybenzyloxy)pent-3-en-2-ol (0.520 g, 1.47 mmol) in benzene (30 mL) was added pyruvic acid (0.164 mL, 2.36 mmol), triethylamine (0.551 mL, 3.70 mmol), 4-dimethylaminopyridine (0.224 g, 1.84 mmol), and 2,4,6-trichlorobenzoylchloride (0.731 mL, 2.99 mmol). The reaction mixture was then stirred for 16 h, diluted with 5% aqueous citric acid, and extracted with EtOAc (3 × 25 mL). The organic layers were combined, dried with Na2SO4, and concentrated in vacuo. The resultant oil was purified by column chromatography (10:1 hexanes:EtOAc) to provide 13 (0.617 g, 99%) as a yellow oil: [α]23D 8.2 (c 2.8, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.31 – 7.23 (m, 2H), 6.93 (d, J = 8.6, 2H), 6.01 (dt, J = 15.6, 5.3, 1H), 5.81 (dd, J = 15.7, 6.9, 1H), 5.53 (dd, J = 12.1, 6.2, 1H), 4.52 (s, 2H), 4.07 (d, J = 5.0, 2H), 3.90 – 3.76 (m, 5H), 2.52 (d, J = 5.4, 2H), 0.91 (s, 9H), 0.10 (t, J = 4.2, 6H); 13C NMR (100 MHz, CDCl3) δ 192.3, 167.3, 159.7, 132.9, 132.5, 130.2, 130.0, 114.3, 77.4, 72.5, 69.6, 64.9, 55.7, 27.2, 26.2, 18.7, -4.98; IR (thin film) 3021, 2933, 1733, 1216, 755 cm-1; HRMS (ESI) m / z calcd for C22H34NaO6Si (M + Na)+ 445.2022, found 445.2022.
3.3 (2R,3R,E )-6-(tert-Butyldimethylsilyloxy)-2-hydroxy-3-((4-methoxybenzyloxy)methyl)-2-methylhex-4-enoic acid (14)
To 13 (0.318 g, 0.752 mmol) in toluene (10 mL) was added silacyclopropane 2 (0.215 g, 1.09 mmol) and the reaction mixture was then cooled to −25 °C. After stirring for 0.5 h, silver tosylate (0.021 g, 0.075 mmol) was added and the mixture was allowed to warm to room temperature. Upon stirring for 3 h, the solution was concentrated in vacuo and the resultant oil was purified by column chromatography (5:2 hexanes:EtOAc with 1% AcOH) to provide 14 (0.254 g, 80%) as a pale yellow oil: [α]23D − 12.7 (c 3.68, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.24 (d, J = 8.6, 2H), 6.97 − 6.70 (m, 2H), 5.77 (t, J = 5.8, 2H), 4.42 (s, 2H), 4.23 (d, J = 3.5, 2H), 3.83 (d, J = 3.9, 3H), 3.61 (dd, J = 7.2, 5.5, 2H), 2.99 − 2.76 (m, 1H), 1.40 (s, 3H), 0.96 (d, J = 2.6, 9H), 0.18 − 0.09 (m, 6H).13C NMR (100 MHz, CDCl3) δ 180.4, 159.7, 134.9, 129.9, 129.9, 129.9, 128.8, 126.1, 114.3, 76.3, 73.6, 71.4, 64.1, 55.6, 49.4, 27.7, 27.4, 26.4, 25.5, 18.8, -4.66, -4.68; IR (thin film) 3434, 3018, 2933, 2859, 1770, 1722 cm-1; HRMS (ESI) m / z calcd for C22H36NaO6Si (M + Na)+ 447.2179, found 447.2169.
3.4 (R,E)-5-(tert-Butyldiphenylsilyloxy)-1-(4-methoxybenzyloxy)pent-3-en-2-yl 2-oxopropanoate (15)
α-Keto ester 15 was isolated as a yellow oil (1.15g, 59%) using a procedure identical to the one outlined in 3.3: [α]23D −17.1 (c 9.42, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.84 − 7.70 (m, 4H), 7.56 − 7.11 (m, 8H), 7.01 − 6.91 (m, 2H), 6.09 − 5.96 (m, 1H), 5.90 (ddt, J = 15.4, 7.0, 1.7, 1H), 5.77 − 5.65 (m, 1H), 4.62 (d, J = 5.0, 2H), 4.34 (ddd, J = 4.7, 3.8, 2.3, 2H), 3.88 (s, 3H), 3.71 (ddd, J = 14.7, 11.0, 5.7, 2H), 2.54 (s, 3H), 1.16 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 192.2, 160.3, 159.6, 136.6, 135.8, 135.3, 133.6, 132.8, 130.1, 130.1, 129.8, 129.8, 129.7, 128.0, 128.4, 128.0, 128.0, 123.2, 114.2, 75.6, 73.2, 71.0, 63.6, 55.5, 27.1, 27.0, 19.50; IR (thin film) 3016, 2933, 2858, 1735, 1581, 1513 cm-1; HRMS (ESI) m / z calcd for C32H38NaO6Si (M + Na)+ 569.2335, found 569.2335.
