

Abstract
Many therapeutic antibodies act as antagonists to competitively block cellular signaling pathways. We describe here an approach for the therapeutic use of monoclonal antibodies based on context-dependent attenuation to reduce pathologically high activity while allowing homeostatic signaling in biologically important pathways. Such attenuation is achieved by modulating the kinetics of a ligand binding to its various receptors and regulatory proteins rather than by complete blockade of signaling pathways. The anti-interleukin-1β (IL-1β) antibody XOMA 052 is a potent inhibitor of IL-1β activity that reduces the affinity of IL-1β for its signaling receptor and co-receptor but not for its decoy and soluble inhibitory receptors. This mechanism shifts the effective dose response of the cytokine so that the potency of IL-1β bound by XOMA 052 is 20–100-fold lower than that of IL-1β in the absence of antibody in a variety of in vitro cell-based assays. We propose that by decreasing potency of IL-1β while allowing binding to its clearance and inhibitory receptors, XOMA 052 treatment will attenuate IL-1β activity in concert with endogenous regulatory mechanisms. Furthermore, the ability to bind the decoy receptor may reduce the potential for accumulation of antibody·target complexes. Regulatory antibodies like XOMA 052, which selectively modulate signaling pathways, may represent a new mechanistic class of therapeutic antibodies.
Keywords: Antibodies, Cytokine Action, Diabetes, Drug Action, Inflammation, Interleukin, Surface Plasmon Resonance (SPR), IL-1 Receptor, Interleukin-1 Beta, Regulatory Antibody
Introduction
Recombinant monoclonal antibodies have emerged as powerful targeted therapies for severe diseases (1, 2), often acting by blocking activity of dysregulated cellular pathways. However, most pathways that have been linked to disease when abnormally activated have important roles in healthy tissues. This observation is particularly true for the immune system, where highly potent cytokines such as IL-1β,3 tumor necrosis factor α, and IL-6 drive inflammation in pathological contexts but also have important beneficial roles in the control of infections (3). Successful treatment of some diseases may therefore require attenuation rather than complete inhibition of signaling pathways to restore a normal physiological state with acceptable side-effect profiles. We describe here an antibody that regulates activity of its target antigen by reducing the affinity of the antigen binding to its signaling receptor. This strategy has not been addressed previously with therapeutic monoclonal antibodies in the clinic.
IL-1β is a highly potent cytokine that drives the acute phase inflammatory response and has an essential role in the innate immune response (4–7). Although high levels of IL-1β have been implicated in inflammatory diseases (3, 8) including type 2 diabetes (9–12), low levels have beneficial effects on pancreatic beta cell function, proliferation, and survival (13–16), intestinal epithelial cell survival (17, 18), and neuronal response to injury (19). As in many receptor-ligand systems, IL-1β signaling is complex, with multiple ligands interacting with membrane-bound and soluble forms of several receptors (20). IL-1β signaling activity is mediated by a single receptor, IL-1 receptor type I (IL-1RI) (21), and its co-receptor IL-1 receptor accessory protein (IL-1RAcP) (22). A second IL-1 family member, IL-1α, signals through the same receptor complex but has not been implicated in inflammatory diseases (3). IL-1β activity is under tight physiological control, with multiple levels of negative regulation including neutralization and endocytosis of IL-1β mediated by the decoy receptor IL-1 receptor type II (IL-1RII) (23–25), inhibition of circulating IL-1β mediated by multiple soluble forms of its receptors (sRI, sRII, and sRAcP) (25, 26), and competitive inhibition by an inhibitory IL-1 homologue, IL-1 receptor antagonist (IL-1Ra) (25, 27). The complexity of this receptor-ligand system presents a challenge for the selection of therapeutic anti-IL-1β antibodies. The optimal antibody would selectively attenuate systemic high-level IL-1β signaling to lower, beneficial levels while allowing very high local concentrations of IL-1β to initiate protective inflammatory responses (in response to infection, for instance). At the same time, the antibody should not interfere with neutralization of IL-1β by soluble receptors, clearance of IL-1β by receptor-mediated pathways, IL-1α signaling, or IL-1Ra activity.
We propose that using an antibody to selectively reduce the affinity of a ligand for its signaling receptor will have the effect of reducing signaling output by reducing receptor occupancy by ligand. Kinetic modulation of a receptor-ligand interaction with an antibody can be described by the following equation (for derivation, see supplemental Fig. S1)
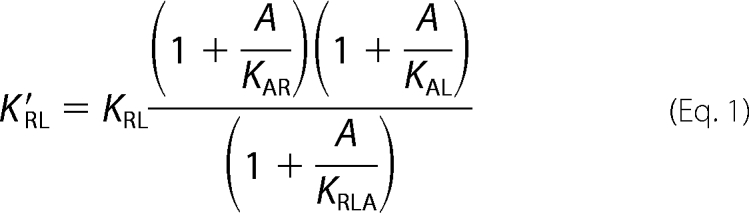 |
where the receptor-ligand equilibrium binding constant in the presence of antibody (K′RL) is a function of receptor-ligand equilibrium constant (KRL), antibody concentration (A), antibody affinity for the complex (KRLA), and antibody affinity for either the receptor (KAR) or the ligand (KAL). A regulatory antibody would bind its target (either the receptor or the ligand) in such a manner that the affinity of the ligand for its receptor is reduced, but not completely eliminated. We refer to antibodies with these characteristics as “regulatory antibodies” to distinguish them from previously described antagonist, agonist, and catalytic antibodies. One important implication of this model is that the degree of signaling attenuation is constant and is independent of antibody concentration once all target is bound by antibody. At that point, signaling will depend on the concentration of the ligand, its signaling receptor, and other endogenous regulatory mechanisms. In contrast, a traditional blocking antibody will reduce signaling in a concentration-dependent manner until reaching complete neutralization when the target is saturated. As the degree of attenuation by the regulatory antibody depends instead on the ratio of K′RL to KRL, which is an inherent property of the attenuating antibody, it is possible to produce multiple antibodies that provide various degrees of attenuation of a particular interaction. The theory behind this model is generally applicable to any biological effect that is mediated by interaction of two or more components, independent of target class. We have generated a number of regulatory antibodies to multiple targets,4 demonstrating that this class of antibodies is not limited to a single target or system.
