

Abstract
Feline immunodeficiency virus (FIV) OrfA is an accessory protein that is critical for productive viral replication and infection in T cells. Here, we show that OrfA acts to markedly reduce cell surface expression of the FIV primary binding receptor. Downregulation does not occur at the transcriptional or translational level in that the amounts of CD134 mRNA and protein in total cell lysates are not altered between parental 104-C1 T cells and the same cell line stably expressing OrfA (104-C1-OrfA). Analysis by confocal microscopy revealed significant accumulation of CD134 in the Golgi apparatus of 104-C1 cells expressing OrfA. OrfA does not cause a generalized disruption of membrane trafficking in that surface expression of CD9 is unaffected by OrfA overexpression. Consistent with the above observations, OrfA-negative FIV-34TF10 productively infects CrFK (CD134-negative) and 104-C1-OrfA (CD134 downregulated by OrfA) cells but fails to productively infect either 104-C1 (CD134-positive) cells or GFox (CrFK cells overexpressing CD134) cells. FIV-34TF10 in which the OrfA reading frame is open (OrfArep) productively infects CrFK, GFox, 104-C1, and 104-C1-OrfA cells. We hypothesize that reduced surface expression of the receptor, a hallmark of retrovirus infections, may facilitate an increase in virus release from the infected cell by minimizing receptor interactions with budding virus particles.
Feline immunodeficiency virus (FIV), a member of the Lentivirus genus of retroviruses, induces a disease in cats similar to human AIDS, characterized by a progressive depletion of CD4+ T lymphocytes and immunologic decompensation in the domestic cat (27). Although evolutionarily diverse, FIV resembles human immunodeficiency virus (HIV) in many respects, including the general structure of the lentivirus genome, the target cells infected in vivo, and the course of virus infection and disease state. Accessory and regulatory gene products encoded by both viruses aid in facilitating infection in the face of host cell defenses (3, 11, 18, 19, 23). FIV encodes a protein termed OrfA that has been implicated in a number of actions that positively influence virus infection. OrfA is a 77-amino-acid protein with a molecular mass of approximately 8 kDa, encoded by a short open reading frame between vif and env. Early studies suggested that OrfA was a transactivator, with data indicating a net increase in translation of gene products whose transcription is driven by the FIV long terminal repeat (LTR) (8, 34, 37). However, increases in transcription/translation were small (3- to 5-fold), and later studies indicated that the mechanism was distinct from other transactivators (2). Other studies failed to show an increase in LTR-driven transcripts, although OrfA can localize to the nucleus using a nuclear localization signal (NLS) and may induce G2 cell cycle arrest (14). Additional studies suggested that OrfA affects a later step in virus replication, such as virus particle formation and release from the cell, or possibly early steps, such as virus binding and entry (13). These observations suggest that OrfA might exhibit functions homologous to those ascribed to HIV Vpr, Vpu, or Nef (22) and might thus represent a multifunctional accessory protein.
FIV and HIV share similar modes of receptor utilization. Feline CD134 acts as the primary binding receptor for FIV entry into host cells, a function that parallels use of CD4 as a primary receptor in HIV infection (33). Binding of CD134 alters the conformation of FIV envelope protein SU and promotes high-affinity binding to the chemokine receptor CXCR4, the entry receptor for domestic cat FIVs (5, 30, 38). In the scenario of HIV infection, the virus attaches via a high-affinity interaction with CD4, resulting in the conformational change in the envelope glycoprotein gp120 and exposure of the binding sites for the chemokine receptor CXCR4 or CCR5 (17). Besides making full use of host cell receptors for viral invasion, HIV also has evolved mechanisms to limit the expression of CD4 on the surfaces of or within cells to facilitate viron release and to avoid superinfection. Extensive studies have been conducted to demonstrate that HIV accessory proteins Vpu and Nef are involved in this process and utilize two distinct pathways for CD4 downregulation on T cells and macrophages (10, 12, 19, 24). To date, no evidence has been presented to show the similar virus host interaction in FIV.
In the present report, we performed extensive analyses of receptor expression and virus infection on the CD134+ continuous T-cell line 104-C1 and 104-C1 cells engineered to express OrfA (104-C1-OrfA) in attempts to further define the function(s) of FIV OrfA. The results showed that OrfA expression is associated with a marked reduction in CD134 expression on the cell surface and is essential in generating a productive viral infection.
MATERIALS AND METHODS
Cell lines and viruses.
The continuous T-cell line 104-C1 was isolated from feline peripheral blood mononuclear cells (PBMC) by limiting dilution. 104-C1 cells were maintained in RPMI 1640 (Gibco, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Innovative Research), 2 mM l-glutamine (Sigma), 1 mM sodium pyruvate (Sigma), 10 mM HEPES (Sigma), 1× minimal essential medium (MEM)-vitamins (Mediatech), 1× nonessential amino acids (Sigma), 1× β-mercaptoethanol (Gibco-BRL), 2.5 μg/ml concanavalin A (ConA; Sigma), 50 U/ml of human recombinant interleukin 2 (IL-2; Hoffman-La Roche), and 50 μg/ml gentamicin (Gemini Bioproducts). 104-C1-OrfA was a construction of 104-C1 cells expressing FIV-PPR OrfA and a green fluorescent protein (GFP), which has been described previously (2). Crandell feline kidney (CrFK) cells were obtained from the American Type Culture Collection. GFox cells are CrFK cells stably transfected with the feline primary binding receptor CD134 (6). The IL-2-independent feline lymphoma cell line 3201 was obtained from W. Hardy (Sloan-Kettering Memorial Hospital, New York, NY). The FIV strains used in this study were FIV-34TF10, a molecular clone derived from the Petaluma isolate (35), and FIV-PPR, a molecular clone derived from San Diego isolates (29). OrfArep is a molecular clone of 34TF10 in which the premature stop codon in the OrfA gene has been changed to a tryptophan codon through site-directed mutagenesis (37).
Infection and RT activity assay.
Virus with reverse transcriptase (RT) values above 100K cpm were used in all infection assays. For 104-C1 or 104-C1-OrfA cells, 105 cells were seeded in a 96-well microtiter plate and the cells were infected with either 100 μl (FIV OrfArep) or 50 μl (FIV-PPR) virus stock and then incubated at 37 οC in 5% CO2. For CrFK or GFox cells, 2 × 104 cells were seeded in a 12-well plate and 100 μl of virus was used to infect the cells. Virus production was measured over time using a micro-RT assay. To do the RT activity assay, 50 μl of cell-free supernatant was incubated for 10 min at room temperature with 10 μl of lysis buffer (0.75 M KCl, 20 mM dithiothreitol, 0.5% Triton X-100). Forty microliters of enzyme cocktail containing 125 mM Tris-HCl (pH 8.1), 12.5 mM MgCl2, 1.25 μg poly(rA)-poly(dT)12-18 (Amersham Biosciences, Piscataway, NJ), and 1.25 μCi of [3H]dTTP (DuPont, Boston, MA) was added to the sample, followed by 2 h of incubation at 37°C. RT activity was measured as previously described (9).
