

Abstract
Peroxynitrite is formed in macrophages by the diffusion-limited reaction of superoxide and nitric oxide. This highly reactive species is thought to contribute to bacterial killing by interaction with diverse targets and nitration of protein tyrosines. This work presents for the first time a comprehensive analysis of transcriptional responses to peroxynitrite under tightly controlled chemostat growth conditions. Up-regulation of the cysteine biosynthesis pathway and an increase in S-nitrosothiol levels suggest S-nitrosylation to be a consequence of peroxynitrite exposure. Genes involved in the assembly/repair of iron-sulfur clusters also show enhanced transcription. Unexpectedly, arginine biosynthesis gene transcription levels were also elevated after treatment with peroxynitrite. Analysis of the negative regulator for these genes, ArgR, showed that post-translational nitration of tyrosine residues within this protein is responsible for its degradation in vitro. Further up-regulation was seen in oxidative stress response genes, including katG and ahpCF. However, genes known to be up-regulated by nitric oxide and nitrosating agents (e.g. hmp and norVW) were unaffected. Probabilistic modeling of the transcriptomic data identified five altered transcription factors in response to peroxynitrite exposure, including OxyR and ArgR. Hydrogen peroxide can be present as a contaminant in commercially available peroxynitrite preparations. Transcriptomic analysis of cells treated with hydrogen peroxide alone also revealed up-regulation of oxidative stress response genes but not of many other genes that are up-regulated by peroxynitrite. Thus, the cellular responses to peroxynitrite and hydrogen peroxide are distinct.
Keywords: Bacterial Transcription, Microarray, Oxidative Stress, Reactive Oxygen Species, Transcription, Nitration, Peroxynitrite, S-Nitrosylation
Introduction
In response to infection, mammalian immune cells form high levels of nitric oxide and superoxide by the activity of inducible nitric-oxide synthase and NADPH oxidase, respectively. Peroxynitrite is generated by the subsequent diffusion-controlled reaction of nitric oxide with superoxide. Peroxynitrite is a highly reactive species that gives rise to both oxidative (1–3) and nitrosative (4, 5) stresses in bacteria (reviewed in Ref. 6). The half-life of this species is thought to be as little as <10 ms in biological systems (6); however, this is sufficient time for interaction with both intracellular bacteria and those in the vicinity of the phagocyte (7). Once at the site of a bacterial membrane, peroxynitrite can enter the cell either by passive diffusion (i.e. as peroxynitrous acid, ONOOH) or by anion channels (as peroxynitrite anion, ONOO−). Here, it is able to react with a variety of targets causing protein modification (5), lipid oxidation (8), and DNA damage (9), either directly or indirectly via its breakdown products (the term peroxynitrite hereafter is used to refer to the sum of ONOO− and ONOOH).
Oxidative stress caused by peroxynitrite can arise from the direct oxidation reactions of peroxynitrous acid or from the formation of oxidizing radicals (e.g. •OH) and can result in the oxidation of lipids, thiols, and heme proteins, the hydroxylation of aromatic centers, and damage to iron-sulfur clusters (6). Nitrosative stress may be caused by the nitration of phenols (5) and the nitrosylation of thiols. Peroxynitrite has been shown to react with and alter the function and/or stability of proteins, including lysozyme and phosphatase (10, 11).
Previous studies into the bacterial detoxification of peroxynitrite have implicated, among others, the oxidative stress response enzymes alkyl hydroperoxide reductase (AhpCF) from Salmonella enterica sv. Typhimurium (2, 3), catalase-hydroperoxidase I (KatG) from Mycobacterium tuberculosis (12), and S. enterica sv. Typhimurium (13) and the truncated hemoglobin GlbO from Mycobacterium leprae (14). These enzymes were identified as peroxynitritases using in vitro assays with purified components, suggesting a role for peroxiredoxins and heme-containing proteins in peroxynitrite detoxification. Another route of peroxynitrite breakdown in biological systems is thought to be via its reaction with carbon dioxide (7, 15–17), which can rapidly react with peroxynitrite to form carbonate and nitrogen dioxide radicals. This fast reaction can limit the toxicity of peroxynitrite to bacterial cells not in the immediate vicinity of its formation and in a proximity-dependent manner (7).
Although much work has been reported on the interaction of peroxynitrite with discrete molecules in vitro, there is surprisingly little evidence in the literature concerning the global bacterial targets of this highly reactive species in vivo. Many in vivo studies investigate the overall effects of peroxynitrite on an organism as a function of growth rate and viability without an attempt to identify specific bacterial interactions (18).
The reactions of peroxynitrite are complex and their effects are wide ranging; this work is an attempt to define under carefully controlled conditions the bacterial targets of this highly reactive species and the mechanisms employed by the bacteria to combat them.
EXPERIMENTAL PROCEDURES
E. coli Growth Conditions
The strain used was an E. coli K12 derivative, MG1655. Cells were grown in defined minimal medium with glycerol as the sole carbon source, as described previously (19). For batch culture, cells were grown at 37 °C and 200 rpm in 20 ml of defined medium. The 250-ml flasks used were fitted with side arms for measurements of optical density using a Klett-Summerson photoelectric colorimeter (Klett Manufacturing Co.) fitted with a number 66 red filter. Optical densities are given as standard Klett units without correction. For continuous culture, cells were grown in an Infors Labfors-3 fermentor as described previously (19) with defined medium containing 2 mm glycerol as the limiting carbon source.
