

Abstract
Described herein are several unique analytical applications utilizing mass spectrometry and the selective modification of the free thiol form of cysteine in both peptides and proteins by various quinones. This simple modification can be used to quantify the number of free or disulfide bound cysteines in a protein. In addition, quinone modification can also be used to easily probe the solvent accessibility of cysteine residues, which provides information about protein structure or folding state. Furthermore, the chromophoric properties of the quinone moiety can be leveraged for site specific photodissociation of the backbone. The photodissociation reveals both the presence and location of modified cysteine residues. For example, cleavage of the protein backbone of alpha Hemoglobin is observed selectively at a single cysteine out of 140 residues in the whole protein. This selective backbone fragmentation is accompanied by a parent ion mass loss which is unique to the modifying quinone. When combined, this information can be used to determine both the presence and site of modification generated by naturally occurring molecules, such as dopamine, which can harness quinone chemistry to modify proteins.
Introduction
Cysteine is unique among the canonical amino acids for several reasons. For one, cysteine is the only residue which routinely defines protein structure with covalent bonds coupled through the side chain.1,2 The typical arrangement requires two cysteine side chains, which are oxidized and linked together to form a cystine disulfide bridge. Cysteine also plays an important role in metal ion coordination, serving as a metal ligand in numerous metalloproteins.3–6 Cysteine is also among the most reactive amino acids,7,8 which facilitates post translational modifications and leads to active participation in redox chemistry within cells.9–12 These unique chemical properties of cysteine make it an important analytical target. Mass spectrometry (MS) is well suited for the analysis of biological molecules, including peptides and proteins; however, cysteine frequently interferes with mass spectral analysis. Disulfide bridges form undesirable cross links which prevent linear dissociation of the molecule and are therefore frequently reduced and capped to avoid interference.13 Additionally, the oxidation of cysteine is a common post translational modification which is not always of biological origin, but can complicate the analysis of spectra and must be given due consideration.14,15
Another approach which can be employed to avoid these problems is to directly target cysteine or disulfide bonds in proteins with site specific chemistry. For example, disulfide bonds can be targeted with some degree of selectivity in the gas phase via electron capture dissociation16,17 or the use of 157nm light.18 In solution, there are various strategies which can be employed to selectively modify cysteine.19–26 Of particular relevance presently, it has been shown previously that thiol functional groups will react with quinones via a Michael type addition in aqueous solution.27 This modification can be implemented to yield selective modification of free cysteine residues in peptides and proteins. Due to the selectivity of this modification, only cysteine residues are modified to any significant extent.28 An example of this type of modification is shown in Scheme 1 below. The semiquinone formed from the Michael addition can revert back to the quinone form depending on the redox potential of the solution. The quinone form is primarily observed in the experiments described below. The mass shift after reaction with quinone can effortlessly be used to quantify the number of cysteines present in a peptide or protein. This easily attainable information has been used previously to improve protein identification through database searching.29
Scheme 1.
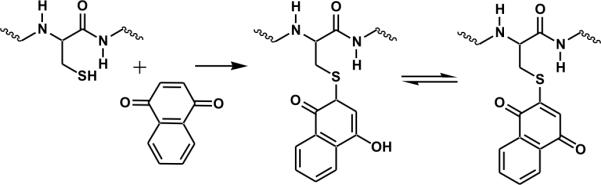
Modification of cysteine by quinone
In addition to causing a mass shift, quinone modification of cysteine introduces a chromophore which is adjacent to the Cβ-S bond. Recent experiments on modified phosphopeptides have demonstrated that chromophores adjacent to a Cβ-S bonds can be used to selectively fragment the peptide backbone following photoactivation with ultraviolet (UV) light.30 Photodissociation (PD) occurs due to dissociative excited state chemistry which allows bonds to be broken homolytically prior to significant energy redistribution.31–33 This contrasts with the situation which occurs with radicals generated by collisional activation which tend to yield less selective fragmentation.34–39 When beta radicals are generated by PD, specific backbone fragmentation at the modified residue is highly favored. Similar chemistry should be accessible by photoexcitation of quinone modified cysteine residues, which is the subject of the present manuscript.
It is demonstrated herein that quinone modified cysteine residues undergo direct dissociation following excitation by 266nm light. The dissociation yields a radical on the remaining peptide or protein at the beta position of cysteine. This beta radical facilitates selective backbone dissociation, typically yielding d-type ions at the modified cysteine residues for both peptides and whole proteins. In addition, quinone modification can be harnessed to monitor protein structure via solvent accessibility moderated differential reactivity. Site selective backbone dissociation can be used to identify the sites of modification in these structure probing experiments, if desired. Finally, PD can be also be used to characterize biologically relevant post translational modification of cysteine by quinones. For example, dopamine can adopt a quinone intermediate structure capable of reacting with free cysteine residues. It is demonstrated that site specific PD can be utilized to identify both the presence and location of dopamine modifications. Interestingly, a novel double modification state is identified for dopamine which can crosslink backbone strands in a manner similar to a disulfide bond.