3.5 (2S,3S,E)-3-((tert-Butyldiphenylsilyloxy)methyl)-2-hydroxy-6-(4-methoxybenzyloxy)-2-methylhex-4-enoic acid (16)
To 15 (0.188 g, 0.344 mmol) in toluene (5 mL) was added silacyclopropane 2 (0.095 g, 0.48 mmol) and the reaction mixture was cooled to −25 °C. After stirring for 0.5 h, silver tosylate (0.009 g, 0.03 mmol) was added and the mixture was allowed to warm to ambient temperature. After stirring for 2 h, the solution was concentrated in vacuo and the resultant oil was purified by column chromatography (5:2 hexanes:EtOAc with 1% AcOH) to provide 16 (0.099 g, 53%) as a pale yellow oil: [α]23D −0.7 (c 5.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.66 (d, J = 7.5, 4H), 7.54 −7.31 (m, 6H), 7.27 (d, J = 8.5, 2H), 6.89 (d, J = 8.5, 2H), 5.89 (dd, J = 15.5, 9.5, 1H), 5.74 (dt, J = 15.5, 5.8, 1H), 4.46 (s, 2H), 4.02 (d, J = 5.6, 2H), 3.97 − 3.66 (m, 6H), 2.77 (dt, J = 9.1, 4.4, 1H), 1.42 (s, 3H), 1.06 (s, 10H); 13C NMR (100 MHz, CDCl3) δ 180.1, 159.5, 135.9, 135.8, 132.5, 132.4, 132.2, 130.5, 130.3, 129.7, 129.0, 128.1, 128.1, 114.1, 71.9, 70.3, 66.3, 55.5, 50.4, 27.0, 25.4, 19.3; IR (thin film) 3502, 2933, 2858, 1724, 1513 cm-1; HRMS (ESI) m / z calcd for C32H40NaO6Si (M + Na)+ 571.2492, found 571.2505.
3.6 (R)-((S,E)-Hex-4-en-3-yl) 3-(benzyloxy)-2-oxobutanoate (17)
To (2S,3R)-((S,E)-hex-4-en-3-yl) 3-(benzyloxy)-2-hydroxybutanoate (0.152 g, 0.520 mmol) in CH2Cl2 (10 mL) was added pyridine (0.25 mL) followed by Dess-Martin periodinane (0.330 g, 0.779 mmol). The reaction mixture was stirred for 16 h and then diluted with saturated aqueous Na2SO3 and extracted with CH2Cl2 (3 × 15 mL). The organic layers were then combined, dried over Na2SO4, and concentrated in vacuo. The crude oil was purified by column chromatography (10:1 hexanes:EtOAc) to afford 17 (0.114 g, 76%) as a yellow oil: [α]23D 16.0 (c 1.66, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.44 − 7.26 (m, 5H), 5.86 (dq, J = 13.1, 6.5, 1H), 5.58 − 5.42 (m, 1H), 5.33 (q, J = 6.9, 1H), 4.73 (d, J = 11.5, 1H), 4.63 − 4.50 (m, 2H), 2.05 − 1.61 (m, 5H), 1.48 (d, J = 6.9, 3H), 0.95 (t, J = 7.4, 3H); 13C NMR (100 MHz, CDCl3) δ 196.3, 162.8, 137.7, 131.6, 128.9, 128.6, 128.4, 79.50, 77.9, 73.0, 27.8, 18.2, 17.0, 9.9; IR (thin film) 2971, 2939, 2879, 1726, 1214 cm-1; HRMS (ESI) m / z calcd for C17H22NaO4 (M + Na)+ 313.1416, found 313.1415. Anal. Calcd for C17H22O4: C, 70.32; H, 7.64. Found: C, 70.14; H, 7.68.