Viability Assays
Viability was measured after serial dilution of samples in phosphate-buffered saline by plating 10-μl aliquots on Luria-Bertani agar and incubation overnight at 37 °C.
Peroxynitrite Treatment
Peroxynitrite preparations were supplied by Calbiochem. The reagent was added directly to growing cultures in log phase, determined by a Klett value of ∼50 or an A600 of ∼0.6 measured on a Beckman DU® 650 spectrophotometer when grown in fermentor vessels.
Microarray and Real Time PCR Analysis
Cells were grown aerobically in continuous culture as described above. Following 25 h of continuous growth, 10 ml of culture was added to RNAprotect (Qiagen) immediately before and 5 min after exposure to 300 μm peroxynitrite or hydrogen peroxide. RNA extraction was achieved using the RNeasy minikit from Qiagen. Each condition included three biological repeats of control and stressed cells. Other procedures for microarray analysis and RT-PCR4 were as described previously (19). The GEO accession number for the microarray dataset is GSE19370 and can be viewed at www.ncbi.nlm.nih.gov.
Modeling Transcription Factor Activities
Transcriptomic data were analyzed using a probabilistic model as described previously (20), which adopts a log-linear approximation to the transcriptional reaction to changes in transcription factor activity.
Assay of S-Nitrosothiol Levels
E. coli cultures were grown in defined media to log phase and incubated in the presence or absence of 1 mm peroxynitrite for 1 h before lysis by sonication. Lysates were cleared by centrifugation and treated, or not, with HgCl2 to remove S-nitrosothiols (SNOs). SNO concentrations were measured by chemiluminescence on a Sievers model 280i nitric oxide analyzer (21). SNO concentrations were calculated from the integral of the detected signal over time and were compared with a series of standards using S-nitrosoglutathione. Data were normalized to total protein content as determined by the Bradford assay (22).
Cloning and Purification of Recombinant ArgR
The argR gene was PCR-amplified from E. coli genomic DNA and ligated into pre-digested pTrcHis A vector (Invitrogen) by standard techniques. The plasmid was used to transform chemically competent E. coli JM109 cells. Cultures were grown in 1 liter of Circlegrow® medium supplemented with ampicillin (150 μg/ml) in 2-liter baffled flasks for 20 h at 30 °C and 250 rpm. Cells were harvested by centrifugation and resuspended in 50 mm Tris/MOPS, pH 8.0, and 300 mm NaCl (binding buffer) plus a mixture of protease inhibitors (Sigma, 1 ml/20 g cell weight).
Cells were lysed by sonication, on ice and cell debris was removed by centrifugation at 20,000 × g at 4 °C for 30 min. The supernatant was loaded onto a pre-equilibrated Talon metal affinity column (Clontech) and washed with binding buffer followed by wash buffer (binding buffer plus 10 mm imidazole). The protein was eluted with 200 mm imidazole and subsequently desalted on a PD-10 desalting column (Bio-Rad) with 50 mm Tris/MOPS, pH 8.0.
SDS-PAGE and Western Blot Analysis of ArgR Nitration
Aliquots of 60 μm ArgR protein were incubated at room temperature with varying concentrations of peroxynitrite before analysis by SDS-PAGE on 4–20% polyacrylamide gels. Protein degradation was monitored by staining with Coomassie Brilliant Blue R-250 (Bio-Rad). Nitration was confirmed by transfer to nitrocellulose membranes and subsequent incubation with monoclonal antibodies to 3-nitrotyrosine (Axxora Ltd.) followed by washing and incubation with horseradish peroxidase-conjugated goat anti-mouse IgG (Sigma). Membranes were exposed to x-ray film following reaction with ECL reagents.
RESULTS
Toxicity of Peroxynitrite Is Cell Density-dependent
At very low cell densities (106 bacteria/ml), peroxynitrite exerts significantly greater toxicity to Escherichia coli than nitric oxide (18). Because of the inherent reactivity of peroxynitrite, we wanted to test whether the toxicity of peroxynitrite was dependent on culture turbidity, i.e. the concentration of cell targets. E. coli cells were grown in defined medium to 5, 25, or 50 Klett units (4 × 107, 2 × 108, or 4 × 108 colony-forming units/ml, respectively) and stressed with various concentrations of peroxynitrite (0–800 μm). The cell density was monitored for several hours further to assess the effect upon growth. Fig. 1(A–C) shows that E. coli resistance to peroxynitrite increases with cell density, presumably due to a larger number of targets with which the stressor reacts thus lessening the impact on individual cells. The corresponding viable count assays, taken 1 h post-treatment, provided further evidence that higher cell densities reduced the toxic effect of peroxynitrite with only the highest concentration (800 μm) showing a significant decrease in cell viability (Fig. 1, D–F). At the lowest cell density (5 Klett units), a significant decrease in viability was seen at much lower peroxynitrite concentrations with a reduction in viability from the time of injection being evident at 800 μm peroxynitrite. This is in contrast to a previous study, which showed that peroxynitrite toxicity was independent of culture turbidity over the range 106 to 1010 colony-forming units/ml (23).
FIGURE 1.