Here we describe XOMA 052, a recombinant monoclonal antibody with long circulating half-life (28), high affinity (29), and high specificity for IL-1β (supplemental Fig. S2), that simultaneously reduces the affinity of IL-1β for IL-1RI (K′RL > KRL) while leaving affinity for IL-1RII and the soluble inhibitory receptors largely unaffected by antibody binding. XOMA 052 is a potent inhibitor of IL-1β activity both in vitro and in vivo (30–32).
EXPERIMENTAL PROCEDURES
Antibodies and Recombinant Proteins
XOMA 052 is a Human EngineeredTM IgG2 kappa antibody with 97% human sequence and affinity for IL-1β of 300 fm (29). Control blocking antibodies 5 and 6 were an IgG1 lambda and IgG2 kappa, respectively, synthesized by fusing the variable region sequences reported for receptor blocking antibodies (33, 34) to the appropriate human constant regions. The IL-1 receptor-blocking activities of the control blocking antibodies were verified by SPR analysis (not shown). The isotype control antibody was an anti-keyhole limpet hemocyanin (KLH) human IgG2 lambda antibody (clone KLH8.G2, generated at XOMA). Recombinant human IL-1Ra (catalog number 280-RA), sRI (catalog number 269-1R-100/CF), sRII (catalog number 263-2R-050/CF), and sRAcP·Fc chimera (catalog number 676-CP-100) were purchased from R&D Systems. Recombinant human IL-1β was purchased from Peprotech (catalog number 200-001B) or R&D Systems (catalog number 201-LB).
In Vitro Signal Complex Assembly
Stepwise formation of the sRI·IL-1β·RAcP ternary complex bound to XOMA 052 was performed on a multi-SPR array system (ProteOn XPR 36TM, Bio-Rad) at 25 °C using HEPES-buffered saline running buffer (0.01 m HEPES, pH 7.4, 0.15 m NaCl, and 0.05% surfactant P20). A ProteOn GLM sensor chip (Bio-Rad, catalog number 176-5012) was prepared by amine coupling NeutrAvidin (Thermo Scientific, catalog number 31000) in 0.01 m sodium acetate (pH 4.5) to an activated sensor chip surface at high density (≥10,000 response units (RU)). XOMA 052 and Blocking Ab 5 were biotinylated by reacting with 5–10 molar excess of NHS-PEO12-Biotin reagent (Thermo Scientific, catalog number 21329) according to the vendor's instructions. Excess free biotin was removed by centrifugation of proteins through a desalting column (Thermo Scientific, ZebaTM desalting spin column, catalog number 89882). Biotinylated antibodies were captured on different channels of the NeutrAvidin-coated sensor chip at densities of 500–600 RU. A reference channel was prepared in the same manner without injection of antibody. Binding to captured antibodies was tested by successive injections of 50 nm IL-1β, 100 nm sRI, and 200 nm sRAcP·Fc. The SPR binding responses were double-referenced using the ProteOn data manager program to subtract buffer injections and signal from reference surfaces.
KinExA Equilibrium Measurements
The affinities of IL-1β ± XOMA 052 binding to soluble IL-1 receptors sRI and sRII were determined in solution using KinExA technology (Sapidyne, Inc.). Equilibrium experiments were conducted by serially diluting soluble receptors from 150 nm to 4 pm in PBS (0.01 m phosphate, pH 7.4, 0.15 m NaCl, 0.02% azide) with 1% bovine serum albumin sample buffer into a constant binding site concentration of IL-1β alone or mixed with XOMA 052. To obtain KD-controlled data, the binding site concentration was no more than 2-fold above the KD (IL-1β = 1 nm and IL-1β + XOMA 052 = 5 nm). For all experiments in which XOMA 052 was present, the antibody concentration was maintained at a 100-fold molar excess over IL-1β to ensure that all of the cytokine was bound by XOMA 052. The IL-1β ± XOMA 052 plus receptor mixtures were incubated at room temperature (∼22 °C) for 12–24 h prior to assay initiation to allow complex formation to reach equilibrium. Following the incubation period, the IL-1β receptor complexes were drawn through a solid phase, consisting of receptor-blocking anti-IL-1β antibody-coupled beads, to capture unbound IL-1β ± XOMA 052. The captured IL-1β ± XOMA 052 was detected using a polyclonal anti-IL-1β antibody (R&D Systems) followed by a phycoerythrin-conjugated anti-goat IgG secondary antibody (Jackson ImmunoResearch Laboratories). The bound signals were converted into relative values as a proportion of control in the absence of receptors. Two replicates of each sample were measured for all equilibrium experiments. The equilibrium titration data were fit to a 1:1 binding model using KinExA software (Version 2.4; Sapidyne Inc.). These measurements were repeated a total of five times for sRI and two times for sRII. Results from experiments using SPR (see Table 1 and data not shown)5 are consistent with KinExA results.
TABLE 1.
