

Abstract
Prestin is a member of the SLC26 family of anion transporters that is responsible for outer hair cell (OHC) electromotility. Measures of voltage-evoked charge density (Qsp) of prestin indicated that the protein is highly expressed in OHCs, with single cells expressing up to 10 million molecules within the lateral membrane. In contrast, charge density measures in transfected cells indicated that they express, at best, only a fifth as many proteins on their surface. We sought to determine whether associations with other OHC-specific proteins could account for this difference. Using a yeast two-hybrid technique, we found microtubule-associated protein 1S (MAP1S) bound to prestin. The interaction was limited to the STAS domain of prestin and the region connecting the heavy and light chain of MAP1S. Using reciprocal immunoprecipitation and Forster resonance energy transfer, we confirmed these interactions. Furthermore, co-expression of prestin with MAP1S resulted in a 2.7-fold increase in Qsp in single cells that was paralleled by a 2.8-fold increase in protein surface expression, indicating that the interactions are physiological. Quantitative PCR data showed gradients in the expression of prestin and MAP1S across the tonotopic axis that may partially contribute to a previously observed 6-fold increase in Qsp in high frequency hair cells. These data highlight the importance of protein partner effects on prestin.
Keywords: Anion Transport, Membrane Proteins, Membrane Trafficking, Microtubules, Protein Structure, Cochlea Amplifier, MAP1S, Outer Hair Cell, Prestin
Introduction
Prestin is a member of the SLC26 family of anion transporters that is responsible for outer hair cell (OHC)3 electromotility, the basis of mammalian cochlear amplification (1–4). Prestin is found in the lateral membrane of the OHC (5, 6) and has piezoelectric properties, deriving from reciprocal voltage and mechanical sensitivity (7–10). The voltage sensor of the protein, which is integral to electromotility, generates charge movement that can be detected as a nonlinear capacitance (NLC) (11, 12). NLC parallels electromotility and has been established as an excellent surrogate marker for electromotility (11). There are three commonly described attributes of NLC as follows: Vh, the voltage of peak capacitance; z, an estimate of charge carried by a single motor; and Qsp the charge moved across a unit of membrane. Vh reflects the steady state energy profile of the protein, and Qsp provides an estimate of the density of the protein in the plasma membrane.
Prestin in OHCs exists in very high density. It is estimated that OHCs contain up to 10 million of these molecules in the lateral membrane of a given cell (13, 14). Measures of NLC in transfected cells, however, have not yielded this high density of prestin. Measures of Qsp can reach 5 fC/pF in transiently transfected CHO cells (15) but contrasts with a Qsp of 220 fC/pF in the mature OHC (16). Although differences in transcription could account for this change, we have been unable to induce OHC-like prestin expression levels in the membrane in transfected cells using the best available promoters, including cytomegalovirus and the tetracycline-inducible promoter. Moreover, mRNA expression in OHCs is not as robust as would be expected for a protein with such high levels of expression (17), indicating low turnover of prestin that is stable in the membrane. In contrast, exogenous expression of prestin using a cytomegalovirus promoter in chick hair cells results in NLC measures that were significantly increased compared with CHO cells.4 These data have led us to speculate on the existence of post-translational mechanisms or protein associations in hair cells that are absent in transfected cells. We have other reasons to hypothesize post-translational mechanisms or associations that modulate prestin activity. For instance, whereas prestin possesses many of the major features of the OHC motor, there were notable discrepancies in the behavior of the OHC motor and that of prestin in transfected cells. For example, the magnitude of the effects of prior (prepulse) voltage and membrane tension is somewhat less than found with the native OHC motor (18). These data prompted us to seek other additional mechanisms that would modify prestin activity.
It is evident over the last 15 years that many membrane proteins, including ion channels and receptors, interact with other proteins to form large complexes. The components of these complexes affect the delivery of these proteins to the surface as well as their function. For instance, there is substantial literature showing the effect of ancillary proteins on ion channels and receptors (19–21). In view of these findings, we sought to find proteins that interacted with prestin. We used the yeast two-hybrid technique to identify proteins that interacted with prestin. We used the intracellular C terminus of prestin in a Gal-4-based yeast two-hybrid technique to probe a brain cDNA library. We identified several proteins that interacted with prestin. Here, we describe the interaction between one of these proteins, microtubule-associated protein 1-S (MAP1S), and prestin, and we explore its physiological consequences.
EXPERIMENTAL PROCEDURES
Yeast Two-hybrid Experiments
Yeast two-hybrid experiments were done as described previously (49). Briefly, the C terminus of prestin (amino acids 491–744) was subcloned into pGBKT7 and used as bait to interrogate a rat brain cDNA library in pGAD T7 (Clontech). AH109 cells were serially transfected with both constructs and plated on drop out medium lacking histidine, adenine, tryptophan, and leucine. Single colonies were isolated and serially re-plated in similar conditions on plates containing 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal). Cultures from single isolated colonies were used for yeast mini preps. Plasmid was electroporated into DH101 Escherichia coli, grown in ampicillin and plasmid DNA isolated for sequencing.
DNA Constructs and Transfection
Gerbil prestin and MAP1S sequences used were as described previously (1, 38). For NLC recording, transient co-transfection of prestin (0.8 μg per well of 24-well plate) and MAP1S-CFP (or CFP) (0.8 μg per well of 24-well plate) into CHO cells was achieved with Lipofectamine (Invitrogen) in accordance with the manufacturer's recommendations. We identified MAP1S-CFP (or CFP)-transfected cells with CFP fluorescence for electrophysiological recording, and only cells that showed NLC were analyzed. We were thus able to determine with certainty that these cells contained both plasmids. For co-immunoprecipitation, FLAG-MAP1S-CFP was transfected into a prestin-Myc stable line in human embryonic kidney (HEK) cells established in our laboratory5 or HEK cells with Lipofectamine.
Prestin Surface Expression and Western Blots
Surface expression of prestin alone or with MAP1S was determined using a surface biotinylation assay (Thermo Scientific/Pierce, Rockford, IL). Transiently transfected CHO cells were washed with PBS and incubated in the presence of sulfo-NHS-biotin (sulfosuccinimidyl-2-(biotinamido) ethyl-1,3-dithiopropionate) for 30 min at 4 °C. Free sulfo-NHS-biotin was quenched by washing cells in 140 mm Tris-Cl, and the cells were lysed in lysis buffer containing the following (in mm): 20 Tris, pH 8.0, 137 NaCl, 5 NaEDTA, 5 NaEGTA, 10% glycerol, 0.5% Triton X-100, 0.2 phenylmethylsulfonyl fluoride, 50 NaF, 20 benzamidine. The lysates were cleared by centrifugation, and its protein concentration was assayed (Bio-Rad) and equalized with sample buffer. Streptavidin-agarose was added, and the mixtures were incubated for 1 h at room temperature with constant agitation. After centrifugation at 1000 × g, the beads were washed with washing buffer. The bound surface proteins were released by the addition of 50 mm dithiothreitol and analyzed by SDS-PAGE and Western blotting. Lysates or eluates of surface-labeled proteins were separated on a precast 4–15% Tris-HCl SDS-polyacrylamide gel (Bio-Rad). Proteins were transferred by wet transfer to polyvinylidene fluoride membrane (Roche Applied Science). Western blots were probed with anti-prestin N20 (Santa Cruz Biotechnology, Santa Cruz, CA) at a 1:500 dilution and then with horseradish peroxidase-conjugated bovine anti-goat secondary antibody (Santa Cruz Biotechnology) at 1:5000 dilution with five washes in TBST between each step. The presence of horseradish peroxidase-conjugated antibody was detected using SuperSignal® West Dura extended duration Substrate (Thermo Scientific/Pierce).