Cell density dependence on growth and viability of E. coli in response to peroxynitrite stress. Cells were stressed with 0 (●), 200 (▼), 400 (■), or 800 μm (♦) peroxynitrite at 5 (A), 25 (B) and 50 (C), Klett units (indicated by arrows). Data are representative of three biological replicates. Viability counts were determined for cells stressed with 0, 200, 400 or 800 μm peroxynitrite at 5 (D), 25 (E), and 50 (F) Klett units after incubation for 1 h. * and # denote a statistically significant reduction in viable cell counts relative to unstressed cells after 1 h and to cell numbers at the time of stress (horizontal line), respectively. CFU, colony-forming units.
Transcriptomic Changes in Response to Peroxynitrite
Transcriptomic analysis was undertaken on samples derived from chemically defined medium in continuous culture under aerobic conditions. Under these conditions, E. coli were grown under highly controlled conditions of pH, aeration, and medium feed rate, allowing changes in gene expression to be attributed solely to peroxynitrite and not to other factors, for example changes in growth rate or differences in nutrient levels. Cell density dependence data show that cultures growing at ∼50 Klett units (A600 ∼0.6) were able to withstand a stress of 300 μm peroxynitrite without significant loss of viability, and the reduction in growth rate was such that a dilution rate of 0.2 h−1 (equivalent to a doubling time of 3.47 h) could be used during continuous culture without risk of washout upon exposure to the stressor (Fig. 1).
Cells were grown continuously for 5 culture volumes prior to stressing in defined medium with 2 mm glycerol as the limiting carbon source to maintain a cell density of A600 ∼0.6. The cultures were stressed with 300 μm peroxynitrite, and sampling took place immediately prior to and 5 min after treatment. Samples were immediately removed into RNAprotect to stabilize the RNA for transcriptomic analysis. Microarrays showed a number of genes up-regulated in response to peroxynitrite stress, which could be classified into several key areas (Fig. 2). A number of oxidative stress response genes were up-regulated, most notably katG and ahpCF. KatG has been proposed to act as a peroxynitritase in M. tuberculosis (12) as have KatG and AhpCF in S. enterica sv. Typhimurium (2, 13). Interestingly, the expression of none of the recognized nitrosative stress response genes (hmp, norVW etc.) was altered after peroxynitrite exposure, indicating that this reactive species does not act as a classical nitrosative stress, as do nitric oxide or GSNO (19, 24). Both the cysteine and arginine biosynthesis pathways were up-regulated, indicating possible targets of attack by peroxynitrite as well as genes involved in iron-sulfur cluster assembly/repair, the high affinity phosphate transport system (pst), and the (gsi) glutathione import system. The transcription levels of several genes encoding membrane and transport proteins were also altered both positively and negatively (supplemental Tables 1 and 2), which is suggestive of interactions between peroxynitrite and the membrane during passage into the cell.
FIGURE 2.

Microarray analysis of the differential expression of genes involved in the response to peroxynitrite exposure. The mean fold change in individual gene expression after exposure to 300 μm peroxynitrite compared with unstressed controls grown aerobically is indicated by the color scale bar. Unless otherwise stated, p values were <0.05; * indicates a p value of between 0.05 and 0.1.
Transcriptomic Changes in Response to Hydrogen Peroxide Stress
Commercially available preparations of peroxynitrite may contain hydrogen peroxide and nitrite as unused substrates from synthesis (25). These contaminants clearly have the potential to influence the transcriptomic data obtained from the microarray analysis of peroxynitrite-treated cells. As no nitrosative stress response genes were up-regulated, it could be concluded that no effect from nitrite contamination was evident. However, due to the up-regulation of oxidative stress response genes after peroxynitrite exposure, further transcriptomic analysis was undertaken using 300 μm hydrogen peroxide as the stressor to compare and contrast the transcriptomic response to hydrogen peroxide treatment with that of peroxynitrite preparations. This is the first assessment of the transcriptomic effects of hydrogen peroxide upon E. coli cells in defined medium and continuous culture conditions. As expected, oxidative stress response genes were up-regulated along with genes involved in hexuronate utilization and dps, which protects DNA from the oxidative damage caused by hydrogen peroxide (Fig. 3). However, the overall response to hydrogen peroxide treatment under these conditions was mild compared with that for peroxynitrite stress, with the transcription levels of fewer genes being significantly altered.
FIGURE 3.

Microarray analysis of the differential expression of genes involved in the response to hydrogen peroxide treatment. The mean fold change in individual gene expression after exposure to 300 μm hydrogen peroxide compared with unstressed controls grown aerobically is indicated by the color scale bar. p values were <0.05.
Real Time PCR Analysis of Transcriptomic Changes
RT-PCR was used to corroborate the microarray data and provide further evidence for the implication of specific transcriptional changes in response to peroxynitrite. RT-PCR of peroxynitrite-treated cells utilized peroxynitrite that was pre-treated with MnO2, which is known to remove contaminating hydrogen peroxide (26) by catalyzing its breakdown into oxygen and water. This was used as a further control to ensure that the transcriptomic responses seen in the microarray data were due to peroxynitrite (or its breakdown products) alone.