Fluorescence-activated cell sorting (FACS) analysis.
The evaluation of CD134 or CD9 surface expression on 104-C1 and 104-C1-OrfA cells was done by flow cytometry analysis. Cells (3 × 105) were incubated with rabbit anti-cat/feline CD134 (1:100) or mouse anti-cat/feline CD9 (1:100) for 45 min at room temperature on a rocker tray. The cells were then washed twice in 1× phosphate-buffered saline (PBS)-2% FBS to remove unbound antibody. A 1:600 dilution of phycoerythrin-conjugated goat anti-rabbit IgG or a 1:1,000 dilution of phycoerythrin-conjugated goat F(ab′)2 anti-mouse IgG was then incubated with the cells for 45 min, at room temperature, in the dark. The cells were then again washed twice and analyzed by flow cytometry with a FACSCanto flow cytometer (Becton Dickinson) using the FLOWJO (Tree Star) software program. We also evaluated SU binding to CD134 by incubating FIV-PPR SU-Fc adhesion (1 μg/ml) (7) with cells, followed by detection with 1:1,000 phycoerythrin-conjugated goat anti-human IgG1 Fc antibody (MP Biomedicals, Aurora, OH).
siRNA knockdown assays.
Small interfering RNA (siRNA) targeting orfA (Ambion, Inc., Foster, CA) in 104-C1-OrfA cells was delivered through electroporation. Cells were grown into log phase, and 4 × 105 cells were suspended in 200 μl 1× Dulbecco's phosphate-buffered saline (DPBS) and transferred into a 4-mm electroporation cuvette (Bio-Rad Laboratories, Inc., Hercules, CA). Next, 4 μl siRNA (10 μM) was added into the cell suspension to make a final concentration of 200 nM siRNA. Cells were electroporated using optimal conditions of 420 V and 25-μF capacity, followed by 10 min of incubation at 37°C. After incubation, cells were transferred into 48-well plates in 1 ml fresh medium. The samples were then incubated at 37°C in a humidified 5% CO2 incubator for 72 h before analysis. Two sets of siRNA (Applied Biosystems, Foster City, CA) targeting different regions of OrfA mRNA were tested: siRNA_OrfA_1 (positions 83 to 105 [sense, 5′ UUAGAGAGGGAUAAAUUGATT 3′, and antisense, 5′ UCAAUUUAUCCCUCUCUAATT 3′]) and siRNA_OrfA_2 (positions 74 to 96 [sense, 5′ GCACAUCAAUUAGAGAGGGTT 3′, and antisense, 5′ CCCUCUCUAAUUGAUGUGCTT 3′]).
Western blots.
104-C1 and 104-C1-OrfA total cell lysates and plasma membrane protein were prepared by following the instructions for a membrane protein extraction kit (BioVision, Mountain View, CA). Briefly, about 5 × 107 cells were pelleted and washed once with 1 ml of ice-cold PBS. Cells were then resuspended in 1 ml of homogenization buffer and homogenized on ice for 30 times. The homogenate was then centrifuged at 700 × g for 10 min at 4°C, and the supernatant containing the total cell lysate proteins were collected. To further extract plasma membrane protein, the supernatant obtained as described above was centrifuged at 10,000 × g for 30 min at 4°C and the pellet containing the total cellular membrane protein (proteins from both a plasma membrane and a cellular organelle membrane) was collected. Plasma membrane protein was then extracted using the upper- and lower-phase solutions according to instructions provided by the manufacturer. Next, the protein concentration was measured using a detergent-compatible protein assay protocol (Bio-Rad Laboratories, Hercules, CA). Samples of total cellular protein (7 μg) or plasma membrane protein (2 μg) were subjected to 10 to 20% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and separated protein bands were transferred onto a nitrocellulose membrane. Rabbit antisera against feline CD134 or a mouse monoclonal antibody against feline CD9 (VPG16, a gift from Brian Willett) was diluted 1:100 in blocking buffer (5% milk in PBS), and incubation was performed overnight at 4°C with agitation. After the primary antibody incubation, the membrane was washed three times with 1× PBS plus 0.05% Tween 20, followed by three washes with 0.5 M lithium chloride plus 1% NP-40, and then incubated with goat anti-rabbit or goat anti-mouse horseradish peroxidase-conjugated secondary antibodies at a 1:10,000 dilution for 2 h at room temperature. For protein band detection, membranes were treated with SuperSignal West Dura enhanced chemiluminescent substrate (Pierce Biotechnology, Inc.) and then exposed to X-ray film (Genesee Scientific Corp., San Diego, CA). The signals were quantified with a Molecular Dynamics densitometer and Image Quant version 1.2 (Molecular Dynamics, Sunnyvale, CA).
Quantitative real-time PCR.
Cellular RNA was extracted using an Aurum total RNA minikit (Bio-Rad Laboratories, Hercules, CA) and quantified using a NanoDrop spectrophotometer (NanoDrop Technologies, Inc., Wilmington, DE). The real-time reverse transcriptase PCR (RT-PCR) was performed by using Bio-Rad iScript one-step RT-PCR technology, and the PCR was run on an iCycler (Bio-Rad Laboratories, Hercules, CA). Each reaction mixture contained 12.5 μl of commercial 2× RT-PCR mix, 200 nM (each) forward and reverse primer, 200 nM 6-carboxyfluorescein (FAM)-labeled TaqMan probe, 1 μl of iScript reverse transcriptase for one-step RT-PCR, and ∼0.1 to ∼0.5 μg of RNA template. The PCR was performed using the following conditions: 10 min at 50°C and 5 min at 95°C, followed by 40 cycles of 10 s at 95°C and 30 s at 53°C. The mRNA quantity from each sample was calculated by using a standard curve constructed by analyzing a serially diluted (range, 102 to 1010 copies/μl) plasmid DNA, which contained a cloned copy of feline CD134 or FIV orfA gene.
