

Abstract
The E3 ubiquitin ligase Nedd4-2 regulates several ion transport proteins, including the epithelial Na+ channel (ENaC). Nedd4-2 decreases apical membrane expression and activity of ENaC. Although it is subject to tight hormonal control, the mechanistic basis of Nedd4-2 regulation remains poorly understood. To characterize regulatory inputs to Nedd4-2 function, we screened for novel sites of Nedd4-2 phosphorylation using tandem mass spectrometry. Three of seven identified Xenopus Nedd4-2 Ser/Thr phosphorylation sites corresponded to previously identified target sites for SGK1, whereas four were novel, including Ser-293, which matched the consensus for a MAPK target sequence. Further in vitro and in vivo phosphorylation experiments revealed that Nedd4-2 serves as a target of JNK1, but not of p38 MAPK or ERK1/2. Additional rounds of tandem mass spectrometry identified two other phosphorylated residues within Nedd4-2, including Thr-899, which is present within the catalytic domain. Nedd4-2 with mutations at these sites had markedly inhibited JNK1-dependent phosphorylation, virtually no ENaC inhibitory activity, and significantly reduced ubiquitin ligase activity. These data identify phosphorylatable residues that activate Nedd4-2 and may work together with residues targeted by inhibitory kinases (e.g. SGK1 and protein kinase A) to govern Nedd4-2 regulation of epithelial ion transport.
Keywords: E3 Ubiquitin Ligase, JNK, Mass Spectrometry (MS), Protein Phosphorylation, Sodium Channels, Nedd4-2, SGK1
Introduction
Nedd4-2, a member of the HECT (homology to the E6-associated protein C terminus) family of E3 ubiquitin ligases (1, 2), is a physiologically important regulator of the epithelial Na+ channel (ENaC)3 (1, 3). ENaC likely exists as an αβγ-heterotrimer at the apical membrane in a variety of epithelial tissues, including the kidney collecting duct, where it controls renal Na+ reabsorption (4). Nedd4-2 decreases ENaC cell-surface expression, and inactivation of Nedd4-2 in cultured cells increases surface expression and Na+ current (5), whereas genetic deletion of Nedd4-2 in mice causes salt-sensitive hypertension (6). In humans, the clinical manifestations of Liddle syndrome underscore the physiological importance of Nedd4-2. This severe inherited form of hypertension results from ENaC mutations that abolish interaction with Nedd4-2 and cause renal Na+ retention (7, 8). Potential effects of Nedd4-2-mediated ENaC ubiquitination include increased endocytosis from the plasma membrane and enhanced proteasomal and/or lysosomal degradation (9, 10). The effects of Nedd4-2 on ENaC trafficking are mediated in part through direct ubiquitination of ENaC and possibly through ubiquitination of components of the trafficking machinery. Furthermore, Nedd4-2 may decrease ENaC activity by limiting its cell-surface residence time and thus cleavage of α- and γ-ENaC subunits by activating proteases (11).
Phosphorylation is an important mechanism for the regulation of Nedd4-2 and other E3 ligases. For example, Nedd4-2 phosphorylation by SGK1 (serum- and glucocorticoid-regulated kinase 1) and protein kinase A (PKA) has been reported at three residues (12–14). SGK1 expression and activity are stimulated by aldosterone, whereas PKA is activated by vasopressin. Therefore, Nedd4-2 is a convergence point for hormonal regulation of epithelial Na+ transport in the distal nephron. The inhibitory effects of SGK1 and PKA on Nedd4-2 are mediated by Nedd4-2 phosphorylation-induced interaction with 14-3-3 scaffolding proteins, which prevent interaction with and ubiquitination of ENaC (15–17). Mutation of SGK1 target sites inhibits Nedd4-2 and leads to enhanced ENaC cell-surface expression and activity. Other Nedd4-2 inhibitory kinases have been suggested, including Akt, which is related to SGK1 (18). Furthermore, we have shown recently that IκB kinase-β stimulates ENaC through phosphorylation of Nedd4-2 (19), whereas AMP-activated protein kinase negatively regulates ENaC through a stimulatory phosphorylation of Nedd4-2, which enhances the interaction of β-ENaC with Nedd4-2 (20, 21).
Other E3 ligases are regulated by phosphorylation as well. Notably, Itch/AIP4 (atrophin 1-interacting protein 4), a related HECT E3 ligase, is phosphorylated at three residues by JNK1 (c-Jun N-terminal kinase 1), which disrupts an intramolecular interaction between the catalytic HECT domain and a proline-rich N-terminal domain of the ligase. This conformational change activates the ligase and increases auto-ubiquitination and ubiquitination of Itch targets (22). Phosphorylation by SGK3/cytokine-independent survival kinase (a close relative of SGK1) and Fyn negatively regulate Itch/AIP4 (23, 24). Thus, phosphorylation has been described as a mechanism for modulation of its catalytic function in a context-dependent manner. With these observations in mind, we sought to identify novel phosphorylation-dependent regulatory inputs to Nedd4-2 using tandem mass spectrometry coupled with in vitro phosphorylation assays and site-directed mutagenesis. We then confirmed the effects on Nedd4-2 and ENaC function through biochemical and electrophysiological assays. Using this approach, we have identified several novel sites of phosphorylation on Nedd4-2 and quantified SGK1-mediated phosphorylation. Our data show that Ser-293 and Thr-899 are novel sites of phosphorylation and support the idea that specific kinases (possibly JNK1) may activate Nedd4-2 and inhibit ENaC.