Co-immunoprecipitation
Reciprocal immunoprecipitation was done with anti-FLAG (MAP1S-CFP) antibody and anti-Myc (prestin-YFP) antibody using immunoprecipitation kits purchased from Sigma. In brief, membrane-enriched protein lysates were generated 48 h after transfection. 60 μl of anti-FLAG M2-agarose or 60 μl of anti-c-Myc-agarose was added to the lysates. The mixtures were incubated with constant agitation for 2 h at 4 °C. Following this binding step, the beads were washed seven times with washing buffer. To elute the FLAG fusion protein (MAP1S) or Myc fusion protein (prestin), 100 μl of 5 μg/ml FLAG peptide or c-Myc peptide (Sigma) was added to the resin. The samples were incubated with constant agitation for 30 min at 4 °C and then eluted by centrifugation. The eluted samples were boiled with loading buffer and 6% β-mercaptoethanol for 5 min and then were separated on a precast 4–15% Tris-HCl SDS-polyacrylamide gel (Bio-Rad). Proteins were transferred by wet transfer to polyvinylidene fluoride membrane (Roche Applied Science). Western blots were probed with anti-c-Myc antibody-peroxidase conjugate (1:3000) or anti-FLAG M2 antibody-peroxidase conjugate (1:4000) (Sigma) for 1 h at room temperature. Immunoreactive proteins were detected using SuperSignal® West Dura extended duration substrate (Thermo Scientific/Pierce).
Isolation of Individual OHCs from Guinea Pig Cochlea
Hartley albino guinea pigs (∼200 g) were sacrificed with halothane (2-bromo-2-chloro-1,1,1-trifluoroethane, Halocarbon Labs, River Edge, NJ), and the temporal bones were dissected in PBS. Four turns of the cochleae were separated and hair cells dislodged by mechanical trituration in PBS and dispersed in Petri dishes to allow cells to settle.
Individual OHCs were captured using a pipette (∼30-μm tip) attached to a micro-manipulator (Scientifica, Uckfield, UK) with the cells visualized using a Nikon Eclipse 600 FN microscope (Nikon, Melville, NY). The tips were silanized with trimethylchlorosilane (Fluka, St. Louis, MO) to prevent cell adhesion to the pipette wall. Cells were aspirated into the pipette using gentle suction. Isolated cells were expelled into Eppendorf tubes and stored in a −80 °C freezer.
Quantitative Immunofluorescence
Quantitative immunofluorescence was done as described previously (50), with modifications. In particular, we used a confocal microscope to obtain images and used the attendant Zeiss LSM software to analyze and extract data. Cochlea were isolated from mice euthanized by CO2 asphyxiation. Cochlea were dissected from these animals and fixed in 4% paraformaldehyde, PBS for 1 h. The cochlea were washed in PBS (three times) and placed in blocking solution (PBS, 1% bovine serum albumin, 5% horse serum, 0.1% Tween 20). Tissue was incubated in 1:500 anti-prestin antibody in blocking solution overnight (N20 prestin) (Santa Cruz Biotechnology) at 4 °C. After washing in PBS, 0.1% Tween 20 (three times), the tissue was incubated with Alexa 648-conjugated horse anti-goat antibody (1:1000) for 1 h at room temperature. The tissue was washed again in wash buffer (three times) and incubated with a 1:50 dilution of mouse anti-MAP1S antibody (Abnova, 00055201B01) for 1 h at room temperature. Alexa 488 goat anti-mouse antibody (1:1000) was added after washing the tissue. The tissue was dissected, and the three turns of the cochlea were separated after a further three washes. Tissue was then mounted using Vectorshield, and viewed using a Zeiss 510 meta confocal microscope. Sixteen bit images were acquired using a 40× water immersion lens (N.A. 1.2), with fixed laser settings, a scan rate of 6.4 μs/pixel, a pinhole aperture of 1.0 Airy units, and fixed detector gain. Regions of interest identified as horizontal sections of circumscribed hair cells in the most basal and apical ends of the cochlea were identified, and fluorescence data were extracted. We established that the fluorescence intensity was within the linear range and used mean fluorescence density as a measure of protein concentration. Surrounding supporting cells, where there is minimal prestin and MAP1S expression, were used to subtract background fluorescence. Cells from four individual cochlea were used for these analyses. The specificity of the antibodies was established by Western blots of cell lysates from cells with the respective constructs. Both antibodies identified bands of the expected size with minimal additional bands.
Single Cell Nested PCR from OHC
cDNAs from each cell were synthesized as described previously in a total volume of 20 μl (51). Nested PCR was performed in two steps. 35 cycles of PCR amplification were performed (94 °C for 30 s, 55 °C for 1 min, and 68 °C for 3 min) using 5 μl of cDNA and MAP1S outer primers (MAP1SOUTF, AACTTCTTCCTGCGTGTGCG; MAP1SOUTR, ATGCGTCTCCTCATACCACCTGTG). A second nested PCR amplification was then done using 5 μl of the initial PCR as template. This second PCR step involved 30 cycles of amplification using MAP1S inner primers as follows: MAP1SINF, GTGTGCGTGCACTCTGCTAT; MAP1SINR, TCACTGCAGAGTCGAAGGTG. Both steps used Expand High Fidelity PCR enzyme (Roche Applied Science). PCR fragments were analyzed on a 1% agarose gel. These primers spanned an intron of 501 bp. Control PCRs included aliquots from isolated cells to which no reverse transcriptase was added (no cDNA).
Quantitative (q) PCR
Organs of Corti were peeled off of each turn of the cochlea, and total RNA was isolated using the RNAqueous kit (Ambion, Austin, TX) following the manufacturer's instructions. cDNAs was synthesized using oligo(dT) primers and random hexamers as described previously (51). Triplicate qPCR amplifications using 1 μl of cDNA from each turn of the cochlea were performed using the IQ SYBR Green super mix (Bio-Rad). The reaction mixtures were set up in 96-well thin wall plates (Bio-Rad) and run on either C1000 thermal cycler with a CFX96 optical reaction module (Bio-Rad) or Mx3000P QPCR system (Stratagene, La Jolla, CA). The parameters were 95 °C for 30 s, 55 °C for 1 min, and 72 °C for 30 s for 35 cycles. Amplification data were analyzed using CFX Manager software (Bio-Rad) and normalized to 18 S RNA.