Materials and methods
Peptides SKGKSKRKKDLRISCNSK, SLRRSSCFGGR, RLCRIVVIRVCR, DYMGWMDF, MEHFRWG, KWDNQ, IARRHPFL, and KKRAARATS-NH2 were purchased from American Peptide Company. AEAEYEK and Ac-CLKKLsGK were purchased from QCB. Beta lactoglobulin, and lysozyme were purchased from MP Biomedicals. DRVYIHPF, human Hemoglobin, Alpha Lactalbumin, the oxidized beta chain of insulin, dopamine, Benzoquinone (BQ), 1,4 Naphthoquinone (NQ), dithiothreitol (DTT), Guanidine HCl, and Trifluoroacetic acid (TFA) were purchased from Sigma-Aldrich. Tris(2-carboxyethyl)phosphine (TCEP), and acetonitrile (ACN) were purchased from Fisher Scientific. Urea and acetic acid were from EMD and 1,4 Anthraquinone (AQ) was from Alfa Aesar. All were used as received without further purification. Water was purified by Millipore Direct-Q (Millipore,Billerica, MA). A protein MacroTrap and trap holder consisting of a polymeric reversed-phase packing with retention similar to C4 was purchased from Michrom Bioresources, Inc. (Auburn, CA)
Quinone modification (under non-reducing conditions)
The quinones examined in this study are shown in Chart 1 below. Quinone stocks were prepared in ACN at mM concentrations and stored in the dark to reduce degradation and prepared fresh daily. Quinone stock was added to peptide solutions in 0.5 to 4 times excess of peptide concentration. Solutions were diluted with 50:50 ACN: water to peptide concentrations of 10μM for analysis.
Chart 1.
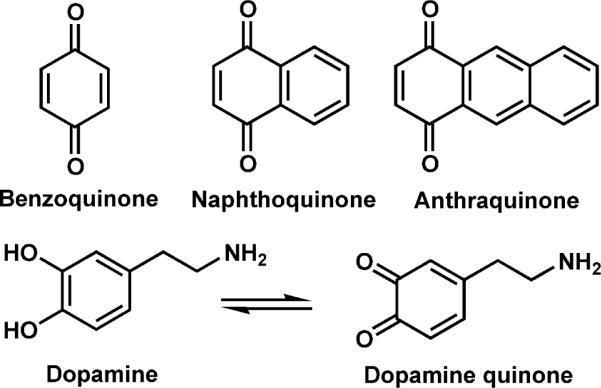
Quinones examined herein.
Hemoglobin was diluted to concentrations of 5μM protein and 40μM of NQ in 50:50 ACN: water. Acid was not added to samples in order to favor the formation of lower charge states which facilitates charge state assignments of the fragments generated.
Modification of the free cysteine of Beta lactoglobulin was performed in non reducing conditions as follows: 5μl of 20mM NQ stock in ACN was added to 10ul of 87uM protein stock along with an additional 5μl of ACN to give 50:50 water: ACN solution. The reaction was allowed to sit at room temperature for 4 hours before excess NQ was removed with a protein MacroTrap according to manufacturer's instructions. Final protein concentration after cleanup was ~4μM for analysis by MS.
Modification of disulfide containing proteins
Disulfide bonds were reduced prior to addition of quinone with either DTT or TCEP. Alpha Lactalbumin (7μM) was combined with 100μM DTT and allowed to sit at RT for 15min prior to addition of Naphthoquinone (40μM). Lysozyme (0.732mM, 2μl of 1.83mM) was reduced with TCEP (50mM, 1μl of 250umM) for 2 hours at RT. Guanidine (~2M, 2μl of ~5M stock) was added to the sample to denature the protein. NQ (10μl of 1mM stock in ACN) was added to the reduced protein solution. Excess reactants were removed by desalting with a protein MacroTrap and diluted to ~7μM protein prior to analysis by MS.
Probing solvent accessibility of cysteines in Hemoglobin
Hemoglobin stock solution was diluted into each of the three following solutions with a final protein concentration of 8.6μM: 20mM PBS (phosphate buffered saline), 8M urea, and 50% ACN. 20mM Ammonium bicarbonate was added to each sample to maintain a constant pH. To each sample 1μl of 5mM BQ or AQ stock was added (50uM final conc). The samples were allowed to react at room temperature for 15min at which point the reaction was stopped by addition of TFA to reach a pH of 3–4. Protein samples were desalted by protein Macrotrap, eluding the protein from the trap in 200μl of 90% ACN and 0.1%TFA. Stopping the reactions by addition of acid allows cleanup of the samples to be performed, and all solutions were electrosprayed directly from the trap eluent.
Dopamine modification
Peptide SKGKSKRKKDLRISCNSK (10μM) was mixed with dopamine (50μM) and 0.0033% hydrogen peroxide to promote the formation of dopamine quinone. The solution was diluted to the listed peptide concentration in 50:50 water: ACN and electrosprayed. Hemoglobin (4μM) was mixed with dopamine (100μM) alone, then diluted and electrosprayed in 50:50 water: ACN.