Effect of XOMA 052 on affinities of IL-1β interactions
Measurements by SPR are consistent with KinExA and demonstrate that XOMA 052 reduces the affinity of IL-1β binding to IL-1 sRI and sRAcP, but not IL-1 sRII. Affinities are the average of multiple (n) independent experiments. Methods for SPR experiments are described in the supplemental data. NA, not applicable.
| Interaction by SPR | Immobilized on SPR chip | Injected | No Antibody | +XOMA 052 | n |
|---|---|---|---|---|---|
| nm | nm | ||||
| IL-1β + sRI ↔ IL-1β·sRI | sRI | IL-1β ± XOMA 052 | 0.7 ± 0.1 | 6.1 ± 0.2 | 5 |
| sRI·biotin | IL-1β ± XOMA 052 | 0.5 ± 0.2 | 4.3 ± 0.3 | 3 | |
| sRI·Fc chimera | IL-1β | 0.5 ± 0.2 | NA | 3 | |
| XOMA 052·biotin + IL-1β | sRI | NA | 10 ± 4 | 5 | |
| IL-1β + sRII ↔ IL-1β·sRII | sRII | IL-1β ± XOMA 052 | 7 ± 2 | 8 ± 3 | 5 |
| sRII·biotin | IL-1β ± XOMA 052 | 7.3 ± 0.6 | 6 ± 1 | 5 | |
| XOMA 052·biotin + IL-1β | sRII | NA | 2 ± 1 | 3 | |
| IL-1β·sRI + sRAcP-Fc ↔ IL-1β·sRI·sRAcP-Fc | sRAcP·Fc·biotin | sRI/IL-1β ± XOMA 052 | 25 ± 4 | 70 ± 20 | 3 |
| IL-1β·sRII + sRAcP-Fc ↔ IL1β·sRII·sRAcP-Fc | sRII·biotin + IL-1β ± XOMA 052 | sRAcP·Fc | 28 ± 12 | 22 ± 6 | 2 |
Kinetic Analysis of the Interaction between sRAcP and sRI·IL-1β
A NeutrAvidin-coated sensor chip was prepared as described above. Soluble RAcP·Fc was biotinylated as described above and captured on different flow channels of the sensor chip (400 RU). Reference channels (NeutrAvidin only) were prepared similarly except that no sRAcP·Fc was captured. Using the Co-inject mode, sRI (1:3 dilution series of sRI starting from 100 nm in a buffer containing 500 nm of either IL-1β or IL-1β precomplexed with a 4-fold molar excess of XOMA 052) was injected for 2 min at a flow rate of 30 μl/min followed by a second analyte injection of either 500 nm IL-1β or 500 nm IL-1β precomplexed with a 4-fold molar excess of XOMA 052. Injection of cytokine ± XOMA 052 during the dissociation phase was required to maintain saturation of receptor with cytokine. This allows measurement of the dissociation of the sRI·cytokine complex from the immobilized sRAcP·Fc independently from dissociation of the cytokine from sRI. Reference sensorgrams were subtracted from binding sensorgrams using the Scrubber analysis program. Kinetic parameters were obtained by global fitting of the data to 1:1 interaction model.
IL-1β ± XOMA 052 Binding to Membrane-bound IL-1 Receptors
IL-1β binding to full-length IL-1RI and IL-1RAcP was measured on human embryonic kidney cells expressing the Epstein-Barr virus nuclear antigen-1 (293-EBNA, Invitrogen, catalog number R620-07) maintained in Hyclone SFM4Tx (catalog number SH30860.02), 1% fetal bovine serum (Hyclone catalog number SH30070.03), 0.25 mg/ml Geneticin® (Invitrogen, catalog number 10131), 4 mm l-glutamine, and transiently transfected with genes encoding IL-1RI and RAcP (Origene, catalog numbers TC119624 and SC109339). IL-1β was labeled with Alexa Fluor 647 microscale protein labeling kit (Invitrogen, catalog number A30009) according to the manufacturer's procedure. The binding of the labeled IL-1β to sRI and XOMA 052 was tested by SPR and did not appear to be significantly altered by the label (data not shown). 293-EBNA cells were transfected using LipofectamineTM 2000 transfection reagent (Invitrogen, catalog number 11668-019) according to the manufacturer's instructions. The cells were collected after 4 days of incubation at 37 °C, 5% CO2 on a shaker, and both transgene expression and IL-1β binding were verified by flow cytometry (data not shown). Growth medium supernatants were removed and replaced with binding buffer (0.01 m phosphate, pH 7.4, 0.15 m NaCl, 1% bovine serum albumin, 0.02% azide) containing increasing amounts of either IL-1β Alexa Fluor 647 or IL-1β Alexa Fluor 647 plus 20-fold molar excess of XOMA 052 at the concentrations indicated. After three washes with binding buffer, the cells were analyzed by a FACScan flow cytometer (BD Biosciences), and the mean fluorescence was calculated for each sample. Data were analyzed using Prism software (GraphPad, Inc.).
MRC-5 IL-6 Release Assay
MRC-5 human lung fibroblast cells (ATCC, Manassas, VA) were seeded into a sterile 96-well tissue culture plate at 5000 cells/well in minimal essential medium, Earle's (Invitrogen, catalog number 10370-021) with 10% fetal bovine serum (HyClone). After an overnight incubation at 37 °C with 5% CO2, supernatants were removed and replaced with growth medium containing recombinant human IL-1β plus control Blocking Ab 6, anti-KLH isotype control antibody, or XOMA 052 at the concentrations indicated. For the antibody potency assay, IL-1β was preincubated with the indicated antibody for 1 h at 37 °C prior to addition to the cells and added at a final concentration of 100 pg/ml IL-1β. For the IL-1β dose-response assay, increasing amounts of IL-1β were preincubated overnight at room temperature with a 100-fold molar excess of antibody prior to addition to the MRC-5 cells. Following a 20-h incubation at 37 °C with 5% CO2, cell supernatants were removed and diluted according to estimated IL-6 concentration and assayed for human IL-6 by ELISA (Quantikine human IL-6 ELISA, R&D Systems, catalog number D6050) according to the manufacturer's instructions. All samples were set up and assayed in duplicate or triplicate. Data were analyzed using Prism software (GraphPad, Inc.).