3.7 1-(Benzyloxy)ethyl)-2-hydroxy-3-methylhept-4-enoic acid (18)
To 17 (0.150 g, 0.514 mmol) in toluene (5 mL) was added silacyclopropane 2 (0.147 g, 0.745 mmol) and the reaction mixture was cooled to −25 °C. After stirring for 0.5 h, silver tosylate (0.014 g, 0.05 mmol) was added and the mixture was allowed to warm to ambient temperature. Upon stirring for 2 h, the solution was concentrated in vacuo and the resultant oil was purified by column chromatography (5:2 hexanes:EtOAc with 1% AcOH) to provide a 1:1 mixture of diastereomers of 18 (0.108 g, 72%) as a pale yellow oil: 1H NMR (400 MHz, CDCl3) δ 7.40 − 7.14 (m, 5H), 5.60 − 5.49 (m, 2H), 4.73 (dd, J = 11.3, 5.8, 2H), 4.11 − 3.88 (m, 1H), 3.00 − 2.70 (m, 1H), 2.18 − 1.97 (m, 2H), 1.34 (d, J = 5.9, 3H), 1.04 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 177.1, 138.4, 135.1, 128.9, 128.8, 128.7, 128.5, 128.2, 83.0, 78.8, 72.0, 41.9, 26.0, 16.5, 15.3, 14.1, 13.1; IR (thin film) 3020, 2973, 1756, 1708, 1216 cm-1; HRMS (ESI) m / z calcd for C17H24NaO4 (M + Na)+ 315.1572, found 315.1572.
3.8 (3R)-((E)-2,2-Dimethylhex-4-en-3-yl) 3-(benzyloxy)-2-oxobutanoate (19)
To (±)(2S,3R)-((E)-2,2-dimethylhex-4-en-3-yl) 3-(benzyloxy)-2-hydroxybutanoate (0.007 g, 0.02 mmol) in CH2Cl2 (1 mL) was added pyridine (0.2 mL) followed by Dess-Martin periodinane (0.014 g, 0.032 mmol). The reaction mixture was stirred for 16 h and then diluted with saturated aqueous Na2SO3, extracted with CH2Cl2 (3 × 5 mL). The organic layers were then combined, dried over Na2SO4, and concentrated in vacuo. The crude oil was purified by column chromatography (10:1 hexanes:EtOAc) to afford 19 (0.069 g, 99%) as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.46 − 7.32 (m, 5H), 5.91 − 5.79 (m, 1H), 5.61 − 5.46 (m, 1H), 5.15 (t, J = 8.1, 1H), 4.74 (d, J = 11.5, 1H), 4.65 − 4.45 (m, 2H), 1.79 − 1.71 (m, 2H), 1.67 − 1.56 (m, 1H), 1.50 (d, J = 4.0, 3H), 0.98 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 196.2, 162.5, 137.2, 132.5, 128.5, 128.1, 128.0, 125.3, 84.9, 72.6, 34.5, 29.8, 25.8, 17.9, 16.7; IR (thin film) 3091, 3018, 2969, 1724, 1479 cm-1; HRMS (ESI) m / z calcd for C19H26NaO4 (M + Na)+ 341.1729, found 347.1738.
3.9 1-(Benzyloxy)ethyl)-2-hydroxy-3,6,6-trimethylhept-4-enoic acid (20a-c)
To 19 (0.0623 g, 0.196 mmol) in toluene (2 mL ) was added 1,1-di-tert-butyl-2,2-dimethylsilirane 2 (0.0587 g, 0.296 mmol) and the reaction mixture was cooled to −25 °C. After stirring for 0.5 h, silver tosylate (0.006 g, 0.02 mmol) was added and the mixture was allowed to warm to room temperature. After stirring for 12 h, HF·Pyr (0.20 mL, 1.75 mmol) was added to the solution. The reaction mixture was then diluted with saturated aqueous NaHCO3, the organic layer was rinsed with saturated aqueous NaHCO3 (3 × 25 mL), the aqueous layers were then combined and acidified with 1M HCl (ca 100 mL) until pH 2. After the desired pH was reached, the aqueous layer was extracted with CH2Cl2 (3 × 25 mL), the organic layers were combined, dried with Na2SO4, and concentrated to provide a 2:1:1 mixture of diastereomers of 20 (0.042 g, 67%) as a pale yellow oil. Major diastereomer: 1H NMR (400 MHz, CDCl3) δ 7.53 − 7.28 (m, 5H), 5.70 − 5.58 (m, 1H), 5.33 (m, 1H), 4.81 − 4.65 (m, 2H), 4.59 − 4.45 (m, 2H), 4.05 − 3.92 (m, 1H), 2.82 − 2.74 (m, 1H), 1.32 −1.24 (m, 3H), 1.11 − 0.96 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 176.5, 144.89, 137.2, 129.2, 128.8, 128.3, 124.7, 81.2, 77.1, 72.5, 41.5, 33.6, 30.2, 16.1, 14.1; IR (thin film) 3551, 3020, 2962, 2865, 1756, 1710 cm-1; HRMS (ESI) m / z calcd for C19H28NaO4 (M + Na)+ 343.1885, found 343.1882.
