

Abstract
The genetic basis of most of dilated cardiomyopathy (DCM) cases remains unknown. A recent study indicated that mutations in a highly localized five amino acid hotspot in exon 9 of RBM20, a gene encoding a ribonucleic acid‐binding protein, caused aggressive DCM. We undertook this study to confi rm and extend the nature of RBM20 mutations in another DCM cohort. Clinical cardiovascular data, family histories, and blood samples were collected from patients with idiopathic DCM. DNA from 312 DCM probands was sequenced for nucleotide alterations in exons 6 through 9 of RBM20, and additional family members as possible. We found six unique RBM20 rare variants in six unrelated probands (1.9%). Four mutations, two of which were novel (R634W and R636C) and two previously identified (R634Q and R636H), were identified in a five amino acid hotspot in exon 6. Two other novel variants (V535I in exon 6 and R716Q in exon 9) were outside of this hotspot. Age of onset and severity of heart failure were variable, as were arrhythmias and conduction system defects, but many subjects suffered severe heart failure resulting in early death or cardiac transplantation. This article concludes that DCM in patients with RBM20 mutations is associated with advanced disease. Clin Trans Sci 2010; Volume 3: 90–97
Keywords: dilated cardiomyopathy, heart failure, genetics, RNA‐binding protein, mutation
Introduction
Dilated cardiomyopathy (DCM; OMIM 115200) is a debilitating primary cardiac muscle disease with a 5‐year mortality approaching 50% following diagnosis. 1 , 2 Whether familial or sporadic, DCM shows remarkably high genetic heterogeneity. 3 , 4 To date, the molecular basis of most of DCM cases remains unknown despite the fact that mutations in more than 30 genes have been shown to be disease causing or disease associated. 5 , 6 Because of this marked locus heterogeneity, the fraction of DCM patients who have a mutation in any one gene is small, and ranged from 0.3% to 5.9% in our recent resequencing studies. 7 , 8 , 9 , 10 Most of the genes with mutations causing DCM encode for sarcomeric proteins involved in contraction, or cytoskeletal proteins important for cell structure or force transduction. 5 , 6 Exceptions to this include a mutation identified in the eye absent transcription factor 4 (EYA4) in a family with both DCM and sensorineural hearing loss, 11 and mutations in the genes encoding presenilin 1 and 2 (PSEN1 and PSEN2) that we recently identified in DCM families. 10 Although the pathogenic mechanisms of EYA4 and the presenilins in DCM remain to be def ned, both are known to play regulatory roles in the heart even though they are neither essential structural components nor contractile proteins.
A recent genetic linkage and gene‐mapping study demonstrated that mutations in RBM20, a ribonucleic acid (RNA)‐binding protein gene, cause DCM. 12 This discovery is intriguing in several aspects. To our knowledge, this is the first report to suggest that a genetic abnormality of an RNA‐binding protein can lead to cardiomyopathy. 13 It is also noteworthy that RBM20 mutations were associated with severe cases of DCM with high mortality and patients needing heart transplantation. Moreover, the five mutations identified in the first report were concentrated in a small region of RBM20, spanning only five amino acids, and encoded by a single exon. 12
To further evaluate the role of RBM20 in DCM pathogenesis and the DCM clinical characteristics caused by RBM20 mutations, we genetically screened a cohort of 312 DCM probands, and their family members when a mutation was identified. We found six unique mutations in six unrelated probands, four of which were novel. These results expand the RBM20 mutation spectrum in DCM and further emphasize the importance of RBM20 in the myocardial disease.
Materials and Methods
Clinical evaluation
Written, informed consent was obtained from all subjects, and the Institutional Review Boards at the Oregon Health & Science University and the University of Miami approved the study. The investigation included 312 probands (290 Caucasians, of whom 7 were of Hispanic descent; 16 African‐Americans, 3 Asians and 3 Native Americans/Alaskan Natives) and used methods of clinical categorization of DCM as previously described. 14 Clinical data were obtained through our own evaluations, which included minimally a history and physical examination, an electrocardiogram (ECG) and an echocardiogram as previously described 15 or through medical record or death certif cate review. DCM was def ned as lefiventricular enlargement (lefiventricular end‐diastolic dimension [LVEDD]≥95th percentile or Z‐score >1.65) of a gender‐ and height‐matched Framingham population 16 with systolic dysfunction (lefiventricular ejection fraction [LVEF] less than or equal to 50%), and the exclusion of other possible causes of cardiomyopathy, meeting diagnostic criteria consistent with idiopathic dilated cardiomyopathy (IDC). 14 , 17 Familial dilated cardiomyopathy (FDC) was assigned in cases with a documented diagnosis of IDC in two or more family members. 14 , 17 Family members with some cardiovascular abnormality but not meeting criteria for DCM were categorized as unknown.