NFκB Luciferase Assay
HeLa cervical adenocarcinoma cells (ATCC, Manassas VA) were stably transfected with an NFκB-luciferase reporter plasmid (Biomyx, catalog number P2120), and a tumor necrosis factor α-responsive clonal derivative was selected. The reporter cell line was maintained in Dulbecco's modified Eagle's medium/F12 medium (Invitrogen, catalog number 11330-032) with 10% fetal bovine serum (HyClone) and 0.25 mg/ml hygromycin (Invitrogen). One day prior to treatment, cells were transferred into opaque white 96-well plates (Corning, catalog number 3917) at 20,000 cells/well in hygromycin-free growth medium. For the IL-1β titration assay, increasing amounts of IL-1β were preincubated overnight at room temperature with a 1000-fold molar excess of antibody prior to addition to the reporter cells. For the antibody potency assay, recombinant human IL-1β was preincubated with increasing concentrations of antibody for 1 h at 37 °C prior to addition to the cells and added at a final IL-1β concentration of 25 pg/ml. IL-1β plus antibody was incubated with the HeLa reporter cells for 16 h at 37 °C in 5% CO2. To assess luciferase expression levels, Bright-Glo (Promega) was added to the cells according to manufacturer's instructions, and samples were read on a FlexStation 3 (Molecular Devices). All samples were set up and assayed in duplicate or triplicate. Data were analyzed using Prism software (GraphPad, Inc.).
IL-1β Signal Transduction
A549 cells were obtained from ATCC and maintained in minimal essential medium, Earle's (Invitrogen, catalog number 10370-021) supplemented with 10% fetal bovine serum (HyClone). Subconfluent monolayers were washed twice with room temperature PBS and then incubated overnight at 37 °C in serum-free medium containing 0.5% bovine serum albumin. XOMA 052 at a 100-fold molar excess was incubated with the diluted IL-1β overnight at 4 °C in serum-free medium. Following 18 h in serum-free medium, the cells were incubated for 15 min in prewarmed serum-free media containing IL-1β with or without 100-fold molar excess of XOMA 052. Cells were cooled on ice, rinsed twice with ice-cold Tris-buffered saline, and then lysed with radioimmune precipitation assay buffer (Pierce, catalog number 89901) containing protease inhibitor mixture (Pierce, catalog number 78429) and phosphatase inhibitor mixture (Pierce, catalog number 78426). The monolayer was scraped and transferred to a microcentrifuge tube, incubated for 15–45 min on ice, and then spun at 4 °C in a microcentrifuge for 20 min at 15,000 relative centrifugal force to clear. Total protein concentration was measured by a BCA assay (Pierce, catalog number 23227). This study was carried out twice, and samples were analyzed in duplicate.
Analysis of Phosphoproteins
Phosphorylation of p38, ERK, JNK kinases, and total IκB was assessed by Western blotting (n = 2). An equal amount of protein in each sample (25 μg of whole cell lysate/well) was separated by 4–12% SDS-PAGE and transferred to polyvinylidene difluoride membrane. Membranes were blocked in Tris-buffered saline buffer containing 0.1% Tween 20 and 5% bovine serum albumin and probed with mouse anti-p-ERK-1, anti-p-JNK-1, anti-p-p38, and anti-IκB or anti-ERK, anti-JNK, and anti-p38 polyclonal antibody (Cell Signaling Technologies). Following incubation with peroxidase-conjugated sheep anti-mouse or donkey anti-rabbit secondary antibody (GE Healthcare, catalog number NA931V and catalog number NA934V, respectively), proteins were visualized with chemiluminescence detection system.
Phosphoprotein Quantification
Phosphorylated ERK and JNK were quantified using the MULTI-SPOT assay system (MesoScale Discovery, MAP kinase whole cell lysate kit, MSD number K11101D-1) according to the manufacturer's instructions. Data were analyzed using Prism software (GraphPad, Inc.).
In Vivo Assays
For this study, we used control Blocking Ab 5, which completely blocks IL-1β binding to IL-1 sRI and sRII (not shown). Complexes of IL-1β with XOMA 052 or the control blocking antibody were prepared by mixing 0.4 μg/ml rhIL-1β with a 100-fold molar excess of antibody and incubating overnight at 25 °C. A third solution was prepared using anti-KLH-IgG2 as a non-binding isotype control antibody. All proteins were formulated in sterile PBS. rhIL-1β either complexed or mixed with the isotype control antibody was diluted 2-fold with PBS and administered intravenously to 8-week-old female C57BL/6 mice at a dose volume of 100 μl to give a dosage of 1 mg/kg (20 ng of rhIL-1β/mouse). In two independent experiments, four mice per group were dosed, and blood was collected from all mice by postmortem cardiac puncture at 48 h after intravenous injection. Samples were frozen at −70 °C for subsequent analysis.
Assay of Total rhIL-1β in Mouse Serum
Human IL-1β levels were measured by ELISA (BD OptEIATM human IL-1β ELISA set, BD Biosciences, catalog number 557953) as per kit instructions. The non-interference of the capture and detection antibodies by XOMA 052 and control Blocking Ab 5 were verified both by ELISA and by SPR (not shown). In brief, ELISA plates (Corning Costar, catalog number 9018) were coated overnight at 4 °C with anti-human IL-1β capture antibody provided (BD Biosciences, catalog number 51-9002512) in 0.1 m NaHCO3. The following day, the plates were washed with PBS-Tween 20 and blocked for 2 h with blocking buffer (10% fetal bovine serum in PBS). Samples were prepared in duplicate with eight serial 2-fold dilutions. Samples and standards (recombinant human IL-1β standard, BD Biosciences catalog number 51-9004477) were prepared in 10% mouse serum in PBS, added to the wells, and incubated for 2 h at room temperature. The plates were then washed four times with PBS. Detection antibody was added (anti-human IL-1β-biotin, BD Biosciences, catalog number 51-9002516), and plates were incubated for 1 h at room temperature. The plates were subsequently washed six times with PBS-Tween 20. Streptavidin-labeled horseradish peroxidase (BD Biosciences, catalog number 51-90002813) was added and incubated for 30 min. Plates were washed six times, and 3,3′,5,5′-tetramethylbenzidine solution (Calbiochem, catalog number 613544) was added and incubated for 30 min in the dark. Stop solution (0.2 n H2SO4) was added, and plates were read at 450 nm on a microtiter plate spectrophotometer within 30 min of stopping the reaction. The sensitivity of the ELISA was from 250 to 3.9 pg/ml.