The primer sequences were as follows: prestin, PRQ96F, CAGCAGTTGACTGCCCTGTA, and PRQ276R, ACGTGGTACTTCTGGGTTGC; MAP1S, MAP1Q2945F, GAGTTCTAGCCCCACACTGC, and MAP1Q3105R, TCTGCCTCTCCAACCTGAGT; and 18 S, 18 S344F, AGAAACGGCTCCACATCCAAG, and 18 S493R, TCAAAGTCCCTCCAATGGTCC.
Electrophysiological Recording
Whole-cell patch clamp recordings were performed at room temperature using an Axon 200B amplifier (Axon Instruments) as described previously (15). Cells were recorded 48 h after transfection to allow for stable measurement of nonlinear capacitance. Ionic blocking solutions were used to isolate capacitive currents. The bath solution contained the following (in mm): tetraethyl ammonium 20, CsCl 20, CoCl2 2, MgCl2 1.47, Hepes 10, NaCl 99.2, CaCl2·2H2O 2, pH 7.2, and the pipette solution contained the following (in mm): CsCl 140, EGTA 10, MgCl2 2, Hepes 10, pH 7.2. Osmolarity was adjusted to 300 ± 2 mosm with dextrose. Command delivery and data collections were carried out with a Windows-based whole-cell voltage clamp program, jClamp (Scisoft, CT), using a Digidata 1322A interface (Axon Instruments). Capacitance was evaluated with a continuous high resolution 2-sine wave technique fully described elsewhere (52, 53). Capacitance data were fitted to the first derivative of a two-state Boltzmann function to extract Boltzmann parameters (11); a two-state Boltzmann model adequately describes charge movement of prestin (54, 55) as shown in Equations 1 and 2,
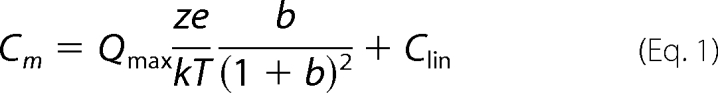 |
where
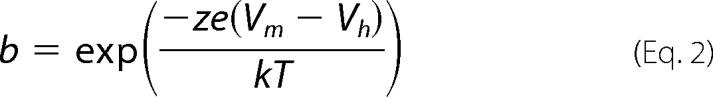 |
where Qmax is the maximum nonlinear charge transfer; Vh is the voltage at peak capacitance or half-maximal nonlinear charge transfer; Vm is the membrane potential; Clin is linear capacitance; z is the unitary charge movement or valence (also a metric of voltage sensitivity); e is electron charge; k is the Boltzmann constant, and T is absolute temperature. Qmax is reported as Qsp, the specific charge density, i.e. total charge moved normalized to linear capacitance. A Student's t test was used to evaluate the effects of mutants on the different parameters of NLC.
RESULTS
Yeast Two-hybrid Results Show That Prestin Interacts with MAP1S
In seeking to find binding partners to prestin, we used the C terminus of prestin as bait in a Gal-4-based yeast two-hybrid screen. The Gal-4-based method requires soluble (nonmembrane-bound) protein, and we used the intracellular hydrophilic C terminus of prestin for this experiment. We subcloned cDNA encoding amino acids 488–744 of prestin into the pGBK-T7 vector and screened a rat brain cDNA library. Of several clones identified, one encoded the C terminus of MAP1S from amino acids 720–972. The interaction was detectable even in the most stringent conditions. Thus, yeast containing both of these constructs grew in the absence of histidine and adenine and expressed β-galactosidase.
Interactions between MAP1S and Prestin Is Limited to Specific Regions
We attempted to isolate the regions of MAP1S that interacted with prestin (Fig. 1A). We divided the MAP1S amino acid sequence from 720 to 972 into approximately three equal segments and used these three segments (encoding amino acids 720–802, 803–886, and 887–972) to test if they interacted with prestin using the yeast two-hybrid assay. In this instance, MAP1S cDNA fragments were subcloned into pGADT7 and used in a complementation assay with the C terminus of prestin (amino acids 488–744) in pGBK-T7. Interactions were detected using the middle segment encoding amino acids 802–886. This region was further subdivided into five segments of ∼16 amino acids each, and interactions with the C terminus of prestin were assayed using the yeast two-hybrid assay. This third more focused complementation assay (again using MAP1S fragments in pGAD-T7 and amino acids 488–744 of prestin in pGBK-T7) revealed that MAP1S interacted with a short segment limited to amino acids 819–835. These 17 residues are not predicted to contain a specific secondary structure, but rather they contain a large number of proline residues. Moreover, these 17 amino acids lie in a region between the purported heavy and light chains of the protein. The heavy and light chains of the better studied MAP1B has been defined as extending from residues 1 to 2185 and 2210 to 2459, respectively. The corresponding residues in MAP1S are 1–821 and 840–972. A search using the FingerPRINTScan revealed two other proteins containing a similar sequence, Kv3.3 and Atrophin.
FIGURE 1.

Yeast two-hybrid experiments detect interactions between prestin and MAP1S, which is restricted to specific domains. Initial yeast two-hybrid screening using the C terminus of prestin as bait revealed MAP1S as a binding partner. This clone extended from residues 720 to 972. Subsequently, we used several truncations of these constructs in a yeast two-hybrid assay to clarify the specific domains involved in this interaction. A, initial screening using three equal parts of the protein revealed the interacting domain to lie within the middle fragment (amino acids (AA) 803–886). Subsequent yeast two-hybrid assays (data not shown) using 15–16-amino acid fragments within this region confirmed that the interaction with prestin was mediated by amino acids 819–835 in MAP1S, which lacks predicted secondary structure, contains a large number of proline residues, and lies between the heavy (HC) and light chains (LC) of the protein. Also indicated are the heavy chain (red) and light chain (green) regions of MAP1S relative to the region in MAP1S identified in the yeast two-hybrid screen. B, in a similar approach, we used three equal fragments of the C terminus of prestin in a yeast two-hybrid assay to determine areas that were important in interactions with MAP1S(720–972). We determined that the interacting domains in prestin extended across residues 491–655, which includes parts of the STAS domain and included the entire IVS subdomain within it. We have also shown in schematic form the transmembrane region (blue) of the prestin, its C terminus (black), and the STAS domain (orange) within the C terminus.
We next attempted to determine the interacting sites within prestin responsible for binding to MAP1S (Fig. 1B). Here too we used a similar strategy of using the yeast two-hybrid assay to ascertain interactions of three equal regions of the C terminus of prestin with amino acids 720–972 of MAP1S. In these experiments, these smaller fragments of cDNA were subcloned into pGBKT7. As shown in Fig. 1B, regions of prestin that are important for interacting with MAP1S extended across amino acids 491–660. This region includes parts of the STAS domain (525–712) and encompasses almost the entirety of the IVS region of the STAS domain.
MAP1S Is Expressed in OHC and Shows a Tonotopic Gradient
To determine whether the interaction between MAP1S and prestin is physiologically significant, we sought to ascertain if MAP1S existed in OHCs from guinea pig cochlea (the species in which there is the most amount of physiological data). A schematic of the guinea pig cochlea along with the tonotopic axis is shown in Fig. 2. We isolated individual OHCs and performed single cell PCRs from these cells. As evident in Fig. 3, OHCs contained MAP1S. Single cell nested PCR from OHCs from the three turns of the cochlea all revealed MAP1S. We then attempted to quantify the expression of MAP1S and prestin across the tonotopic axis using qPCR. As shown in Fig. 3, prestin mRNA expression is increased 1.2-fold in the middle turn compared with the 3rd turn and 2.8-fold in the basal turn compared with the 3rd turn. In contrast, MAP1S expression is increased 7-fold in the middle turn compared with the third turn and 13-fold in the basal turn compared with the third turn.