Genetic studies
Genomic DNA was extracted from whole blood according to a standard salting out procedure. 18 Prior to the current study, we sequenced 14 FDC genes in genomic DNA from these probands. 7 , 8 , 9 , 10 The gene encoding RBM20 maps to chromosome 10q25 and has 1,227 amino acids encoded by 14 exons. A region in the center of the protein, approximately between residue 500 and 800, is conserved from chicken and mouse to chimpanzee. Both the RNA recognition motif region 1 (RRM‐1) and the arginine/serine (RS) rich domains are also located within this region. A U1 zinc f nger region in exon 14 is also conserved to some degree (human, chimpanzee, and rat). The protein sequences outside of these regions are less conserved, and significant portions are not conserved even among mammals. We presumed that any genetic variants in less‐conserved regions may have reduced functional consequence, and further, variants identified in these regions would be dif cult to interpret. We accordingly focused our study on the most conserved regions of RBM20. Bidirectional DNA sequencing was conducted for the region of RBM20 showing strong conservation across species, including exon 6 through exon 9. Primers were designed with Primer3 to amplify the exons and their flanking intronic regions. Polymerase chain reaction (PCR) products were cleaned with ExoI‐SAP enzymatic digestion (USB Corp., Cleveland, OH, USA). Sequencing reactions were performed with BigDye 3.1 and run on a 3130XL Genetic Analyzer (Applied Biosystem, Foster City, California, USA) as per the manufacturer's instructions. A total of 450 control DNAs were sequenced for any identified nonsynonymous variants. Sequencher (Gene Codes, Ann Arbor, MI, USA) was used to facilitate data analysis and mutation identif cation followed by visual inspection of individual sequencing traces. Genetic variants were annotated according to the reference sequence (accession NM_001134 and NP_001127835). The standard Basic Local Alignment Search Tool for Proteins (BLASTP) search was conducted via NCBI search launcher to identify proteins homologous to human RBM20. The CLUSTALW2 algorithm (http://www.ebi.ac.uk/Tools/clustalw2/index.html) was used to perform multiple sequence alignment with the protein sequences of significant homology to human RBM20.
Results
Genetic studies
The bidirectional sequencing of exon 6 through exon 9 encoding the conserved region of RBM20 was completed for DNA specimens from 312 unrelated subjects with DCM. Six dif erent protein‐altering variants were identified in six unrelated probands ( Figure 1 ); four of them (V535I, R634W, R636C, and R716Q) were novel. T eir positions within cDNA (NM_001134363) and genomic DNA (UCSC hg18), nucleotide, and predicted amino acid changes are summarized ( Table 1 ). These variants were predicted to alter highly conser ved amino acids ( Figure 2 ), none of which were present in dbSNP or 450 Caucasian control DNAs (900 chromosomes).
Figure 1.
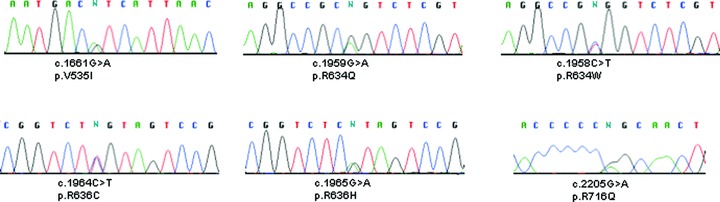
Detection of six different missense mutations of RBM20 in six unrelated DCM probands by direct sequencing. The observed single nucleotide substitutions and the predicted amino acid changes are indicated underneath each electropherogram. cDNA numbering is based on the reference sequence NM_001134. Amino acid numbering is according to protein sequence NP_001127835.
Table 1.
RBM20 mutation identified in DCM probands.
| cDNA position (NM_001134363) | UCSC genomic position (hg18):Chr10 | Exon | Nucleotide change | Codon change | Amino acid change | Pedigree letter and DCM proband | Comment |
|---|---|---|---|---|---|---|---|
| 1661 | 112547331 | 6 | G > A | GTC > ATC | V535I | A II‐2 | Novel |
| 1959 | 112562046 | 9 | G > A | CGG > CAG | R634Q | B IV‐1 | Prior report 12 |
| 1958 | 112562045 | 9 | C > T | CGG > TGG | R634W | C II‐1 | Novel |
| 1964 | 112562051 | 9 | C > T | CGT > TGT | R636C | D III‐2 | Novel |
| 1965 | 112562052 | 9 | G > A | CGT > CAT | R636H | E II‐2 | Prior report 12 |
| 2205 | 112562292 | 9 | G > A | CGG > CAG | R716Q | F II:6 | Novel |
Figure 2.

Alignment of conserved region among orthologous RBM20 proteins from different species. The amino acids predicted to be altered by mutations are indicated by a box. The predicted residue changes are also noted. Protein sequences were obtained from GenBank with the following accession numbers: NP_001127835 for human; XP_508032 for chimpanzee; NP_001101081 for rat; XP_001481318 for mouse; XP_421755 for chicken; and XP_683222.