The primers and probes (IDT Integrated DNA Technologies, San Diego, CA) used to detect CD134 and FIV OrfA mRNAs were as follows (5′ to 3′): fCD134-f, CCGTGAACTACGAGCCTTGC; fCD134-r, GTGGGTGTGCATCTCTGCTT; fCD134 dual probe, 56-FAM/AGTGCAACCAGAGAAGTGGGAGCGAGC/3BHQ_1; OrfA_F, TGAAAGATTAGACAAGGAAG; OrfA_R, TGCAGAAGTCTAATCAATTT; and OrfA dual probe, 56-FAM/CATCAGGATATTTGTATTAGCACATCA/3BHQ_1.
Primers and probes were designed using Primer3 (32).
Virus entry assay.
The assay for FIV-PPR or FIV-34TF10 Env-mediated virus entry was conducted by following a protocol previously described (5). Briefly, cytomegalovirus (CMV)-FIV hybrid vector (pCFIV) pseudotyped with the FIV-PPR or FIV-34TF10 Env gene was cotransfected with a β-galactosidase (β-Gal) transgene, pTFIVCβ (16), in 293T cells. Three days after transfection, viral supernatants were collected and reverse transcriptase activity was adjusted to 50,000 cpm/ml. One milliliter was used to transduce 2 × 105 104-C1 or 104-C1-OrfA cells in triplicate, and β-Gal activity was measured 48 h later with the TropixGalacto-Star chemiluminescent reporter gene assay (Applied Biosystems, Foster City, CA) according to the manufacturer's guidelines. For GFox and CrFK cells, 2 × 104 cells were used in the transduction in accordance with the same procedure.
Confocal microscopy and image analysis.
Immunofluorescence staining was performed on permeabilized 104-C1 and 104-C1-OrfA cells on eight-well chamber slides (Nalge Nunc International, Naperville, IL). CD134 receptors were labeled with 1:50 rabbit anti-cat CD134 and 1:100 phycoerythrin (red)-conjugated goat anti-rabbit IgG. Golgi complexes were stained with 1:200 GM130 antibody (Novus Biologicals, LLC, Littleton, CO) plus goat anti-rabbit IgG Alexa Fluor 647 (far red), and the cell nuclei were stained with DAPI (4′, 6′-diamidino-2-phenylindole) (blue) (Invitrogen Corporation, Carlsbad, CA). Confocal images were captured using a Zeiss 710 laser confocal scanning microscope (LCSM) running the Zen 2009 software program (Carl Zeiss, Inc., Thornwood, NY). Eight-bit optical image sections (0.5-μm interval step slices) were scanned at 512 by 512 and then were imported for analysis with Image Pro Plus (Media Cybernetics, Inc., Bethesda MD), Image J (NIH, Bethesda, MD), and LSM Examiner (Carl Zeiss, Inc., Thornwood, NY) software for quantitative analysis. Quantitative colocalization between two fluorescently labeled reagents was quantified by obtaining the threshold range of real over background signal and then using the average real threshold range to calculate the correlation coefficients (M values) for at least 60 cells in three separate experiments. Confocal fluorescence image composites all have an additional image panel representing only colocalized zones between two fluorophores.
RESULTS
OrfA is essential for infection of CD134+ feline cell lines.
The OrfA-negative FIV-34TF10 isolate and a version where the stop codon in the orfA gene has been changed to tryptophan (OrfArep) (37) were used to infect Crandell feline kidney (CrFK) cells, GFox cells, and the primary feline T-cell line 104-C1 (Fig. 1). Unlike 104-C1 and GFox cells, CrFK cells lack the binding receptor CD134 and express only the entry receptor CXCR4 (7). FIV-34TF10 is a laboratory-adapted, CD134-independent isolate and so can productively infect CrFK, whereas CD134-dependent laboratory strains cannot (25, 29). Both FIV-34TF10 and OrfArep productively infected CrFK (Fig. 1A). In contrast, no RT activity could be detected when 34TF10 was used to infect CD134+ 104-C1 (Fig. 1B) or GFox (Fig. 1C) cells, whereas OrfArep was able to generate a fast and productive infection in the same cell lines. Thus, OrfA is an essential factor for FIV infection in thymocytes (40), in which CD134 is involved as a binding receptor.
FIG. 1.

CrFK, GFox, and 104-C1 cell infection assay using FIV-34TF10 and OrfArep strains. Virus growth in cells was evaluated by a reverse transcriptase activity assay over time. (A) Both 34TF10 and OrfArep strains can productively infect CrFK cells, and the RT activity level is significantly above that of the cell-only control at 14 days postinfection. (B and C) 104-C1 and GFox cells can be productively infected with the OrfArep strain but not with the 34TF10 strain.
FACS analysis revealed CD134 expression as a function of OrfA.
To further investigate a possible interaction between FIV OrfA and CD134 receptors, we ran FACS assays to evaluate CD134 expression levels on the surfaces of 104-C1 and 104-C1-OrfA cells. 104-C1-OrfA is a stable cell line generated in a previous study (2). This cell line can efficiently express orfA and a downstream gfp gene from an integrated retro-vector. Surprisingly, our data showed a significant difference in CD134 surface expression between the two cell lines, with 104-C1 cells expressing about 10 times more CD134 than 104-C1 expressing OrfA (Fig. 2A, left panel). To further assess the amounts of CD134 expression on both cells, we also used an Fc-labeled FIV SU protein to bind CD134 and then used a fluorescence-conjugated goat anti-human IgG1 Fc to detect the Fc-SU-CD134 complexes on the cell surface. The results correlated with the previous assay in showing that the level of SU binding to 104-C1 via surface-expressed CD134 was substantially greater than that of binding to 104-C1-OrfA cells (Fig. 2A, middle panel). In addition, unlike for CD134, 104-C1 CD9 surface molecules were not downregulated in the presence of OrfA, indicating the specific regulatory effect of OrfA on CD134 (Fig. 2A, right panel). Next, we examined CD134 expression on infected 104-C1 cells (Fig. 2B). Both FIV-PPR and FIV-34TF10/OrfArep were used to infect 104-C1 cells. It has been reported that CD134 and CXCR4 are downregulated following infection by FIV of various origins, including FIV-PPR (39). Consistent with previous studies, CD134 expression on 104-C1 cells was reduced 2-fold 2 days after infection with FIV-PPR (Fig. 2B, left panel). Furthermore, CD134 expression on FIV-34TF10/OrfArep-infected 104-C1 cells was reduced 4- to 5-fold (Fig. 2B, right panel) compared to the level for the uninfected cell group or cells infected with OrfA-negative 34TF10.
FIG. 2.