3.10 Furan-2-ylmethyl 2-oxo-2-phenylacetate (21)
To a solution of benzoylformic acid (0.500 g, 3.33 mmol) in CH2Cl2 (25 mL) was added furfuryl alcohol (0.217 g, 2.22 mmol), N,N-dimethylaminopyridine (0.026 g, 0.22 mmol), and dicyclohexylcarbodiimide (0.687 g, 3.33 mmol). After stirring for 6 h, the solution was filtered, diluted with saturated aqueous NaHCO3, extracted with CH2Cl2 (3 × 25 mL). The organic layers were combined, dried with Na2SO4, and concentrated in vacuo. The resultant oil was purified via column chromatography to give 21 (0.20 g, 45%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 8.0, 2H), 7.69 (t, J = 7.4, 1H), 7.60 − 7.47 (m, 3H), 6.59 (d, J = 2.6, 1H), 6.45 (s, 1H), 5.42 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 186.2, 163.8, 148.5, 144.2, 135.4, 132.8, 130.5, 129.3, 112.4, 111.2, 59.7; IR (thin film) 2955, 2877, 1736, 1689, 1451 cm-1; HRMS (ESI) m / z calcd for C13H10NaO4 (M + Na)+ 253.0477, found 253.0470.
3.11 (S,E)-3-(2,2-Dimethyl-1,3-dioxolan-4-yl)allyl 2-oxo-2-phenylacetate (23)
To a solution of benzoylformic acid (0.500 g, 3.33 mmol) in CH2Cl2 (25 mL) was added (S,E)-3-(2,2-dimethyl-1,3-dioxolan-4-yl)prop-2-en-1-ol (0.351 g, 2.22 mmol), N,N-dimethylaminopyridine (0.026 g, 0.22 mmol), and dicyclohexylcarbodiimide (0.687 g, 3.33 mmol). After stirring for 6 h, the solution was filtered, diluted with saturated aqueous NaHCO3, extracted with CH2Cl2 (3 × 25 mL). The organic layers were combined, dried with NaSO4, and concentrated in vacuo. The resultant oil was purified by column chromatography (5:1 hexanes:EtOAc) to give 23 (0.25 g, 39%) as a yellow oil: [α]23D 15.2 (c 1.77, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.02 (dt, J = 8.5, 1.5, 2H), 7.73 − 7.62 (m, 1H), 7.58 − 7.48 (m, 2H), 6.01 (ddd, J = 7.6, 6.3, 0.6, 1H), 5.92 (ddd, J = 15.5, 8.1, 3.8, 1H), 4.90 (d, J = 5.8, 2H), 4.58 (q, J = 6.8, 1H), 4.14 (dd, J = 8.2, 6.3, 1H), 3.64 (dd, J = 8.2, 7.4, 1H), 1.43 (d, J = 16.5, 6H); 13C NMR (100 MHz, CDCl3) δ 186.3, 163.7, 135.4, 134.0, 130.4, 129.3, 126.4, 110.00, 109.99, 76.3, 69.6, 65.8, 27.0, 26.2; IR (thin film) 3020, 1739, 1691, 1527, 1220 cm-1; HRMS (ESI) m / z calcd for C16H18NaO5 (M + Na)+ 313.1052, found 313.1046. Anal. Calcd for C16H18O5: C, 66.19; H, 6.25. Found: C, 66.41; H, 6.34.
3.12 3-Methylbut-2-enyl 2-oxo-2-phenylacetate (25)
To benzoylformic acid (0.500 g, 3.33 mmol) in CH2Cl2 (25 mL) was added 3-methyl-2-buten-1-ol (0.191 g, 2.22 mmol), N,N-dimethylaminopyridine (0.026 g, 0.22 mmol), and dicyclohexylcarbodiimide (0.687 g, 3.33 mmol). After stirring for 6 h, the solution was filtered, diluted with saturated aqueous NaHCO3 and extracted with CH2Cl2 (3 × 25 mL). The organic layers were combined, dried with NaSO4, and concentrated in vacuo. The resultant oil was purified by column chromatography to give 25 (0.516 g, 96%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 7.9, 2H), 7.67 (t, J = 7.4, 1H), 7.52 (t, J = 7.5, 2H), 5.49 (t, J = 6.9, 1H), 4.91 (d, J = 7.4, 2H), 1.81 (d, J = 6.1, 6H); 13C NMR (100 MHz, CDCl3) δ 186.9, 164.3, 141.5, 135.3, 132.9, 130.4, 129.3, 117.8, 63.4, 26.2, 18.5; IR (thin film) 2973, 2936, 2859, 1912, 1747 cm-1; HRMS (ESI) m / z calcd for C13H14NaO3 (M + Na)+ 241.0841, found 241.0842. Anal. Calcd for C13H14O3: C, 71.54; H, 6.47. Found: C, 71.68; H, 6.64.