FIGURE 2.

Anatomy of the cochlea. OHCs are arranged tonotopically along the three turns of the cochlea. A, schematic of the external appearance of the cochlea shows its three turns. Hair cells in these turns are arranged tonotopically with cells at the base responding to high frequency (Freq) sounds and cells at the apex responding to low frequency sound. Notably, hair cells in the 1st, 2nd, and 3rd turns of the cochlea respond to sound of decreasing frequency. B, section across the cochlea reveals the organ of Corti to be compartmentalized. An expanded view of the organ of Corti reveals it to contain three rows of OHCs and one row of inner hair cells. TM, transmembrane; IHC, inner hair cell; BM, basilar membrane.
FIGURE 3.

MAP1S is expressed in individual hair cells, and its mRNA shows a marked gradient in expression across the tonotopic axis. A, MAP1S was detected in 12 individual OHCs by nested PCR using cDNA from single cells. The product of 147 bp is the expected product of the inner primer pair. The products were separated on a 2% agarose gel. Also included are DNA size markers (M); a positive control (+, using plasmid containing MAP1S cDNA as template), and negative control (−, no reverse transcriptase). The sizes of the markers indicated in blue are 506, 398, 356, 298, 220, 203, 154, and 134 bp. B, shown in graph form are the fold change of prestin and MAP1S mRNA determined by qPCR across the organ of Corti from the three turns of the cochlea compared with the 3rd turn of the cochlea. The expression of each mRNA species was normalized to the expression of the 18 S subunit of ribosomal RNA. As is evident, although prestin shows an almost 3-fold increase in expression from the third to the basal (first) turn of the cochlea, MAP1S shows a more dramatic 13-fold increase in expression from the third to the basal (first) turn of the cochlea. The error bars are ± S.E., n = 4.
We attempted to confirm our protein data using antibodies to these proteins in the guinea pig. However, because of poor antibody recognition of MAP1S in this species, we were unable to confirm or refute our qPCR data. We were able, however, to use mouse cochlea to confirm our PCR data from the guinea pig cochlea. As shown in Fig. 4, mouse OHCs demonstrate co-localization of these two proteins. Moreover, there is an apical to basal gradient in the expression of prestin and MAP1S. Thus, prestin showed a 7-fold increase in expression in the most basal hair cells, whereas MAP1S increased 3.5-fold in these cells compared with those from the apex.
FIGURE 4.

MAP1S co-localizes with prestin in mouse OHC and, along with prestin, shows a gradient in expression across the tonotopic axis. A, prestin co-localizes with MAP1S. Shown are mouse OHCs labeled consecutively with antibodies against MAP1S (left) and prestin (middle). There is co-localization of the two proteins in these cells (right). B, OHCs from the most apical (left) and basal (right) portions of the mouse cochlea were labeled with antibodies to prestin (upper) and MAP1S (lower). As is evident, these two proteins are abundant in OHCs of the basal turn. Contrast in the right panel was increased post hoc to better show the expression of MAP1S and prestin. C, these findings were confirmed using quantitative immunofluorescence. OHCs from the most basal and apical portions of the mouse cochlea together with cells from the midpoint between these two extremes were tested for the expression of prestin and MAP1S. The mean fluorescence density (± S.E.) of MAP1S in the apical, mid, and basal hair cells are 88 (±14, n = 6), 183 (±28, n = 12), and 371 (±48, n = 6). Similarly, values for prestin are 257 (±53, n = 6), 744 (±60, n = 12), and 1723 (±130, n = 6). These quantitative data confirm a 7-fold increase in prestin expression and a 3.5-fold increase in MAP1S expression from the most apical to basal ends of the mouse cochlea.
In Vivo Interactions between MAP1S Are Confirmed by Immunoprecipitation
Although the interaction between prestin and MAP1S was suggested by the yeast two-hybrid experiments done under stringent conditions, it is widely believed that the yeast two-hybrid assay can be erroneous, yielding false positives from nonspecific interactions. To confirm interactions between prestin and MAP1S, we performed a reciprocal immunoprecipitation using prestin and MAP1S in its entirety tagged with c-Myc and FLAG tags, respectively. FLAG-MAP1S was transfected into cells constitutively expressing prestin-YFP-c-Myc. Immunoprecipitation and wash steps were stringent and included high salt and detergent (0.05% Triton X-100). As shown in Fig. 5, prestin and MAP1S could reciprocally immunoprecipitate the other protein. Together with our data showing interactions between prestin and MAP1S using the yeast two-hybrid system, these data argue for a robust interaction between the two proteins. Interestingly, the form of MAP1S that was immunoprecipitated by prestin was the entire coding sequence and not the heavy chain, although there was evidence of the protein being cleaved into heavy and light chains in these cells. Consistent with these data, our yeast two-hybrid assay identified the interacting domain within MAP1S as restricted to an area between the heavy and light chains of the protein.
FIGURE 5.

Reciprocal immunoprecipitations confirm the interaction between prestin and MAP1S. A permanent cell line expressing prestin-YFP-myc was transfected with FLAG-MAP1S-CFP. A, immunoprecipitations were performed with anti-FLAG (MAP1S) antibody and the presence of prestin in the immunoprecipitate detected on Western blots with anti-Myc antibody. Lanes 1–3 are control crude lysates of untransfected HEK cells, cells expressing prestin-YFP-myc, and cells expressing prestin-YFP-myc and FLAG-MAP1S-CFP, respectively. Lane 4 is an immunoprecipitate with FLAG antibody of lysates from HEK cells expressing prestin-YFP-myc together with FLAG-MAP1S-CFP. Prestin-YFP-myc is indicated by the thick arrow. Lane 5 is an anti-FLAG immunoprecipitate of HEK cells expressing prestin-YFP-myc. The absence of prestin in this lane confirms the specificity of the immunoprecipitating antibody. B, reciprocal experiment was performed to further confirm the interactions. Immunoprecipitation of cell lysates was done using anti-Myc antibody, and the presence of MAP1S in the immunoprecipitate was detected by Western blotting using anti-FLAG antibody. Lanes 1 and 2 are crude cell lysates of HEK cells expressing FLAG-MAP1S-CFP and HEK cells expressing prestin-YFP-myc together with FLAG-MAP1S-CFP, respectively. Lane 3 is the Myc immunoprecipitate of cells expressing prestin-YFP-myc and FLAG-MAP1S-CFP. FLAG-MAP1S-CFP is indicated by the thin arrow. Lane 4 is a Myc immunoprecipitate of cells expressing FLAG-MAP1S-CFP alone. The absence of prestin in lane 4 confirms the specificity of the immunoprecipitating Myc antibody. Although lanes 1–4 were separated on a single gel and transferred to the same blot, lanes 1 and 2 have been separated from lanes 3 and 4 in the figure for reasons of clarity. Lanes 1 and 2 are a shorter exposure and lanes 3 and 4 a longer exposure of the same blot. The experiments were repeated three times.