FACS assays for detection of cell surface receptors. (A) Detection of CD134 and CD9 receptors on 104-C1 and 104-C1-OrfA cells. (Left) Detection of CD134 using rabbit anti-CD134 antibody. Red, background control; blue, 104-C1-OrfA cells; green, 104-C1 cells. (Middle) Detection of CD134 using SU-Fc adhesion. Red, background control; blue, 104-C1-OrfA cells; green, 104-C1 cells. (Right) Detection of CD9 using mouse anti-CD9 antibody. Red, background control; blue, 104-C1-OrfA cells; green, 104-C1 cells. (B) FACS analysis of infected 104-C1 cells using rabbit anti-CD134 antibody. (Left) Detection of CD134 on FIV-PPR-infected 104-C1 cells. Red, background control; blue, FIV-PPR-infected 104-C1 cells; green, uninfected 104-C1cells. (Right) Detection of CD134 on 34TF10-OrfArep-infected 104-C1 cells. Red, background control; blue, OrfArep-infected 104-C1 cells; green, uninfected 104-C1 cells.
CD134 surface expression increases when OrfA is knocked down by siRNA.
To further confirm our observation that OrfA reduces the surface expression of CD134, we used siRNA to knock down OrfA in the 104-C1-OrfA cell line and assess the subsequent changes in CD134 expression using FACS analysis. To best optimize siRNA assays, two sets of siRNA duplexes targeting different regions of OrfA mRNA were pooled and transfected into cells by electroporation. Since gfp was placed downstream of orfA on the integrated retro-vector (2), knockdown of OrfA also affected GFP expression. Using quantitative real-time PCR, we detected a significant reduction in OrfA mRNA in 104-C1-OrfA cells after transfection with OrfA siRNA (Fig. 3A). No such change was observed when a scrambled siRNA control was used. Seventy-two hours after transfection, cells were collected and subjected to FACS analysis to evaluate the CD134 expression on cells. The results showed that CD134 was increased about 2-fold on the surfaces of siRNA-treated 104-C1-OrfA cells compared to the level for nontransfected cells (Fig. 3B) or cells treated with scrambled siRNA.
FIG. 3.
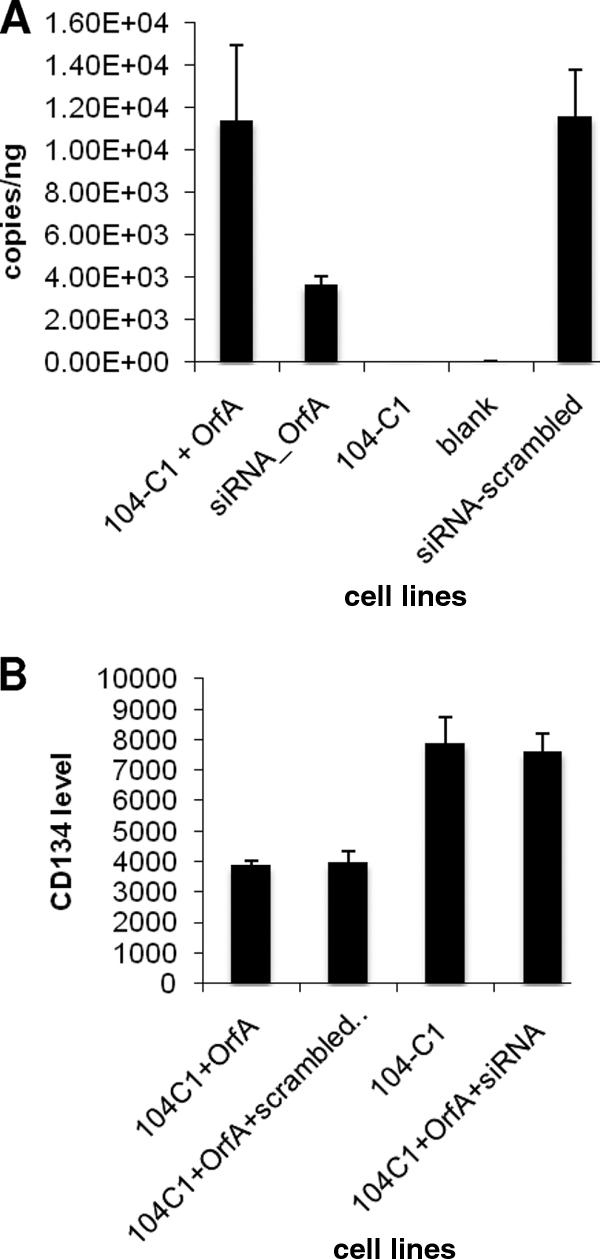
OrfA mRNA quantitation and FACS assays on CD134 surface levels for siRNA-treated OrfA knockdown cells. (A) Quantitative real-time PCR showed that OrfA mRNA level was greatly reduced in siRNA-transfected 104-C1-OrfA cells. Scrambled siRNA had no effect on the turnover rate of OrfA mRNA. (B) siRNA knockdown assay for confirming OrfA as a function of CD134 downregulation. FACS analysis for detecting CD134 on siRNA-treated OrfA transfected 104-C1 cells. The CD134 level on transfected cells is upregulated 2-fold compared to the level for 104-C1-OrfA. Scrambled siRNA had no effect on the regulation of CD134.
Western blot analysis and mRNA quantitation on CD134.
To investigate the mechanisms of OrfA influence on CD134 expression, we directed our efforts to explore different levels of gene regulation that might be accomplished by OrfA. First, we quantified the CD134 mRNA in 104-C1-OrfA cells using real-time PCR. Interestingly, our results showed that the CD134 mRNA level is not altered in this cell line in comparison to the level for parental 104-C1 cells (Fig. 4A). As expected, the GFox cells overexpressing CD134 are high in cellular CD134 mRNA expression. CrFK as a negative control had no CD134 amplification, as determined by PCR. Western blot analysis was then performed to detect CD134 and CD9 in both total cell lysates and in preparations of cellular plasma membrane (Fig. 4B). There was no significant difference in the quantities of CD134 and CD9 in the total cell lysates for the two cell lines (Fig. 4B, compare lanes 1 and 2 for both upper and lower panels). However, quantitative analysis by densitometry revealed that the CD134 level in the plasma membrane preparation for 104-C1 was 3.5-fold higher than that in 104-C1-OrfA cells (Fig. 4B, compare lanes 3 and 4 in upper panel), which correlates with FACS analyses. In contrast, no significant difference was detected in the levels of CD9 in the two plasma membrane preparations (Fig. 4B, compare lanes 3 and 4 in lower panel), again consistent with FACS analysis. Thus, surface CD134 expression is reduced in 104-C1-OrfA cells, but total transcription and translation of receptor remain the same with OrfA expression. No CD134 or CD9 was detected in plasma membrane preparations from either CrFK or 3201 cells, used as negative controls (Fig. 4B, lanes 5 in upper and lower panels).