3.13 (±)-2-Ethyl-2-hydroxy-3-vinylheptanoic acid (27)
To (E)-oct-2-enyl 2-oxobutanoate (0.861 g, 4.34 mmol) in toluene (20 mL) was added silacyclopropane 2 (1.41 g, 6.30 mmol) and the reaction mixture was cooled to −25 °C. After stirring for 0.5 h, silver tosylate (0.121 g, 0.434 mmol) was added and the mixture was allowed to warm to room temperature. After stirring for 12 h, HF·Pyr (0.50 mL, 4.4 mmol) was added to the solution. The reaction mixture was then diluted with saturated aqueous NaHCO3, the organic layer was rinsed with saturated aqueous NaHCO3 (3 × 45 mL), the aqueous layers were then combined and acidified with 1 M HCl (300 mL) until pH 2. After the desired pH was reached, the aqueous layer was extracted with CH2Cl2 (3 × 50 mL), the organic layers were combined, dried with Na2SO4, and concentrated to provide 27 (0.519 g, 60%) as a white powder: mp 57 °C; 1H NMR (400 MHz, CDCl3) δ 5.81 − 5.54 (m, 1H), 5.25 (d, J = 10.1, 1H), 5.14 (d, J = 17.1, 1H), 2.37 (t, J = 10.3, 1H), 2.13 − 1.70 (m, 2H), 1.59 − 1.11 (m, 6H), 0.92 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 181.5, 137.1, 119.2, 80.8, 52.2, 31.1, 29.8, 29.2, 22.8, 14.4, 8.16; IR (thin film) 3535, 3020, 2959, 1706, 1220 cm-1; HRMS (ESI) m / z calcd for C11H20NaO3 (M + Na)+ 199.1334, found 199.1332. Anal. Calcd for C11H20O3: C, 65.97; H, 10.07. Found: C, 65.71; H, 9.96.
3.14 3-Hydroxy-5-(iodomethyl)-3-phenyldihydrofuran-2(3H)-one (28a)
To (±)-2-hydroxy-2-phenylpent-4-enoic acid (0.023 g, 0.12 mmol) in acetonitrile (3 mL) at 0 °C was added NaHCO3 (0.030 g, 0.36 mmol) followed by iodine (0.091 g, 0.36 mmol). After stirring for 16 h the reaction mixture was diluted with water, and extracted with CH2Cl2 (3 × 15 mL). The organic layers were then combined, dried with Na2SO4, and concentrated in vacuo. The crude mixture was then purified via column chromatography (3:1 hexanes:EtOAc) to afford a 3:2 mix of diastereomers of 28a (0.034 g, 91%) as a pale oil. Major diastereomer: 1H NMR (500 MHz, CDCl3) δ 7.69 − 7.16 (m, 5H), 4.82 − 4.75 (m, 1H), 3.55 − 3.48 (m, 1H), 3.43 − 3.33 (m, 1H), 3.18 (s, 1H), 2.92 (m, 1H), 2.51 (dd, J = 13.1, 9.4, 1H); 13C NMR (125 MHz, CDCl3) δ 177.0, 139.7, 129.3, 128.9, 125.4, 79.2, 76.1, 45.7, 6.3; IR (thin film) 3020, 2957, 1738, 1690 cm-1; HRMS (ESI) m / z calcd for C11H11INaO3 (M + Na)+ 340.9651, found 340.9651.
3.15 (3S,4R)-4-Butyl-3-hydroxy-5-(1-iodoethyl)-3-methyldihydrofuran-2(3H)-one (28b)
To (2S,3R)-2-hydroxy-2-methyl-3-((E)-prop-1-enyl)heptanoic acid (0.027 g, 0.14 mmol) in acetonitrile (3 mL) at 0 °C was added NaHCO3 (0.034 g, 0.040 mmol) followed by iodine (0.101 g, 0.401 mmol). The reaction mixture was stirred for 10 h, diluted with H2O (3 mL), and extracted with CH2Cl2 (3 × 15 mL). The organic layers were then combined, dried with Na2SO4, and concentrated in vacuo. The crude mixture was then purified by column chromatography (3:1 hexanes:EtOAc) to provide 28b (0.021 g, 48%) as a 4:1 mixture of diastereomers. Major diastereomer: 1H NMR (500 MHz, CDCl3) δ 4.98 (dq, J = 12.3, 6.2, 1H), 3.96 (t, J = 10.4, 1H), 3.10 (s, 1H), 2.41 (dt, J = 9.1, 4.5, 1H), 1.81 − 1.69 (m, 3H), 1.66 (s, 1H), 1.63 − 1.51 (m, 3H), 1.48 (s, 3H), 1.46 − 1.34 (m, 2H), 0.98 (t, J = 7.3, 3H); 13C NMR (125 MHz, CDCl3) δ 175.7, 81.9, 73.8, 50.6, 33.3, 32.2, 30.5, 23.3, 22.9, 21.4, 14.0; IR (thin film) 3020, 2958, 2876, 1734, 1690, 1215 cm-1; HRMS (ESI) m / z calcd for C11H19INaO3 (M + Na)+ 349.0277, found 349.0269.