Physiological Interaction between MAP1S Confirmed by Forster Resonance Energy Transfer (FRET)
Our immunoprecipitation data strongly argue for an interaction between prestin and MAP1S. We next sought to determine that such an interaction took place in vivo (and prior to cell lysis in the immunoprecipitation assay). We chose to use FRET to demonstrate these interactions. Prestin and MAP1S were tagged at their C termini with YFP and cyan fluorescent protein (CFP). cDNA encoding these two fused proteins were inserted into an internal ribosome entry site vector and expressed in CHO cells. As evident in Fig. 6, cells transfected with this construct and expressing both these fused proteins show FRET. FRET efficiency was measured after acceptor photobleaching and showed values similar to what we have previously found in prestin-prestin interactions (15). FRET efficiency after photobleaching in Prestin-YFP/MAP1S-CFP was 2.8 (±0.9 S.E.) and contrasts with −36 (±11 S.E.) in prestin-YFP/CFP. In contrast, the FRET efficiency of a construct in which CFP and YFP were in tandem separated by three amino acids was 35 (±10 S.E.). These results argue that the distance between MAP1S and prestin is proximate to that between two molecules of prestin. These results also argue that interactions between prestin and MAP1S are likely physiological.
FIGURE 6.

FRET experiments using acceptor photobleaching confirm in vivo interactions between prestin and MAP1S. A, photomicrographs of a CHO cell expressing prestin-YFP and MAP1S-CFP in an internal ribosome entry site vector are shown. The cell was excited with an argon laser at 458 nm, and the CFP emission in the bandwidth from 476 to 485 nm was recorded with a meta detector (upper two panels). The cell was also excited at 514 nm, and YFP emission at 536–545 nm was recorded (lower two panels). YFP was photobleached by continuous excitation at 514 nm for 1 min. Emission of CFP after photobleaching (right upper panel) shows an increase compared with CFP emission before photobleaching (left upper panel). In contrast, YFP emission (left lower panel) shows a decrease after photobleaching (right lower panel) compared with YFP emission before photobleaching (left lower panel). B, CFP FRET efficiency was measured after photobleaching as described previously (15). The MAP1S-CFP and prestin-YFP pair shows an increase in FRET efficiency after photobleaching (mean 11.02 ± 2.0 S.E., n = 8) and contrasts with MAP1S-CFP and YFP pair that shows a decrease in FRET efficiency (mean −40.0 ± 4.6 S.E., n = 7). Also included is a positive control that has CFP and YFP in tandem separated by one amino acid and fused to the C terminus of the membrane-spanning voltage-sensing phosphatase from Nematostella vectensis. This construct yielded higher FRET efficiency (mean 37.87 ± 6.26 S.E., n = 4).
MAP1S Increases NLC
Having established a likely physiological interaction between prestin and MAP1S, we sought to ascertain if MAP1S affected prestin function. CHO cells were transfected with 0.8 μg of prestin and 0.8 μg of MAP1S fused to CFP per well in a 24-well plate. The control group was transfected with 0.8 μg of prestin and 0.8 μg of CFP. We assayed the physiological function of prestin by determining NLC in cells from these two groups. As shown in Fig. 7, cells transfected with prestin and MAP1S showed a 2.7-fold excess in NLC compared with cells that were transfected with prestin and CFP together. Prestin and MAP1S-transfected cells showed Qsp of 6.38 fC/pF, which compared with 2.34 fC/pF in cells transfected with prestin and CFP alone (Table 1). In contrast, MAP1S did not affect other aspects of NLC function. Thus, cells transfected with prestin and MAP1S had mean Vh and z values of −120 mV and 0.63e and contrasted with values of −119 mV and 0.6e in cells transfected with prestin and CFP alone (Table 1). It should be noted that the Qsp values obtained in these experiments were 50% of those that we normally achieve in CHO cells. Most likely, this results from the use of half the relative amount of prestin plasmid in each transfection that we normally use, necessitated by the need to co-transfect MAP1S at the same time.
FIGURE 7.

MAP1S increases Qsp. A, CHO cells transiently transfected with prestin and MAP1S-CFP showed a statistically significant (p < 0.05, t test) increase in Qsp (mean 6.38 ± 1.89 S.E.) compared with cells transfected with prestin and CFP (mean 2.34 ± 0.47 S.E.). B, shown are two representative NLCsp (defined as NLC (pF)/linear capacitance (pF) to correct for effects of cell size) traces from two comparable cells transfected with the combination of plasmids prestin/CFP and prestin/MAP1S-CFP. There is a notable increase in peak NLCsp in the cell transfected with the plasmid combination prestin/MAP1S-CFP compared with the cell transfected with the plasmid combination prestin/CFP.
TABLE 1.
MAP1S increases Qsp but does not affect other measures of NLC (Vh and z)
Shown are values of NLC in cells transfected with the combination of plasmids prestin/MAP1S-CFP or prestin/CFP. Although Qsp is significantly increased, both the voltage of peak capacitance (Vh) and estimates of the charge carried by a single motor z are comparable.
| Qsp | Vh | z | n | |
|---|---|---|---|---|
| fC/pF | mV | |||
| 0.8 μg of normal prestin + 0.8 μg of CFP | 2.34 ± 0.47 | −119.29 ± 3.17 | 0.60 ± 0.02 | 11 |
| 0.8 μg of normal prestin + 0.8 μg of MAP1S-CFP | 6.38 ± 1.89 | −120.47 ± 3.61 | 0.63 ± 0.04 | 10 |
MAP1S Increases Surface Expression of Prestin
Our data showing increased charge movement in the membrane of cells expressing MAP1S and prestin could be due to an increased number of molecules delivered to the membrane or as a result of an allosteric effect on each molecule at the surface of the cell. Our estimates of z, the charge carried by individual motors, were unchanged in cells transfected with prestin and MAP1S and argue against an allosteric effect. Rather our electrophysiological data argue for an increased number of motors delivered to the surface of the cell. To test this possibility, we assayed prestin expression in the membrane using a surface biotinylation assay. As shown in Fig. 8, there was a 2.8-fold increase in surface expression of prestin induced by MAP1S. This increase corresponded well with our electrophysiological findings that showed a 2.7-fold increase in specific charge, Qsp. Because MAP1S binds tubulin, and because there is evidence that microtubule-associated mechanisms are important for surface expression of cell surface proteins, we sought to determine whether tubulin disruption affected prestin surface expression. In several experiments, cells were treated with and without 10 μm colchicine soon after transfection with prestin-YFP and 10 μm colchicine maintained in culture for 48 h. NLC in these cells was assayed at 48 h and showed no difference in the two groups (Table 2).
FIGURE 8.