Consistent with the first report, 12 four of the six mutations were located in the same mutation hotspot in the RS‐rich region in exon 9 and modif ed two amino acids (R634 and R636), of which both were previously reported to be altered. Two of these missense mutations were novel, R634W and R636C. The other two, R634Q and R636H, had been identified in the first report. 12
The two other novel mutations (V535I and R716Q) were found in domains outside of the hotspot region. V535I was found in exon 6 in the strictly conserved RRM‐1. R716Q was located in exon 9 approximately 60 residues downstream of the RS‐rich region (residues 632–654).
All available DNA specimens from family members of the probands with RBM20 mutations were also sequenced for the family mutation. Seventeen family members from three unrelated probands (pedigrees D–F, Figure 3 ) tested positive for the family mutations. Three individuals in family F who had no DNA available for genetic analysis were considered to be obligate mutation carriers.
Figure 3.
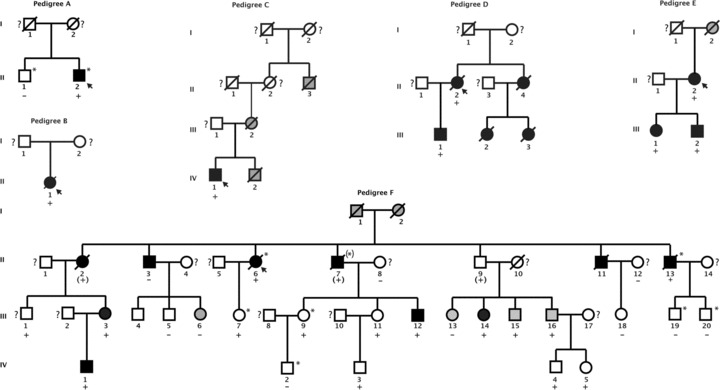
Pedigrees of DCM families carrying RBM20 mutations (family A with V535I, family B with R634Q, family C with R634W, family D with R636C, family E with 636H, and family F with R716Q). Squares indicate males; circles, females; open symbols, unaffected verifi ed by medical records; solid symbols, affected with DCM verifi ed by medical records; gray symbols, individuals with a history of DCM from whom medical records were not available or who have some cardiovascular abnormality but not meeting criteria for being affected with DCM (available data shown in Table 2 ). Because DCM can be present in asymptomatic individuals, question marks denote unaffected subjects by family history from whom medical records were unavailable. Slanted bars indicate deceased family members. A plus sign indicates the presence of an RBM20 mutation; minus signs, the absence of the mutation. Asterisk signs (pedigree A and pedigree F) indicate the presence of an LDB3 mutation (A698T) or LMNA mutation (R388H; as previously published 9 ), respectively.
Clinical studies
The general clinical profile of the probands in our DCM cohort has been previously summarized. 14 Among six probands identified to have RBM20 mutations, by self report five were Caucasian with European ancestry, and the proband from pedigree E had Caucasian and Native American ancestry. Pedigrees for the six probands are shown ( Figure 3 ).
Twenty‐one family members from four families (C, D, E, and F) also exhibited mild‐to‐severe DCM, including heart failure, or needing heart transplantation or an implantable cardiac def brillator (ICD) ( Table 2 ). All affected family members (except for one, pedigree F, II‐3) tested positive for their respective RBM20 mutations ( Figure 3 ). Clinical data and cardiac function of these family members are summarized in Table 2 .
Table 2.
Clinical characteristics of subjects.
| Pedigree name and position | Age at evaluation or DCM diagnosis | DCM | Age of death | ECG/Arrhythmia | LVEDD, mm (Z‐score) | LVEDD Framingham percentile | EF | RBM20 mutation | Comment |
|---|---|---|---|---|---|---|---|---|---|
| Idiopathic dilated cardiomyopathy (sporadic) | |||||||||
| A: II‐1 | 71 | No | PVCs | 48 (−1.06) | < 95 | 55% | No | ||
| A: II‐2 | 50 | Yes | 1AVB, AF, paced rhythm | 60 (2.5) | > 99 | 25% | V535I | ICD, HF, stroke, heart transplantation | |
| B: II‐1 | 46 | Yes | 53 | LAFB, LAE | 73 (6.8) | > 99 | 20% | R634Q | Pacemaker, HF, stroke |
| Familial dilated cardiomyopathy | |||||||||
| C: II‐3 | NA | No | NA | NA | NA | NA | NA | Unknown | Reported death during third decade from myocardial disease. No medical records. |
| C: III‐2 | 53 | Yes | 63 | NA | NA | NA | NA | Unknown | Reported affected with normal angiogram; no medical records |
| C: IV‐1 | 49 | Yes | Normal | 62 (2.8) | > 99 | 44% | R634W | LVE | |
| C: IV‐2 | NA | Yes | 21 | NA | NA | NA | NA | Unknown | Reported affected. Died awaiting heart transplantation; no medical records |
| D: II‐2 | 43 | Yes | 64 | SSS, AF, VT, VF | 71 (6.8) | > 99 | 10% | R636C | HF, syncope, ICD/pacemaker, LVAD |
