

Abstract
Scytalidoglutamic peptidase (SGP) is the prototype of fungal glutamic peptidases that are characteristically pepstatin insensitive. These enzymes have a unique catalytic dyad comprised of Gln53 and Glu136 that activate a bound water molecule for nucleophilic attack on the carbonyl carbon atom of the scissile peptide bond. The hydrolysis by SGP at peptide bonds with proline in the P1′ position is a rare event among peptidases that we investigated using the series of fluorescence resonance energy transfer peptides, Abz-KLXPSKQ-EDDnp, compared with the series Abz-KLXSSKQ-EDDnp. The preference observed in these two series for Phe and His over Leu, Ile, Val, Arg, and Lys, seems to be related to the structure of the S1 subsite of SGP. These results and the pH profiles of SGP activity showed that its S1 subsite can accommodate the benzyl group of Phe at pH 4 as well as the positively charged imidazolium group of His. In the pH range 2 to 7, SGP maintains its structure and activity, but at pH 8 or higher it is irreversibly denatured. The intrinsic fluorescence of the Trp residues of SGP were sensitive to the titration of carboxyl groups having low pK values; this can be attributed to the buried Asp57 and/or Asp43 as described in SGP three-dimensional structure. The solvent kinetic isotope effects and the proton inventory experiments support a mechanism for the glutamic peptidase SGP that involves the nucleophilic attack of the general base (Glu136) activated water, and establish a fundamental role of the S1 subsite interactions in promoting catalysis.
Keywords: Enzyme Kinetics, Enzyme Mechanisms, Isotope Effects, Kinetics, Peptidases, Protease, Proteolytic Enzymes
Introduction
The recently established glutamic peptidase family (Family G1 in MEROPS, also known as the Eqolisins) is a novel group of acid peptidases having structures and mechanistic features distinct from the canonical peptidase families (1, 2), and so far, found only in the filamentous fungi (3) where they play key roles in fungal growth (4). Glutamic peptidases were first differentiated from aspartic peptidases as being insensitive to pepstatin and the absence of sequence similarity to the well characterized pepsin-like and retroviral aspartic peptidases (5, 6).
The Scytalidoglutamic peptidase (SGP),2 isolated from the wood-degrading fungus Scytalidium lignicolum, is the prototype of glutamic peptidases. Gln53 and Glu136 (in SGP numbering) were proposed to participate in the catalytic process because the single point mutants E136A, Q53A, and Q53E lost both the autoprocessing and the enzymatic activities (7). Site-directed mutagenesis studies on Aspergillus niger glutamic peptidase established that these same residues constitute the catalytic dyad in this peptidase (8). The molecular structure of SGP was determined in its unbound native form, in complex with hydrolytic products and transition state peptide analogues (1, 9, 10). SGP has a unique β-strand tertiary structure among the known peptidases, and each layer of the sandwich is formed by seven antiparallel β-sheets. The catalytic residues Glu136 and Gln53 are both located in the same layer of the sandwich structure. The crystal structure of A. niger glutamic peptidase has also been determined and found to be very similar to the molecular structure of SGP (10, 11). By analogy with the aspartic peptidases and the structural and site-directed mutagenesis information, catalysis by SGP involves a mechanism in which the Glu136 residue activates a nucleophilic water molecule and the Gln53 residue stabilizes the tetrahedral intermediate on the course of hydrolytic reaction (1, 9). In this model, the catalytic glutamate acts as a general base in the first step of catalysis (water activation). However, from the structural data obtained with A. niger glutamic peptidase, a mechanism in which the catalytic Glu acts as a general acid was proposed (11). Because a similar catalytic mechanism is expected to occur within this family, the biochemical characterization of its catalytic mechanism will be of great value. The substrate specificity of SGP has been earlier reported (7) and the S1 subsite (Schechter and Berger nomenclature (12)) has higher preference for Phe, Tyr, and His, and the subsites S3 and S3′, display a greater affinity for basic amino acids. A noteworthy observation was the hydrolysis of angiotensin II at the His-Pro peptide bond (1), because peptide bonds with the imino group of Pro usually make that bond resistant to enzymatic hydrolysis.
Solvent kinetic isotope effects (SKIEs) have been used to establish the presence of proton bridges in the rate-determining transitions state(s) of general acid-base-catalyzed reactions (13–15). The proton inventory method comprises SKIEs in a series of mixtures of light and heavy water in which the reaction parameter k is expressed as various kn(n) functions of deuterium atom fractions n that is present in the isotopic solvent. The shape of the resulting function, the magnitude of solvent isotopic effect, and the number of protons transferred are diagnostic of the reaction mechanism (14, 15). Both methodologies have been used widely as probes of the mechanism of action of proteolytic enzymes. The data obtained have been crucial for the interpretation of mechanistic proposals for the catalytic activity of serine (16–18), cysteine (19–21), aspartic (22–24), and metallopeptidases (25–27).