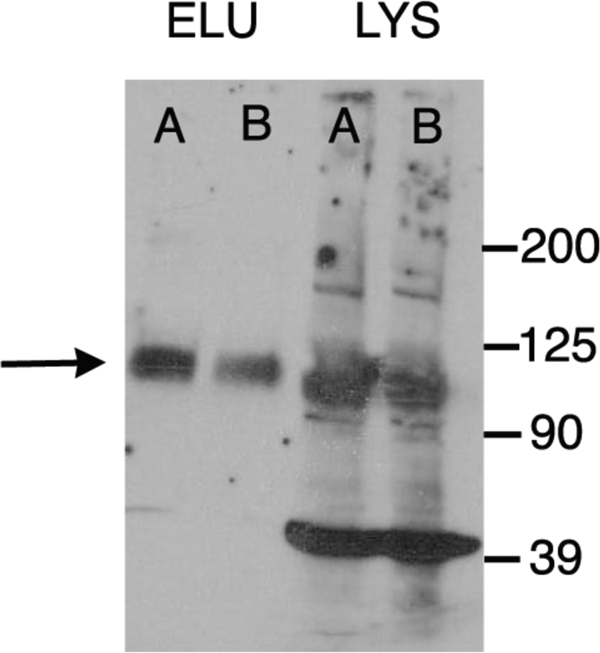
MAP1S increases surface expression of prestin. CHO cells were transfected with prestin/MAP1S CFP and separately prestin/CFP plasmids. Proteins on the surface of the cell were labeled with sulfosuccinimidyl-2-(biotinamido)ethyl-1,3-dithiopropionate and isolated using a Neutravidin resin. The surface proteins were eluted using 50 mm dithiothreitol and separated by SDS-PAGE, and the presence of prestin was detected by Western blotting (left two lanes). The amount of surface protein loaded in each lane has been normalized to the total protein in the cell lysate. As is evident, cells expressing prestin and MAP1S-CFP (A) demonstrate an increase (2.8-fold ±0.9 S.E., n = 4) in surface expression of prestin when compared with cells expressing prestin and CFP (B). The comparable expressions of prestin in the corresponding cell lysates are shown in the two right lanes confirming that the total amounts of prestin in these cells were equivalent. The prestin band is shown by an arrow. The numerous other bands in the lysates (for example, the band at 39 kDa) are nonspecific and detected in untransfected HEK cells probed with the secondary antibody alone (data not shown). ELU, eluate; LYS, lysate.
TABLE 2.
Colchicine has no effect on the Qsp
The table shows the effects of treating cells with 10 μm colchicine for 48 h after transfection. Measures of NLC, including Qsp, Vh, and z, were unchanged by treatment with this drug. It should be noted that because treatment with this drug is cytotoxic, we incorporated in our data analysis only those cells that showed good seal resistance, using it as a surrogate marker for the well being of cells.
| Qsp | Vh | z | n | |
|---|---|---|---|---|
| fC/pF | mV | |||
| Prestin | 7.35 ± 0.42 | −110.03 ± 3.00 | 0.71 ± 0.02 | 5 |
| Prestin + 10 μm colchicine | 7.33 ± 1.88 | −107.70 ± 10.24 | 0.69 ± 0.03 | 5 |
DISCUSSION
Our data show for the first time an interaction between prestin and MAP1S and evinces a physiological role for this interaction. The initial yeast two-hybrid data showing an interaction between these proteins are substantiated by the immunoprecipitation data. Subsequently, we demonstrate that the interaction is physiological with FRET, and electrophysiological recordings show an enhanced amount of prestin on the surface of the cell in the presence of MAP1S. The latter finding is further confirmed by biochemical means. Finally, we show that there is a gradient in the expression of prestin and MAP1S along the tonotopic axis. This gradient may partially explain the increase in charge density in high frequency OHCs.
Interaction between MAP1S and Prestin Is Restricted to Specific Regions of the Proteins
The interaction between these two proteins occurs in the region between the heavy and light chain in MAP1S and the proximal portion of the STAS domain in prestin. Although it is possible that interactions with prestin may extend into the loops connecting consecutive transmembrane domains, our current approaches limit our exploration of this possibility. To date there has been no evidence that the region between the heavy and light chain of MAP1S is involved in interactions with other proteins (22). Consistent with these results, our immunoprecipitation data show that prestin interacts with the holoprotein and not its component heavy and light chains. The cleavage site between the heavy and light chains of MAP1S lies in the region between them and coincides with the prestin-binding site (22). The light and heavy chains of the molecule have been shown to bind different elements of the cytoskeleton (22). Thus, the N terminus of the light chain binds tubulin, and the more C-terminally placed MH3 domain binds actin. In contrast, the heavy chain interferes with the interaction between the light chain and tubulin. In this respect, our data that show no effect on surface expression of prestin by colchicine is consistent with the result that prestin surface expression is mediated by the holoprotein, which does not interact with tubulin.
Other data from the yeast two-hybrid experiments show that amino acids involving the STAS domain of prestin are involved in the interaction with MAP1S. Specifically, the sequence extends across the first three β-strands, the first α-helix, and the intervening IVS region of the purported STAS domain in prestin determined by molecular modeling (22, 23). The STAS domain resembles the bacterial SPOIIAA transcription factor, the crystal and NMR structure of which has been determined (24, 25). The SPOIIAA transcription factor is involved in protein-protein interactions with the anti-σ factor SPOIIAB. Binding of these two proteins results in σ factor release from SPOIIAB, which in turn triggers transcription (26–28). Mutations within the STAS domain of SLC26A2, SLC26A3, and SLC26A4 and the Arabidopsis thaliana sulfate transporter Sultr 1.2 have been shown to alter anion transport function (29–34). Moreover, the STAS domain in SLC26A3 also interacts with cystic fibrosis transmembrane regulator, and this interaction reciprocally modulates cystic fibrosis transmembrane regulator channel function as well as SLC26A3 transporter function (35). In contrast, cystic fibrosis transmembrane regulator interacts with the STAS domain of SLC26A9 and inhibits Cl− currents as well as its Cl−-HCO3− exchange (36). Given these data, it was our expectation that MAP1S would modulate NLC. However, we did not note an alteration in measures of prestin “kinetics.” Both Vh and z, reflecting the steady state energy profile and unitary charge movement, were unchanged in cells co-transfected with MAP1S. In contrast, we noted increased delivery of prestin to the surface of the cell. Several mutations in the STAS domain resulted in altered delivery of SLC26A3, a related protein, to the surface of the cell (32). Taken together, these data suggest that modulation of the STAS domain may affect surface delivery of SLC26 proteins (prestin), in addition to affecting the kinetics of the protein (SLC26A3). Efforts are currently underway in our laboratory to determine how MAP1S affects prestin anion transport (37), especially given the data that mutations in the STAS domain have been shown to affect anion transport in members of the SLC26 family of proteins. Furthermore, given the sequence homology in this region between the different members of the SLC26 family of proteins, and the ubiquitous distribution of MAP1S, we would anticipate that these anion transporters also will interact with MAP1S (38). In this regard, the yeast two-hybrid experiments identifying residues 491–572, which is conserved between prestin and other SLC26 members, could suggest homologous regions from other SLC26 members as also interacting with MAP1S. In contrast, it is possible that residues 573–660, which has little homology in other SLC26 family members, confers specificity to the interactions with prestin.