3.16 4-Butyl-3-ethyl-3-hydroxy-5-(hydroxymethyl)dihydrofuran-2(3H)-one (28c)
To (±)-2-ethyl-2-hydroxy-3-vinylheptanoic acid (0.042 g, 0.19 mmol) in CH2Cl2 (5 mL) was added N-methylmorpholine-N-oxide (0.037 g, 0.24 mmol) followed by OsO4 (2.5% in t -BuOH, 0.007 mL). The mixture was stirred for 56 h, diluted with Na2SO3, and extracted with EtOAc (3 × 10 mL). The organic layers were then combined, dried with Na2SO4, and concentrated in vacuo. The resulting viscous oil was then purified via column chromatography (1:1 hexanes:EtOAc) to provide 28c (0.040 g, 73%) as a pale oil. 1H NMR (400 MHz, CDCl3) δ 4.14 (ddd, J = 9.7, 4.4, 2.2, 1H), 4.04 (d, J = 13.0, 1H), 3.72 (dd, J = 12.9, 4.2, 1H), 3.13 (s, 1H), 2.72 − 2.17 (m, 2H), 1.96 − 1.68 (m, 3H), 1.63 (d, J = 8.5, 1H), 1.55 − 1.34 (m, 4H), 1.07 (t, J = 7.5, 3H), 0.97 (t, J = 7.1, 3H); 13C NMR (100 MHz, CDCl3) δ 178.7, 82.9, 78.1, 62.8, 46.6, 29.9, 26.1, 25.8, 23.3, 14.3, 7.6; IR (thin film) 3434, 3020, 2935, 1772, 1214 cm-1; HRMS (ESI) m / z calcd for C11H20NaO4 (M + Na)+ 239.1259, found 239.1266.
3.17 4-Butyl-3-ethyl-3-hydroxy-5-(iodomethyl)dihydrofuran-2(3H)-one (28d)
To a solution of (±)-2-ethyl-2-hydroxy-3-vinylheptanoic acid (0.020 g, 0.10 mmol) in THF (3 mL) at 0 °C was added N-iodosuccinimide (0.024 g, 0.11 mmol). After stirring for 0.5 h, the reaction mixture was concentrated in vacuo and the crude mixture was purified by column chromatography to give 28d (0.029 g, 92%) as a 3:1 mixture of diastereomers. Major diastereomer: 1H NMR (400 MHz, CDCl3) δ 3.95 (ddd, J = 9.2, 5.9, 3.3, 1H), 3.65 (dd, J = 11.4, 3.3, 1H), 3.36 (dd, J = 11.4, 5.9, 1H), 2.82 (s, 1H), 2.38 (td, J = 8.8, 5.4, 1H), 1.90 − 1.32 (m, 7H), 1.12 (t, J = 7.4, 1H), 1.06 (t, J = 7.5, 3H), 0.98 (t, J = 7.0, 3H); 13C NMR (100 MHz, CDCl3) δ 177.4, 128.8, 80.5, 78.4, 52.2, 29.7, 26.0, 23.3, 14.3, 7.5, 6.6; IR (thin film) 3480, 2960, 2932, 2862, 1780 cm-1; HRMS (ESI) m / z calcd for C11H19INaO3 (M + Na)+ 349.0277, found 349.0268.
3.18 5-(Bromomethyl)-4-butyl-3-ethyl-3-hydroxydihydrofuran-2(3H)-one (28e)
To a solution of (±)-2-ethyl-2-hydroxy-3-vinylheptanoic acid (0.020 g, 0.10 mmol) in THF (3 mL) at 0 °C was added N-bromosuccinimide (0.019 g, 0.11 mmol). After stirring for 0.5 h, the reaction mixture was concentrated and the crude mixture was purified by column chromatography (5:1 hexanes:EtOAc) to give 28e (0.021 g, 76%) as a 5:1 mixture of diastereomers. Major diastereomer: 1H NMR (400 MHz, CDCl3) δ 4.23 (d, J = 5.7, 1H), 3.80 (d, J = 11.7, 1H), 3.55 (dd, J = 11.6, 5.4, 1H), 2.90 (s, 1H), 2.49 (d, J = 5.8, 1H), 1.90 − 1.34 (m, 8H), 1.07 (t, J = 7.3, 3H), 0.98 (t, J = 6.2, 3H); 13C NMR (100 MHz, CDCl3) δ 177.6, 128.6, 80.5, 78.2, 50.1, 33.1, 29.8, 26.0, 23.3, 14.2, 7.5; IR (thin film) 3471, 3020, 2960, 2933, 2873, 1778 cm-1; HRMS (ESI) m / z calcd for C11H20BrO3 (M + H)+ 279.0596, found 279.0593.
