

Abstract
Purpurogallin (PPG, 2) and poloxin (3) have been reported as inhibitors of the polo-box domain (PBD) of human polo-like kinase 1. However, our experimental results demonstrated that PPG, but not poloxin, binds to the phospho-binding pocket of the PBD, suggesting that their modes of PBD inhibition are distinct. Induced fit docking analyses led us to propose that PPG fills the SpT pocket via π−π stacking and hydrogen-bonding interactions, thus providing a rationale for designing novel PBD inhibitors. In contrast, poloxin may fill a different site present near the SpT pocket by forming a covalent bond with a nucleophilic Cys residue.
Keywords: Polo-like kinase 1, polo-box domain, induced fit docking, binding mode, cancer therapeutics
Polo-like kinase 1 (Plk1) belongs to a subfamily of Ser/Thr protein kinases, collectively termed polo-like kinases (Plks). A growing body of evidence suggests that Plk1 plays pivotal roles during multiple stages of M-phase progression and, as a result, in cell proliferation.1 Not surprisingly, Plk1 is overexpressed in a wide spectrum of cancers in humans.2 Furthermore, recent studies revealed that Plk1 is critically required for the viability of activated Ras- or p53 deletion-containing cancer cells.3,4 Thus, Plk1 is an attractive target for the development of anticancer therapeutics. Plk1 offers, within one molecule, two functionally different drug targets with distinct properties: the N-terminal catalytic domain (NCD) made of ∼250 amino acid residues and the C-terminal noncatalytic, but functionally essential, polo-box domain (PBD) consisting of two polo-box motifs (PB1 and PB2) with ∼80 residues in each motif.
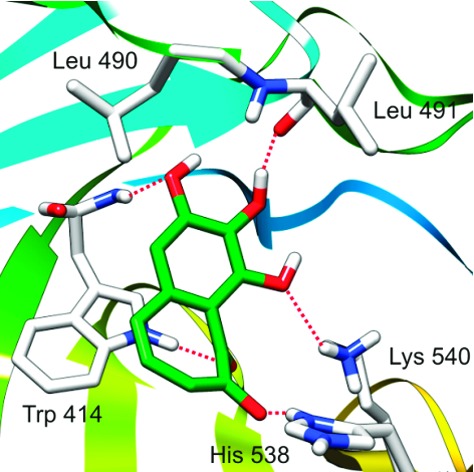
Over the years, efforts to generate Plk1-specific inhibitors by targeting the catalytic activity of Plk1 have encountered significant difficulties, mainly because of the high degree of structural similarities among the ATP-binding pockets of the protein kinase superfamily. Therefore, development of novel inhibitors that target the structurally unique PBD of Plk1 may serve as an alternative strategy.5−7 To date, at least seven X-ray crystal structures of the human Plk1 PBD have been resolved and reported by several groups.8−11 Results show that the PB1 and PB2 contain identical folds of β6α (a six-stranded antiparallel β-sheet and an α-helix) and form a heterodimeric phosphopeptide-binding module. Diverse phosphopeptides, including PLHSpT (compound 1; Figure 1) from the T78 motif of a centromere protein PBIP1,12 are shown to bind to the PBD in a cleft formed between the PB1 and the PB2 motifs by forming direct and bridged hydrogen bonds (Figure 1). The His 538 and Lys 540 residues from the PB2 are essential for electrostatic interactions with the negatively charged phosphate group of the phosphorylated Thr (pThr) residue, whereas the Trp 414 residue from the PB1 is central for the selection of the Ser residue at the pThr-1 position (−1 indicates the relative position of the Ser residue from the pThr residue) by engaging in hydrogen bonding and van der Waals interactions.8,9
Figure 1.
The binding mode of PLHSpT with the Plk1 PBD.
Besides various PBD-binding phosphopeptides, only three compounds, purpurogallin (PPG; compound 2), poloxin (compound 3), and thymoquinone (compound 4), have been reported as small drug-like Plk1 PBD inhibitors. PPG, identified from an in vitro high-throughput screen designed to isolate potential anti-PBD inhibitors, is a benzotropolone-containing natural compound derived from nutgall.6 PPG delocalizes Plk1 from specific subcellular structures and induces a spindle checkpoint-dependent mitotic arrest in HeLa cells,6 which would be expected if it disrupts the function of PBD in vivo. Interestingly, PPG-like compounds lacking the 4-hydroxyl group (see Figure 2 for atom numbering) fail to inhibit the Plk1 PBD, suggesting that this group is required for the observed inhibitory activity.6 Similarly, poloxin and thymoquinone, isolated from another high-throughput screen for PBD inhibitors, have been reported to inhibit the function of Plk1 PBD in vitro and interfere with the function of Plk1 PBD in vivo.7
Figure 2.