| D: II‐4 | 24 | Yes | 25 | RAD, sinus tach | NA | NA | NA | Unknown | HF, DCM by medical records |
| D: III‐1 | 23 | Yes | IVCD, NSSTT | 58 (2.1) | 97.5–98.9 | 35% | R636C | ||
| D: III‐2 | 16 | Yes | 16 | Normal | 81.5 (9.0) | > 99 | 21% | Unknown | HF, heart transplant |
| D: III‐3 | NA | Yes | 14 | NA | 75 (7.6) | > 99 | 31% | Unknown | HF |
| E: I‐2 | NA | Unknown | 53 | Yes | NA | NA | NA | Unknown | Death certifi cate with cardiac arrhythmia and focal myocardial necrosis |
| E: II‐2 | 51 | Yes | 56 | PVCs | NA | NA | 40% | R636H | ICD, LVE, and DCM by medical records |
| E: III‐1 | 30 | Yes | NSVT, PVC, bigemeny | 52 (2.5) | > 95 | 25% | R636H | ICD, HF, syncope | |
| E: III‐2 | 25 | Yes | NSSTT | NA | NA | 28% | R636H | LVE and DCM by medical records | |
| F: I‐1 | NA | Unknown | 70 | NA | NA | NA | NA | Unknown | Reported affected with HF |
| F: I‐2 | NA | Unknown | 70 | NA | NA | NA | NA | Unknown | Reported with edema and enlarged heart |
| F: II‐2 | 51 | Yes | Sinus tach, LAE, NSSTT | 55 (NA) | NA | 33% | R716Q | LVE, HF, ICD | |
| F: II‐3 | 58 | Yes | RBBB, bradycardia, NSSTT | 60 (2.8) | > 99 | 48% | No | SSS, pacemaker, borderline DCM; remote history of easily treated heart failure. | |
| F: II‐6 | 37 | Yes | 56 | Normal | 54 (2.7) | > 99 | 25% | R716Q | Syncope/presyncope |
| F: II‐7 | 31 | Yes | 31 | NA | NA | NA | NA | (R716Q) | SCD. DCM at autopsy |
| F: II‐9 | NA | NA | NA | NA | NA | NA | (R716Q) | No records | |
| F: II‐11 | 35 | Yes | 43 | LAE, NSSTT, SVT, VT | 70 (4.4) | > 99 | 32% | Unknown | ICD, died awaiting heart transplant |
| F: II‐13 | 44 | Yes | 51 | NSSTT, NSCD, Anterior MI | 76 (6.1) | > 99 | 23% | R716Q | Anterior MI 5 years after IDC diagnosis |
| F: III‐1 | NA | Unknown | NA | NA | NA | NA | R716Q | No records, reported healthy | |
| F: III‐3 | 32 | Yes | NSSTT | 50 (1.6) | ∼95 | 40% | R716Q | Mild DCM | |
| F: III‐4 | 32 | No | Normal | 56 (1.5) | < 95 | 69% | Unknown | No sequencing data | |
| F: II1‐5 | 20 | No | Normal | 48 (−0.7) | < 95 | 66% | No | ||
| F: II1‐6 | 27 | No | Bradycardia | 55 (3.1) | > 99 | 66% | No | Asymptomatic | |
| F: II‐7 | 22 | No | Normal | 46 (0.1) | < 95 | 62% | R716Q | ||
| F: II1‐9 | 36 | No | Normal | 47 (0.6) | < 95 | 65% | R716Q | ||
| F: lll‐l 1 | 26 | No | Normal | 50 (1.3) | < 95 | 63% | R716Q | ||
| F:III‐12 | 33 | Yes | NSSTT | 57 (1.7) | > 95 | 15% | R716Q | HF | |
| F: III‐13 | 21 | No | Normal | 53 (2.0) | 97.7–98.9 | 65% | No | Asymptomatic | |
| F: III‐14 | 39 | Yes | Normal | 54 (2.3) | 97.7–98.9 | 49% | R716Q | Borderline DCM | |
| F: III‐15 | 40 | No | NSCD | 58 (1.8) | > 95 | 63% | R716Q | ||
| F: III‐16 | 37 | No | Q in III, F | 61 (2.6) | > 99 | 58% | R716Q | LVE | |
| F: 111‐18 | 24 | No | Normal | 51 (1.4) | < 95 | 58% | No | ||
| F: III‐19 | 30 | No | NSSTT | 53 (0.7) | < 95 | 61% | No | ||
| F: III‐20 | 18 | No | IRBBB | 53 (0.4) | < 95 | 61% | No | ||
| F: IV‐1 | 18 | Yes | NSSTT | 61.4 (2.9) | > 99 | 28% | R716Q | Asymptomatic presentation | |
| F: IV‐2 | 20 | No | Normal | 55 (0.3) | < 95 | 59% | No | ||
| F: IV‐3 | 6 | No | Normal | 37.5 (−1.2) | < 95 | 72% | R716Q | ||
| F: IV‐4 | 12 | No | Normal | 46 (0.04) | < 95 | 62% | R716Q | ||
| F: IV‐5 | 9 | No | Normal | 43 (0.1) | < 95 | 65% | R716Q | ||
*LVEDD is left ventricular end‐diastolic dimension measured by echocardiography; the Z‐score is the number of standard deviations of the LVEDD above a height and gender‐based population mean. LVEDD Framingham percentile is the LVEDD percentile from a height and gender‐based population (see methods). AF = atrial fibrillation; AVB = atrioventricular block; HF = heart failure; ICD = implantable cardiac defibrillator; IRBBB = incomplete right bundle branch block; IVCD = intraventricular conduction delay; LAE = left atrial enlargement; LAFB = left anterior fascicular block; LVAD = left ventricular assistance device; LVE = left ventricular enlargement; LVEF = left ventricular ejection fraction; MI = myocardial infarction; NA = not applicable; NSCD = nonspecific conduction delay; NSSTT = nonspecific ST‐T changes; NSVT = nonsustained ventricular tachycardia; PVCs = premature ventricular contractions; RAD = right axis deviation; RBBB = right bundle branch block; SCD = sudden cardiac death; SSS = sick sinus syndrome; VF = ventricular fibrillation; VT = ventricular tachycardia.