3.19 (3R,4S)-5-(1,2-Dihydroxyethyl)-3-hydroxy-4-((4-methoxybenzyloxy)methyl)-3-methyldihydrofuran-2(3H)-one (29)
To (2R,3R,E)-2,6-dihydroxy-3-((4-methoxybenzyloxy)methyl)-2-methylhex-4-enoic acid (0.17 g, 0.53 mmol) in CH2Cl2 (10 mL) was added N-methylmorpholine-N-oxide (0.084 g, 0.72 mmol) followed by OsO4 (2.5% in t-BuOH, 0.13 mL). The mixture was stirred for 16 h, diluted with saturated aqueous Na2SO3, and then extracted with CHCl3:i-PrOH (3:1, 3 × 15 mL). The organic layers were combined, dried with NaSO4, and concentrated in vacuo. The resultant oil was purified by column chromatography (CHCl3:MeOH 10:1) to give a 5:1 mixture of diastereomers of 29 (0.086 g, 50%) as a pale yellow oil. Major diastereomer: 1H NMR (400 MHz, CDCl3) δ 7.25 (d, J = 8.4, 2H), 6.91 (d, J = 8.5, 2H), 5.04 (s, 1H), 4.78 (s, 1H), 4.45 (d, J = 6.5, 2H), 4.33 (d, J = 6.7, 1H), 3.98 − 3.37 (m, 7H), 2.79 (d, J = 5.8, 1H), 1.33 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 179.4, 159.8, 129.9, 128.8, 114.3, 80.5, 74.3, 73.5, 71.8, 67.2, 64.0, 55.7, 47.5, 19.4; IR (thin film) 3398, 3020, 2937, 1776, 1513 cm-1; HRMS (ESI) m / z calcd for C16H22NaO7 (M + H)+ 349.1263, found 349.1267.
3.20 (3R,4S)-3-Hydroxy-4-((4-methoxybenzyloxy)methyl)-3-methyl-5-vinyldihydrofuran-2(3H)-one (30)
To a solution of 29 (0.19 g, 0.58 mmol) in THF (60 mL) was added triphenylphosphine (0.46 g, 1.8 mmol), imidazole (0.24 g, 3.5 mmol), and iodine (0.44 g, 1.8 mmol). The reaction mixture was heated to reflux for 2 h and then concentrated. The resultant oil was taken up in MTBE (20 mL) and washed with saturated aqueous NaHCO3, saturated aqueous Na2SO3, and brine. The organic fraction was dried over Na2SO4 and concentrated in vacuo to provide a crude oil that was purified by column chromatography (1:1 hexanes:EtOAc) to give 30 as a yellow oil (0.17 g, 95%): 1H NMR (400 MHz, CDCl3) δ 7.35 − 7.11 (m, 2H), 6.90 (dd, J = 6.4, 4.9, 2H), 5.89 (ddd, J = 17.2, 10.4, 6.8, 1H), 5.40 (d, J = 17.1, 1H), 5.32 (d, J = 10.4, 1H), 4.58 (dd, J = 9.7, 7.0, 1H), 4.52 − 4.36 (m, 2H), 3.82 (s, 3H), 3.60 (d, J = 5.8, 2H), 2.81 (m, 1H), 2.51 (dt, J = 9.9, 5.8, 1H), 1.40 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 178.6, 159.6, 134.5, 129.6, 119.6, 114.1, 79.7, 74.9, 73.4, 65.4, 55.5, 52.0, 20.3; IR (thin film) 3440, 3020, 2935, 1778, 1513 cm-1; HRMS (ESI) m / z calcd for C16H20NaO5 (M + Na)+ 315.1208, found 315.1204.
Scheme 7.

Two potential transition states for compound 17.
Scheme 12.

Silylene transfer products can be converted to γ-lactones (Table 3).