In this work we sought to characterize the chemical mechanism of glutamic peptidases by the use of pH rate profiles and SKIEs on SGP hydrolysis of several fluorescence resonance energy transfer (FRET) peptides MCA-KLFSSK-Q-EDDnp, MCA-KLFPSK-Q-EDDnp, MCA-KLHSSK-Q-EDDnp, and MCA-KLHPSKQ-EDDnp, where the MCA (4-methylcoumarin-7-amide) is the fluorescence donor and glutaminyl-[N-(2,4-dinitrophenyl)-ethylenediamine] (Q-EDDnp) is the fluorescence quencher. These peptides were chosen based on the substrate specificity of SGP described above (1, 7) and from the analysis of kinetic parameters for the hydrolysis of the two series of FRET peptides Abz-KLXSSK-Q-EDDnp and Abz-KLXPSK-Q-EDDnp, where the fluorescence donor is the ortho-aminobenzoic acid (Abz). The aim of this limited substrate specificity analysis was to verify whether the hydrolysis at the amino side of Pro depends on the nature of the particular amino acid that contributed its carboxyl group to the scissile peptide bond. Another question was how SGP can accommodate equally well the hydrophobic side chain of Phe or Tyr and the hydrophilic positively charged imidazolium side chain of His. For the pH profile analysis, the MCA was used as fluorescent group because its fluorescence is insensitive to pH variation.
EXPERIMENTAL PROCEDURES
Materials
Anhydrous dimethyl sulfoxide (dimethyl sulfoxide), heavy water with 99.9% deuterium content, and anhydrous methanol were purchased from Sigma. All buffer salts were reagent-grade and purchased from Fisher Scientific (Pittsburgh, PA) or Sigma.
Peptides
FRET peptides containing Abz or MCA were synthesized by solid-phase synthesis (28, 29). An automated bench-top simultaneous multiple solid-phase peptide synthesizer (PSSM 8 system, Shimadzu, Japan) was used to synthesize peptides using the Fmoc (N-(9-fluorenyl)methoxycarbonyl) procedure. The molecular mass and purity of the peptide was checked by analytical HPLC and by matrix-assisted laser desorption ionization time-of-flight using the mass spectrometer Microflex-LT (Bruker-Daltonics, Billerica, MA). Stock solutions of peptides were prepared in dimethyl sulfoxide, and the concentration was measured spectrophotometrically using the molar extinction coefficient of 17,300 m−1 cm−1 at 365 nm.
Solutions
Buffer solutions for the proton inventory studies were prepared gravimetrically by mixing appropriate quantities of buffers made in H2O or D2O at the required pH or pD. The pD of deuterium oxide solution was obtained from pH meter readings according to the relationship pD = pH (meter reading) +0.4 (30). The fraction of deuterium in each buffer (n) was calculated accounting for density, mass of the enzyme stock solution, and presence of buffer salts.
Enzyme
SGP was obtained as described in Ref. 31. Briefly, 42 mg of SGP were dissolved in 42 ml of buffer A (100 mm sodium acetate, pH 4.8) just before handling and applied to a Mono Q HR5/5 1-ml column (Amersham Biosciences) previously equilibrated with buffer A. Samples were eluted with a 50-ml linear gradient of buffer B (100 mm sodium acetate, pH 4.8, 1 m sodium chloride) and the fractions containing hydrolytic activities were analyzed by 10% SDS-PAGE. Single band active fractions were pooled together and the protein concentration was determined by the bovine serum albumin method. The 0.2 mg/ml sample was concentrated in Centricon 3000 MW to a final concentration of 1 mg/ml. The active enzyme concentration was determined with the SGP transition state mimic inhibitor Ac-FKF(3S,4S)-phenylstatinyl-LR-NH2 (designated as TA1) (7), and stored at −80 °C.
Determination of the Substrate Cleavage Sites
The scissile bond of hydrolyzed peptides were identified by the isolation of the fragments using analytical HPLC followed by determination of their molecular mass by LCMS-2010 equipped with an ESI-probe (Shimadzu, Japan).
Intrinsic Fluorescence
Intrinsic fluorescence of SGP (5.0 μg/ml) was measured in the standard buffer (75 mm Tris-HCl, 25 mm MES, 25 mm acetic acid, and 25 mm glycine) at pH values from 0.7 to 10.1. Fluorescence was monitored at 30 °C for each sample with λex = 295 nm and λem from 300 to 450 nm.
Circular Dichroism
CD spectra were recorded on a Jasco J-810 spectropolarimeter with a Peltier system for controlling cell temperature. The absorbance spectra of SGP (80 μg/ml) were collected in the far UV range (190–260 nm) using a 1-cm path length cell in standard buffer at different pH conditions: pH 2.0, 4.0, and 9.0. The system was routinely calibrated with an aqueous solution of twice crystallized d10 camphorsulfonic acid. Ellipticity was recorded as the mean residue molar ellipticity [θ] (degree cm2 dmol−1). The spectrometer conditions typically included a sensitivity of 100 mdeg, a resolution of 0.5 nm, a response time of 4 s, a scan rate of 20 nm/min, and 4 accumulations at 30 °C. The control baseline was obtained with all buffer components prepared without the proteins.