EXPERIMENTAL PROCEDURES
Cell Culture
Human embryonic kidney (HEK293) cells and mouse polarized kidney cortical collecting duct (mpkCCDc14) cells were maintained and cultured as described previously (15).
Generation of Recombinant Nedd4-2
Wild-type or mutant N-terminal glutathione S-transferase (GST)-tagged Xenopus Nedd4-2 (xNedd4-2; homologous to human Nedd4-2) subcloned in pGEX4T1 (gift of Dr. Olivier Staub) was expressed in Escherichia coli Rosetta 2 BL21 cells (EMD Biosciences). Individual colonies were grown overnight at 30 °C to A600 = 0.6, induced with 40 μm isopropyl β-d-thiogalactopyranoside at room temperature overnight, and isolated using glutathione-Sepharose 4B beads (Amersham Biosciences) following the manufacturer's protocol. Uniform expression was confirmed by SDS-PAGE and Coomassie Blue staining and quantified by the Bradford assay.
In Vitro Phosphorylation Assays
Assays of Nedd4-2 in vitro phosphorylation were performed with either recombinant Nedd4-2 or FLAG-xNedd4-2 (20) transfected and immunopurified from HEK293 cells. In vitro phosphorylation assays were performed using 0.8, 1.6, 0.8, and 0.2 ng of purified active JNK1α1, p38 MAPKα, ERK1 (extracellular signal-regulated kinase 1), and SGK1, respectively (Upstate) or control buffer and with 10 μCi of [γ-32P]ATP following the manufacturer's instructions. For recombinant Nedd4-2, phosphorylated samples were blotted onto phosphocellulose P81 squares (Whatman), washed, and counted in a scintillation counter. The respective positive control peptides supplied by the manufacturer were used to ensure adequate intrinsic kinase activity. For cell-expressed Nedd4-2, the reaction product was analyzed as described previously (20).
JNK1 Phosphorylation of Nedd4-2 in Cultured Cells
HEK293 cells were transiently transfected with FLAG-Nedd4-2 and either green fluorescent protein (GFP; pGreenLantern, Invitrogen) or human JNK1β1 (gift of Dr. Bing Ye) 1 day prior to experimentation. In vivo [32P]orthophosphate labeling assays of Nedd4-2 were performed as described previously (20).
Xenopus Oocyte Coexpression Assay and Two-electrode Voltage Clamp
Maintenance of Xenopus laevis frogs, surgical extraction of ovaries, and collagenase treatment of oocytes were carried out as described (25). cRNAs for all proteins (mouse α-, β-, and γ-ENaC subunits and wild-type (WT) and mutant Nedd4-2) were synthesized using the mMESSAGE mMACHINE kit (Ambion) and were injected into Stage V-VI oocytes. Two-electrode voltage clamp (TEV) measurements of ENaC currents in oocytes were performed as described previously (21). Lysis and immunoblotting of oocytes for expression of FLAG-tagged Nedd4-2 were performed using SDS-PAGE, anti-FLAG antibody M2 (Sigma), and anti-β-actin antibody (Sigma) as described previously (26).
ENaC Ubiquitin Assays
Ubiquitin assays were performed as described previously (15), except that HEK293 cells grown on 6-cm2 dishes were transfected using the FuGENE HD (Roche Applied Science) technique with 1.5 μg of plasmid DNA encoding differentially C-terminal epitope-tagged mouse ENaC subunits (hemagglutinin (HA)-tagged α-ENaC, V5-tagged β-ENaC, and Myc-tagged γ-ENaC) and 2 μg of FLAG-tagged WT or mutant Nedd4-2. Samples were analyzed by SDS-PAGE and immunoblotted with anti-ubiquitin antibody (Covance) or anti-FLAG- or anti-HA-horseradish peroxidase antibody (Roche Applied Science). Densitometry was measured using NIH ImageJ software, and values for ubiquitinated ENaC were normalized to vector or WT Nedd4-2 as indicated.
Auto-ubiquitin Assay of Nedd4-2
Auto-ubiquitination of FLAG-Nedd4-2 was performed as described previously (27). HEK293 cells were transfected with GFP or FLAG-tagged WT or mutant Nedd4-2. Immunoprecipitated FLAG-Nedd4-2 was then incubated with yeast E1, ATP, and HA-tagged ubiquitin with or without rabbit E2 (Boston Biochem) for 1 h at room temperature. Samples were analyzed by SDS-PAGE.