Increase in Qsp Is Likely Brought About by Increased Amounts of Prestin on the Surface of the Cell
As noted above, our data on measures of NLC show that MAP1S affects Qsp but not other measures of NLC, including z and Vh. Theoretically, Qsp could be increased by increasing the number of functional motors in the membrane or by increasing the net charge carried by each motor. However, given that z, our best estimate of charge carried by an individual motor, was unchanged in the presence of MAP1S, it is most likely that the number of motors delivered to the surface is increased. This conclusion is borne out by our determination of surface expression of prestin in the presence and absence of MAP1S. The biotinylation assay revealed a 2.8-fold increase in surface expression of prestin that compared with a 2.7-fold increase in Qsp. Thus, there was good concordance between our biochemical assays of surface expression and electrophysiological determination of surface expression. How is this increase in surface expression brought about? Three possibilities are suggested as follows: increased delivery of newly synthesized protein to the surface, decreased degradation of the protein, or increased recycling of the protein that is endocytosed from the surface. Although we have no experimental data to discern these possibilities at present, we expect that MAP1S will likely deliver more newly synthesized protein to the surface. Previous experiments in our laboratory indicate that turnover rates of prestin may be slow; in the presence of brefeldin A, a blocker of newly synthesized protein delivery into the surface membrane, OHCs in culture maintained robust NLC for up to 7 days.6
MAP1S has been shown to interact with the NR3A N-methyl-d-aspartic acid receptor subunit, the fibroblast growth factor-associated protein LRPPRC, the tumor suppressor protein RASSF1A, and the sperm protein VCY2 that has been implicated in azoospermia (39–43). However, in all these instances there were no clearly observed consequences of the interactions. Thus, the functional effect of MAP1S on prestin expression appears to be the first direct demonstration of any functional effect by MAP1S. In the case of the NR3A receptor and MAP1S, there was sharp co-localization of the two proteins in dendritic spines that led the authors to speculate that MAP1S was important for localizing NR3A receptors to dendritic spines (41). However, there were no direct tests of this possibility. In the related MAP1B protein, early data suggested that MAP1B was important for targeting γ-aminobutyric acid type C receptors to specific subcellular locales (44). These two proteins co-localize in retinal bipolar cells (44). Heterologous expression of the two proteins in COS cells resulted in an altered distribution of the γ-aminobutyric acid type C receptor. However, subsequent MAP1B knock-out experiments were confounding with normal γ-aminobutyric acid type C clustering in bipolar cells (45).
There are a number of reports suggesting a role for the microtubule-associated motors in directing organelles and proteins to subcellular sites, including dendritic shafts (46–48). Given the known associations between MAP1S and tubulin, an obvious mechanism for prestin delivery to the surface of the cell would be through its association via MAP1S with microtubules and their motors. However, we show that continued disruption of microtubules (after transfection) with 10 μm colchicine had no effect on Qsp.
Interactions between Prestin and MAP1S Likely Has Physiological Significance
Our data showing a graded tonotopic expression of prestin and MAP1S have bearing on physiological findings in the cochlea. Prior work has compared Qsp in guinea pig OHCs isolated from regions along the tonotopic axis and found an ∼5–6-fold increase in Qsp in the lateral membrane of OHC responding to high frequency sound compared with OHC responding to low frequency sound (13). However, prestin mRNA expression increases 1.2-fold from the 3rd turn to the 2nd turn and 2.8-fold from the third turn to the basal turn. Thus, the prestin expression increase in high frequency cells is less than the increase in Qsp. Whether the even greater increase in MAP1S expression that we find contributes to the excess increase in Qsp remains to be investigated. In the mouse where physiological data are unavailable, but antibodies to MAP1S and prestin are available, there is a 7-fold increase in prestin protein expression from the most apical to the most basal hair cells. Similarly, there is also a smaller 3.5-fold increase in protein expression of MAP1S. This is an important issue, because the increase in Qsp in high frequency cells has been proposed to be a mechanism that provides a constant electrical energy to these cells, which may help circumvent the low pass filter effects of the cell membrane (13).
In conclusion, we show for the first time an interaction between prestin and the microtubule-associated protein MAP1S. The interaction is confirmed by immunoprecipitation and FRET. Our data from FRET suggest that the relative distance between MAP1S and prestin is proximate to the distances between two molecules of prestin. The interaction results in an increased amount of prestin on the surface of the cell that accounts for the increase in Qsp brought about by MAP1S. We demonstrate the presence of MAP1S in individual OHCs and confirm a gradient in the expression of prestin and MAP1S along the tonotopic axis. This gradient of both proteins could explain the increase in charge movement density (Qsp) seen in OHCs from higher frequencies.
Acknowledgment
We thank Dr. Fritz Probst for the MAP1S constructs and for critical reading of this manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants DC 007894, DC 000273, and DC 0081130 from NIDCD.
J.-P. Bai and D. Navaratnam, unpublished observations.
S. Bian, manuscript in preparation.
E. Navarrete and J. Santos-Sacchi, unpublished data.
- OHC
- outer hair cell
- PBS
- phosphate-buffered saline
- FRET
- Forster resonance energy transfer
- CHO
- Chinese hamster ovary
- NLC
- nonlinear capacitance
- CFP
- cyan fluorescent protein
- YFP
- yellow fluorescent protein
- qPCR
- quantitative PCR
- fC
- femtocoulomb
- pF
- picofarad.