Photodissociation of quinone modified peptides and proteins
Solutions were analyzed in cation mode by an LTQ linear ion trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA) with a standard electrospray source. The posterior plate of the LTQ was modified with a quartz window to transmit fourth harmonic (266 nm) laser pulses from a flashlamp-pumped Nd:YAG laser (Continuum, Santa Clara,CA). Pulses were synchronized to the end of the isolation step of a typical MS2 experiment by feeding a TTL trigger signal from the mass spectrometer to the laser via a digital delay generator (Berkeley Nucleonics, San Rafael, CA). This allowed photodissociation (PD) to be carried out analogous to collision induced dissociation (CID). Multi shot experiments were carried out with a burst pulse sequence where each of the pulses were separated by 100msec and the activation time was adjusted in the instrument software to accommodate adequate time for multiple pluses prior to scanning.
Results and Discussion
Selective cysteine modification by quinones
The following peptides were treated with either 1,4-naphthoquinone (NQ) or anthraquinone (AQ): KWDNQ, IARRHPFL, MEHFRWG, DRVYIHPF, KKRAARATS, AEAEYEK, DYMGWMDF, SKGKSKRKKDLRISCNSK, SLRRSSCFGGR, RLCIVVIRVCR (containing a disulfide bond), and the beta chain of insulin with oxidized cysteines. These peptides were chosen because they contain collectively all 20 natural amino acids and three common forms of cysteine (free, oxidized, and disulfide bound). The full mass spectrum of peptide SLRRSSCFGGR is shown in Figure 1a after reaction with NQ. Abundant addition of a single quinone to the peptide is observed in all three charge states. No unmodified peptide is observed to remain after the reaction. Modification at the cysteine side chain was confirmed by MS/MS (shown below in Figure 3b). SKGKSKRKKDLRISCNSK was the only other peptide to mass shift due to attachment of NQ or AQ. A single modification at cysteine was also observed for this peptide (data not shown). These results demonstrate the selectivity for quinone addition to free cysteine and suggest that in the absence of structural interference or steric hindrance, modification can easily be driven to completion. In Figure 1b the mass spectrum of Hemoglobin treated with NQ is shown. Hemoglobin consists of four protein chains, two alpha and two beta. In the human variant, the alpha chain contains a single cysteine residue and the beta chain contains two, none of which are involved in disulfide bonds and are expected to exist in the free thiol form. A single distribution of peaks is observed in Figure 1b for the alpha chain of Hemoglobin (labeled as #). Deconvolution of the data yields a single molecular weight corresponding to loss of the noncovalently bound heme and addition of a single quinone. The beta chain yields two distributions of peaks, both of which result from loss of heme and addition of either one (labeled with *) or two (**) NQ. Little or no unmodified protein was observed in any of the charge states for either protein chain. Nevertheless, structural interference likely prevents complete modification of the beta chain. As was observed for the peptide in Figure 1a, the maximum number of quinones attached to the protein is consistent with the number of free cysteines in both systems.
Figure 1.

a) Full mass spectrum of SLRRSSCFGGR after reaction with NQ. b) Full mass spectrum of Hemoglobin modified with NQ. Alpha chain (#) modified by one NQ. Beta chain single (*) and double (**) NQ modifications. c) Beta Lactoglobulin after NQ modification. Two natural variants A and B (both containing five cysteines and two disulfide bonds) are observed each with a single NQ modification. d) NQ modification of lysozyme after partial reduction of all four disulfide bonds.
Figure 3.

a) Photodissociation of the peptide SLRRSSCFGGR modified with NQ. Homolytic cleavage of the Cβ-S bond of the modified cysteine side chain is labeled C-S. b) CID of the NQ modified peptide. c) CID of the unmodified peptide for comparison.
Modification of beta lactoglobulin with disulfide bonds intact is shown in Figure 1c. Beta lactoglobulin contains five cysteine residues, four of which are bound by two disulfide bonds. Two variants of beta lactoglobulin are observed in the full mass spectrum. These two common variants, A (labeled #) and B (labeled *) differ in sequence by two amino acids D64G and V118A. Beta lactoglobulin contains a disulfide bond between cysteines 66 and 160 and a second disulfide bond between cysteine 106 and either cysteine 119 or 122 (both have been cited in literature).40,41 Both variants A and B are observed to have shifted mass due to modification with a single quinone. This mass shift relative to the unmodified protein allows easy determination of the number of free cysteines present in beta lactoglobulin.
Cysteines are often used to stabilize protein structure through the formation of disulfide bridges. Determining the presence of a disulfide bond by mass alone can be difficult, especially in large proteins where it can be difficult to detect a two dalton mass difference. Modification of free cysteines yields a much larger mass difference which is easily distinguishable. Cysteine modification before and after disulfide reduction can be utilized to determine the number of disulfide bonds present in a protein. For example, lysozyme contains eight cysteine residues that are all involved in disulfide bonds. Addition of quinone to the native protein results in no change in mass because there are no free cysteines to react with the quinone. After treatment with TCEP to partially reduce disulfide bonds, attachment of quinone is evident as shown in Figure 1d. (TCEP is used in place of DTT because of its ability to reduce disulfide bonds without reacting with NQ.) A distribution of zero to eight NQ modifications of lysozyme is observed, indicative that some fraction of all four disulfide bonds was reduced. It is also interesting to note that modification peaks follow a statistical pattern favoring an even number of modifications. It is likely that once a disulfide bond is reduced, attaching quinone to both cysteines is favored over attaching quinone to only one site. These observations are in agreement with the known high reactivity of the cysteine side chain.42 It is also clear in Figure 1d that the protein observed without modification decreases in relative intensity as the charge state of the protein increases. It is well known that unfolded proteins in ESI spectra are present in the higher charge states.43,44 A protein with larger surface area, due to unfolding, is able to accommodate a larger number of charges than a more compact form of the same protein. It follows that the protein observed at higher charge states originated from a more open conformation. It is also likely that protein with a disulfide bond reduced would take on a more open conformation since the structure is no longer rigidly held in place. The increased amount of quinone modification at higher charge states is consistent with a change in the protein structure to a more open conformation.