Transient Transfection and Electrophysiological Measurements in mpkCCDc14 Cells
Approximately 2 × 106 mpkCCDc14 cells at 80–90% confluency were trypsinized and then resuspended in 200 μl of Nucleofector T solution (Amaxa Biosystems) containing 4 μg of pGreenLantern (GFP), pcDNA3-JNK1α1-WT, pcDNA3-JNK1α1-T183E/Y185E (active mutant), or pcDNA3-JNK1α1-K55M (dominant-negative mutant) plasmid cDNA/transfection. Cells were electroporated with the plasmid DNA in the Nucleofector electroporation device according to the manufacturer's instructions using Amaxa Program K-29. An equal volume of warm culture medium was then added, and the cell suspension was plated onto a plastic 6-well plate containing 2 ml of culture medium. The following day, the cells were trypsinized and plated onto 0.33-cm2 Transwells (Corning Costar 3413) at superconfluency. The cells were used for electrophysiological study 2–4 days later when the cell monolayers reached transepithelial resistances >1000 ohm-cm2. Transepithelial resistance and potential difference across monolayers were measured using an epithelial volt-ohm meter as described previously (21). Equivalent short-circuit currents were calculated by Ohm's law (28).
Statistical Analysis
TEV data generated from different oocyte batches were pooled and analyzed using an analysis of variance factorial model to account for batch-to-batch variability in amiloride-sensitive ENaC currents. For other experiments, statistics were performed using paired Student's t tests. In all cases, p values <0.05 were considered significant. Error bars indicate S.E.
RESULTS
Initial Identification of Phosphorylated Residues in Nedd4-2
Nedd4-2 contains 70 Ser, 60 Thr, and 32 Tyr residues. To determine which of these potential target sites are phosphorylated at base line in cells, Nedd4-2 was expressed in HEK293 cells, purified by immunoprecipitation and elution, separated by gel electrophoresis, enzymatically digested, and then analyzed by tandem mass spectrometry. This initial screen revealed seven phosphorylation sites in Nedd4-2, all of which are conserved from Xenopus to human Nedd4-2 but interestingly are not present in the related isoform Nedd4-1 (Fig. 1A). Through efficient elution of Nedd4-2 and the use of multiple enzymes for peptide digestion, 90% sequence coverage (873 of 971 amino acids) was obtained for analysis (Fig. 1B), although in some cases, the exact phosphorylated residue could not be unambiguously identified (supplemental Fig. 1).
FIGURE 1.

Phosphorylation of Nedd4-2. A, schematic of Nedd4-2 domains (C2, WW1–4, and HECT). Arrows indicate relative positions of phosphorylated residues. Ser-338, Thr-363, and Ser-444 are previously described targets of SGK1 and PKA. B, sequence of Nedd4-2. Phosphorylated residues identified by mass spectrometry are circled. Only one of the serines of Ser-179–Ser-181 is phosphorylated, and two of the three serines Ser-471, Ser-475, and Ser-483 are phosphorylated. Underlined residues indicate sequence coverage by mass spectrometry analysis, 873 of 971 (90%). C, ratio of phosphorylated peptide to non-phosphorylated peptide (%) for each of the three known SGK1/PKA-phosphorylated residues (Ser-338, Thr-363, and Ser-444) under different conditions. White bars indicate treatment of HEK293 cells with 25 μm LY294002 (general PI3K inhibitor and hence nonspecific SGK1 inhibitor) for 30 min prior to harvest of Nedd4-2. Gray bars indicate cotransfection of HEK293 cells with constitutively active SGK1 (S422D) and Nedd4-2.
As controls for Nedd4-2 phosphorylation, we modulated the cellular activity of SGK1 either by treating Nedd4-2-transfected cells with a phosphatidylinositol 3-kinase (PI3K) inhibitor (LY294002), which inhibits PI3K-dependent activation of SGK1 (29), or by cotransfecting with constitutively active SGK1. We then compared the ratio of peak intensities of phosphorylated and the equivalent non-phosphorylated peptides by mass spectrometry under these conditions. Three residues showed a higher ratio of phosphorylated to non-phosphorylated peptide in samples from cells cotransfected with active SGK1 versus those treated with LY294002: Ser-338, Thr-363, and Ser-444 (Fig. 1C). Each of these residues had been identified previously by mutation analysis as a site of SGK1/PKA-mediated phosphorylation (12, 14). The SGK1-dependent increase in phosphorylation of Ser-338 and Thr-363 was quite robust (>4.5-fold), whereas the increase at Ser-444 was only modest (∼1.5-fold) and showed higher basal (PI3K-independent) phosphorylation.
Ser-293 was one of four novel phosphorylated residues identified by mass spectrometry (supplemental Fig. 1). Ser-293 is embedded within a canonical target motif for the proline-directed Ser/Thr MAPK kinase family, Pro-X-(Ser/Thr)-Pro (Fig. 1B) (30). MAPKs have been shown previously to negatively regulate ENaC (31). The related E3 ligase Itch/AIP4 is also activated by MAPK (JNK1) phosphorylation in a similar proline-rich domain (22). These observations prompted us to further characterize the role of Ser-293 in regulation of Nedd4-2.