FIG. 4.
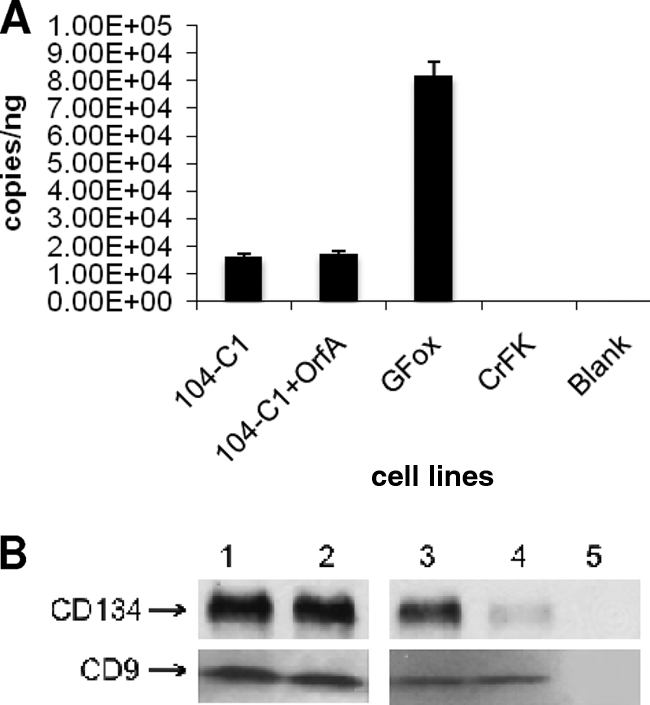
Quantitative real-time PCR and Western blot analysis on CD134 and CD9 expression. (A) Quantitative real-time PCR assessment on CD134 mRNA levels in various cell lines. The amount of CD134 mRNA in 104-C1-OrfA cells is approximately the same as that in 104-C1 cells. (B) Western blot against CD134 and CD9 in 104-C1 and 104-C1-OrfA cells. Lane 1, 104-C1 total cell lysate (7 μg); lane2, 104-C1-OrfA total cell lysate (7 μg); lane3, 104-C1 plasma membrane protein (2 μg); lane 4, 104-C1-OrfA plasma membrane protein (2 μg); lane 5, CrFK (upper panel) and 3201 (lower panel) plasma membrane protein (2 μg) as negative controls.
104-C1-OrfA cells can be infected with either FIV-34TF10 or OrfArep.
FIV-34TF10 cannot productively infect CD134+ 104-C1 cells (Fig. 1B). Since CD134 surface expression is greatly reduced in OrfA-transfected 104-C1 cells, we hypothesized that the transfected cell line might become susceptible to infection by FIV-34TF10. Data from the RT activity assay provided supportive evidence for this assumption in that FIV-34TF10 and FIV-34TF10-OrfArep were infectious on 104-C1-OrfA cells (Fig. 5).
FIG. 5.

Infection assays on 104-C1-OrfA cells by FIV-34TF10 and OrfArep showing that the cells were infected by both viral strains.
Influence of cell surface receptor on viral entry.
To show that downregulation of the CD134 surface receptor by OrfA can change the pattern of viral entry, we conducted a β-Gal assay to assess the degree of viral entry after a single round of infection. FIV hybrid vector (pCFIV) pseudotyped with the FIV-PPR or FIV-34TF10 Env gene was used to generate virons, which were then used to infect GFox, CrFK, 104-C1, and 104-C1-OrfA cells (Fig. 6A). The results showed that FIV-PPR Env-based pseudovirons entered the GFox cells much more efficiently than CrFK cells. In addition, FIV-PPR Env facilitated entry into 104-C1 cells approximately two times more than into 104-C1-OrfA cells, which have reduced expression of cell surface CD134. The results are consistent with CD134 dependence for FIV-PPR Env-directed since 104-C1 and GFox cells have more surface CD134 receptors than 104-C1-OrfA and CD134− CrFK cells. The 34TF10 Env-based pseudovirons were not as efficient for entry into GFox cells as they were for entry into CrFK cells, suggesting that the overexpression of CD134 molecules on the GFox cell surface might negatively influence the process (Fig. 6B). However, no significant difference was observed for 34TF10 Env-mediated entry into 104-C1 versus 104-C1-OrfA cells.
FIG. 6.
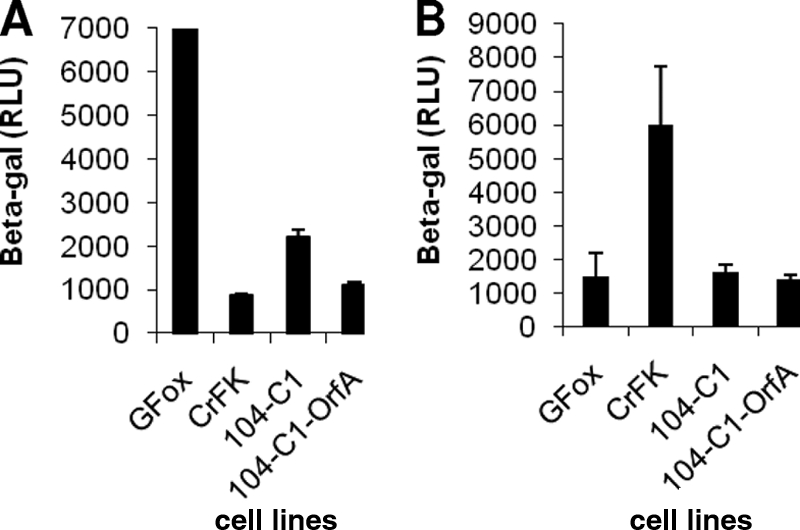
β-Gal assay for measurement of the entry of FIV-PPR or FIV-34TF10 envelope-pseudotyped virons. (A) pCFIV vectors pseudotyped with FIV-PPR Env were used to assess the degree of viral entry. A β-Gal assay performed at 48 h posttransduction shows that pCFIV pseudotyped with PPR Env infected GFox and 104-C1 cells more efficiently than it did CrFK and 104-C1-OrfA cells. RLU, relative luminescence units. (B) pCFIV vectors pseudotyped with 34TF10 Env were used to assess the viral entry for the same cell lines as those used for the above-described assay. The results showed that the viron infected CrFK cells more readily than it did GFox cells. On the other hand, no significant difference was observed for the 34TF10 Env-mediated viral entry between 104-C1 and 104-C1-OrfA cells.
Confocal microscopy analysis.