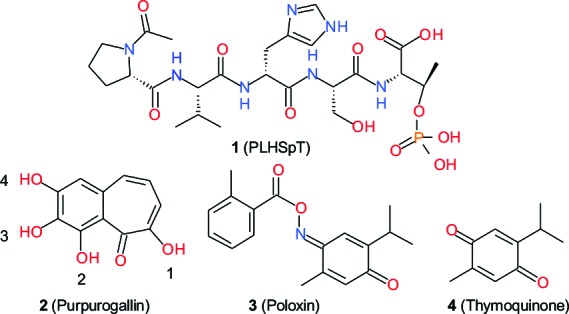
Structures of compounds 1−4.
To quantitatively determine the efficiency of PBD-binding inhibition by these compounds, an enzyme-linked immunosorbent assay (ELISA)-based inhibition assay that utilizes the specific interaction between the Plk1 PBD and a ligand from the p-T78 motif of PBIP112 was performed (see the Supporting Information for details). The addition of the PLHSpT peptide yielded a dose-dependent Plk1 PBD inhibition (Figure 3) with a Kd of 0.445 μM.11 Under the same conditions, PPG exhibited a moderate level of PBD inhibition with an estimated Kd of 2.7 μM (based on the observation that the inhibitory activity of PPG is ∼6 times lower than that of PLHSpT in Figure 3). Surprisingly, however, poloxin failed to display any significant level of PBD inhibition even at a concentration 1000 times higher than that of the p-T78 ligand. Relative activities of the compounds 1−3 in the inhibition of the Plk1 PBD are summarized in Table 1. Because the assay is specific for the presence of phospho-dependent PBD interaction,11 these results suggest that, unlike PPG, poloxin may not directly bind to the phospho-recognition module of the Plk1 PBD.
Figure 3.
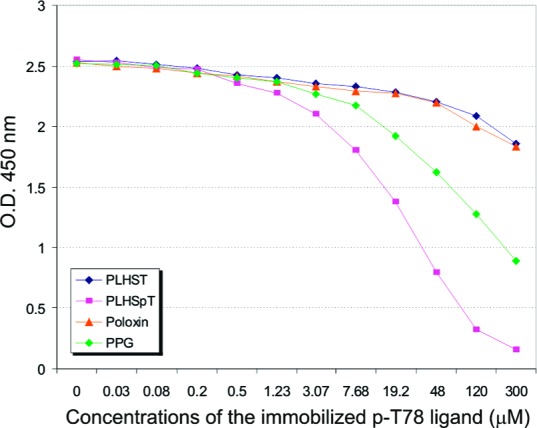
Inhibition of Plk1 PBD binding by PLHSpT, PLHST, PPG, and poloxin. O.D., optical density.
Table 1. Inhibition of Plk1 PBD by Compounds 1−4.
| compound | PBD inhibitory activitya | Kd (μM) |
|---|---|---|
| 1 | high | 0.445 |
| 2 | moderate | 2.7 (estimated) |
| 3 | no activity | |
| 4 | not determined |
Measured by an ELISA-based Plk1 PBD competition assay.