JNK1 Phosphorylates Nedd4-2 in Vitro and in Cultured Cells
To further explore the potential role of Ser-293 in Nedd4-2, we sought to identify kinase(s) that could phosphorylate this residue. We tested whether different MAPK family members phosphorylate Nedd4-2 at Ser-293 by incubating purified GST-Nedd4-2 and [γ-32P]ATP with one of each of the subfamilies of MAPKs: JNK1, p38 MAPKα, and ERK1. Purified constitutively active SGK1 served as a positive control for Nedd4-2 phosphorylation. Both SGK1 and JNK1, but not p38 MAPKα or ERK1, significantly enhanced phosphorylation of GST-Nedd4-2 in vitro (Fig. 2A). JNK1-dependent phosphorylation of the GST-tagged S293A Nedd4-2 mutant was also significantly reduced by ∼30% (Fig. 2B, left panel), but SGK1-mediated phosphorylation was comparable between WT and S293A Nedd4-2. Similar amounts of Nedd4-2 fusion proteins were used in these assays (Fig. 2B, right panel).
FIGURE 2.

JNK1 phosphorylates Nedd4-2 at Ser-293. A, in vitro kinase assay with recombinant kinase incubated with recombinant GST-WT Nedd4-2 (N42) and [γ-32P]ATP. Specific activity is relative to GST alone, as quantified by nanomoles of incorporated [γ-32P]ATP/mg of active kinase. *, p < 0.002 (in triplicate). B, left panel, in vitro kinase assay comparing targets GST, GST-WT Nedd4-2, and GST-S293A Nedd4-2. *, p < 0.0001, versus GST; #, p < 0.002, versus WT (in triplicate). Right panel, Coomassie Blue-stained gel of recombinant proteins. Lane M, molecular mass markers. C, in vivo phosphorylation of Nedd4-2 by JNK1. Upper panel, representative one-dimensional PAGE analysis showing phosphopeptide screen image of immunoprecipitated FLAG-tagged WT versus S293A Nedd4-2 cotransfected with JNK1 versus GFP and incubated with [32P]orthophosphate. Lower panel, immunoblot of the same membrane with anti-FLAG antibody. D, densitometry of phospho-screen image intensity of Nedd4-2 band corrected for the corresponding immunoblot band intensity. *, p < 0.003, WT versus S293A; #, p ≤ 0.001, JNK1 versus GFP (n = 3).
Relative to expression of GFP (control), expression of wild-type JNK1 along with Nedd4-2 in HEK293 cells increased [32P]orthophosphate in vivo labeling of Nedd4-2 by >150% (Fig. 2, C and D). JNK1 coexpression enhanced phosphorylation of S293A Nedd4-2 by only ∼40% (Fig. 2D). These results indicate that Ser-293 serves as a site for JNK1-mediated Nedd4-2 phosphorylation in vitro and in cells. However, these results suggest that additional JNK1 phosphorylation site(s) exist within Nedd4-2.
Ser-293 Potentiates Inhibition of ENaC by Nedd4-2
Using a Xenopus oocyte coexpression assay and the TEV technique to measure whole-cell currents, we found that the S293A mutation in Nedd4-2 modestly but significantly reduced the ability of Nedd4-2 to inhibit amiloride-sensitive ENaC currents. Compared with oocytes not expressing exogenous Nedd4-2, there was an ∼90% ENaC current inhibition with coexpression of WT Nedd4-2. However, there was a modest but significant reduction in the inhibition of ENaC current (by only ∼80%) with S293A Nedd4-2 coexpression (supplemental Fig. 2). However, this mutation did not affect regulation of Nedd4-2 through previously known mechanisms, including interaction with 14-3-3, ENaC regulation by AMP-activated protein kinase, interaction with ENaC channels, and ubiquitination of ENaC (supplemental Fig. 3).
JNK1 phosphorylates Nedd4-2 partially at Ser-293, and the S293A mutation had only a modest effect on Nedd4-2 function. Therefore, we sought to identify the additional JNK1 phosphorylation site(s) within Nedd4-2.
Mass Spectrometry Identifies Additional JNK1 Phosphorylation Sites within Nedd4-2
We performed in vitro phosphorylation of Nedd4-2 in the presence or absence of recombinant JNK1. Using phosphopeptide enrichment followed by tandem mass spectrometry, two new putative phosphorylation sites were identified. On the basis of this screen, we quantitated JNK1-mediated phosphorylation of Nedd4-2 with or without mutations to Ala at Ser-293, Thr-408, and Thr-899. The S293A and T899A mutants were phosphorylated significantly less than WT. The T408A mutation alone or with S293A did not further decrease phosphorylation, but the triple mutant (S293A/T408A/T899A) was phosphorylated ∼70% less than WT and was significantly attenuated compared with S293A alone (Fig. 3). SGK1 was still able to phosphorylate the triple mutant with efficacy comparable to that of WT (supplemental Fig. 4).
FIGURE 3.

JNK1 phosphorylates Nedd4-2 at three sites. A, FLAG-Nedd4-2 was transfected and immunoprecipitated from HEK293 cells, followed by an in vitro phosphorylation reaction with or without recombinant JNK1. A representative phospho-screen image (upper panel) and immunoblot (IB; lower panel) are shown. B, shown is the densitometry of relative phosphorylation. *, p < 0.0005, versus no kinase; #, p < 0.003, versus WT; †, p < 0.02, versus WT or S293A (n = 4–11 experiments).