RESULTS
Activation of the IL-1β signaling pathway is initiated by binding of IL-1β to IL-1RI followed by recruitment of IL-1RAcP into a ternary signaling complex. A standard blocking antibody binds to IL-1β and competes for binding to IL-1RI, completely blocking signaling by the antibody·ligand complex. However, when XOMA 052 binds to IL-1β, the antibody·IL-1β complex is still capable of binding IL-1RI, and the complex of XOMA 052, IL-1β, and sRI can bind to sRAcP in vitro, suggesting that binding of XOMA 052 to IL-1β does not block assembly of the signaling complex (Fig. 1). In control experiments (not shown), we demonstrated that assembly of this complex occurs in an obligate stepwise manner and that the antibody does not bind to either receptor in the absence of IL-1β.
FIGURE 1.

Binding of IL-1β by XOMA 052 allows assembly of IL-1 signaling complex. XOMA 052 and control Blocking Ab 5 were immobilized on different flow cells of an SPR sensor surface, and IL-1β, sRI, and sRAcP·Fc fusion protein were injected at the points indicated by arrows 1, 2, and 3, respectively. The extracellular domains of both IL-1RI and IL-1RAcP are able to bind to IL-1β bound by XOMA 052. IL-1β bound by Blocking Ab 5 does not allow complex assembly.
Kinetics of binding of IL-1β to IL-1RI and IL-1RII are complex, perhaps reflecting the conformational flexibility of the receptors and the multiple binding interfaces between the cytokine and its receptors (35, 36). For this reason, we used equilibrium dissociation constants (KD) to compare the effects of XOMA 052 on the affinity of binding of IL-1β to these receptors. In-depth kinetic characterization of this system and the effect of XOMA 052 will be described in a separate publication.5 Equilibrium and kinetic analyses revealed that XOMA 052 reduces the affinity of IL-1β binding to sRI by 5–10-fold (Fig. 2a and Table 1) while having no effect on the affinity of IL-1β for sRII (Fig. 2b and Table 1). In addition, XOMA 052 reduces affinity of subsequent binding of sRAcP to the IL-1β·sRI complex by another 2–3-fold (Fig. 2, c and d, and Table 1).
FIGURE 2.

Effect of XOMA 052 on IL-1β binding to receptors. XOMA 052 reduces the affinity of IL-1β binding to sRI measured by KinExA from 2 to 10 nm (n = 5) (a) but has no effect on the affinity of IL-1β binding to sRII of 2 nm (n = 2) (b). XOMA 052 reduces the affinity of sRAcP binding to sRI·IL-1β complex from 25 (c) to 69 nm (d), as measured by SPR. XOMA 052 shifts the apparent affinity of Alexa Fluor 647-labeled IL-1β binding to HEK293 cells expressing IL-1RI and IL-1RAcP from 2 to 9 nm (n = 2) (e).
Binding of XOMA 052·IL-1β complexes to the membrane forms of the receptors was verified by flow cytometry with HEK293 cells transfected with full-length IL-1RI and IL-1RAcP (Fig. 2e). The shift in the binding of fluorescently labeled IL-1β to cell surface receptors is consistent with a decrease in the affinity caused by XOMA 052.
Although XOMA 052 binding to IL-1β does not abrogate in vitro signal complex formation at high concentrations of IL-1β, it nonetheless is a potent inhibitor of IL-1β activity at physiological and pathologically relevant concentrations. XOMA 052 completely neutralizes 100 pg/ml IL-1β in an MRC-5 cytokine release assay with an observed IC50 in the low pm range. This is comparable with neutralization observed with a control blocking antibody (Fig. 3a) and ∼10-fold more potent than recombinant IL-1Ra (supplemental Fig. S3).
FIGURE 3.

Effect of XOMA 052 on activity of IL-1β in cell-based activity assays. a, XOMA 052 neutralizes IL-1β stimulation of IL-6 release by MRC-5 cells with an IC50 of ∼2 pm at the EC50 for this assay (100 pg/ml IL-1β). This activity is comparable with that of control Blocking Ab 6, with an IC50 of ∼6 pm. b, XOMA 052 attenuates the dose response of MRC-5 cells to IL-1β ∼50-fold (EC50 values of 12 and 815 pm with an anti-KLH antibody and XOMA 052, respectively), whereas Blocking Ab 6 almost completely inhibits IL-1β activity. c, XOMA 052 neutralizes IL-1β stimulation of NFκB activation in HeLa cells stably expressing an NFκB-luciferase reporter construct with an IC50 of ∼1 pm at the EC50 for this assay (25 pg/ml IL-1β). Under these conditions, the potency of the control blocking antibody is 36-fold lower, with an IC50 of 36 pm. RLU, relative light units. d, XOMA 052 attenuates the dose response of HeLa cells stably expressing an NFκB-luciferase reporter construct to IL-1β stimulation ∼20-fold, from an EC50 of 1.7 pm in the presence of a non-binding anti-KLH antibody to 36 pm with XOMA 052. For a, c, and d, error bars show S.D. of three replicates for test samples and duplicates for the anti-KLH antibody control. For b, error bars show S.D. between duplicate samples. All experiments were run at an n = 2.