The average age of D CM ons et for the RBM20 mutation carriers was 37.1 years, and the mean LVEF was 31%. Six mutation carriers also had an ICD or pacemaker implanted, and several subjects had supraventricular and/or ventricular arrhythmias, suggesting that RBM20 mutations in addition to disturbing cardiac contraction may also adversely impact cardiac conduction and rhythm. No patient who carried an RBM20 mutation had lefiventricular hypertrophy by echocardiography, with lefiventricular septal and posterior wall measurements within normal limits (data not shown).
Two RBM20 mutations (V535I, pedigree A and R634Q, pedigree B) had sporadic DCM, while the other four (R634W, R636C, R636H, and R716Q) had familial DCM (pedigrees C, D, E, F, Figure 3 ) consistent with autosomal dominant inheritance. We were able to evaluate the segregation of RBM20 mutations within three families (pedigree D, E, and F) with multiple affected members. As shown in these three pedigrees, all of the affected family members were positive for their respective RBM20 mutation, except for subject II‐3 in pedigree F, whose DCM may have arisen from environmental factors or another unknown genotype.
The clinical presentations of the probands carrying RBM20 mutations and their affected family members are provided ( Table 2 and Figure 3 ):
Pedigree A
The proband (II‐2) had been hospitalized at the age of 39 for arrhythmia. He later presented with shortness of breath and was diagnosed with DCM at the age of 50 ( Table 2 ). At 59 years, his condition worsened with symptomatic heart failure requiring heart transplantation. Minimal family history was available. His father had atrial f brillation at the age of 60. His brother had a normal echocardiogram, but his ECG showed premature ventricular contractions. In addition to the novel RBM20 variant V535I identified in the current study, the proband was known to carry an LDB3 variant A698T, assigned as a possibly DCM‐causing variant, as previously reported. 7 Even though the newly identified RBM20 variant V535I changed a highly conserved amino acid in its RNA‐binding motif, this proband had no other affected family members to assess the cosegregation of this variant with DCM. T us, the degree to which this variant contributed to the proband's DCM, particularly in the setting of the LDB3 variant, remains unknown.
Pedigree B
The proband (II‐1) presented with a flu‐like illness that prompted a chest X‐ray examination, revealing remarkable cardiomegaly. A diagnosis of DCM was made at the age of 46. Seven years later, the patient was admitted with decompensated heart failure and died that same year. Her family history was noncontributory. No relatives were available for further evaluation of the R634Q variant. Of note, this proband had the same mutation and a similar clinical course as that identified in a large linkage family (DC‐35) described in the first RBM20 report. 12 DCM in that family was aggressive with disease onset at 18 years, and seven family members died prematurely from DCM. Collectively, the data suggest that this mutation is malignant whether present in sporadic or familial DCM.
Pedigree C
The proband (IV‐1) was diagnosed with DCM af er cardiovascular screening due to his family history, remarkable for his brother (IV‐2), considered for heart transplantation and deceased at age of 21, their mother (III‐2), also reported affected, and her maternal uncle (II‐3), suspected to be affected with DCM, who died in his 30s attributed to myocardial disease. A novel RBM20 mutation (R634W) was identified from this proband. Unfortunately, no additional DNAs were available from family members to assess its segregation with DCM. Since the RBM20 mutation R634Q was initially identified as causative of DCM in a large linkage family, 12 the RBM20 mutation (R634W) at the same residue in the RS‐rich region likely contributed to the DCM in this family.