Probing Protein Structure
The possibility for probing protein structure as a function of the accessibility of cysteine residues was examined in greater detail by modification of Hemoglobin with benzoquinone (BQ) and anthraquinone (AQ) under different solvent conditions. BQ and AQ were chosen for the comparison as they encompass the two extremes of the quinone sizes utilized. Three different solvent conditions were employed to produce different structural states of Hemoglobin: 20mM phosphate buffered saline (PBS), 8M urea, and 50% ACN. The PBS solution should maintain the native protein conformation while the 8M urea and 50% ACN solution are likely to yield denatured states. A representative full mass spectrum is shown in Figure 2a for modification with BQ in 50% ACN. In order to facilitate comparison of the results, the relative amount of modification observed for each protein chain summed over all charge states is shown in Figures 2b and c.
Figure 2.

a) Full mass spectrum for Hemoglobin modified by BQ in 50%ACN, subscripts indicate number of modifications. b) The relative extent of BQ modification of Hemoglobin over all charge states is summarized for three solvent systems: phosphate buffered saline (PBS), 8M urea, and 50% ACN. The x-axis labels indicate the chain and corresponding number of modifications. c) Anthraquinone modifications are shown in plots analogous to those in b).
As seen in Figure 2b, the alpha chain of Hemoglobin is only ~40% modified with BQ in PBS, but almost completely modified in urea and 50%ACN. These results suggest that denaturing of the protein structure enables more complete quinone attachment. Similar results are obtained for the beta chain, which has two cysteine residues. In PBS a single modification is dominant, whereas two modifications are favored in the urea and ACN denaturing environments. Interestingly, for the beta chain the extent of reactivity is not identical in the two denaturing environments, suggesting that different structures or structural ensembles are present. In Figure 2c, modification with the larger AQ is shown. The alpha chain is not completely modified under any solvent condition for AQ, which contrasts the results obtained with BQ. The most straightforward explanation for this observation is that the larger size of AQ reduces the accessibility of cysteine and the amount of modification observed. With AQ, the extent of modification is different for both the alpha and beta chains in all solvent environments, suggesting that AQ is a more sensitive probe of protein structure.
Fragmentation at modified cysteine residues
Photodissociation of the +3 charge state of the SLRRSSCFGGR peptide modified with a single NQ is shown in Figure 3a. Two major fragment peaks are observed. The first (labeled C-S) is due to homolytic cleavage of the Cβ-S bond of cysteine. This characteristic loss of 190Da is a unique indicator that the selected ion contains a cysteine residue modified by NQ. The second most abundant peak observed is the d7 fragment. Generic fragment structures are shown in Scheme 2. This ion results from selective backbone fragmentation at the modified cysteine. Also observed, although in smaller abundance, is fragmentation of the peptide backbone on the N terminal side of the modified cysteine to yield an a6 fragment. The mechanism of fragment formation is similar to previously described radical directed dissociation.30,45 PD of the unmodified peptide was also performed (data not shown). No fragmentation was observed from PD of the unmodified peptide, indicating that the fragments in Figure 3a are generated due to absorption at the quinone.
Scheme 2.
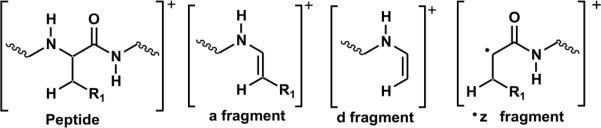
Structures of fragments formed by photodissociation
Collision induced dissociation (CID) of the +3 charge state of NQ modified peptide SLRRSSCFGGR is shown in Figure 3b. For comparison, CID of the +3 charge state of the unmodified peptide is shown in Figure 3c. Fragmentation by CID of the modified and unmodified peptides is very similar. Nearly all fragmentation pathways are analogous between the two, and nearly identical relative abundances are observed as well. Fragments including the modified cysteine are shifted in mass due to the retained quinone modification. The primary difference between the two CID spectra is the loss of modified cysteine side chain (labeled M-C*). This fragment is generated by cleavage of the carbon sulfur bond. However, fragmentation does not occur homolytically as in PD, thus a radical is not generated. Overall, the modification does not seem to significantly affect the fragmentation behavior of the peptide by CID. Comparison of Figures 3a and b, which are PD and CID of the modified peptide, reveals the selectivity afforded by photodissociation. The site of modification can be determined with either CID or PD. However this process is simpler by PD due to the selectivity and simplicity of the fragmentation. While the value of this fragmentation may not be apparent in determining the site of cysteine in peptides, the utility becomes more apparent in larger systems.