Mutations of Additional Sites Modulate ENaC Current and Disrupt ENaC Ubiquitination
We next tested whether the phosphorylation-deficient triple mutant of Nedd4-2 altered ENaC function using the TEV technique in oocytes. Expression of the Nedd4-2 triple mutant abrogated the inhibition of ENaC currents that occurred with expression of WT Nedd4-2 to the extent that the currents were similar to those measured with expression of ENaC alone (Fig. 4A). Comparable WT and triple mutant Nedd4-2 protein expression levels in the oocytes were confirmed for these experiments (Fig. 4B). Taken together, our results suggest that these three residues can be phosphorylated by JNK1 and are important for the functional regulation of ENaC by Nedd4-2. We predicted that this robust inactivation of Nedd4-2 function via the triple mutant would allow us to study the mechanism more clearly.
FIGURE 4.

Decreased inhibition of ENaC by the Nedd4-2 triple mutant. α-, β-, and γ-ENaC subunits were coexpressed either alone (white bar) or with N-terminal FLAG-tagged WT (gray bar) or triple mutant (black bar) Nedd4-2 in Xenopus oocytes (2 ng of each cRNA). A, using the TEV technique, amiloride-sensitive ENaC currents with WT Nedd4-2 were significantly decreased, but currents with triple mutant Nedd4-2 were not statistically different from ENaC alone. *, p < 0.002, versus ENaC alone (N = four batches, n = 39 eggs). B, equivalent expression levels of FLAG-Nedd4-2 in oocytes were verified by Western blotting, and currents shown were normalized for protein expression as quantitated by densitometry of the bands relative to β-actin. IB, immunoblot.
We first tested whether the triple mutant affected the strength of interaction between Nedd4-2 and any of the ENaC subunits (supplemental Fig. 5). As measured by reciprocal co-immunoprecipitation assays in HEK293 cells cotransfected to express ENaC subunits and Nedd4-2, we did not observe any significant differences in the apparent binding affinity of triple mutant versus WT Nedd4-2 for the α-, β-, and γ-ENaC subunits. However, immunoblots of the whole-cell lysates revealed substantially reduced protein expression of all three ENaC subunits with coexpression of WT Nedd4-2 relative to the triple mutant (supplemental Fig. 5A). Together, these results suggest that expression of the Nedd4-2 triple mutant dampens the inhibition of cellular ENaC abundance found with expression of WT Nedd4-2 without affecting its apparent binding affinity for ENaC subunits. We next performed ubiquitination assays in HEK293 cells to test whether these residues affect the ability of Nedd4-2 to ubiquitinate α-ENaC (Fig. 5). Expression of the triple mutant significantly decreased α-ENaC ubiquitination. Interestingly, stepwise inclusion of each of the mutations at these phosphorylatable residues progressively decreased ubiquitinated ENaC versus vector control, but mutation of Thr-899 alone was sufficient to demonstrate the effect. Ubiquitination of α-ENaC was attenuated by 57% with coexpression of triple mutant Nedd4-2 compared with WT.
FIGURE 5.

α-ENaC ubiquitination assay in HEK293 cells. Vector (V) or FLAG-tagged Nedd4-2 constructs (WT, phosphorylation-deficient mutants, and ligase-dead C938S) were cotransfected with differentially tagged αβγ-ENaC. A, cell dissociation followed by immunoprecipitation (IP) of the HA-tagged α-ENaC subunit and immunoblotting (IB) with anti-ubiquitin (upper panel) and anti-HA (middle panel) antibodies. Also shown is an immunoblot of the whole-cell lysate (WCL) with anti-FLAG antibody (lower panel). B, densitometry comparing the signals of ubiquitinated adducts of α-ENaC. *, p < 0.02, versus WT Nedd4-2 (n = 3).
Mutation of Thr-899 Is Sufficient to Disrupt Ubiquitin Ligase Activity of Nedd4-2
Transfection of Nedd4-2 or mutants in HEK293 cells was followed by immunoprecipitation and an in vitro auto-ubiquitination assay using recombinant E1, E2, ATP, and HA-ubiquitin (Fig. 6). WT Nedd4-2 exhibits potent auto-ubiquitination activity, but the Nedd4-2 triple mutant did not significantly ubiquitinate itself compared with the vector control (GFP). Abolition of Thr-899 was again sufficient to reproduce this effect.
FIGURE 6.

Auto-ubiquitination of Nedd4-2. FLAG-tagged Nedd4-2 constructs (WT, triple mutant, and T899A) were transfected into HEK293 cells. Following immunoprecipitation (IP) with anti-FLAG beads, samples were incubated with the E1 and E2 enzymes, ATP, and HA-tagged recombinant ubiquitin in vitro and analyzed by SDS-PAGE, followed by immunoblotting with anti-HA (upper panel) and anti-FLAG (lower panel) antibodies successively. As a negative control, GFP was substituted for FLAG-Nedd4-2 (upper panel, first lane), or E2 was not included (fifth lane). A representative experiment (n = 3) is shown.
JNK1 Inhibits ENaC-mediated Na+ Transport
To probe the effect of JNK1 on ENaC currents in epithelial cells that endogenously express Nedd4-2 and ENaC (13), we overexpressed WT, constitutively active (T183E/Y185E), or kinase-dead (K55M) JNK1 in immortalized mpkCCDc14 cells (28). Relative to WT JNK1, expression of kinase-dead JNK1 increased ENaC-dependent equivalent short-circuit currents by 31 ± 15%, whereas constitutively active JNK1 decreased currents by 38 ± 11% (Fig. 7). These results suggest that JNK1 (a candidate kinase for these novel residues) modulates ENaC currents in polarized kidney epithelial cells.