In the case of peroxynitrite, we selected genes (Fig. 4A) whose up-regulation had been observed to be significant in the microarray data, namely argI (ornithine transcarbamylase), cysD and cysJ (sulfate and sulfite metabolism), ahpC (alkyl hydroperoxide reductase), and katG (catalase-hydroperoxidase). All were significantly up-regulated (4–8-fold) when studied by RT-PCR, in broad agreement with the transcriptome data. Two genes were also chosen to confirm the lack of up-regulation of the nitrosative stress response (hmp and norV). RT-PCR was also undertaken using peroxynitrite not treated with MnO2 as a true control for the microarray data. The transcriptomic changes were in agreement with that of the MnO2-treated RT-PCR and microarray figures (data not shown). In the case of hydrogen peroxide, we selected three genes (Fig. 4B) whose expression was marginally elevated in the microarrays (2–4-fold; Fig. 3), i.e. entA, fumC, and uxaC. In addition, we studied katG, whose expression is up-regulated in both arrays but to a lesser extent in the hydrogen peroxide dataset and argI as an example of a gene that is 4–6-fold up-regulated in the microarrays of peroxynitrite cells. The katG gene was up-regulated ∼3-fold, which is approximately half that seen in the peroxynitrite RT-PCR. The other four genes showed changes of about 2-fold or less (Fig. 4B), which is the minimal threshold normally applied to transcriptome data. Thus, expression of entA, fumC, and uxaC (none of which is regulated by OxyR) is not markedly influenced by hydrogen peroxide under these conditions, and hydrogen peroxide contaminating the peroxynitrite appears not to be responsible for the changes in arg gene expression. In all cases, genes were normalized to the control gene fixA.
FIGURE 4.

RT-PCR was used to monitor the expression of genes involved in the response to MnO2-treated peroxynitrite (A) or hydrogen peroxide (B). Cells grown in batch culture were exposed to 300 μm peroxynitrite pretreated with MnO2 or 300 μm hydrogen peroxide, respectively, for 5 min before aliquots of culture were removed to RNAprotect and processed for RT-PCR analysis. The mean fold change in individual gene expression compared with unstressed cells was calculated (n = 3 ± S.E.). C, growth of the oxyR mutant was monitored in the presence and absence (●) of 300 μm peroxynitrite (▼) or hydrogen peroxide (○). Where appropriate, cells were stressed at 50 Klett units (indicated by arrow). Values are mean ± S.E. of three biological repeats.
Statistical Analysis of Transcriptomic Data
Probabilistic modeling of the transcriptomic data were focused upon a set of five regulators as follows: OxyR, ArgR, CysB, PhoB, and ExuR based on gene changes in the microarray data. A list of the genes making up the regulons of these transcription factors, as well as information on the sign of the regulation (if available), was obtained from regulonDB (available on line). Analysis of the data was based on the probabilistic state-space model developed by Sanguinetti et al. (20), which has been utilized previously to identify transcription factor activity changes of E. coli upon exposure to carbon monoxide (27). This uses a log-linear approximation to the dynamics of transcriptional regulation, which are then modeled as shown in Equation 1,
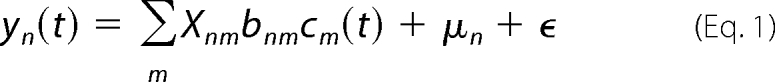 |
Here, yn(t) is the mRNA log fold change for gene n at time t; Xnm is a binary matrix whose nm entry is 1 if and only if gene n is bound by transcription factor m; bnm is a constant representing the sensitivity of gene n to transcription factor m; cm(t) is the log activity of transcription factor m at time t; μn represents the base-line expression for each gene, and ϵ is an error term. The sensitivities bnm are given a zero-mean Gaussian prior distribution, whereas the activities are modeled using a stationary, unitary variance Gaussian Markov process. In this case, the yn(0) = 0 for all genes, so that the time course consists of only 2 points (0 and 5 min).
The estimated change in transcription factor activity (with associated uncertainty) for these regulators revealed that peroxynitrite causes a significant change in OxyR, ArgR, CysB, and PhoB after 5 min of exposure, although the same concentration of hydrogen peroxide causes small but statistically significant changes in the activity of OxyR and ExuR only (Fig. 5). Because the transcription factor activities are given the same prior distribution with unitary variance, a direct comparison of the activities obtained from the two data sets, as presented in Fig. 5, is meaningful. To empirically ascertain the statistical significance of the results, we reran the model with 100 independent randomizations of the network structure. The null hypothesis is that with a random network there should be no signal, so that only very few if any of the transcription factors would change significantly. Using this analysis, we can estimate that the probability of obtaining significant changes in the activity of four transcription factors from random data is ∼10−5.
FIGURE 5.

Probabilistic modeling of transcriptomic data for the effects of peroxynitrite and hydrogen peroxide treatment. Predicted activities (which may be positive or negative) in response to treatment with 300 μm peroxynitrite (black) or hydrogen peroxide (gray) are shown for the transcription factors OxyR, ArgR, CysB, PhoB, and ExuR.
Deletion of OxyR Changes the Cellular Response to Peroxynitrite
A striking response of E. coli to peroxynitrite is up-regulation of katG and ahpCF (Fig. 2 and Fig. 4, A and B), and so we explored the basis of this response by RT-PCR analysis of an E. coli strain lacking OxyR (ΔoxyR, GSO77) (28). Deletion of this transcription factor eliminated the up-regulation of ahpC and katG in response to both peroxynitrite and hydrogen peroxide stress (Fig. 4). This suggests that these genes are up-regulated by OxyR in response to peroxynitrite and hydrogen peroxide in wild type cells, and this highlights a common response to these two oxidative stresses.