Pedigree D
Because of a markedly positive family history of cardiovascular disease, the proband (II‐2) that underwent clinical screening was diagnosed with asymptomatic DCM, frequent ventricular premature beats, and ventricular tachycardia ( Table 2 ). Her family history was positive for early deaths in her sister (II‐4) and nieces (III‐2 and III‐3), from arrhythmias and/or postheart transplant complications. The proband died at the age of 64 due to strokes related to a left ventricular assist device while awaiting heart transplantation. A novel RBM20 mutation (R636C) was identified, which was also present in her affected son (III‐1).
Pedigree E
The proband (II‐2) was diagnosed with DCM af er complaints of chest tightness, difficulty in breathing, and irregular heart rate ( Table 2 ). Both the proband's daughter (III‐1), who had ICD resuscitation from sudden cardiac death, and son (III‐2) carried an RBM20 mutation (R636H). The proband's mother (I‐2) died in 1967 with symptoms of heart failure, and her death certificate listed cardiac arrhythmia as the cause of death. The R636H mutation, the same mutation identified in family DC‐49 of the initial report, 12 was found to segregate with DCM in this family, which is of Caucasian and Native American ancestry. Both were small families with two and three affected individuals, respectively. DCM started in young adulthood in each family and presented mostly with severe systolic dysfunction and heart failure.
Pedigree F
This is a non‐Hispanic white pedigree with an extensive family history of DCM. The proband (II‐6) presented with signs of heart failure and arrhythmia. A diagnosis of DCM was made at the age of 37. Four family members suf ered from premature death due to DCM (II‐2, II‐7, II‐11, and II‐13) as shown in pedigree F ( Figure 3 , Table 2 ). One family member (IV‐1) at the age of 18 was found to have a significantly dilated heart with depressed systolic function. The R716Q variant was identified in the proband and 17 family members (including three obligate carriers). While 8 of the 17 carrying the variant were affected, two additional mutation‐positive subjects (III‐15 and III‐16) have mild left ventricular enlargement (LVEDD Z ‐score 1.8 and 2.6, respectively) and minor ECG changes with normal systolic function. Notably, eight members in family F did not exhibit signs of DCM but tested positive or were obligate carriers for the R716Q mutation, indicating the incomplete penetrance of this mutation. The average age of those bearing the R716Q mutation and showing no sign of DCM is 23.5 years, which is younger than the average age of onset (37.1 years as noted above) of those expressing the DCM phenotype. Subject II‐3, who has borderline DCM with a remote history of easily treated heart failure and sick sinus syndrome requiring a pacemaker, tested negative for the RBM20 variant. As indicated by an asterisk in pedigree F, several members of the family F were also known to harbor a LMNA variant R388H, as previously reported. 9
We previously sequenced the coding sequences and intron/ exon junctions of the following DCM genes: LMNA, PSEN1, PSEN2, MYH7, TNNT2, SCN5A, CSRP3, TCAP, LBD3, MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 for the probands in our DCM cohort. 7 , 8 , 9 , 10 No mutations in these genes were identified in the six RBM20 mutation positive probands reported here except for the LMNA and LDB3 variants noted above.
Discussion
RBM20 has recently been established as a DCM causative gene by its strong genetic linkage in two extended FDC families and the mutations that were found in six additional families. 12 The six RBM20 mutations identified in our present report may also cause DCM for the following reasons: they were all predicted to substitute a highly conserved amino acid, five of them were located within predicted functional domains, two (R634Q and R636H) had been previously reported in association with DCM, 12 and none of the variants were present in normal controls. Three variants showed cosegregation with DCM in their respective families (pedigrees D, E, and F), strengthening evidence that these mutations were causative of DCM. Three variants were identified in pedigrees with no other affected subjects (pedigrees A and B) or no DNA available from other affected subjects (pedigree C), obviating our ability to ascertain if the variants segregated with DCM in family members. In one of these (pedigree A, V535I mutation), the proband also harbored a mutation in LDB3, 7 another gene reported in association with DCM, confounding the degree of certainty that the V535I was causative of disease. The mutation R716Q (family F) is unique and is located approximately 60 residues downstream of the RS‐rich region with conservation from chicken to human. The mutation was found in a large DCM family and segregated with DCM in multiple affected family members (except for one individual, noted above, either a phenocopy or a genocopy), suggesting that the R716Q likely contributed to the DCM pathogenesis in this family.