Our model predicts that the reduction in the affinities of signal complex components will cause a shift in the cellular dose response to IL-1β. When XOMA 052 is maintained in molar excess of the concentration of IL-1β, the antibody increases the EC50 of the IL-1β dose-response curve up to 70-fold in the MRC-5 cell IL-6 release assay relative to that of IL-1β in the presence of a control antibody while still permitting maximum activity in the presence of very high concentrations of ligand. Under the same conditions, an IL-1β blocking antibody nearly ablates cellular response across a broad range of IL-1β concentrations (Fig. 3b). Similar dose-response shifts with XOMA 052 are seen in stimulation of IL-8 expression in whole blood (supplemental Fig. S4) and experiments using an NFκB reporter construct (Fig. 3, c and d) in transfected HeLa cells. Although a similar dose-response shift in NFκB activation was observed in the presence of XOMA 052, the control antibody also appeared to incompletely block signaling at extremely high concentrations of IL-1β. However, we believe that the loss of complete neutralization at the higher concentrations results from the 20-fold lower affinity of the control antibody as compared with XOMA 052 in combination with the high sensitivity of this reporter assay. The calculated amount of free IL-1β at the highest concentration of complex at equilibrium is 0.1 pm, which can account for the observed level of NFκB activation. The affinity of XOMA 052, on the other hand, would allow a free IL-1β concentration of only 0.005 pm at equilibrium, which is well below the level required for detectable NFκB activity in this assay. NFκB activation observed for XOMA 052·IL-1β complexes therefore cannot be explained by free IL-1β in these conditions. Comparable results were obtained in additional cell assays (not shown), demonstrating that this effect is not unique to a particular cell line or assay system.
IL-1β has been shown to activate a number of specific protein kinases, including the NFκB-inducing kinase (NIK) and three distinct mitogen-activated protein (MAP) kinase cascades (37). To determine whether binding by XOMA 052 might have differential effects on signaling through the different pathways, we measured phosphorylation of JNK, ERK 1/2, and p38, as well as total IκB levels, by increasing concentrations of IL-1β in the presence and absence of excess XOMA 052 (Fig. 4 and data not shown). Consistent with our model and with previous cell-based studies, XOMA 052 shifted the dose response of IL-1β. For two cell lines, A549 and HepG2, XOMA 052 increases the EC50 (i.e. decreases the potency) for phosphorylation of JNK and ERK ∼65–130-fold (Fig. 4, c and d, and supplemental Table S1). Quantitation of the phosphorylation of JNK, ERK 1/2, and p38, as well as total IκB levels, showed that the relative activities of the different pathways are reduced proportionally by XOMA 052, indicating that there is no differential change in the downstream signaling through these pathways.
FIGURE 4.

Evaluation of p-JNK, p-p38, and total IκB in IL-1β- and IL-1β + XOMA 052-treated A549 cells. Cell lysates of A549 cells treated with increasing concentration of IL-1β ± XOMA 052 for 15 min were separated by SDS-PAGE followed by Western blot of p-JNK (a), p-p38 (b), and IκB (c) as described under “Experimental Procedures” (n = 2). phos, phosphorylated. Quantification of phosphoprotein levels by the MULTI-SPOT assay system shows that XOMA 052 shifts the dose response of JNK and ERK phosphorylation in IL-1β-stimulated A549 cells, changing the EC50 from 10 to 770 pm (d) and from 15 to 990 pm (e) respectively (n = 2, error bars show S.D. between duplicate samples).
Although reducing IL-1β affinity for IL-1RI causes attenuation of signaling, maintenance of efficient binding to IL-1RII is important because IL-1RII functions as a decoy receptor on responding cells to attenuate sensitivity to IL-1β (24, 38–40). In addition, IL-1RII-mediated internalization of IL-1β is believed to be an important pathway for clearance of IL-1β (23). When binding of a therapeutic antibody to its target interrupts physiological clearance pathways, the prolonged half-life and high affinity of the antibody can cause the accumulation of antibody·antigen complexes (41). Although such complexes are typically inactive, it is necessary to maintain excess levels of antibody to ensure that any antigen that dissociates from the antibody is rapidly rebound by antibody.
To test whether the ability of IL-1β·XOMA 052 complexes to bind IL-1 receptors affects clearance of IL-1β·antibody complexes, we injected mice with recombinant human IL-1β, either free (incubated with anti-KLH isotype control) or precomplexed with either XOMA 052 or control Blocking Ab 5, and measured the amount of total measurable IL-1β remaining in serum after 48 h. Although free IL-1β was undetectable at this time point, an average of 4.2-fold less IL-1β bound to XOMA 052 was detected in the serum as compared with IL-1β bound to Blocking Ab 5 (Fig. 5). At the same time, the serum concentrations of both antibodies were similar (not shown), indicating that the difference in the amount of IL-1β bound to XOMA 052 as compared with IL-1β bound to Blocking Ab 5 did not result from differential clearance of antibodies themselves.
FIGURE 5.

Comparison of the effects of XOMA 052 and Blocking Ab 5 on persistence of recombinant human IL-1β in mouse serum in vivo. In two independent experiments, an average of 4-fold less IL-1β bound to XOMA 052 was detected in the serum after 48 h as compared with IL-1β bound to Blocking Ab 5. Error bars show S.D. of duplicate samples from four animals in each treatment group. Significance was determined using a paired (two-tailed) t test.
DISCUSSION
IL-1β has been described as a master cytokine involved in initiating the innate immune response in vertebrates. Although inappropriate expression of IL-1β has been implicated in the pathogenesis of multiple diseases, several examples of the beneficial effects of this cytokine have been described. For example, pretreatment with IL-1β protects against infection, radiation, hyperoxia, arthritis, antigen-induced histamine release, inflammatory bowel disease, and contact dermatitis. Thus, as was recognized decades ago, it is possible that a small amount of IL-1 is beneficial, whereas larger amounts of the cytokine are detrimental (42). The dual nature of its role in both protecting against and causing tissue damage or disease makes IL-1β a complex target for optimal therapeutic intervention.
In recent years, the number of therapeutic agents directed against IL-1 that are advancing through clinical trials has grown substantially, which has led either to drug approval or to advancement to phase II/III trials. To date, results of clinical trials on rheumatoid arthritis, cryopyrin-associated periodic syndromes, and type 2 diabetes have been very promising. Several neutralizing therapeutic antibodies with high affinity have been described (40, 41). Although it may be perceived that most of these antibodies exert their activity through similar mechanisms such as ligand blocking and therefore should have similar results, subtle differences among these antibodies with regard to epitope and mechanism of inhibition may result in differences in activity. Thus, studies elucidating the structure-function relationship of inhibitory anti-IL-1β antibodies may provide some understanding of mechanism of action and predict various outcomes and safety profiles in the clinic.