Better understanding of the binding mode of PPG to the PBD may allow new insights into the design and development of potent small-molecule inhibitors against the PBD. Traditional virtual docking methods assume a receptor to be rigid. However, this assumption often leads to misleading results and erroneous conclusions because, in reality, many proteins undergo side chain or backbone movements or both upon ligand binding. Although the overall structure of the PBD appears to be largely the same in the presence or absence of a binding peptide,11 the hinge-bending motions of the two polo-box motifs relative to each other suggest that the PBD binding site is somewhat flexible. Consistent with this view, close alignment of the crystal structures of 3HIK11 (complexed with PLHSpT) and 1Q4O8 (no ligand) revealed that some side chains clearly move in response to phosphopeptide binding (see Figure S1 of the Supporting Information). Thus, Schrödinger's Induced Fitting Docking (IFD) protocol13 was used to explore the binding modes of PPG to the PBD. Because PPG has one enolized and three phenolic hydroxyl groups, one needs to address the protonation state of all four hydroxyl groups under physiological conditions. To this end, ADME Boxes 4.914 and Marvin 5.1.4,15 two programs for predicting pKa values for pharmaceutical substances16 from Pharma Algorithms, Inc., and ChemAxon Ltd., respectively, were employed to calculate the pKa values of each hydroxyl group. Both methods yielded similar pKa values (Table 2): Hydroxyl groups 1 and 4 had calculated pKa values of ∼6.0 and ∼8.0, respectively, whereas the other two hydroxyl groups had higher values. These results suggest that, at pH 7.4, hydroxyl group 1 is likely to be deprotonated; hydroxyl group 4 might be deprotonated, while the other two might not. Figure 4 shows the microspecies distribution of PPG calculated by Marvin 5.1.4: At pH 7.4, 76.5, 15.0, and 3.0% of the PPG would be present as the microspecies 2-2, 2-3, and 2-1, respectively.
Table 2. Calculated pKa Values for Hydroxyl Groups in PPG.
| hydroxyl group | ADME boxes 4.9 | Marvin 5.1.4 |
|---|---|---|
| 1 | 6.3 | 5.98 |
| 2 | 15.8 | 10.11 |
| 3 | 12.0 | 13.84 |
| 4 | 8.1 | 7.99 |
Figure 4.
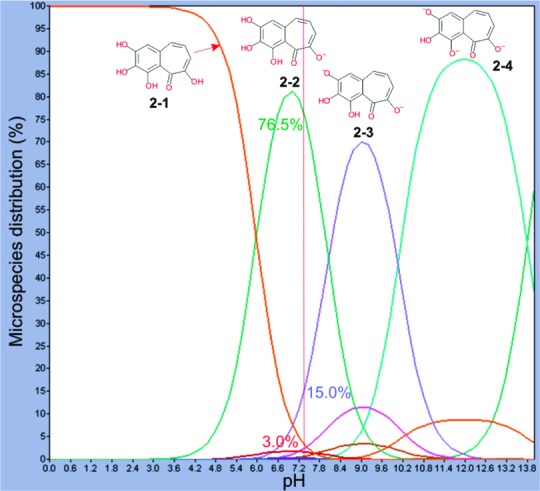
Microspecies distribution of PPG calculated by Marvin 5.1.4.
The predicted major microspecies at physiological conditions, 2-2, which has one negative charge, was docked into the active site of the PBD when examined by IFD. One of the docked conformations exhibiting an IFDScore of −8.47 kcal/mol is shown in Figure 5, which demonstrates that PPG can fill the SpT pocket of the PBD well by using almost every part of the molecule. The negatively charged enolized hydroxyl group of the bound PPG forms an electrostatic interaction with the positively charged His 538; the other three phenolic hydroxyl groups and the carbonyl group form four hydrogen bonds with the positively charged Lys 540, the backbone NH of Trp 414, the indole ring of Trp 414, and the backbone carbonyl of Leu 491; the phenyl ring and one double bond in the tropolone ring of PPG form π−π stacking interactions with the pyrrole ring and the phenyl ring (both embedded in the indole ring) of Trp 414, respectively. We hypothesize that microspecies 2-3 would have similar interactions with higher affinity, because the hydroxyl group 4 would be negatively charged; thus, the hydrogen bond formed between this group and the backbone NH of Trp 414 would be much stronger than the one formed between the undeprotonated hydroxyl group 4 and the backbone NH of Trp 414. Our additional IFD results support this assumption with a better IFDScore of −9.24 kcal/mol. The analogue of PPG lacking the hydroxyl group 4 was also used in the docking study. However, it did not form reasonable docking poses. In good agreement with the observations made by Watanabe et al.,6 this finding strongly suggests that the hydroxyl group 4 is critical for PBD inhibition.
Figure 5.