Fragmentation at cysteine residues in whole proteins
This methodology can also be extended to whole proteins. In Figure 4a, the PD spectrum for the +20 charge state of alpha Hemoglobin modified with NQ is shown. Homolytic cleavage of the Cβ-S bond is clearly present. The characteristic loss of the quinone-thiol at M-190Da is labeled as C-S. A single backbone fragment is observed at Cys104 yielding the +15 and +16 d104 ions. The complementary z fragment (z37) is also observed, although with smaller intensity compared to d104. The z37 fragment is also present in two charge states (+4 and +5). The charge states of the two complementary fragments equal the parent ion charge state of +20, also indicating that the d and z fragments are formed from a single initial backbone fragmentation. The single cysteine in alpha Hemoglobin is located at residue 104, the only site at which backbone fragmentation is observed. This demonstrates the ability of RDD to selectively cleave a single bond in a whole protein. This level of selectivity has not been achieved in any previous experiments and even supersedes previous PD experiments with whole proteins where iodination at tyrosine was observed to yield semi-selective fragmentation.32
Figure 4.

a) Photodissociation of the alpha chain of Hemoglobin modified with a single NQ. The NQ modification is lost due to homolytic cleavage of the Cβ-S bond of cysteine (labeled as C-S). Backbone fragmentation at cysteine is observed generating d104 and z37. b) Photodissociation of the beta chain of Hemoglobin with two NQ modifications.
Photodissociation of the beta chain with two NQ was also performed as shown in Figure 4b. Loss of a single naphthoquinone-thiol is primarily observed although a very small amount of the second quinone is also lost. Fragmentation at the two cysteine residues, positions 93 and 112, is observed. Fragments d93 and d112 are generated just C terminal to the two cysteines although the amount of fragmentation at cysteine 93 is significantly larger in abundance. Three laser pulses were used in the spectrum shown, thus it is possible that absorption and fragmentation will occur at a single cysteine or at both sites. The d112 fragment can only be observed if absorption does not occur or result in fragmentation at cysteine 93. However d93 can be observed regardless of what occurs at cysteine 112. Therefore the fragmentation observed at d112 is smaller in abundance than at d93. If absorption and fragmentation occurred at both positions simultaneously, an internal fragment could theoretically be observed. No such fragment was identified.
Probing protein structural changes
Alpha Lactalbumin (ALA) is a 123 residue protein containing eight cysteines involved in four disulfide bonds. Partial reduction by DTT and subsequent addition of NQ yields the full mass spectrum shown in Figure 5a. Peaks are labeled with the charge state and the number of quinone modifications determined by mass (11-1 denotes the +11 charge state with a single quinone). The majority of the protein either has one or two NQ modifications. The number and relative abundance of NQ varies depending on the charge state, with higher charge states having a larger number of NQ modifications. Again, this observation is likely correlated with a difference in protein structure.
Figure 5.

a) Full mass spectrum of Alpha Lactalbumin (ALA) following modification with NQ. Peaks are labeled with the charge state and the number of NQ modifications. b) Photodissociation of the +11 charge state of ALA with a single NQ modification. Cβ-S bond cleavage is labeled (C-S). Two backbone fragments are observed, z4 and d6, at the N and C terminal cysteines. c) Photodissociation of the +12 charge state of ALA with two NQ modifications.
Photodissociation of the 11-1 peak is shown in Figure 5b. Fragmentation of the Cβ-S bond of cysteine (labeled C-S) is noted. Two peaks are observed due to backbone fragmentation, z4 and d6. These fragments occur at the two cysteines (Cys6 and Cys120) located near the N and C termini of the protein. These two cysteines have previously been shown to be linked through a disulfide bond. Thus the protein in the 11-1 peak most likely has a single disulfide bond reduced between Cys6 and Cys120. Photodissociation of the higher charge state (+12) of protein with two quinones attached is shown in Figure 5c. Fragments z4 and d6 are observed as before. Additional backbone fragmentation is observed in comparison to Figure 5b. One of these new fragments (z95-c119) corresponds to an internal fragment between residues 28 and 119. More specifically, the internal fragment is due to a z-type fragmentation at Cys28 and a c-type fragmentation at Cys120. Additional nonselective dissociation is observed at Tyr103, which is known to be a favorable radical dissociation pathway.32 These results illustrate that site specific identification of cysteine will probably be limited to the two or three most reactive sites in proteins containing multiple disulfide bonds.