Acknowledgments
This research was supported by the National Institute of General Medical Sciences of the National Institutes of Health (GM-54909). K. A. W. thanks Amgen and Lilly for awards to support research. We thank Dr. Phil Dennison (UCI) for assistance with NMR spectroscopy and Dr. John Greaves and Ms. Shirin Sorooshian (UCI) for mass spectrometry.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Coppola GM, Schuster HF. Hydroxy Acids in Enantioselective Synthesis. Weinheim: Germany: 1997. [Google Scholar]
- 2.Hannessian S. Total Synthesis of Natural Products The Chiron Approach. Pergamon Press; New York: 1983. p. Chaper 2. [Google Scholar]
- 3.Ball M, Andrews SP, Wierschem F, Cleator E, Smith MD, Ley SV. Org Lett. 2007;7:663–666. doi: 10.1021/ol062947x. [DOI] [PubMed] [Google Scholar]
- 4.Goldup SM, Pilkington CJ, White AJP, Burton A, Barrett AGM. J Org Chem. 2006;71:6185–6191. doi: 10.1021/jo060931e. [DOI] [PubMed] [Google Scholar]
- 5.Bunte JO, Cuzzupe AN, Daly AM, Rizzacasa MA. Angew Chem, Int Ed. 2006;45:6376–6380. doi: 10.1002/anie.200602507. [DOI] [PubMed] [Google Scholar]
- 6.Queffélec C, Bailly F, Mbemba G, Mouscadet JF, Debyser Z, Witvrouw M, Cotelle P. Eur J Med Chem. 2008;43:2268–2271. doi: 10.1016/j.ejmech.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 7.Andrus MB, Sekhar SBBV, Meredith EL, Dalley NK. Org Lett. 2000;2:3035–3037. doi: 10.1021/ol0002166. [DOI] [PubMed] [Google Scholar]
- 8.Crimmins MT, Emmitte KA, Katz JD. Org Lett. 2000;2:2165–2167. doi: 10.1021/ol006091m. [DOI] [PubMed] [Google Scholar]
- 9.He L, Byun HS, Bittman R. J Org Chem. 2000;65:7627–7633. doi: 10.1021/jo001226n. [DOI] [PubMed] [Google Scholar]
- 10.Jung JE, Ho H, Kim HD. Tetrahedron Lett. 2000;41:1793–1796. [Google Scholar]
- 11.Nam J, Lee SK, Park YS. Tetrahedron. 2003;59:2397–2401. [Google Scholar]
- 12.Tang L, Deng L. J Am Chem Soc. 2002;124:2870–2871. doi: 10.1021/ja0255047. [DOI] [PubMed] [Google Scholar]
- 13.Yu H, Ballard E, Boyle PD, Wang B. Tetrahedron. 2002;58:7663–7679. [Google Scholar]
- 14.Howard BE, Woerpel KA. Org Lett. 2007;9:4651–4653. doi: 10.1021/ol702148x. [DOI] [PubMed] [Google Scholar]
- 15.Driver TG, Woerpel KA. J Am Chem Soc. 2004;126:9993–10002. doi: 10.1021/ja0306563. [DOI] [PubMed] [Google Scholar]
- 16.Heinicke J, Gehrhus B. J Organomet Chem. 1992;423:13–21. [Google Scholar]
- 17.Ando W, Hagiwara K, Sekiguchi A. Organometallics. 1987;6:2270–2271. [Google Scholar]
- 18.Calad SA, Woerpel KA. J Am Chem Soc. 2005;127:2046–2047. doi: 10.1021/ja042893r. [DOI] [PubMed] [Google Scholar]
- 19.Hiersemann M, Nubbemeyer U. The Claisen Rearrangement: Methods and Applications. WILEY-VCH; Weinheim: 2007. [Google Scholar]
- 20.Wood JL, Moniz GA, Pflum DA, Stoltz BM, Holubec AA, Dietrich HJ. J Am Chem Soc. 1999;121:1748–1749. [Google Scholar]
- 21.Yamazaki T, Shinohara N, Kitazume T, Sato M. J Org Chem. 1995;60:8140–8141. [Google Scholar]
- 22.Nubbemeyer U. Synthesis. 2003:961–1008. [Google Scholar]
- 23.Singh P, Mittal A, Kaur P, Kumar S. Tetrahedron. 2006;62:1063–1068. [Google Scholar]
- 24.Bartlett PA, Barstow JF. J Org Chem. 1982;47:3933–3941. [Google Scholar]
- 25.Liu H, Siegel DR, Danishefsky SJ. Org Lett. 2006;8:423–425. doi: 10.1021/ol052618p. [DOI] [PubMed] [Google Scholar]
- 26.Pattarozzi M, Zonta C, Broxterman QB, Kaptein B, De Zorzi R, Randaccio L, Scrimin P, Licini G. Org Lett. 2007;9:2365–2368. doi: 10.1021/ol070764k. [DOI] [PubMed] [Google Scholar]
- 27.Trost BM, Lee C. J Am Chem Soc. 2001;123:12191–12201. doi: 10.1021/ja0118338. [DOI] [PubMed] [Google Scholar]
- 28.Yvin JC, Chevolot-Magueur AM, Chevolot L. J Am Chem Soc. 1982;104:4498–4500. [Google Scholar]
- 29.Garegg PJ, Samuelsson B. Synthesis. 1979:469–470. [Google Scholar]