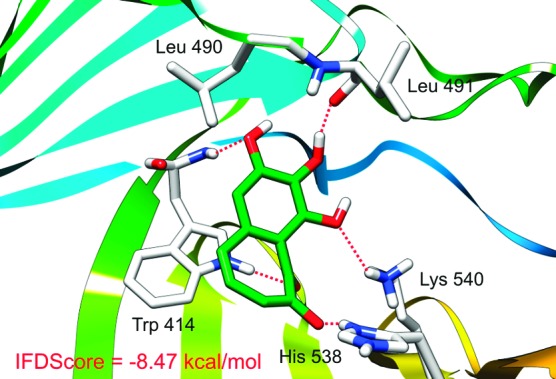
Proposed binding mode of PPG to the Plk1 PBD generated by Schrödinger's IFD protocol (see the text for details).
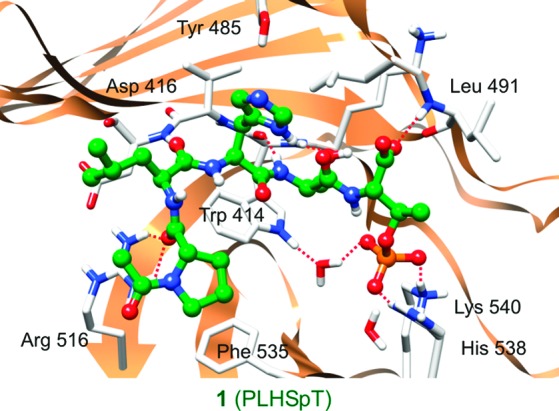
In a previous study, we showed that PLHSpT and LHSpT exhibit high and moderate levels of binding to the PBD, respectively, whereas both HSpT and SpT fail to display a detectable level of affinity.11 These observations suggest the importance of the N-terminal residues for the stability of the interaction between the phosphopeptides and the PBD. However, unlike PPG, PLHSpT failed to exhibit π−π stacking interaction with the Trp 414 residue (Table 3). Furthermore, the indispensability of the hydroxyl group 4 in PPG for the PBD binding also suggests that the hydrogen bond between this group and the backbone NH of Trp 414 is critical for the interaction. These findings suggest that exploitation of the interactions with Trp 414 is likely important to achieve a high affinity binding of small drug-like molecules to the PBD.
Table 3. Differences in the PBD Binding between PLHSpT and PPG.
| interaction | PLHSpT | PPG |
|---|---|---|
| electrostatica | His 538, Lys 540 | His 538 |
| hydrogen bonding | Trp 414 (2), Asp 416, Leu 491, Arg 516 | Trp 414 (2), Leu 491, Lys 540 |
| hydrophobic | Phe 535 | |
| π−π stacking | no | Trp 414 |
Electrostatic interaction can be considered as a strong hydrogen bond interaction.
Poloxin failed to dock into the phospho-binding pocket of the PBD using IFD, no matter whether a hydrogen bond to either one of the His 538 and Lys 540 residues crucial for phospho-dependent PBD binding was set as a constraint. This finding suggests that the SpT pocket of PBD is not the direct target of poloxin. This view is in good agreement with the results obtained with the PBD-binding inhibition assay shown in Figure 3.
Then, how can poloxin inhibit the PBD of Plk1 and interfere with the function of Plk1? It has been shown that inhibition of the kinase activity of Plk1 also delocalizes Plk1 by impairing the PBD-dependent subcellular localization through a self-priming and binding mechanism.17,18 We therefore examined whether or not poloxin influences the kinase activity of Plk1 by carrying out in vitro Plk1 kinase assays. However, under the conditions where a Plk1 NCD inhibitor, BI 253617 (see Figure S2 of the Supporting Information), potently inhibited Plk1, both PPG and poloxin failed to exhibit any detectable level of inhibitory activity against Plk1 (see Figure S3 of the Supporting Information).
We then considered two other possible mechanisms for poloxin's inhibitory activity against Plk1 PBD: (1) Poloxin may bind to another crucial site within the PBD, and/or (2) it may form a covalent bond with one of the nucleophilic amino acid side chains of Cys, Lys, Ser, or His residues of the PBD through its highly reactive α,β-unsaturated carbonyl group (a Michael acceptor).19 The latter property has been exploited in the development of irreversible drugs for the treatment of cancer.20
The program SiteMap 2.321 was used to locate other possible active sites of PBD. Besides the site that accommodates the phosphopeptide, two additional pockets (pockets #1 and #2) were found (Table 4). It is interesting to note that a strongly nucleophilic Cys residue (Cys 544 in pocket #1 and Cys 573 in pocket #2; see Table 4 for details) is located on the rim of each of these pockets. This finding led us to speculate that Michael acceptors such as poloxin and thymoquinone could undergo covalent modifications with the Cys residue of the Plk1 PBD and then fill the pocket to inhibit the latter. Consistent with this observation, pocket #1 is a deep slot (Figure 6A) sufficient to allow either poloxin or thymoquinone to be docked into the pocket by IFD easily. In contrast, pocket #2 is shallow and is composed of charged and polar residues; as a consequence, it may be unable to accommodate the overall hydrophobic molecules such as poloxin and thymoquinone to fill this pocket.