Dopamine modified protein
Dopamine is a biologically relevant small molecule neurotransmitter involved in several processes in the brain. Dopamine can also adopt a quinone form and modify cysteine residues.46,47 The PD method described herein was explored as a method for characterizing peptides and proteins modified by dopamine. To confirm that cysteine residues react with dopamine, the cysteine containing peptide SKGKRKKDLRISCNSK was mixed with dopamine and a small amount of H2O2 (also present in cells) to promote formation of dopamine quinone. Subsequent MS of the sample determined that a small amount of the dopamine adduct had formed with the peptide. CID confirmed that a covalent bond had formed between the peptide and dopamine (data not shown). PD of this protein-dopamine adduct is shown in Figure 6a. The largest fragment generated due to PD is a mass loss of 184Da. This unique loss corresponds to the loss of dopamine and the sulfur of cysteine by homolytic cleavage of the Cβ-S bond. The primary backbone fragment observed, although small, is d15 at cysteine. These two fragments yield all the necessary information to determine both the presence and location of the dopamine modification. The peak corresponding to a loss of 184 Da is unique to dopamine and the presence of this peak alone indicates that a dopamine modification is present. Furthermore the additional d-fragment selectively identifies the location of the dopamine modified cysteine residue.
Figure 6.

a) Photodissociation of the peptide SKGKSKRKKDLRISCNSK modified with dopamine. Homolytic cleavage of the Cβ-S bond is observed (C-S). Backbone fragmentation (d15) at cysteine identifies the site of modification. b) Full MS observed after dopamine addition to Hemoglobin. Only the β chain is modified (labeled *). c) PD of the +15 charge state of β chain of Hemoglobin with a single dopamine modification. d) PD of the +15 charge state of β chain of Hemoglobin without modification for comparison.
Figure 6b is the full mass spectrum observed upon spraying a solution containing Hemoglobin incubated with dopamine without the addition of any H2O2. The alpha chain is observed in a single distribution of unmodified apo-protein. The beta chain is observed in two distributions. The larger of the two is the unmodified apo-protein. The second distribution corresponds to apo-protein with addition of a single dopamine modification by mass. Figure 6c is PD performed on the peak at m/z 1068 selected from the spectrum in Figure 6b. By deconvolution of charge states it was determined that the peak corresponded to the +15 charge state of the beta chain of Hemoglobin with a single dopamine modification. PD of the unmodified +15 charge state of the beta chain of Hemoglobin (from a solution without dopamine) is shown in Figure 6d for comparison. PD of both the modified and unmodified protein generates peaks corresponding to fragments y131 and b15 which occur at W15. The side chain of tyrosine is also observed to be lost in both spectra. This fragmentation at tryptophan is atypical and is not observed in CID, thus it is likely due to electronic excitation at tryptophan.48 However this behavior is not due to the quinone modification as it is also present in the unmodified protein. Three peaks observed are unique to PD of the modified protein. The first of these is a loss of 184Da, the same loss observed with the dopamine modified peptide due to homolytic cleavage of the Cβ-S bond of cysteine. The presence of this peak alone confirms that the isolated species contains a dopamine modification at a cysteine residue. The other two unique peaks correspond to selective fragmentation at the modified site to yield z34 and d112+dopamine. These complementary d and z fragments occur due to radical directed fragmentation at cysteine 112; however, beta Hemoglobin contains another cysteine residue at position 93. Fragmentation at the modified residue should be accompanied by loss of the modification. Thus the d112+dopamine fragment is unexpected. Further experiments were performed to understand this unique instance. A subsequent step of CID on the d112+dopamine fragment was performed to determine if the dopamine was covalently or non-covalently attached to the protein fragment. Dopamine was not lost upon CID (data not shown), suggesting it is covalently bound.
Figure 7 shows CID spectra for the unmodified and modified beta chain of Hemoglobin, respectively. As seen in Figure 7a, CID of the unmodified protein yields abundant y111, y96, and y47 fragments, which form due to fragmentation at proline residues 36, 51, and 100 respectively.49,50 In comparison, CID of the modified protein shown in Figure 7b only yields abundant y111 and y96 fragments which are mass shifted by the addition of dopamine. Fragmentation at proline 100, yielding fragment y47 is observed in the modified protein with and without dopamine modification, however the fragmentation at proline 100 is greatly reduced in the modified protein. This indicates that there is something preventing fragmentation from occurring or being observed at proline 100. Due to the structure of dopamine, it is possible for two Michael type additions to occur on a single dopamine. The beta chain of Hemoglobin contains two cysteine residues. Attachment of both cysteines to a single dopamine would cyclize part of the protein and explain this fragmentation behavior. If proline fragmentation at P100 occurs during collisional activation, it will not be observed because both fragments are still held together by dopamine. This is consistent with the large decease in intensity of y47 in the modified protein. The small amount of y47 that is observed can be rationalized by a small amount of uncylcilzed protein with dopamine attached to only one cysteine. Even in this special case of double modification, PD is capable of identifying one of the sites of attachment with ease and the existence of the other is revealed by retention of the dopamine modification.
Figure 7.

a) CID of the +14 charge state of the unmodified beta chain of Hemoglobin. The primary fragments observed are y111, y96 and y47 occurring due to proline fragmentation at residues 36, 51, and 100 b) CID of the +14 charge state of beta Hemoglobin with a single dopamine modification.