FIGURE 7.
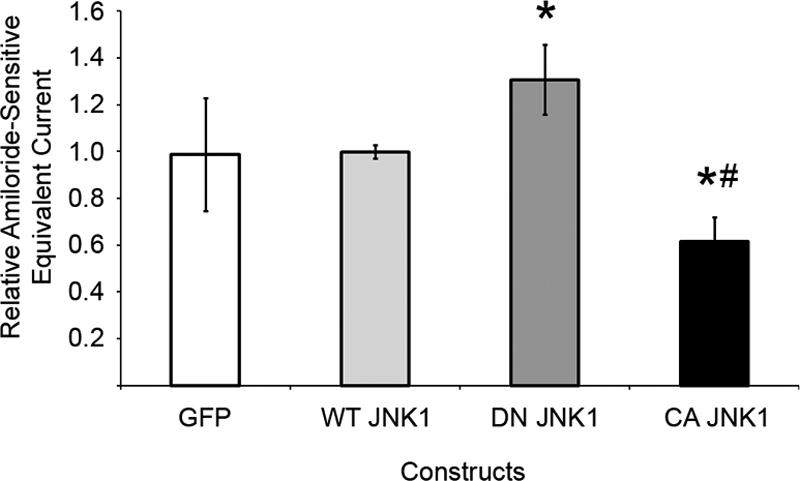
JNK1 inhibits endogenous ENaC-mediated sodium currents. Following electroporation to express the indicated proteins, mpkCCDc14 cells were grown to high resistance on permeable supports. Amiloride-sensitive equivalent currents are shown for cells expressing GFP or dominant-negative (DN) or constitutively active (CA) JNK1 relative to WT JNK1. *, p < 0.02, versus WT; #, p = 0.002, versus dominant-negative JNK1 (n = 3).
DISCUSSION
Nedd4-2 regulates several ion transport proteins in addition to ENaC (32–35), and recent animal studies have confirmed its importance in the pathogenesis of hypertension (6). The only mode of ENaC regulation by Nedd4-2 described to date involves modulation of the Nedd4-2/ENaC interaction. Our identification of additional phosphorylated residues in Nedd4-2 led us to hypothesize that other kinase networks may modulate Nedd4-2 function and thus ion transport. We have presented an unbiased approach toward elucidating these regulatory networks and have discovered a novel regulatory mechanism of Nedd4-2 and Na+ transport. Phosphorylation-deficient Nedd4-2 mutants are unable to either ubiquitinate ENaC or inhibit ENaC currents effectively in cells. These mutants directly alter ubiquitin ligase activity, which implies a broader role for these sites in the scope of Nedd4-2 actions.
Our initial mass spectrometry phosphopeptide screen revealed Ser-293 and at least six other residues as phosphorylation targets in cell-expressed Nedd4-2. Each of these residues is conserved across mammalian species. Although three of these residues had been implicated previously in SGK1- and PKA-mediated stimulation of ENaC (12, 14), Ser-338 and Thr-363 had not been definitively shown to be modified by SGK1 or other PI3K-dependent kinases prior to this study. Nedd4-2 phosphorylation at these sites was dramatically changed by SGK1 modulation in cells and highlights the relevance of SGK1-dependent phosphorylation at these sites in vivo (Fig. 1C). Phosphorylation of Ser-444 is higher at base line and only modestly increased, similar to aldosterone-treated epithelial cells (13). The higher basal phosphorylation may represent SGK1-independent pathways such as Akt, PKA, and IκB kinase-β (14, 18, 19).
Mutation of the previously unrecognized phosphorylation site Ser-293 to Ala modestly reduced the ability of Nedd4-2 to inhibit ENaC. Mutation of two additional putative JNK1 phosphorylation sites more effectively reduced Nedd4-2-dependent ENaC inhibition (Fig. 4) by reducing catalytic activity (Fig. 6) without disrupting other known properties of Nedd4-2 (e.g. phosphorylation by SGK1 and interaction with ENaC). Mutation of Thr-899 in Nedd4-2 appears to be dominant over Ser-293 or Thr-408, but abolition of phosphorylation at Ser-293 and Thr-408 alone also decreased α-ENaC ubiquitination. Neither Thr-408 nor Thr-899 was detected in the initial phosphopeptide screen. For the second mass spectrometry experiment, we phosphorylated Nedd4-2 with JNK1 in vitro and performed phosphopeptide enrichment to identify these additional JNK1 phosphorylation sites. The need for these additional approaches to detect the other two sites might imply a difference in the relative stoichiometry of phosphorylation at these residues. Thr-899 resides within the HECT domain of Nedd4-2 and is conserved from Xenopus to human and across the Nedd4 family. To our knowledge, this is the first identification of a phosphorylation site within a HECT domain. It may thus play an important general role in the regulatory mechanism of HECT E3 ligases.