Table 4. Two Novel Plk1 PBD Pockets Predicted by SiteMap 2.3.
| residues forming the pocket | |
|---|---|
| #1 | Pro 407, Ile 408, Phe 409, Arg 500, Ala 506, Leu 508, Pro 509, Asn 527, Ser 529, Cys 544, Pro 545, Leu 546, Met 547 |
| #2 | Gln 397, Tyr 552, Glu 555, Lys 556, Asp 558, Arg 560, Tyr 562, Cys 573, Glu 575, Arg 579 |
Figure 6.
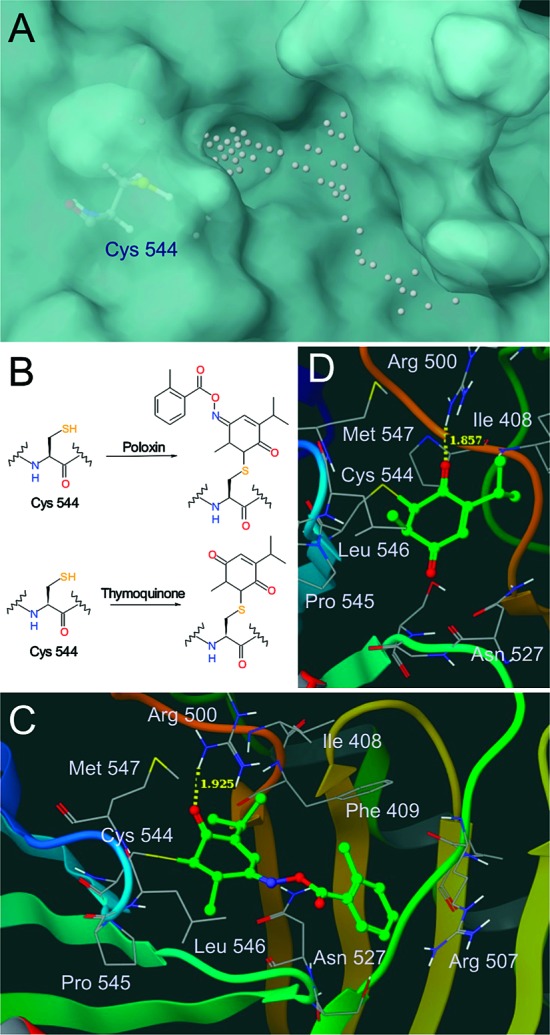
Binding nature of poloxin and thymoquinone to the Plk1 PBD. (A) Pocket #1 identified by SiteMap 2.3. (B) Proposed mechanisms of PBD inhibition by poloxin and thymoquinone. (C and D) Proposed binding modes of poloxin (C) and thymoquinone (D).
To further explore the binding nature of poloxin and thymoquinone to the pocket #1, we then manually connected each of these compounds to the thiol group of Cys 544 on the rim of the pocket. The nucleophilic reaction between the Cys 544 residue and poloxin or thymoquinone may occur in several different ways. We propose that a reaction with the nucleophilic cysteine side chain could occur at the least sterically hindered sp2 carbon atom, yielding the covalent adducts shown in Figure 6B. The complexes were minimized in aqueous solution using MacroModel 9.7.22 These analyses suggested that poloxin and thymoquinone interact with PBD by forming a hydrogen bond with Arg 500 and hydrophobic interactions with Pro 545 and Leu 546 (Figure 6C,D). In addition, the benzyl ring of poloxin could be sandwiched between the carbon chains of Asn 527 and Arg 507. The mechanism involving covalent bond formation between poloxin and the PBD is consistent with the time-dependent inhibition observed by Reindle et al.7
On the basis of the results obtained from the PBD-binding inhibition assay and the IFD, we therefore propose that poloxin inhibits Plk1 PBD through a mechanism that is distinct from binding to the SpT pocket of the PBD. This view may help explain why poloxin, which failed to exhibit Plk1 PBD inhibition activity in our ELISA-based competition assay (Figure 3), inhibits the Plk1 PBD in a different in vitro binding assay and induces Plk1 delocalization in vivo.7 On the other hand, PPG can easily fill the SpT pocket of the PBD by engaging in a π−π interaction with the indole ring of Trp 414, an electrostatic interaction with His 538, and four hydrogen bondings with Lys 540, Trp 414, and Leu 491. This π−π interaction, which was not observed in the interactions between the phosphopeptides and the PBD, could be important for efficient binding of small drug-like molecules to the PBD.