We used immunofluorescence staining to study the nature of apparent internalization and subcellular accumulation of CD134 as a function of OrfA expression. In 104-C1 cells, there is a distinct, sparsely distributed plasma membrane surface localization of CD134 and some punctate internal colocalization (Fig. 7A, white in last panel) to the Golgi apparatus, as indicated by the GM130 counterstaining (Fig. 7, green). The last two panels (Fig. 7A and B) are 8 bit white pseudocolored images of the colocalized zones between the CD134 fluorescent signal (red) and the GM130 fluorescent signal (green). In 104-C1-OrfA cells, there was a significant diminishing of the peripheral localization of CD134 from the cell surface and a coincident increase in localization to the Golgi apparatus, as indicated in white in Fig. 7B. This novel finding is consistent with the interpretation that FIV OrfA expression is associated with retention and accumulation of CD134 in the Golgi apparatus of feline primary lymphocytes, resulting in reduction of cell surface expression of CD134.
FIG. 7.

Confocal microscopy analysis showing CD134 subcellular localization in 104-C1 and 104-C1-OrfA cells. (A) Confocal imaging of permeabilized 104-C1 cells. (B) The same staining for permeabilized 104-C1-OrfA cells. The panels in the 1st column represent staining for CD134 receptors (red) The panels in the 2nd column represent staining with GM130-Alexa for Golgi complexes (intense red, represented by green). The panels in the 3rd column show nuclear stain with DAPI (4′,6-diamidino-2-phenylindole) (blue) and the colocalization (white) of the two fluorescently labeled reagents. The panels in the 4th column show colocalization only. The results show that significantly more CD134 colocalizes with Golgi complexes in OrfA-expressing cells (B) than in untreated 104-C1 cells (A). PE, phycoerythrin.
DISCUSSION
HIV uses CD4 as the primary binding receptor and either CCR5 or CXCR4 as the chemokine receptor for virus entry, whereas FIV uses CD134/CXCR4 as the dual receptor system to produce productive viral infection in cats. Despite its essential role in viral entry by promotion of efficient coreceptor binding and host cell membrane fusion, the presence of CD4 poses problems for HIV spread in vivo due to the possibility of superinfection, premature binding of CD4 to nascent virus particles, and inhibition of virus release (26, 31). Accordingly, HIV-1 has evolved different mechanisms to downregulate CD4 on the surfaces of or within the host cells, involving both the Env glycoprotein gp160 (4, 15) and viral accessory proteins Nef (1, 3) and Vpu (19).
FIV primary receptor CD134 is a 43-kDa glycoprotein found on the surfaces of CD4+ T cells, the main in vivo target for FIV infection (8). CD134 is specifically upregulated when the host T cells have been activated by treatment of IL-2 and ConA (6). Since FIV utilizes a binding/entry receptor system similar to that of HIV, it is reasonable to perceive the presence of a parallel regulatory mechanism for CD134 to aid in optimizing FIV infection. In the present study, for the first time, we demonstrate that the FIV accessory protein OrfA causes reduction in cell surface expression of CD134 on the host cell surface and by doing so causes a net increase in release of virus from the infected cell. OrfA regulation appears to specifically act on CD134, or at least does not cause a global suppression of cell surface protein turnover, since the level of another feline cell surface molecule, CD9, was not affected by OrfA expression.
FIV-34TF10 is a particularly useful molecular clone for study of OrfA function, in that this Petaluma-derived isolate has a premature stop codon in the OrfA open reading frame (28, 35). There is an overall sequence identity level of 91% between this virus and FIV-PPR, with 71% and 85% homology at the amino acid level for the orfA and env genes. FIV-34TF10 is CD134 independent, and the viral entry assay in this study also showed that 34TF10 Env pseudotyped virons were more efficient for CrFK cell-based entry than for entry into GFox cells. However, it is important to emphasize that 34TF10 SU still binds CD134 strongly, even though it is apparently already in a conformation suitable for binding to CXCR4, thus facilitating infection. The current study indicated that FIV-34TF10 can readily infect CD134-negative CrFK cells and generate high RT activity levels in the culture but does not launch efficient infection on primary T cells, feline T-cell lines, CD134-positive GFox cells, or CrFK cells expressing domain 1 of feline CD134 on the background of human CD134 (fD1-CrFK; data not shown) (5). In contrast, an OrfA-repaired 34TF10 strain, OrfArep, can produce robust infection in CD134-positive 104-C1 cells and GFox cells. This phenomenon implies that there is some direct or indirect interaction between FIV OrfA and CD134 and that this interplay is essential for productive viral infection mediated by CD134. This supposition is borne out by the FACS analysis and Western blot data in this study, which clearly show that OrfA causes reduction in surface CD134 expression in OrfA-transfected 104-C1 cells. Important to the biological relevance of the findings, a drop in CD134 surface expression was observed when 104-C1 cells were experimentally infected with either FIV-PPR or OrfArep viruses in a spreading infection. Although there is only an approximately 2-fold reduction of CD134 upon FIV-PPR infection on 104-C1 cells, we believe it is because the FACS analyses are gated for live cells, which includes uninfected cells plus that group of infected cells that have not undergone apoptosis. With longer infection times, the same results are observed, since 70% of the 104-C1 cells infected with FIV-PPR have already died by 4 days postinfection. Experiments employing siRNA to knockdown OrfA expression in 104-C1-OrfA cells caused reemergence of cell surface CD134 expression, thus confirming the inverse relationship in the expression levels of these two proteins.
Extensive studies have been conducted to explore the molecular and cellular mechanisms of HIV-induced CD4 downregulation in host cells. HIV Nef, a small intracellular trafficking protein, mainly acts on mature CD4 in the endocytic or late secretory pathways (20). Nef is reported to link CD4 to components of clathrin-dependent pathways and deliver CD4 to lysosomes for degradation (3). Whether OrfA acts in a similar manner remains to be determined. Real-time quantitative PCR showed that total CD134 mRNA level is not affected in the presence of OrfA expression, indicating that CD134 is not directly regulated at the transcriptional level. However, translational regulation appears also to be ruled out in that Western blot analyses revealed that the overall CD134 level detected from total cell lysates remains unchanged in OrfA-transfected 104-C1 cell lines relative to the level for 104-C1 cells only. This result correlated with a previous report on HIV Nef downregulation against CD4 (1). Confocal microscopy revealed internal accumulation of CD134 associated with the Golgi complex as a function of OrfA expression. No significant degradation of CD134 was observed in the OrfA-expressing cells, but whether the receptor is destined for eventual degradation remains to be determined.