The ΔoxyR strain was also grown in batch culture to monitor the impact upon growth of 300 μm peroxynitrite or hydrogen peroxide in a strain lacking this major oxidative stress response transcription factor. This strain was more sensitive to both stresses than wild type (Fig. 4C). The ΔoxyR strain was more susceptible to peroxynitrite stress than hydrogen peroxide; this is in agreement with our probabilistic modeling data, which show that the activity of OxyR is increased to a greater degree in response to peroxynitrite (Fig. 5).
Peroxynitrite Increases S-Nitrosothiol Levels in E. coli
S-Nitrosylation describes the covalent attachment of a nitric oxide moiety to the thiol residue of a protein, peptide, or amino acid. It is a post-translational modification, which is thought to provide a signaling mechanism in mammals and can be damaging to bacterial cells via alteration of protein activities (19, 29). Thiols are the functional group of cysteines, and as the biosynthetic pathway of this amino acid was up-regulated in response to peroxynitrite treatment (Fig. 2), we wanted to ascertain whether this was a response by the cell to the peroxynitrite-mediated formation of SNOs in E. coli. Cells were grown aerobically in batch culture to log phase before the addition of 1 mm peroxynitrite, which gives a sufficiently large signal to be reliably monitored in the SNO assay. Cultures were incubated for a further 1 h followed by processing for detection of SNOs by chemiluminescence. In the absence of peroxynitrite, there were virtually undetectable levels of SNOs. These samples did however contain small amounts of other nitroso species (e.g. iron nitrosyls/nitrosamines). When exposed to 1 mm peroxynitrite for 1 h, ∼6 pmol of SNOs per mg of total protein were formed (Table 1). This indicates that peroxynitrite is able to form new species by S-nitrosylation, which may provide an explanation for the up-regulation of cysteine biosynthesis genes in response to peroxynitrite.
TABLE 1.
S-Nitrosothiol accumulation measured in wild type E. coli cells
Cells were grown at 37 °C and 200 rpm to an A600 of ∼0.6 and then stressed, or not, with 1 mm peroxynitrite and grown for a further 1 h. Nitrosylation was detected by photolysis-chemiluminescence.
| Stress | SNO | Hg nondisplaceable |
|---|---|---|
| pmol per mg protein | ||
| None | 0.00 ± 0.06 | 0.33 ± 0.05 |
| 1 mm peroxynitrite | 5.72 ± 2.48 | 0.05 ± 0.05 |
Peroxynitrite Nitrates and Degrades ArgR in Vitro
The ArgR protein is a negative regulator of the arginine biosynthesis pathway and forms stable hexamers with a molecular mass of ∼100 kDa. Upon binding to arginine, the ArgR hexamer is allosterically activated for binding to DNA operator sequences upstream of each of the regulated genes thus repressing their transcription. Abolition of this binding would cause an up-regulation of the arginine biosynthesis pathway.
Our probabilistic modeling data suggest that the activity of the ArgR transcription factor is altered upon exposure to peroxynitrite. To test this hypothesis, first, the argR gene was cloned into an overexpression vector followed by expression in Circlegrow® medium. The protein was purified using cobalt affinity and desalting resins. ArgR was shown to be expressed and pure by N-terminal sequencing and SDS-PAGE analysis. Pure ArgR ran as a hexamer on SDS-polyacrylamide gels (Fig. 6A). Aliquots of this protein were incubated at room temperature with peroxynitrite at different concentrations, and the resulting degradation of ArgR was followed by SDS-PAGE and Coomassie staining. Fig. 6A shows the degradation of ArgR after a 5-min incubation with peroxynitrite over a range of ratios, indicating that a molar ratio of 1:10 is sufficient for protein degradation.
FIGURE 6.
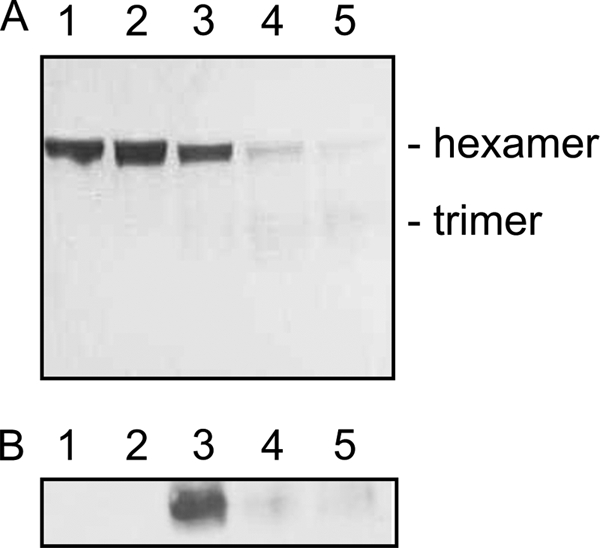
Peroxynitrite causes the nitration and degradation of ArgR in vitro. A, degradation of 60 μm ArgR in the presence of peroxynitrite. Coomassie-stained SDS-polyacrylamide gel of ArgR incubated for 5 min with peroxynitrite in the following molar ratios: lane 1, 1:0; lane 2, 1:1; lane 3, 1:10; lane 4, 1:50, and lane 5, 1:100. B, 20 μm ArgR was incubated with peroxynitrite at a molar ratio of 1:50 for varying lengths of time, followed by SDS-PAGE and Western blot analysis against 3-nitrotyrosine antibodies. Lane 1, peroxynitrite; lane 2, ArgR; lanes 3–5, ArgR incubated with peroxynitrite for 5, 30, and 60 min, respectively.