In this study, we have proposed a binding model for PPG, a drug-like small molecule that inhibits the PBD of Plk1. Our model postulates several crucial interactions with the PBD. In addition to the interactions with the positively charged His 538 and Lys 540 residues in the PB2 motif (preferably electrostatic in nature), several distinct interactions with Trp 414 in the PB1 motif appear to be critical. These include π−π interactions with the indole ring of Trp 414, a hydrogen-bonding interaction with its backbone NH, and another direct or bridged hydrogen-bonding interaction with its pyrrole ring NH. The importance of the His 538, Lys 540, and Trp 414 residues in PBD binding is corroborated by cell-based studies where mutations in these residues greatly impair PBD-dependent subcellular localizations.9,10,23 It should be noted, however, that all three residues (His 538, Lys 540, and Trp 414) critical for the PBD binding are conserved among the PBDs of the three closely related Plk subfamily members (i.e., Plk1, Plk2, and Plk3). Thus, additional interactions with other residues specific to each Plk are likely crucial to achieve the specificity of PBD-dependent interaction. The PBD binding models discussed here may serve as starting points in the generation of pharmacophores that are vitally required for the development of small drug-like molecules against Plk1 PBD.
Acknowledgments
We thank Boehringer Ingelheim (Vienna, Austria) and Wolfgang Reindl (Max Planck Institute of Biochemistry, Martinsried, Germany) for providing BI 2536 and poloxin, respectively. We thank Pharma Algorithms, Inc., and ChemAxon, Ltd., for permitting us access to their programs.
Supporting Information Available
Detailed information on the experimental procedures for molecular modeling, PBD-binding inhibition assay, and Plk1 kinase assay is available free of charge via the Internet at http://pubs.acs.org.
The first two authors equally contributed to this work.
This work was supported in part by the Intramural Research Program of the National Cancer Institute (M.C.N. and K.S.L.) and the Korea Basic Science Institute's International Joint Research Program grant F30601 (J.K.B.).
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Archambault V.; Glover D. M. Polo-like kinases: Conservation and divergence in their functions and regulation. Nat. Rev. Mol. Cell. Biol. 2009, 10, 265–275. [DOI] [PubMed] [Google Scholar]
- Strebhardt K.; Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat. Rev. Cancer 2006, 6, 321–330. [DOI] [PubMed] [Google Scholar]
- Luo J.; Emanuele M. J.; Li D.; Creighton C. J.; Schlabach M. R.; Westbrook T. F.; Wong K. K.; Elledge S. J. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 2009, 137, 835–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur S.; Pagliarini R.; Bunz F.; Rago C.; Diaz L. A. Jr.; Kinzler K. W.; Vogelstein B.; Papadopoulos N. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc. Natl. Acad. Sci. 2009, 106, 3964–3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. S.; Idle J. R. Pinning down the polo-box domain. Chem. Biol. 2008, 15, 415–416. [DOI] [PubMed] [Google Scholar]
- Watanabe N.; Sekine T.; Takagi M.; Iwasaki J.; Imamoto N.; Kawasaki H.; Osada H. Deficiency in chromosome congression by the inhibition of Plk1 polo box domain-dependent recognition. J. Biol. Chem. 2009, 284, 2344–2353. [DOI] [PubMed] [Google Scholar]
- Reindl W.; Yuan J.; Kramer A.; Strebhardt K.; Berg T. Inhibition of polo-like kinase 1 by blocking polo-box domain-dependent protein-protein interactions. Chem. Biol. 2008, 15, 459–466. [DOI] [PubMed] [Google Scholar]