Although our data indicate that JNK1 both regulates ENaC currents in mpkCCDc14 cells (Fig. 7) and phosphorylates Nedd4-2 in vitro and in vivo (Figs. 2 and 3), we were unable to confirm in additional experiments that JNK1-dependent regulation of ENaC requires the phosphorylation of Nedd4-2 at these sites (data not shown). We thus speculate that additional kinase(s), independent of JNK1, also play an important role in phosphorylating these sites (especially Thr-899) that are critical in maintaining Nedd4-2 catalytic function in cells. It is also possible that the lack of measurable effect of JNK1 on Nedd4-2 ubiquitin ligase activity could reflect a limitation of the experimental method. Future experiments will be needed to characterize these novel phosphorylation sites, additional kinases that may phosphorylate these residues, and their relative in vivo stoichiometries.
Phosphorylation of Nedd4-2, and E3 ligases in general, represents a powerful regulatory mechanism to alter the fate of ion channels and other targets of ubiquitination. We have identified novel sites of phosphorylation of Nedd4-2 using mass spectrometry and have characterized the functional importance of phosphorylation at some of these sites on Nedd4-2. The MAPK JNK1 phosphorylates Nedd4-2 and negatively regulates ENaC-mediated Na+ transport. Through these studies, we have described a novel mode of Nedd4-2 regulation via phosphorylation of the HECT domain and have introduced JNK1 as a potential mediator in the regulation of epithelial Na+ transport.
Supplementary Material
Acknowledgments
We thank Thomas Kleyman, Alan Pao, and Rama Soundararajan for suggestions.
This work was supported, in whole or in part, by National Institutes of Health Grants P30 DK079307 (to the Pittsburgh Kidney Research Center), R01 DK075048 (to K. R. H.), K08 DK071648 (to V. B.), and R01 DK056695 (to D. P.). This work was also supported by a grant from the Dutch Kidney Foundation (to M. A. W.). Mass spectrometry analysis was provided by the University of California San Francisco Mass Spectrometry Facility (A. L. B. and R. J. C.), supported by the Biomedical Research Technology Program of the National Center for Research Resources (NCRR) and National Institutes of Health NCRR Grants RR01614 and RR015804.

The on-line version of this article (available at http://www.jbc.org) contains supplemental “Experimental Procedures” and Figs. 1–5.
- ENaC
- epithelial Na+ channel
- PKA
- protein kinase A
- GST
- glutathione S-transferase
- xNedd4-2
- Xenopus Nedd4-2
- MAPK
- mitogen-activated protein kinase
- GFP
- green fluorescent protein
- WT
- wild-type
- TEV
- two-electrode voltage clamp
- HA
- hemagglutinin
- PI3K
- phosphatidylinositol 3-kinase.
REFERENCES
- 1.Kamynina E., Debonneville C., Bens M., Vandewalle A., Staub O. (2001) FASEB J. 15, 204–214 [DOI] [PubMed] [Google Scholar]
- 2.Staub O., Dho S., Henry P., Correa J., Ishikawa T., McGlade J., Rotin D. (1996) EMBO J. 15, 2371–2380 [PMC free article] [PubMed] [Google Scholar]
- 3.Snyder P. M., Steines J. C., Olson D. R. (2004) J. Biol. Chem. 279, 5042–5046 [DOI] [PubMed] [Google Scholar]
- 4.Bhalla V., Hallows K. R. (2008) J. Am. Soc. Nephrol. 19, 1845–1854 [DOI] [PubMed] [Google Scholar]
- 5.Abriel H., Loffing J., Rebhun J. F., Pratt J. H., Schild L., Horisberger J. D., Rotin D., Staub O. (1999) J. Clin. Invest. 103, 667–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi P. P., Cao X. R., Sweezer E. M., Kinney T. S., Williams N. R., Husted R. F., Nair R., Weiss R. M., Williamson R. A., Sigmund C. D., Snyder P. M., Staub O., Stokes J. B., Yang B. (2008) Am. J. Physiol. Renal Physiol. 295, F462–F470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Firsov D., Schild L., Gautschi I., Mérillat A. M., Schneeberger E., Rossier B. C. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 15370–15375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schild L., Lu Y., Gautschi I., Schneeberger E., Lifton R. P., Rossier B. C. (1996) EMBO J. 15, 2381–2387 [PMC free article] [PubMed] [Google Scholar]
- 9.Kabra R., Knight K. K., Zhou R., Snyder P. M. (2008) J. Biol. Chem. 283, 6033–6039 [DOI] [PubMed] [Google Scholar]
- 10.Malik B., Yue Q., Yue G., Chen X. J., Price S. R., Mitch W. E., Eaton D. C. (2005) Am. J. Physiol. Renal. Physiol. 289, F107–F116 [DOI] [PubMed] [Google Scholar]