The mechanisms through which RBM20 mutations cause DCM remain to be elucidated. There are at least three recognizable motifs or domains in RBM20: an RRM‐1 encoded by exons 6 and 7; an RS‐rich region encoded by exon 9, and a U1 zinc f nger encoded by exon 14. 12 The functional significance of these domains can be readily seen through the strict conservation from zebraflsh to human ( Figure 2 ). The RRM‐1 domain is thought to bind the target transcript precursor and regulate splicing. 19 , 20 We speculate that the RBM20 mutation V535I residing in the highly conserved RRM‐1 region may interfere with its RNA‐binding capacity. Four RBM20 mutations (R634Q, R634C, R636C, and R636H) altered two highly conserved arginine residues in its RS‐rich region. Since the RS‐rich domain is predicted to be involved in protein–protein interaction, 19 , 20 those mutations may af ect the ability of RBM20 to interact with other spliceosome proteins, thus disrupting the normal RNA splicing process.
Myocardial DNA undergoes abundant alternative gene splicing events during transcription, and many cardiac genes are tightly regulated at the splicing level. 21 , 22 Some of these have direct relevance to cardiomyopathy and heart failure. For example, aberrant splicing of cardiac troponin T was linked to cardiomyopathy and heart failure. 23 , 24 RNA‐binding proteins including RS‐rich proteins (also known as SR proteins) act as master regulators of gene splicing, and thus, are generally required for cell metabolism and viability. 19 Recent studies, however, indicated that individual SR proteins may each play a def ned role in regulating the splicing of a small set of target genes in a highly tissue‐selective manner. 25 One of the few prototypical SR proteins originally identified to be an essential splicing factor was alternative splicing factor/splicing factor 2 (ASF/SF2). 26 Heart‐specific knockout of the ASF/SF2 dramatically altered the splicing of three critical cardiac genes 27 : cardiac troponin T(TNNT2), the Z‐line protein Cypher (also known as LIM domain‐binding protein 3, LDB3), and Ca2+/calmodulin‐dependent kinase IIδ (CamKII δ) while global gene expression remained unchanged. These splicing changes include TNNT2 switching to its fetal isoform by inclusion of exon 5 and LDB3 switching to its skeletal isoform with maintenance of its exon 4. Another critical and highly relevant alteration was the switch of CamKIIδ (nuclear isoform B and cytoplasmic isoform C) to a neuronal‐specific isoform δA through inclusion of exon 15 and 16. These molecular changes took place approximately 4 weeks before the onset of the DCM phenotype in the knockout mice, raising the possibility that the observed aberrant gene splicing may be the underlying pathogenic mechanism leading to the mouse DCM phenotype. 27 Based on these observations, it is possible that the pathways through which RBM20 mutations cause human DCM may be involved in regulation of a specific set of targeted downstream genes rather than a global disturbance of gene expression. The fact that RBM20 is preferentially expressed in the heart 12 makes such speculation more plausible. Further studies to identify RBM20 target genes are essential for understanding DCM pathogenesis and to explore possible therapy.
In conclusion, we have confirmed that RBM20 mutations may cause DCM that is frequently associated with severe heart failure, arrhythmia, and the need for cardiac transplantation.
Conflict of Interest
There is no conflict of interest of any kind and no relationships with industry.
Acknowledgments
We thank the many families and referring physicians for their participation in the FDC Research Project, without whom these studies would not have been possible. This work was supported by NIH awards RO1‐HL58626 (Dr. Hershberger).
This work was supported by NIH award RO1‐HL58626 (Dr. Hershberger)
References
- 1. Dec GW, Fuster V. Idiopathic dilated cardiomyopathy. N Engl J Med. 1994; 331(23): 1564–1575. [DOI] [PubMed] [Google Scholar]
- 2. Michels VV, Driscoll DJ, Miller FA, Olson TM, Atkinson EJ, Olswold CL, Schaid DJ. Progression of familial and non‐familial dilated cardiomyopathy: long term follow up. Heart. 2003; 89(7) 757–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Seidman J, Seidman C. The genetic basis for cardiomyopathy: from mutation Identification to mechanistic paradigms. Cell 2001; 104(4): 557–567. [DOI] [PubMed] [Google Scholar]
- 4. Burkett EL, Hershberger RE. Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol 2005; 45(7): 969–981. [DOI] [PubMed] [Google Scholar]
- 5. Hershberger RE, Cowan J, Morales A, Siegfried JD. Progress with genetic cardiomyopathies screening, counseling, and testing in dilated, hypertrophic, and arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Heart Fail. 2009; 2(3): 253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Karkkainen S, Peuhkurinen K. Genetics of dilated cardiomyopathy. Ann Med. 2007; 39(2) 91–107. [DOI] [PubMed] [Google Scholar]