- Cheng K. Y.; Lowe E. D.; Sinclair J.; Nigg E. A.; Johnson L. N. The crystal structure of the human polo-like kinase-1 polo box domain and its phospho-peptide complex. EMBO J. 2003, 22, 5757–5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia A. E.; Rellos P.; Haire L. F.; Chao J. W.; Ivins F. J.; Hoepker K.; Mohammad D.; Cantley L. C.; Smerdon S. J.; Yaffe M. B. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell 2003, 115, 83–95. [DOI] [PubMed] [Google Scholar]
- Garcia-Alvarez B.; de Carcer G.; Ibanez S.; Bragado-Nilsson E.; Montoya G. Molecular and structural basis of polo-like kinase 1 substrate recognition: Implications in centrosomal localization. Proc. Natl. Acad. Sci. 2007, 104, 3107–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun S. M.; Moulaei T.; Lim D.; Bang J. K.; Park J. E.; Shenoy S. R.; Liu F.; Kang Y. H.; Liao C.; Soung N. K.; Lee S.; Yoon D. Y.; Lim Y.; Lee D. H.; Otaka A.; Appella E.; McMahon J. B.; Nicklaus M. C.; Burke T. R. Jr.; Yaffe M. B.; Wlodawer A.; Lee K. S. Structural and functional analyses of minimal phosphopeptides targeting the polo-box domain of polo-like kinase 1. Nat. Struct. Mol. Biol. 2009, 16, 876–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y. H.; Park J. E.; Yu L. R.; Soung N. K.; Yun S. M.; Bang J. K.; Seong Y. S.; Yu H.; Garfield S.; Veenstra T. D.; Lee K. S. Self-regulated Plk1 recruitment to kinetochores by the Plk1-PBIP1 interaction is critical for proper chromosome segregation. Mol. Cell 2006, 24, 409–422. [DOI] [PubMed] [Google Scholar]
- Schrödinger Suite 2009 Induced Fit Docking Protocol; Schrödinger, LLC: New York, NY, 2009.; Glide, version 5.5; Schrödinger, LLC: New York, NY, 2009.; Prime, version 2.1; Schrödinger, LLC: New York, NY, 2009.
- ADME/Tox WEB. http://pharma-algorithms.com/webboxes/ (accessed January 19, 2010).
- pKa Plugin ionization equilibrium partial charge distribution. http://www.chemaxon.com/product/pka.html (accessed January 19, 2010).
- Liao C.; Nicklaus M. C. Comparison of nine programs predicting pKa values of pharmaceutical substances. J. Chem. Inf. Model. 2009, 49, 2801–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenart P.; Petronczki M.; Steegmaier M.; Di Fiore B.; Lipp J. J.; Hoffmann M.; Rettig W. J.; Kraut N.; Peters J. M. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr. Biol. 2007, 17, 304–315. [DOI] [PubMed] [Google Scholar]
- Lee K. S.; Park J. E.; Kang Y. H.; Zimmerman W.; Soung N. K.; Seong Y. S.; Kwak S. J.; Erikson R. L. Mechanisms of mammalian polo-like kinase 1 (Plk1) localization: Self- versus non-self-priming. Cell Cycle 2008, 7, 141–145. [DOI] [PubMed] [Google Scholar]
- Chalker J. M.; Bernardes G. J.; Lin Y. A.; Davis B. G. Chemical modification of proteins at cysteine: opportunities in chemistry and biology. Chem. Asian J. 2009, 4, 630–640. [DOI] [PubMed] [Google Scholar]
- Wissner A.; Mansour T. S. The development of HKI-272 and related compounds for the treatment of cancer. Arch. Pharm. (Weinheim) 2008, 341, 465–477. [DOI] [PubMed] [Google Scholar]
- SiteMap, version 2.3; Schrödinger, LLC: New York, NY, 2009. [Google Scholar]
- Macromodel 9.7; Schrödinger, LLC: New York, NY, 2009. [Google Scholar]
- Hanisch A.; Wehner A.; Nigg E. A.; Sillje H. H. Different Plk1 functions show distinct dependencies on Polo-Box domain-mediated targeting. Mol. Biol. Cell 2006, 17, 448–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.