ArgR samples incubated with peroxynitrite at a molar ratio of 1:50 for between 5 and 60 min were electrophoresed and transferred to nitrocellulose membrane for Western blotting against 3-nitrotyrosine monoclonal antibodies. Exposure to x-ray film after treatment with ECL reagents revealed that ArgR is nitrated by peroxynitrite in vitro (Fig. 6B) and that this nitrated species seems to breakdown, which is in agreement with Coomassie-stained protein gels over the same time course (data not shown). As only the hexameric form of ArgR is able to bind the operator DNA (30) and hence repress arginine biosynthetic gene transcription, it is sensible to suggest that this nitration could cause up-regulation of the pathway observed upon exposure to peroxynitrite.
DISCUSSION
The formation rate of peroxynitrite has been estimated at up to 100 μm/min in selected cellular compartments (16). Our own work used a bolus injection of 300 μm peroxynitrite, which should be equivalent to a steady state concentration of 8.4 μm at physiological pH and temperature (31). This level of peroxynitrite is therefore likely to be comparable with levels produced in vivo, as rat alveolar macrophages have been reported to produce up to 1 mm peroxynitrite/min, with concentrations inside the phagocyte likely to be even higher (32). The toxicity of peroxynitrite seems to be more dependent upon the ratio of cell numbers to stressor rather than absolute concentrations (Fig. 1), and so the work presented in this paper attempts to identify bacterial targets and responses when E. coli comes into contact with peroxynitrite rather than to determine what may happen in vivo where a much more complex system exists.
Peroxynitrite is thought to be highly toxic toward cells due to its inherent reactivity; however, a previous study into the effects of nitric oxide upon cell viability utilizing the same conditions as used in this study found that the addition of 200 μm nitric oxide to mid-log phase cultures resulted in a viability drop of ∼30% (24). This is in contrast to the viability of cultures after the addition of 200 μm peroxynitrite, which at mid-log phase (50 Klett units) is not significantly decreased (Fig. 1F). This could be due to the highly reactive nature of peroxynitrite, which is likely to react with extracellular compounds in the media as well as bacterial cells. A second study utilizing cultures transferred to buffer prior to stressing found peroxynitrite toxicity to be much greater than nitric oxide (18).
Transcriptomic analysis of the bacterial response to peroxynitrite indicates that this highly reactive species acts predominantly as an agent of oxidative stress, with targets also including cysteine and arginine biosynthesis and iron-sulfur clusters (Fig. 2). Several oxidative stress response genes are up-regulated, which is in contrast to the classical nitrosative stress response genes that show no change in activity. However, the lack of response by some of the classical nitrosative stress response genes is not surprising as proteins such as Hmp and NorVW specifically detoxify nitric oxide, levels of which would not be expected to increase during peroxynitrite exposure. Furthermore, an increase in Hmp expression during peroxynitrite stress has deleterious effects on S. enterica sv. Typhimurium,5 causing hypersensitivity to the stress. This is probably due to the Hmp-catalyzed production of superoxide in the absence of nitric oxide. In this study, however, the ytfE gene, the product of which is thought to play a role in iron-sulfur assembly and repair in response to both oxidative and nitrosative stresses (33, 34), was mildly up-regulated but not above the threshold of significance for our microarrays. Other genes involved in iron-sulfur cluster assembly and repair are also up-regulated (suf and isc genes). The regulation of other nitrosative stress response genes, including those responsible for nitrite detoxification (nrfA and hcp), was also unaltered by peroxynitrite exposure, suggesting that although undoubtedly levels of nitrite will increase upon exposure to peroxynitrite, levels were either insufficient to up-regulate these genes or detoxification of peroxynitrite was more significant than removal of the comparatively inert nitrite. Some genes that were up-regulated are of unknown function and could play a role in response to the apparent S-nitrosylation of thiols (Table 1) or in response to the nitration of tyrosine residues in proteins (Fig. 6). A significant number of genes encoding membrane proteins were also found to be up-regulated (supplemental Table 1), which suggests that peroxynitrite reacts with proteins in the membrane as well as causing lipid oxidation (35) during its passage into the cell. Nitration and oxidation of tyrosine residues in membrane proteins may be more easily allowed than those in the aqueous phase as key reductants in the cell (e.g. glutathione) are soluble, and so would have no effect in the membrane (36, 37). Also, the peroxynitrite-mediated oxidation and nitration of lipids have been demonstrated (35, 38) and identify another target of this highly reactive species. These changes to the membrane may damage its integrity and/or its functionality, and therefore the up-regulation of membrane proteins is probably a response by the cell to repair any damage caused.
Peroxynitrite, or one of its breakdown products, is able to S-nitrosylate the thiol side chains of cysteine residues within the bacterial cell (39, 40). This post-translational modification is reversible and selective, with the effect of altering activity, localization, and stability of the modified proteins (39, 40). In the human body, S-nitrosylation is thought to be a mechanism by which nitric oxide acts as a signaling molecule (reviewed in Ref. 41). However, in bacterial cells exposed to the mammalian immune response, S-nitrosylation may have deleterious effects causing dysfunction and even death. Interestingly, the glutathione import system (gsiABCD) is also up-regulated in response to peroxynitrite stress (Fig. 2), consistent with previous data showing a decrease in glutathione levels can precede the S-nitrosylation of enzymes (42). However, peroxynitrite per se does not cause S-nitrosylation; the formation of SNOs is more likely due to the production of nitrite during the breakdown of peroxynitrite and its subsequent reactions with nitrite reductases or other enzymes capable of reducing nitrite to nitric oxide.