- 7. Hershberger RE, Parks SB, Kushner JD, Li D, Ludwigsen S, Jakobs PM, Nauman D, Burgess D, Partain J, Litt M. Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin Transl Sci. 2008; 1(1): 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hershberger RE, Norton N, Morales A, Li D, Siegfried J, Gonzalez‐Quintana, J . Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1 And TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ Cardiovasc Genet. 2010; 3: 155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, Peterson A, Li D, Jakobs P, Litt M, Porter CB, Rahko PS, Hershberger RE. Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am Heart J. 2008; 156(1): 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li D, Parks S, Kushner J, Nauman D, Burgess D, Ludwigsen S, Partain J, Nixon RR, Allen CN, Irwin RP, Jakobs PM, Litt M, Hershberger RE. Mutations of presenilin genes in dilated cardiomyopathy and heart failure. Am J Hum Genet. 2006; 79(12): 1030–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schonberger J, Wang L, Shin JT, Kim SD, Depreux FF, Zhu H, Zon L, Pizard A, Kim JB, Macrae CA, Mungall AJ, Seidman JG, Seidman CE. Mutation in the transcriptional coactivator EYA4 causes dilated cardiomyopathy and sensorineural hearing loss. Nat Genet. 2005; 37(4): 418–422. [DOI] [PubMed] [Google Scholar]
- 12. Brauch KM, Karst ML, Herron KJ, De Andrade M, Pellikka PA, Rodeheffer RJ, Michels VV, Olson TM. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J Am Coll Cardiol. 2009; 54(10): 930–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lukong KE, Chang KW, Khandjian EW, Richard S. RNA‐binding proteins in human genetic disease. Trends Genet. 2008; 24(8): 416–425. [DOI] [PubMed] [Google Scholar]
- 14. Kushner JD, Nauman D, Burgess D, Ludwigsen S, Parks SB, Pantely G, Burkett E, Hershberger RE. Clinical characteristics of 304 kindreds evaluated for familial dilated cardiomyopathy. J Card Fail. 2006; 12(6): 422–429. [DOI] [PubMed] [Google Scholar]
- 15. Crispell KA, Wray A, Ni H, Nauman DJ, Hershberger RE. Clinical profiles of four large pedigrees with familial dilated cardiomyopathy: preliminary recommendations for clinical practice. J Am Coll Cardiol. 1999; 34(3): 837–847. [DOI] [PubMed] [Google Scholar]
- 16. Vasan R, Larson M, Levy D, Evans J, Benjamin E. Distribution and categorization of echocardiographic measurements in relation to reference limits. The Framingham heart study: formulation of a height‐ and sex‐specific classification and its prospective validation. Circulation. 1997; 96: 1863–1873. [DOI] [PubMed] [Google Scholar]
- 17. Crispell KA, Hanson EL, Coates K, Toy W, Hershberger RE. Periodic rescreening is indicated for family members at risk of developing familial dilated cardiomyopathy. J Am Coll Cardiol. 2002; 39(9): 1503–1507. [DOI] [PubMed] [Google Scholar]
- 18. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988; 16(3): 1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Long JC, Caceres JF. The SR protein family of splicing factors: master regulators of gene expression. Biochem J. 2009; 417(1): 15–27. [DOI] [PubMed] [Google Scholar]
- 20. Graveley BR. Sorting out the complexity of SR protein functions. RNA. 2000; 6(9): 1197– 1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hodges D, Bernstein SI. Genetic and biochemical analysis of alternative RNA splicing. Adv Genet. 1994; 31: 207–281. [DOI] [PubMed] [Google Scholar]
- 22. Siedner S, Kruger M, Schroeter M, Metzler D, Roell W, Fleischmann BK, Hescheler J, Pfitzer G, Stehle R. Developmental changes in contractility and sarcomeric proteins from the early embryonic to the adult stage in the mouse heart. J Physiol. 2003; 548(Pt 2): 493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Biesiadecki BJ, Elder BD, Yu ZB, Jin JP. Cardiac troponin T variants produced by aberrant splicing of multiple exons in animals with high instances of dilated cardiomyopathy. J Biol Chem. 2002; 277(52): 50275–50285. [DOI] [PubMed] [Google Scholar]
- 24. Philips A V, Timchenko L T, Cooper T A. Disruption of splicing regulated by a CUG‐binding protein in myotonic dystrophy. Science. 1998; 280(5364): 737–741. [DOI] [PubMed] [Google Scholar]
- 25. Ding JH, Xu X, Yang D, Chu PH, Dalton ND, Ye Z, Yeakley JM, Cheng H, Xiao RP, Ross J, Chen J, Fu XD. Dilated cardiomyopathy caused by tissue‐specific ablation of SC35 in the heart. EMBO J. 2004; 23(4): 885–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang J, Takagaki Y, Manley JL. Targeted disruption of an essential vertebrate gene: ASF/SF2 is required for cell viability. Genes Dev. 1996; 10(20): 2588–2599. [DOI] [PubMed] [Google Scholar]
- 27. Xu X, Yang D, Ding JH, Wang W, Chu PH, Dalton ND, Wang HY, Bermingham JR Jr, Ye Z, Liu F, Rosenfeld MG, Manley JL, Ross J Jr, Chen J, Xiao RP, Cheng H, Fu XD. ASF/SF2‐regulated CaMKIIdelta alternative splicing temporally reprograms excitation‐contraction coupling in cardiac muscle. Cell. 2005; 120(1): 59–72. [DOI] [PubMed] [Google Scholar]