How the presence of CD134 receptor negatively affects FIV-34TF10 infection on GFox and 104-C1 cells is still open to discussion. It is important to note that although FIV-34TF10 is CD134 independent for entry, it still binds to CD134 efficiently. Thus, the CD134 independence of this isolate would not preclude a role for CD134 in interfering with virus release by binding to the envelope protein on viral egress from the cell. Viral entry assays showed that overexpression of CD134 on GFox cells restricts the entry of 34TF10 Env-pseudotyped virons while the CD134 on 104-C1 cells did not significantly affect the entry of the same pseudovirion preparations. However, the 104-C1 cell line and variants such as 104-C1-OrfA express much lower levels of CD134 than GFox cells (Fig. 3A), which may explain the difference in level of response. Consistent with a connection between CD134 and OrfA expression, FIV-34TF10 with an open OrfA reading frame (OrfARep) can productively infect both GFox and 104-C1 cells. Whether this is a direct effect between OrfA and CD134 or some indirect mechanism involving up- or downregulation of another cellular component is yet to be determined.
Comparable to HIV CD4, FIV CD134 might inhibit 34TF10 infection mainly by limiting the viral release from host cell surface rather than by blocking the entry of virus. Consistent with this line of reasoning, downregulation of CD134 in 104-C1-OrfA cells resulted in infection by both FIV-34TF10 and OrfArep, effectively eliminating any negative influence of CD134 expression postinfection.
It needs to be mentioned that the orfA gene used to transfect 104-C1 cells in this study was PCR amplified from a FIV-PPR molecular clone and that all the experiments were performed ex vivo. Hence, genetic variation may exist for this gene because of the absence of in vivo selection for OrfA function upon repeated propagation of virus clone ex vivo. In fact, OrfA shows the greatest genetic divergence among all the reported FIV sequences, with divergence levels surpassing even those for the envelope gene. Similar issues were discussed in studies of HIV nef alleles, which lack the function of downregulating CD4 molecules because of the changes of their genetic constitution (21, 36). In this regard, we expect that the function of OrfA for reducing CD134 surface molecules may be greatly amplified under in vivo conditions.
FIV serves as a useful animal model for study of lentivirus infections because of many advantages of the system, including the relative cost of materials and personnel safety in handling viruses and virus-infected tissues. An in-depth study of OrfA functionality as a virus receptor regulator with its connection to viral infection levels and disease progression will aid in understanding the host-pathogen interaction in the setting of natural lentivirus infections, with potential to develop novel broad-based therapeutic strategies. Interfering with the viruses' ability might aid in limiting virus spread and potentially could lead to more-robust targeting of infected cells by the host's immune system.
Acknowledgments
We thank Ying-Chuan Lin for valuable comments, Meaghan Happer for excellent technical assistance, and Gale Sessions for manuscript preparation.
The work was supported in part by from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (award numbers T32 AI007354 and R01 AI25825).
Footnotes
Published ahead of print on 12 May 2010.
REFERENCES
- 1.Benson, R. E., A. Sanfridson, J. S. Ottinger, C. Doyle, and B. R. Cullen. 1993. Downregulation of cell-surface CD4 expression by simian immunodeficiency virus Nef prevents viral super infection. J. Exp. Med. 177:1561-1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chatterji, U., A. de Parseval, and J. H. Elder. 2002. Feline immunodeficiency virus OrfA is distinct from other lentivirus transactivators. J. Virol. 76:9624-9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chaudhuri, R., O. W. Lindwasser, W. J. Smith, J. H. Hurley, and J. S. Bonifacino. 2007. Downregulation of CD4 by human immunodeficiency virus type 1 Nef is dependent on clathrin and involves direct interaction of Nef with the AP2 clathrin adaptor. J. Virol. 81:3877-3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crise, B., L. Buonocore, and J. K. Rose. 1990. CD4 is retained in the endoplasmic reticulum by the human immunodeficiency virus type 1 glycoprotein precursor. J. Virol. 64:5585-5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Parseval, A., U. Chatterji, G. Morris, P. Sun, A. J. Olson, and J. H. Elder. 2005. Structural mapping of CD134 residues critical for interaction with feline immunodeficiency virus. Nat. Struct. Mol. Biol. 12:60-66. [DOI] [PubMed] [Google Scholar]
- 6.de Parseval, A., U. Chatterji, P. Sun, and J. H. Elder. 2004. Feline immunodeficiency virus targets activated CD4+ T cells by using CD134 as a binding receptor. Proc. Natl. Acad. Sci. U. S. A. 101:13044-13049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Parseval, A., and J. H. Elder. 2001. Binding of recombinant feline immunodeficiency virus surface glycoprotein to feline cells: role of CXCR4, cell-surface heparans, and an unidentified non-CXCR4 receptor. J. Virol. 75:4528-4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Parseval, A., and J. H. Elder. 1999. Demonstration that orf2 encodes the feline immunodeficiency virus transactivating (Tat) protein and characterization of a unique gene product with partial rev activity. J. Virol. 73:608-617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Parseval, A., D. L. Lerner, P. Borrow, B. J. Willett, and J. H. Elder. 1997. Blocking of feline immunodeficiency virus infection by a monoclonal antibody to CD9 is via inhibition of virus release rather than interference with receptor binding. J. Virol. 71:5742-5749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doms, R. W., and D. Trono. 2000. The plasma membrane as a combat zone in the HIV battlefield. Genes Dev. 14:2677-2688. [DOI] [PubMed] [Google Scholar]
- 11.Elder, J. H., M. Schnolzer, C. S. Hasselkus-Light, M. Henson, D. A. Lerner, T. R. Phillips, P. C. Wagaman, and S. B. Kent. 1993. Identification of proteolytic processing sites within the Gag and Pol polyproteins of feline immunodeficiency virus. J. Virol. 67:1869-1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia, J. V., and A. D. Miller. 1991. Serine phosphorylation-independent downregulation of cell-surface CD4 by nef. Nature 350:508-511. [DOI] [PubMed] [Google Scholar]
- 13.Gemeniano, M. C., E. T. Sawai, C. M. Leutenegger, and E. E. Sparger. 2003. Feline immunodeficiency virus ORF-Ais required for virus particle formation and virus infectivity. J. Virol. 77:8819-8830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gemeniano, M. C., E. T. Sawai, and E. E. Sparger. 2004. Feline immunodeficiency virus Orf-A localizes to the nucleus and induces cell cycle arrest. Virology 325:167-174. [DOI] [PubMed] [Google Scholar]