We have described a recombinant antibody that differentially tunes the affinity of a ligand for binding to multiple receptors, allowing for context-dependent attenuation of ligand activity. Under physiological conditions where increased levels of IL-1β cause pathology, this mechanism enables XOMA 052 to neutralize excess IL-1β while potentially allowing continued beneficial signaling in response to local inflammatory stimuli. XOMA 052 may therefore allow for better responsiveness of the innate immune system to infection as compared with a complete blockade of IL-1β activity (4–7). Furthermore, the degree of signaling attenuation mediated by XOMA 052 is independent of the concentration of antibody when its concentration is sufficiently high to bind all available IL-1β. Under such saturating conditions, the concomitant signaling output depends on the concentrations of ligand and other components of the native signaling system (e.g. IL-1Ra, IL-1 receptors, and soluble receptors). There is increasing appreciation that many other receptor-ligand systems are comprised of multiple ligands and receptors that generate complex and context-dependent cellular effects. The ability to use antibodies therapeutically as “rheostats” rather than “switches” introduces an additional level of subtlety and sophistication in therapeutic antibody design for regulating the activity of disease-relevant targets.
As a remarkably potent molecule with responses observable at concentrations as low as femtomolar, IL-1β is highly regulated at multiple levels, including transcription, translation, and proteolytic activation, as well as competition for binding to IL-1RI by the receptor antagonist, soluble receptors, and decoy receptor (42). Because IL-1β signal transduction can result from very low receptor occupancy (42) and because IL-1β has positive feedback on its own expression, this finely tuned balance may be perturbed by subtle changes in the relative levels of components of the regulatory mechanism. Reestablishment of homeostatic balance may not require dramatic changes such as complete neutralization, but rather it is possible that attenuation of IL-1β activity will provide more effective treatment than complete blockade of the pathway. The data presented in this study demonstrate that XOMA 052 reduces IL-1β activity in a context-dependent manner that allows regulation by the endogenous competitors. Determination of the advantage of this novel mechanism of action for treatment of IL-1β-mediated diseases is under further investigation in animal models and clinical trials.
Supplementary Material
Acknowledgment
We thank Clint Rogers for critical reviews of the manuscript and insightful comments.

The on-line version of this article (available at http://www.jbc.org) contains supplemental text, Table S1, and Figs. S1–S4.
J. A. Corbin, S. Watson, D. H. Bedinger, M. K. Roell, and M. L. White, manuscript in preparation.
H. Issafras, J. A. Corbin, and M. K. Roell, manuscript in preparation.
- IL-1β
- interleukin-1β
- IL-1α
- interleukin-1α
- IL-1Ra
- interleukin-1 receptor antagonist
- IL-1RI
- IL-1 receptor type I
- IL-1RII
- IL-1 receptor type II
- IL-1RAcP
- IL-1 receptor accessory protein
- sRI
- soluble IL-1 receptor I
- sRII
- soluble IL-1 receptor II
- sRAcP
- soluble IL-1 receptor accessory protein
- KinExA
- kinetic exclusion assay
- SPR
- surface plasmon resonance
- KLH
- keyhole limpet hemocyanin
- PBS
- phosphate-buffered saline
- ELISA
- enzyme-linked immunosorbent assay
- HEK
- human embryonic kidney
- ERK
- extracellular signal-regulated kinase
- JNK
- c-Jun N-terminal kinase
- MAP
- mitogen-activated protein
- rh
- recombinant human
- p
- phosphorylated
- RU
- response units.
REFERENCES
- 1.Liu X. Y., Pop L. M., Vitetta E. S. (2008) Immunol. Rev. 222, 9–27 [DOI] [PubMed] [Google Scholar]
- 2.Trikha M., Yan L., Nakada M. T. (2002) Curr. Opin. Biotechnol. 13, 609–614 [DOI] [PubMed] [Google Scholar]
- 3.Dinarello C. A. (2004) Curr. Opin. Pharmacol. 4, 378–385 [DOI] [PubMed] [Google Scholar]
- 4.Fremond C. M., Togbe D., Doz E., Rose S., Vasseur V., Maillet I., Jacobs M., Ryffel B., Quesniaux V. F. (2007) J. Immunol. 179, 1178–1189 [DOI] [PubMed] [Google Scholar]
- 5.Graves D. T., Chen C. P., Douville C., Jiang Y. (2000) Infect. Immun. 68, 4746–4751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hise A. G., Tomalka J., Ganesan S., Patel K., Hall B. A., Brown G. D., Fitzgerald K. A. (2009) Cell Host Microbe 5, 487–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vonk A. G., Netea M. G., van Krieken J. H., Iwakura Y., van der Meer J. W., Kullberg B. J. (2006) J. Infect. Dis. 193, 1419–1426 [DOI] [PubMed] [Google Scholar]
- 8.Dinarello C. A. (2005) J. Exp. Med. 201, 1355–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maedler K., Sergeev P., Ris F., Oberholzer J., Joller-Jemelka H. I., Spinas G. A., Kaiser N., Halban P. A., Donath M. Y. (2002) J. Clin. Invest. 110, 851–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larsen C. M., Faulenbach M., Vaag A., Vølund A., Ehses J. A., Seifert B., Mandrup-Poulsen T., Donath M. Y. (2007) N. Engl. J. Med. 356, 1517–1526 [DOI] [PubMed] [Google Scholar]