Conclusions
Modification by quinones is selective to the free thiol form of cysteine and was shown to easily quantify the reactive cysteines in both peptides and proteins. Photodissociation of the quinone modified species was utilized to determine the location of modified residues through selective backbone fragmentation. Photo-excitation of the quinone homolytically cleaves the Cβ-S bond of cysteine. This homolytic cleavage results in a radical located at the beta position of cysteine, which facilitates electronic rearrangement to cleave the peptide or protein backbone at the modified cysteine. Photodissociation of modified alpha Hemoglobin was shown to selectively cleave a single bond in the protein backbone at the modified cysteine. This selective modification and fragmentation can be used to probe protein structure. Finally, the technique is amenable for identifying protein modification by naturally occurring quinones such as dopamine.
Acknowledgements
The authors gratefully acknowledge funding from the NSF (CHE-0747481, DGE-0504249) and the NIGMS R01GM084106.
References
- 1.Freedman RB. Curr. Opin. Struct. Biol. 1995;5:85–91. doi: 10.1016/0959-440x(95)80013-q. [DOI] [PubMed] [Google Scholar]
- 2.Darby N, Creighton TE. Method. Mol. Biol. 1995;40:219–252. doi: 10.1385/0-89603-301-5:219. [DOI] [PubMed] [Google Scholar]
- 3.Henehan CJ, Pountney DL, Zerbe O, Vasak M. Protein Sci. 1993;2:1756–1764. doi: 10.1002/pro.5560021019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ngu TT, Stillman MJ. Dalton Trans. 2009;28:5425–5433. doi: 10.1039/b902008j. [DOI] [PubMed] [Google Scholar]
- 5.Duncan KER, Ngu TT, Chan J, Salgado MT, Merrifield ME, Stillman MJ. Exp. Biol. M. 2006;231:1488–1499. doi: 10.1177/153537020623100907. [DOI] [PubMed] [Google Scholar]
- 6.Przybyla AE, Robbins J, Menon N, Peck HDJ. FEMS Microbiol. Rev. 1992;8:109–135. doi: 10.1111/j.1574-6968.1992.tb04960.x. [DOI] [PubMed] [Google Scholar]
- 7.Herring PA, Jackson JH. Microb. Comp. Genomics. 2000;5:75–87. doi: 10.1089/10906590050179765. [DOI] [PubMed] [Google Scholar]
- 8.Rauk A, Yu D, Taylor J, Shustov GV, Block DA, Armstrong DA. Biochemistry. 1999;38:9089–9096. doi: 10.1021/bi990249x. [DOI] [PubMed] [Google Scholar]
- 9.Ghezzi P, Bonetto V, Fratelli M. Antioxid. Redox Sign. 2005;7:7–8. doi: 10.1089/ars.2005.7.964. [DOI] [PubMed] [Google Scholar]
- 10.Barnes S, Shonsey EM, Eliuk SM, Stella D, Barrett K, Srivastava OP, Kim H, Renfrow MB. Biochem. Soc. Trans. 2008;36:1037–1044. doi: 10.1042/BST0361037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ronsein GE, Miyamoto S, Bechara E, Di Mascio P, Martinez GR. Quim. Nova. 2006;29:563–568. [Google Scholar]
- 12.Grimsrud PA, Xie H, Griffin TJ, Bernlohr DA. J. Biol. Chem. 2008;283:21837–21841. doi: 10.1074/jbc.R700019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Witze ES, Old WM, Resing KA, Ahn NG. Nat. Methods. 2007;4:798–806. doi: 10.1038/nmeth1100. [DOI] [PubMed] [Google Scholar]
- 14.Thornalley PJ. Protein Oxidation and Disease. 2006:143–178. [Google Scholar]
- 15.Steen H, Mann M. Adv. Mass Spectrom. 2001;15:527–528. [Google Scholar]
- 16.Chalkley RJ, Brinkworth CS, Burlingame AL. J. Am. Soc. Mass Spectrom. 2006;17:1271–1274. doi: 10.1016/j.jasms.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 17.Zubarev RA, Kruger NA, Fridriksson EK, Lewis MA, Horn DM, Carpenter BK, McLafferty FW. J. Am. Chem. Soc. 1999;121:2857–2862. [Google Scholar]
- 18.Fung YME, Kjeldsen F, Silivra OA, Chan TWD, Zubarev RA. Angew. Chem. Int. Ed. 2005;44:6399–6403. doi: 10.1002/anie.200501533. [DOI] [PubMed] [Google Scholar]
- 19.Chowdhury SM, Munske GR, Ronald RC, Bruce JE. J. Am. Soc. Mass Spectrom. 2007;18:493–501. doi: 10.1016/j.jasms.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cui H, Leon J, Reusaet E, Bult A. J. Chromatogr. A. 1995;704:27–36. [Google Scholar]