REFERENCES
- 1.Zheng J., Shen W., He D. Z., Long K. B., Madison L. D., Dallos P. (2000) Nature 405, 149–155 [DOI] [PubMed] [Google Scholar]
- 2.Liberman M. C., Gao J., He D. Z., Wu X., Jia S., Zuo J. (2002) Nature 419, 300–304 [DOI] [PubMed] [Google Scholar]
- 3.Santos-Sacchi J., Song L., Zheng J., Nuttall A. L. (2006) J. Neurosci. 26, 3992–3998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dallos P., Wu X., Cheatham M. A., Gao J., Zheng J., Anderson C. T., Jia S., Wang X., Cheng W. H., Sengupta S., He D. Z., Zuo J. (2008) Neuron 58, 333–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng J., Madison L. D., Oliver D., Fakler B., Dallos P. (2002) Audiol. Neurootol. 7, 9–12 [DOI] [PubMed] [Google Scholar]
- 6.Belyantseva I. A., Adler H. J., Curi R., Frolenkov G. I., Kachar B. (2000) J. Neurosci. 20, RC116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iwasa K. H. (1993) Biophys. J. 65, 492–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwasa K. H. (2001) Biophys. J. 81, 2495–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gale J. E., Ashmore J. F. (1994) Proc. R. Soc. Lond. B Biol. Sci. 255, 243–249 [DOI] [PubMed] [Google Scholar]
- 10.Kakehata S., Santos-Sacchi J. (1995) Biophys. J. 68, 2190–2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santos-Sacchi J. (1991) J. Neurosci. 11, 3096–3110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ashmore J. F. (1989) in Mechanics of Hearing (Kemp D., Wilson J. P. eds) pp. 107–113, Plenum Publishing Corp., New York [Google Scholar]
- 13.Santos-Sacchi J., Kakehata S., Kikuchi T., Katori Y., Takasaka T. (1998) Neurosci. Lett. 256, 155–158 [DOI] [PubMed] [Google Scholar]
- 14.Gale J. E., Ashmore J. F. (1997) Pflugers Arch. 434, 267–271 [DOI] [PubMed] [Google Scholar]
- 15.Navaratnam D., Bai J. P., Samaranayake H., Santos-Sacchi J. (2005) Biophys. J. 89, 3345–3352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang G., Santos-Sacchi J. (1993) Biophys. J. 65, 2228–2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abe T., Kakehata S., Kitani R., Maruya S., Navaratnam D., Santos-Sacchi J., Shinkawa H. (2007) J. Membr. Biol. 215, 49–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santos-Sacchi J., Shen W., Zheng J., Dallos P. (2001) J. Physiol. 531, 661–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pocklington A. J., Cumiskey M., Armstrong J. D., Grant S. G. (2006) Mol. Syst. Biol. 2, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pocklington A. J., Armstrong J. D., Grant S. G. (2006) Brief. Funct. Genomic Proteomic 5, 66–73 [DOI] [PubMed] [Google Scholar]
- 21.Vacher H., Mohapatra D. P., Trimmer J. S. (2008) Physiol. Rev. 88, 1407–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rouached H., Berthomieu P., El Kassis E., Cathala N., Catherinot V., Labesse G., Davidian J. C., Fourcroy P. (2005) J. Biol. Chem. 280, 15976–15983 [DOI] [PubMed] [Google Scholar]
- 23.Bai J. P., Navaratnam D., Samaranayake H., Santos-Sacchi J. (2006) Neurosci. Lett. 404, 270–275 [DOI] [PubMed] [Google Scholar]
- 24.Seavers P. R., Lewis R. J., Brannigan J. A., Verschueren K. H., Murshudov G. N., Wilkinson A. J. (2001) Structure 9, 605–614 [DOI] [PubMed] [Google Scholar]
- 25.Kovacs H., Comfort D., Lord M., Campbell I. D., Yudkin M. D. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 5067–5071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diederich B., Wilkinson J. F., Magnin T., Najafi M., Erringston J., Yudkin M. D. (1994) Genes Dev. 8, 2653–2663 [DOI] [PubMed] [Google Scholar]
- 27.Clarkson J., Campbell I. D., Yudkin M. D. (2003) Biochem. J. 372, 113–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kroos L., Zhang B., Ichikawa H., Yu Y. T. (1999) Mol. Microbiol. 31, 1285–1294 [DOI] [PubMed] [Google Scholar]
- 29.Taylor J. P., Metcalfe R. A., Watson P. F., Weetman A. P., Trembath R. C. (2002) J. Clin. Endocrinol. Metab. 87, 1778–1784 [DOI] [PubMed] [Google Scholar]
- 30.Karniski L. P. (2001) Hum. Mol. Genet. 10, 1485–1490 [DOI] [PubMed] [Google Scholar]
- 31.Karniski L. P. (2004) Hum. Mol. Genet. 13, 2165–2171 [DOI] [PubMed] [Google Scholar]
- 32.Dorwart M. R., Shcheynikov N., Baker J. M., Forman-Kay J. D., Muallem S., Thomas P. J. (2008) J. Biol. Chem. 283, 8711–8722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shibagaki N., Grossman A. R. (2004) J. Biol. Chem. 279, 30791–30799 [DOI] [PubMed] [Google Scholar]
- 34.Shibagaki N., Grossman A. R. (2006) J. Biol. Chem. 281, 22964–22973 [DOI] [PubMed] [Google Scholar]
- 35.Ko S. B., Zeng W., Dorwart M. R., Luo X., Kim K. H., Millen L., Goto H., Naruse S., Soyombo A., Thomas P. J., Muallem S. (2004) Nat. Cell Biol. 6, 343–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang M. H., Plata C., Sindic A., Ranatunga W. K., Chen A. P., Zandi-Nejad K., Chan K. W., Thompson J., Mount D. B., Romero M. F. (2009) J. Biol. Chem. 284, 28306–28318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bai J. P., Surguchev A., Montoya S., Aronson P. S., Santos-Sacchi J., Navaratnam D. (2009) Biophys. J. 96, 3179–3186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Orbán-Németh Z., Simader H., Badurek S., Tranciková A., Propst F. (2005) J. Biol. Chem. 280, 2257–2265 [DOI] [PubMed] [Google Scholar]
- 39.Liu L., Vo A., Liu G., McKeehan W. L. (2005) Cancer Res. 65, 4191–4201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu L., Vo A., Liu G., McKeehan W. L. (2005) Biochem. Biophys. Res. Commun. 332, 670–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eriksson M., Samuelsson H., Samuelsson E. B., Liu L., McKeehan W. L., Benedikz E., Sundström E. (2007) Biochem. Biophys. Res. Commun. 361, 127–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wong E. Y., Tse J. Y., Yao K. M., Lui V. C., Tam P. C., Yeung W. S. (2004) Biol. Reprod. 70, 775–784 [DOI] [PubMed] [Google Scholar]
- 43.Song M. S., Chang J. S., Song S. J., Yang T. H., Lee H., Lim D. S. (2005) J. Biol. Chem. 280, 3920–3927 [DOI] [PubMed] [Google Scholar]
- 44.Pattnaik B., Jellali A., Sahel J., Dreyfus H., Picaud S. (2000) J. Neurosci. 20, 6789–6796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meixner A., Haverkamp S., Wässle H., Führer S., Thalhammer J., Kropf N., Bittner R. E., Lassmann H., Wiche G., Propst F. (2000) J. Cell Biol. 151, 1169–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Allan V. J., Thompson H. M., McNiven M. A. (2002) Nat. Cell Biol. 4, E236–E242 [DOI] [PubMed] [Google Scholar]
- 47.Murray J. W., Wolkoff A. W. (2003) Adv. Drug Deliv. Rev. 55, 1385–1403 [DOI] [PubMed] [Google Scholar]
- 48.Zheng Y., Wildonger J., Ye B., Zhang Y., Kita A., Younger S. H., Zimmerman S., Jan L. Y., Jan Y. N. (2008) Nat. Cell Biol. 10, 1172–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Navaratnam D. S. (2009) Methods Mol. Biol. 493, 257–268 [DOI] [PubMed] [Google Scholar]
- 50.Blot V., McGraw T. E. (2008) Methods Mol. Biol. 457, 347–366 [DOI] [PubMed] [Google Scholar]
- 51.Navaratnam D. S., Bell T. J., Tu T. D., Cohen E. L., Oberholtzer J. C. (1997) Neuron 19, 1077–1085 [DOI] [PubMed] [Google Scholar]
- 52.Santos-Sacchi J. (2004) Biophys. J. 87, 714–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Santos-Sacchi J., Kakehata S., Takahashi S. (1998) J. Physiol. 510, 225–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Santos-Sacchi J. (1993) Biophys. J. 65, 2217–2227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scherer M. P., Gummer A. W. (2005) Biophys. J. 88, L27–L29 [DOI] [PMC free article] [PubMed] [Google Scholar]