- 11.Donath M. Y., Mandrup-Poulsen T. (2008) Nat. Clin. Pract. Endocrinol. Metab. 4, 240–241 [DOI] [PubMed] [Google Scholar]
- 12.Ehses J. A., Ellingsgaard H., Böni-Schnetzler M., Donath M. Y. (2009) Arch. Physiol. Biochem. 115, 240–247 [DOI] [PubMed] [Google Scholar]
- 13.Maedler K., Schumann D. M., Sauter N., Ellingsgaard H., Bosco D., Baertschiger R., Iwakura Y., Oberholzer J., Wollheim C. B., Gauthier B. R., Donath M. Y. (2006) Diabetes 55, 2713–2722 [DOI] [PubMed] [Google Scholar]
- 14.Spinas G. A., Mandrup-Poulsen T., Mølvig J., Baek L., Bendtzen K., Dinarello C. A., Nerup J. (1986) Acta Endocrinol. 113, 551–558 [DOI] [PubMed] [Google Scholar]
- 15.Spinas G. A., Palmer J. P., Mandrup-Poulsen T., Andersen H., Nielsen J. H., Nerup J. (1988) Acta Endocrinol. 119, 307–311 [DOI] [PubMed] [Google Scholar]
- 16.Donath M. Y., Schumann D. M., Faulenbach M., Ellingsgaard H., Perren A., Ehses J. A. (2008) Diabetes Care 31, Suppl. 2, S161–s164 [DOI] [PubMed] [Google Scholar]
- 17.Waterhouse C. C., Joseph R. R., Stadnyk A. W. (2001) Exp. Cell Res. 269, 109–116 [DOI] [PubMed] [Google Scholar]
- 18.Lebeis S. L., Powell K. R., Merlin D., Sherman M. A., Kalman D. (2009) Infect Immun. 77, 604–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gosselin D., Rivest S. (2007) Brain Behav. Immun. 21, 281–289 [DOI] [PubMed] [Google Scholar]
- 20.Dinarello C. A. (2005) Arthritis Rheum. 52, 1960–1967 [DOI] [PubMed] [Google Scholar]
- 21.Curtis B. M., Gallis B., Overell R. W., McMahan C. J., DeRoos P., Ireland R., Eisenman J., Dower S. K., Sims J. E. (1989) Proc. Natl. Acad. Sci. U.S.A. 86, 3045–3049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hofmeister R., Wiegmann K., Korherr C., Bernardo K., Krönke M., Falk W. (1997) J. Biol. Chem. 272, 27730–27736 [DOI] [PubMed] [Google Scholar]
- 23.Bourke E., Cassetti A., Villa A., Fadlon E., Colotta F., Mantovani A. (2003) J. Immunol. 170, 5999–6005 [DOI] [PubMed] [Google Scholar]
- 24.Bossù P., Visconti U., Ruggiero P., Macchia G., Muda M., Bertini R., Bizzarri C., Colagrande A., Sabbatini V., Maurizi G. (1995) Am. J. Pathol. 147, 1852–1861 [PMC free article] [PubMed] [Google Scholar]
- 25.Burger D., Chicheportiche R., Giri J. G., Dayer J. M. (1995) J. Clin. Invest. 96, 38–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith D. E., Hanna R., Friend D., Moore H., Chen H., Farese A. M., MacVittie T. J., Virca G. D., Sims J. E. (2003) Immunity 18, 87–96 [DOI] [PubMed] [Google Scholar]
- 27.Arend W. P., Malyak M., Guthridge C. J., Gabay C. (1998) Annu. Rev. Immunol. 16, 27–55 [DOI] [PubMed] [Google Scholar]
- 28.Donath M. (2008) Diabetologia 51 (suppl 1), S7 [Google Scholar]
- 29.Masat L., Haak-Frendscho M., Chen G., Horwitz A. H., Roell M. K. (May12, 2009) U. S. Patent 7,531,166
- 30.Owyang A., Gross L., Yin J., Lee S., Shu L., Beergemann J., Jadhav J., Esposito L., Maedler K., Kantak S. (2009) Diabetes 58, 310-OR [Google Scholar]
- 31.Donath M. Y., Weder C., Brunner A., Keller C., Whitmore J., Der K., Zayed H., Scannon P. J., Feldstein J. D., Dinarello C. A., Solinger A. M. (2009) Diabetes 58, 113-OR [Google Scholar]
- 32.Owyang A. M., Maedler K., Gross L., Yin J., Esposito L., Shu L., Jadhav J., Domsgen E., Bergemann J., Lee S., Kantak S. (2010) Endocrinology, in press [DOI] [PubMed] [Google Scholar]
- 33.Green L., Faggioni R., Foord O., Klakamp S., Senaldi G., Schneider A. K. (July28, 2008) U. S. Patent 7, 566, 772
- 34.Gram H., Di Padova F. E. (April1, 2004) U. S. Patent Application 20040063913
- 35.Einstein R., Jackson J. R., D'Alessio K., Lillquist J. S., Sathe G., Porter T., Young P. R. (1996) Cytokine 8, 206–213 [DOI] [PubMed] [Google Scholar]
- 36.Schreuder H., Tardif C., Trump-Kallmeyer S., Soffientini A., Sarubbi E., Akeson A., Bowlin T., Yanofsky S., Barrett R. W. (1997) Nature 386, 194–200 [DOI] [PubMed] [Google Scholar]
- 37.Stylianou E., Saklatvala J. (1998) Int. J. Biochem. Cell Biol. 30, 1075–1079 [DOI] [PubMed] [Google Scholar]
- 38.Lang D., Knop J., Wesche H., Raffetseder U., Kurrle R., Boraschi D., Martin M. U. (1998) J. Immunol. 161, 6871–6877 [PubMed] [Google Scholar]
- 39.Penton Rol G., Polentarutti N., Sironi M., Saccani S., Introna M., Mantovani A. (1997) Eur. Cytokine Netw. 8, 265–269 [PubMed] [Google Scholar]
- 40.Re F., Sironi M., Muzio M., Matteucci C., Introna M., Orlando S., Penton-Rol G., Dower S. K., Sims J. E., Colotta F., Mantovani A. (1996) J. Exp. Med. 183, 1841–1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lachmann H. J., Lowe P., Felix S. D., Rordorf C., Leslie K., Madhoo S., Wittkowski H., Bek S., Hartmann N., Bosset S., Hawkins P. N., Jung T. (2009) J. Exp. Med. 206, 1029–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dinarello C. A. (1994) FASEB J. 8, 1314–1325 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.