- 15.Jabbar, M. A., and D. P. Nayak. 1990. Intracellular interaction of human immunodeficiency virus type 1 (ARV-2) envelope glycoprotein gp160 with CD4 blocks the movement and maturation of CD4 to the plasma membrane. J. Virol. 64:6297-6304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnston, J. C., M. Gasmi, L. E. Lim, J. H. Elder, J. K. Yee, D. J. Jolly, K. P. Campbell, B. L. Davidson, and S. L. Sauter. 1999. Minimum requirements for efficient transduction of dividing and nondividing cells by feline immunodeficiency virus vectors. J. Virol. 73:4991-5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kwong, P. D., R. Wyatt, J. Robinson, R. W. Sweet, J. Sodroski, and W. A. Hendrickson. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lerner, D. L., P. C. Wagaman, T. R. Phillips, O. Prospero-Garcia, S. J. Henriksen, H. S. Fox, F. E. Bloom, and J. H. Elder. 1995. Increased mutation frequency of feline immunodeficiency virus lacking functional deoxyuridine-triphosphatase. Proc. Natl. Acad. Sci. U. S. A. 92:7480-7484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levesque, K., Y. S. Zhao, and E. A. Cohen. 2003. Vpu exerts a positive effect on HIV-1 infectivity by down-modulating CD4 receptor molecules at the surface of HIV-1-producing cells. J. Biol. Chem. 278:28346-28353. [DOI] [PubMed] [Google Scholar]
- 20.Lindwasser, O. W., R. Chaudhuri, and J. S. Bonifacino. 2007. Mechanisms of CD4 downregulation by the Nef and Vpu proteins of primate immunodeficiency viruses. Curr. Mol. Med. 7:171-184. [DOI] [PubMed] [Google Scholar]
- 21.Luria, S., I. Chambers, and P. Berg. 1991. Expression of the type 1 human immunodeficiency virus Nef protein in T cells prevents antigen receptor-mediated induction of interleukin 2 mRNA. Proc. Natl. Acad. Sci. U. S. A. 88:5326-5330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malim, M. H., and M. Emerman. 2008. HIV-1 accessory proteins—ensuring viral survival in a hostile environment. Cell Host Microbe 3:388-398. [DOI] [PubMed] [Google Scholar]
- 23.Mangeat, B., P. Turelli, G. Caron, M. Friedli, L. Perrin, and D. Trono. 2003. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424:99-103. [DOI] [PubMed] [Google Scholar]
- 24.Mariani, R., and J. Skowronski. 1993. CD4 down-regulation by nef alleles isolated from human immunodeficiency virus type 1-infected individuals. Proc. Natl. Acad. Sci. U. S. A. 90:5549-5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyazawa, T., M. Fukasawa, A. Hasegawa, N. Maki, K. Ikuta, E. Takahashi, M. Hayami, and T. Mikami. 1991. Molecular cloning of a novel isolate of feline immunodeficiency virus biologically and genetically different from the original U.S. isolate. J. Virol. 65:1572-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pauza, C. D., J. E. Galindo, and D. D. Richman. 1990. Reinfection results in accumulation of unintegrated viral DNA in cytopathic and persistent human immunodeficiency virus type 1 infection of CEM cells. J. Exp. Med. 172:1035-1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pedersen, N. C. 1993. The feline immunodeficiency virus, p. 181-228. In J. A. Levy (ed.), Retroviridae, vol. 2. Plenum Press, New York, NY. [Google Scholar]
- 28.Phillips, T. R., C. Lamont, D. A. Konings, B. L. Shacklett, C. A. Hamson, P. A. Luciw, and J. H. Elder. 1992. Identification of the Rev transactivation and Rev-responsive elements of feline immunodeficiency virus. J. Virol. 66:5464-5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Phillips, T. R., R. L. Talbott, C. Lamont, S. Muir, K. Lovelace, and J. H. Elder. 1990. Comparison of two host cell range variants of feline immunodeficiency virus. J. Virol. 64:4605-4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poeschla, E. M., and D. J. Looney. 1998. CXCR4 is required by a nonprimate lentivirus: heterologous expression of feline immunodeficiency virus in human, rodent, and feline cells. J. Virol. 72:6858-6866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robinson, H. L., and D. M. Zinkus. 1990. Accumulation of human immunodeficiency virus type 1 DNA in T cells: results of multiple infection events. J. Virol. 64:4836-4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rozen, S., and H. Skaletsky. 2000. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 132:365-386. [DOI] [PubMed] [Google Scholar]
- 33.Shimojima, M., T. Miyazawa, Y. Ikeda, E. L. McMonagle, H. Haining, H. Akashi, Y. Takeuchi, M. J. Hosie, and B. J. Willett. 2004. Use of CD134 as a primary receptor by the feline immunodeficiency virus. Science 303:1192-1195. [DOI] [PubMed] [Google Scholar]
- 34.Sparger, E. E., B. L. Shacklett, L. Renshaw-Gegg, P. A. Barry, N. C. Pedersen, J. H. Elder, and P. A. Luciw. 1992. Regulation of gene expression directed by the long terminal repeat of the feline immunodeficiency virus. Virology 187:165-177. [DOI] [PubMed] [Google Scholar]
- 35.Talbott, R. L., E. E. Sparger, K. M. Lovelace, W. M. Fitch, N. C. Pedersen, P. A. Luciw, and J. H. Elder. 1989. Nucleotide sequence and genomic organization of feline immunodeficiency virus. Proc. Natl. Acad. Sci. U. S. A. 86:5743-5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Terwilliger, E. F., E. Langhoff, D. Gabuzda, E. Zazopoulos, and W. A. Haseltine. 1991. Allelic variation in the effects of the nef gene on replication of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. U. S. A. 88:10971-10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waters, A. K., A. P. De Parseval, D. L. Lerner, J. C. Neil, F. J. Thompson, and J. H. Elder. 1996. Influence of ORF2 on host cell tropism of feline immunodeficiency virus. Virology 215:10-16. [DOI] [PubMed] [Google Scholar]
- 38.Willett, B. J., K. Adema, N. Heveker, A. Brelot, L. Picard, M. Alizon, J. D. Turner, J. A. Hoxie, S. Peiper, J. C. Neil, and M. J. Hosie. 1998. The second extracellular loop of CXCR4 determines its function as a receptor for feline immunodeficiency virus. J. Virol. 72:6475-6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Willett, B. J., E. L. McMonagle, S. Ridha, and M. J. Hosie. 2006. Differential utilization of CD134 as a functional receptor by diverse strains of feline immunodeficiency virus. J. Virol. 80:3386-3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Woo, J. C., G. A. Dean, N. C. Pedersen, and P. F. Moore. 1997. Immunopathologic changes in the thymus during the acute stage of experimentally induced feline immunodeficiency virus infection in juvenile cats. J. Virol. 71:8632-8641. [DOI] [PMC free article] [PubMed] [Google Scholar]