Cysteine can also undergo oxidation caused by peroxynitrite stress (43); this would further increase the requirement of the cells for cysteine and thus enhance up-regulation of the cysteine biosynthetic pathway.
KatG is up-regulated in the peroxynitrite microarrays (Fig. 2), and previous work has found that pure KatG from S. enterica sv. Typhimurium is able to act as a peroxynitritase (13). This suggests that KatG may function in a wider role in response to oxidative stress than previously thought. However, it is reported that KatG does not appear to be as efficient as catalase (44, 45) or AhpC (2) at enhancing the breakdown of peroxynitrite.
This work reveals for the first time an unexpected target of peroxynitrite, namely the arginine biosynthesis pathway. ArgR is the transcriptional regulator for arginine biosynthesis in E. coli. The ArgR polypeptide has a molecular mass of ∼17 kDa, but its functional form is hexameric (30). It is allosterically activated by arginine and acts by binding to operator DNA upstream of arginine biosynthetic genes in the arg regulon causing a repression of transcription.
Peroxynitrite has been shown to nitrate one or more of the three tyrosine residues present in ArgR in vitro (Fig. 6). This nitration appears to result in a degradation of not only the hexameric form of the protein but of the trimer also, which will result in an inability to bind the operator sequences of the arginine biosynthesis genes under its control even in the presence of arginine, resulting in de-repression of transcription. These observations are supported by the transcriptomic data, which show a clear up-regulation in arginine biosynthesis after 5 min of exposure to 300 μm peroxynitrite (Figs. 2 and 5) and by the probabilistic modeling work that predicted the activity change of the regulator ArgR in response to peroxynitrite. Supplementation of the growth medium with excess arginine does not reduce the lag in growth caused by peroxynitrite treatment (data not shown), which also supports the evidence that this is an alteration in the regulation of arginine biosynthesis rather than a response to arginine depletion.
Supplementary Material
Acknowledgment
The GSO77 strain was a kind gift from the laboratory of Dr. Gisela Storz.
This work was supported by the Biotechnology and Biological Sciences Research Council (United Kingdom).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables 1 and 2.
S. McLean and R. K. Poole, unpublished data.
- RT
- real time
- SNO
- S-nitrosothiol
- MOPS
- 4-morpholinepropanesulfonic acid.
REFERENCES
- 1.Beckman J. S., Beckman T. W., Chen J., Marshall P. A., Freeman B. A. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 1620–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bryk R., Griffin P., Nathan C. (2000) Nature 407, 211–215 [DOI] [PubMed] [Google Scholar]
- 3.Master S. S., Springer B., Sander P., Boettger E. C., Deretic V., Timmins G. S. (2002) Microbiology 148, 3139–3144 [DOI] [PubMed] [Google Scholar]
- 4.Pfeiffer S., Gorren A. C., Schmidt K., Werner E. R., Hansert B., Bohle D. S., Mayer B. (1997) J. Biol. Chem. 272, 3465–3470 [DOI] [PubMed] [Google Scholar]
- 5.Ischiropoulos H. (2009) Arch. Biochem. Biophys. 484, 117–121 [DOI] [PubMed] [Google Scholar]
- 6.Ferrer-Sueta G., Radi R. (2009) ACS Chem. Biol. 4, 161–177 [DOI] [PubMed] [Google Scholar]
- 7.Alvarez M. N., Piacenza L., Irigoín F., Peluffo G., Radi R. (2004) Arch. Biochem. Biophys. 432, 222–232 [DOI] [PubMed] [Google Scholar]
- 8.Lee J. B., Hunt J. A., Groves J. T. (1998) J. Am. Chem. Soc. 120, 6053–6061 [Google Scholar]
- 9.Salgo M. G., Bermúdez E., Squadrito G. L., Pryor W. A. (1995) Arch. Biochem. Biophys. 322, 500–505 [DOI] [PubMed] [Google Scholar]
- 10.Curry-McCoy T. V., Osna N. A., Donohue T. M., Jr. (2009) Biochim. Biophys. Acta 1790, 778–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kohr M. J., Davis J. P., Ziolo M. T. (2009) Nitric Oxide-Biology and Chemistry 20, 217–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wengenack N. L., Jensen M. P., Rusnak F., Stern M. K. (1999) Biochem. Biophys. Res. Commun. 256, 485–487 [DOI] [PubMed] [Google Scholar]
- 13.McLean S., Bowman L. A., Poole R. K. (2010) FEBS Lett. 584, 1628–1632 [DOI] [PubMed] [Google Scholar]
- 14.Ascenzi P., Milani M., Visca P. (2006) Biochem. Biophys. Res. Commun. 351, 528–533 [DOI] [PubMed] [Google Scholar]