- 11.Knight K. K., Olson D. R., Zhou R., Snyder P. M. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 2805–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Debonneville C., Flores S. Y., Kamynina E., Plant P. J., Tauxe C., Thomas M. A., Münster C., Chraïbi A., Pratt J. H., Horisberger J. D., Pearce D., Loffing J., Staub O. (2001) EMBO J. 20, 7052–7059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flores S. Y., Loffing-Cueni D., Kamynina E., Daidié D., Gerbex C., Chabanel S., Dudler J., Loffing J., Staub O. (2005) J Am. Soc. Nephrol. 16, 2279–2287 [DOI] [PubMed] [Google Scholar]
- 14.Snyder P. M., Olson D. R., Kabra R., Zhou R., Steines J. C. (2004) J. Biol. Chem. 279, 45753–45758 [DOI] [PubMed] [Google Scholar]
- 15.Bhalla V., Daidie D., Li H., Pao A. C., LaGrange L. P., Wang J., Vandewalle A., Stockand J. D., Staub O., Pearce D. (2005) Mol. Endocrinol. 19, 3073–3084 [DOI] [PubMed] [Google Scholar]
- 16.Ichimura T., Yamamura H., Sasamoto K., Tominaga Y., Taoka M., Kakiuchi K., Shinkawa T., Takahashi N., Shimada S., Isobe T. (2005) J. Biol. Chem. 280, 13187–13194 [DOI] [PubMed] [Google Scholar]
- 17.Liang X., Peters K. W., Butterworth M. B., Frizzell R. A. (2006) J. Biol. Chem. 281, 16323–16332 [DOI] [PubMed] [Google Scholar]
- 18.Lee I. H., Dinudom A., Sanchez-Perez A., Kumar S., Cook D. I. (2007) J. Biol. Chem. 282, 29866–29873 [DOI] [PubMed] [Google Scholar]
- 19.Edinger R. S., Lebowitz J., Li H., Alzamora R., Wang H., Johnson J. P., Hallows K. R. (2009) J. Biol. Chem. 284, 150–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhalla V., Oyster N. M., Fitch A. C., Wijngaarden M. A., Neumann D., Schlattner U., Pearce D., Hallows K. R. (2006) J. Biol. Chem. 281, 26159–26169 [DOI] [PubMed] [Google Scholar]
- 21.Carattino M. D., Edinger R. S., Grieser H. J., Wise R., Neumann D., Schlattner U., Johnson J. P., Kleyman T. R., Hallows K. R. (2005) J. Biol. Chem. 280, 17608–17616 [DOI] [PubMed] [Google Scholar]
- 22.Gallagher E., Gao M., Liu Y. C., Karin M. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 1717–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Slagsvold T., Marchese A., Brech A., Stenmark H. (2006) EMBO J. 25, 3738–3749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang C., Zhou W., Jeon M. S., Demydenko D., Harada Y., Zhou H., Liu Y. C. (2006) Mol. Cell 21, 135–141 [DOI] [PubMed] [Google Scholar]
- 25.Jiang Q., Mak D., Devidas S., Schwiebert E. M., Bragin A., Zhang Y., Skach W. R., Guggino W. B., Foskett J. K., Engelhardt J. F. (1998) J. Cell Biol. 143, 645–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soundararajan R., Zhang T. T., Wang J., Vandewalle A., Pearce D. (2005) J. Biol. Chem. 280, 39970–39981 [DOI] [PubMed] [Google Scholar]
- 27.Bruce M. C., Kanelis V., Fouladkou F., Debonneville A., Staub O., Rotin D. (2008) Biochem. J. 415, 155–163 [DOI] [PubMed] [Google Scholar]
- 28.Bens M., Vallet V., Cluzeaud F., Pascual-Letallec L., Kahn A., Rafestin-Oblin M. E., Rossier B. C., Vandewalle A. (1999) J Am. Soc. Nephrol. 10, 923–934 [DOI] [PubMed] [Google Scholar]
- 29.Park J., Leong M. L., Buse P., Maiyar A. C., Firestone G. L., Hemmings B. A. (1999) EMBO J. 18, 3024–3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Songyang Z., Lu K. P., Kwon Y. T., Tsai L. H., Filhol O., Cochet C., Brickey D. A., Soderling T. R., Bartleson C., Graves D. J., DeMaggio A. J., Hoekstra M. F., Blenis J., Hunter T., Cantley L. C. (1996) Mol. Cell. Biol. 16, 6486–6493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi H., Asher C., Chigaev A., Yung Y., Reuveny E., Seger R., Garty H. (2002) J. Biol. Chem. 277, 13539–13547 [DOI] [PubMed] [Google Scholar]
- 32.Hryciw D. H., Ekberg J., Lee A., Lensink I. L., Kumar S., Guggino W. B., Cook D. I., Pollock C. A., Poronnik P. (2004) J. Biol. Chem. 279, 54996–55007 [DOI] [PubMed] [Google Scholar]
- 33.Jespersen T., Membrez M., Nicolas C. S., Pitard B., Staub O., Olesen S. P., Baró I., Abriel H. (2007) Cardiovasc. Res. 74, 64–74 [DOI] [PubMed] [Google Scholar]
- 34.Lang F., Böhmer C., Palmada M., Seebohm G., Strutz-Seebohm N., Vallon V. (2006) Physiol. Rev. 86, 1151–1178 [DOI] [PubMed] [Google Scholar]
- 35.van Bemmelen M. X., Rougier J. S., Gavillet B., Apothéloz F., Daidié D., Tateyama M., Rivolta I., Thomas M. A., Kass R. S., Staub O., Abriel H. (2004) Circ. Res. 95, 284–291 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.