- 21.Goodlett DR, Bruce JE, Anderson GA, Rist B, Pasa-Tolic L, Fiehn O, Smith RD, Aebersold R. Anal. Chem. 2000;72:1112–1118. doi: 10.1021/ac9913210. [DOI] [PubMed] [Google Scholar]
- 22.Kondo T. Seikagaku. 2004;76:385–390. [PubMed] [Google Scholar]
- 23.Kostic NM. Comments Inorg. Chem. 1988;8:137–162. [Google Scholar]
- 24.Thevis M, Loo RRO, Loo JA. J. Proteome Res. 2003;2:163–172. doi: 10.1021/pr025568g. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Vivekananda S, Men L, Zhang Q. J. Am. Soc. Mass Spectrom. 2004;15:697–702. doi: 10.1016/j.jasms.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 26.Zhou H, Boyle R, Aebersold R. Method. Mol. Biol. 2004;261:511–518. doi: 10.1385/1-59259-762-9:511. [DOI] [PubMed] [Google Scholar]
- 27.Dayon L, Roussel C, Girault HH. Chimia. 2004;58:204–207. [Google Scholar]
- 28.Mason DE, Liebler DC. Chem. Res. Toxicol. 2000;13:976–982. doi: 10.1021/tx0000670. [DOI] [PubMed] [Google Scholar]
- 29.Roussel C, Dayon L, Lion N, Rohner TC, Josserand J, Rossier JS, Jensen H, Girault HH. J. Am. Soc. Mass Spectrom. 2004;15:1767–1779. doi: 10.1016/j.jasms.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 30.Diedrich JK, Julian RR. J. Am. Chem. Soc. 2008;130:12212–12213. doi: 10.1021/ja8023719. [DOI] [PubMed] [Google Scholar]
- 31.Izquierdo JG, Amaral GA, Ausfelder F, Aoiz FJ, Banares L. ChemPhysChem. 2006;7:1682–1686. doi: 10.1002/cphc.200600232. [DOI] [PubMed] [Google Scholar]
- 32.Ly T, Julian RR. J. Am. Chem. Soc. 2008;130:351–358. doi: 10.1021/ja076535a. [DOI] [PubMed] [Google Scholar]
- 33.Sun Q, Nelson H, Ly T, Stoltz BM, Julian RR. J. Proteome Res. 2009;8:958–966. doi: 10.1021/pr800592t. [DOI] [PubMed] [Google Scholar]
- 34.Bagheri-Majdi E, Ke Y, Orlova G, Chu IK, Hopkinson AC, Siu KWM. J. Phys. Chem. B. 2004;108:11170–11181. [Google Scholar]
- 35.Hodyss R, Cox HA, Beauchamp JL. J. Am. Chem. Soc. 2005;127:12436–12437. doi: 10.1021/ja052042z. [DOI] [PubMed] [Google Scholar]
- 36.Laskin J, Yang Z, Lam C, Chu IK. Anal. Chem. 2007;79:6607–6614. doi: 10.1021/ac070777b. [DOI] [PubMed] [Google Scholar]
- 37.Masterson DS, Yin H, Chacon A, Hachey DL, Norris JL, Porter NA. J. Am. Soc. Mass Spectrom. 2004;126:720–721. doi: 10.1021/ja038615u. [DOI] [PubMed] [Google Scholar]
- 38.O'Hair RAJ, Styles ML, Reid GE. J. Am. Soc. Mass Spectrom. 1998;9:1275–1284. [Google Scholar]
- 39.Ryzhov V, Lam AKY, O'Hair RAJ. J. Am. Soc. Mass Spectrom. 2009;20:985–995. doi: 10.1016/j.jasms.2008.12.026. [DOI] [PubMed] [Google Scholar]
- 40.Kuwata K, Hoshino M, Forge V, Era S, Batt CA, Goto Y. Protein Sci. 1999;8:2541–2545. doi: 10.1110/ps.8.11.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McKenzie HA, Ralston GB, Shaw DC. Biochemistry. 1972;11:4539–4547. doi: 10.1021/bi00774a017. [DOI] [PubMed] [Google Scholar]
- 42.Roussel C, Rohner TC, Jensen H, Girault HH. ChemPhysChem. 2003;4:200–206. doi: 10.1002/cphc.200390031. [DOI] [PubMed] [Google Scholar]
- 43.Ferguson PL, Kuprowski MC, Boys BL, Wilson DJ, Pan J, Konermann L. Curr. Anal. Chem. 2009;5:186–204. [Google Scholar]
- 44.Kaltashov IA, Abzalimov RR. J. Am. Soc. Mass Spectrom. 2008;19:1239–1246. doi: 10.1016/j.jasms.2008.05.018. [DOI] [PubMed] [Google Scholar]
- 45.Ly T, Julian RR. J. Am. Soc. Mass Spectrom. 2009;20:1148–1158. doi: 10.1016/j.jasms.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 46.LaVoie MJ, Hastings TG. J. Neurosci. 1999;19:1484–1491. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nicolis S, Zucchelli M, Monzani E, Casella L. Chem. Eur. J. 2008;14:8661–8673. doi: 10.1002/chem.200801014. [DOI] [PubMed] [Google Scholar]
- 48.Perot M, Lucas B, Barat M, Fayeton JA, Jouvet C. J. Phys. Chem. A. 2009;ASAP doi: 10.1021/jp908937s. [DOI] [PubMed] [Google Scholar]
- 49.Loo JA, Edmonds CG, Smith RD. Anal. Chem. 1993;65:425–438. doi: 10.1021/ac00052a020. [DOI] [PubMed] [Google Scholar]
- 50.Vaisar T, Urban J. J. Mass Spectrom. 1996;31:1185–1187. doi: 10.1002/(SICI)1096-9888(199610)31:10<1185::AID-JMS396>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]