- 15.Lymar S. V., Hurst J. K. (1995) J. Am. Chem. Soc. 117, 8867–8868 [Google Scholar]
- 16.Trujillo M., Ferrer-Sueta G., Radi R. (2008) Antioxid. Redox. Signal. 10, 1607–1620 [DOI] [PubMed] [Google Scholar]
- 17.Zhang H., Squadrito G. L., Uppu R. M., Lemercier J. N., Cueto R., Pryor W. A. (1997) Arch. Biochem. Biophys. 339, 183–189 [DOI] [PubMed] [Google Scholar]
- 18.Brunelli L., Crow J. P., Beckman J. S. (1995) Arch. Biochem. Biophys. 316, 327–334 [DOI] [PubMed] [Google Scholar]
- 19.Flatley J., Barrett J., Pullan S. T., Hughes M. N., Green J., Poole R. K. (2005) J. Biol. Chem. 280, 10065–10072 [DOI] [PubMed] [Google Scholar]
- 20.Sanguinetti G., Lawrence N. D., Rattray M. (2006) Bioinformatics 22, 2775–2781 [DOI] [PubMed] [Google Scholar]
- 21.Laver J. R., Stevanin T. M., Read R. C. (2008) Globins and Other Nitric Oxide-Reactive Proteins, Pt. A 436, 113–127 [Google Scholar]
- 22.Bradford M. M. (1976) Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 23.Hurst J. K., Lymar S. V. (1997) Chem. Res. Toxicol. 10, 802–810 [DOI] [PubMed] [Google Scholar]
- 24.Pullan S. T., Gidley M. D., Jones R. A., Barrett J., Stevanin T. M., Read R. C., Green J., Poole R. K. (2007) J. Bacteriol. 189, 1845–1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saha A., Goldstein S., Cabelli D., Czapski G. (1998) Free Radic. Biol. Med. 24, 653–659 [DOI] [PubMed] [Google Scholar]
- 26.Floris R., Piersma S. R., Yang G., Jones P., Wever R. (1993) Eur. J. Biochem. 215, 767–775 [DOI] [PubMed] [Google Scholar]
- 27.Davidge K. S., Sanguinetti G., Yee C. H., Cox A. G., McLeod C. W., Monk C. E., Mann B. E., Motterlini R., Poole R. K. (2009) J. Biol. Chem. 284, 4516–4524 [DOI] [PubMed] [Google Scholar]
- 28.Zheng M., Wang X., Templeton L. J., Smulski D. R., LaRossa R. A., Storz G. (2001) J. Bacteriol. 183, 4562–4570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stamler J. S., Lamas S., Fang F. C. (2001) Cell 106, 675–683 [DOI] [PubMed] [Google Scholar]
- 30.Szwajkajzer D., Dai L., Fukayama J. W., Abramczyk B., Fairman R., Carey J. (2001) J. Mol. Biol. 312, 949–962 [DOI] [PubMed] [Google Scholar]
- 31.Zhu L., Gunn C., Beckman J. S. (1992) Arch. Biochem. Biophys. 298, 452–457 [DOI] [PubMed] [Google Scholar]
- 32.Ischiropoulos H., Zhu L., Beckman J. S. (1992) Arch. Biochem. Biophys. 298, 446–451 [DOI] [PubMed] [Google Scholar]
- 33.Justino M. C., Almeida C. C., Teixeira M., Saraiva L. M. (2007) J. Biol. Chem. 282, 10352–10359 [DOI] [PubMed] [Google Scholar]
- 34.Todorovic S., Justino M. C., Wellenreuther G., Hildebrandt P., Murgida D. H., Meyer-Klaucke W., Saraiva L. M. (2008) J. Biol. Inorg. Chem. 13, 765–770 [DOI] [PubMed] [Google Scholar]
- 35.Radi R., Beckman J. S., Bush K. M., Freeman B. A. (1991) Arch. Biochem. Biophys. 288, 481–487 [DOI] [PubMed] [Google Scholar]
- 36.Bartesaghi S., Ferrer-Sueta G., Peluffo G., Valez V., Zhang H., Kalyanaraman B., Radi R. (2007) Amino Acids 32, 501–515 [DOI] [PubMed] [Google Scholar]
- 37.Bartesaghi S., Valez V., Trujillo M., Peluffo G., Romero N., Zhang H., Kalyanaraman B., Radi R. (2006) Biochemistry 45, 6813–6825 [DOI] [PubMed] [Google Scholar]
- 38.Szabó C., Ischiropoulos H., Radi R. (2007) Nat. Rev. Drug Discov. 6, 662–680 [DOI] [PubMed] [Google Scholar]
- 39.Stamler J. S., Toone E. J., Lipton S. A., Sucher N. J. (1997) Neuron 18, 691–696 [DOI] [PubMed] [Google Scholar]
- 40.Greco T. M., Hodara R., Parastatidis I., Heijnen H. F., Dennehy M. K., Liebler D. C., Ischiropoulos H. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 7420–7425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hess D. T., Matsumoto A., Kim S. O., Marshall H. E., Stamler J. S. (2005) Nat. Rev. Mol. Cell Biol. 6, 150–166 [DOI] [PubMed] [Google Scholar]
- 42.Beltrán B., Orsi A., Clementi E., Moncada S. (2000) Br. J. Pharmacol. 129, 953–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Radi R., Denicola A., Alvarez B., Ferrer-Sueta G., Rubbo H. (2000) Nitric Oxide Biology and Pathobiology (Ignarro L. ed) pp. 57–82, Academic Press, Inc., San Diego [Google Scholar]
- 44.Gebicka L., Didik J. (2009) J. Inorg. Biochem. 103, 1375–1379 [DOI] [PubMed] [Google Scholar]
- 45.Sahoo R., Bhattacharjee A., Majumdar U., Ray S. S., Dutta T., Ghosh S. (2009) Biochem. Biophys. Res. Commun. 385, 507–511 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.