Kinetic Parameter Determination
Pseudo first-order rate constants were measured at [S]≪Km and calculated by nonlinear regression data analysis, using the GraFit software version 5.0 (Erithacus Software, Horley, Surrey, United Kingdom). The specificity rate constants (kcat/Km) were obtained by dividing the first-order rate constant by the active enzyme concentration present in the reaction mixture. The kinetic parameters, kcat and Km, were determined from the initial rate measurements at 8–10 substrate concentrations between 0.15 and 15 Km. The enzyme concentrations were chosen such that less than 5% of the substrate was hydrolyzed over the course of the assay. The data were fitted with respective standard errors to the Michaelis-Menten equation using GraFit software. The reaction rate was converted into micromoles of substrate hydrolyzed per minute based on a calibration curve obtained from the complete hydrolysis of each peptide. In all assays, data were collected at least in duplicate, and the error values were less than 10% for each of the obtained kinetic parameters. Hydrolysis of FRET-MCA peptides were assayed in a Hitachi F-2500 spectrofluorimeter. Fluorescence changes were monitored continuously at λex = 325 nm and λem = 395 nm. When using Abz as fluorescence donor, the assay condition was modified to λex = 320 nm and λem = 420 nm. The hydrolysis conditions for the determination of kinetic parameters of hydrolysis by SGP was 100 mm sodium acetate buffer (pH 4.5), 0.005% Triton X-100 at 37 °C.
The pH and pD Dependence of the Specificity Constant (kcat/Km)
The pH dependence of the SGP catalyzed hydrolysis of MCA-KLFSSKQ-EDDnp was followed either under pseudo first-order rate constants or under Michaelis-Menten kinetics conditions (8–10 substrate concentrations between 0.15 and 15 Km). The data were fitted with the GraFit software according to the equation,
 |
where, k = kcat/Km or kcat. Equation 1 fits the data when the pH activity profile depends upon two ionizing groups (double pKa) but does not assume that the activity is 0 at extreme pH values. k(Limit1) represents the limit of the k value at acid limb (low pH), k(Limit2) is the pH-independent maximum for the k value, and k(Limit3) is the limit of the k value at alkaline limb (high pH). The pK1 and pK2 values are related to the dissociation constants of a catalytically competent base and acid, respectively, which were estimated from the pH versus kcat/Km curves, and were identified as pKe1 and pKe2, respectively. pKe indicates that we are dealing with the dissociation constant of free enzyme.
Temperature Effect on pH Dependence
The pH dependence of SGP-catalyzed hydrolysis of MCA-KLFSSKQ-EDDnp was followed by pseudo first-order kinetics in citric acid/phosphate buffer mixtures with constant ionic strength I = 0.1 (32) at the temperatures of 10, 20, 30, and 40 °C. The pKe1 and pKe2 values were obtained as described above and the effect of temperature calculated using the van't Hoff equation (2),
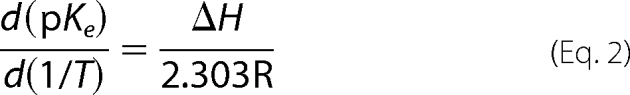 |
where R is the gas constant (1.987 cal mol−1 K−1), T is the absolute temperature, and ΔH is the heat of ionization.
Temperature Dependence of the Specificity Constant (kcat/Km)
Activation parameters were calculated from the linear plot of ln[(kcat/Km)/T] versus 1/T (Equation 3),
where R is the gas constant (8.314 J mol−1 K−1), T is the absolute temperature, NA is Avogadro's number, h is Plank's constant, the enthalpy of activation ΔH* = −(slope) × 8.314 J mol−1, the entropy of activation ΔS* = (intercept − 23.76) × 8.314 J mol−1 K−1. The free energy of activation ΔG*, was calculated from Equation 4.
SKIE
The SKIE analyses were performed on pH and pD independent values of kcat and kcat/Km. For that, pH or pD profiles of SGP hydrolysis of MCA-KLFSSKQ-EDDnp were obtained as described above over pH or pD ranges of 3 to 7 at five different isotopic water mixtures of deuterium atom fractions (n = 0, 0.25, 0.50, 0.75, and 0.99). The experiments were performed at least in duplicate in the standard buffer. Data for all pH or pD profiles were fitted to Equation 1 and the pH (or pD) independent values k(Limit1) and k(Limit2) with the respective error for each value of n were obtained.
Proton Inventory
The proton inventory methodology was performed as described in Refs. 15, 18, and 33, based on the fitting of the experimental data with simplified forms of the Gross-Butler (Equation 5) that relates the dependence of a particular rate parameter (kcat or kcat/Km) to the atom fraction of deuterium, n, in the solvent mixtures,
 |
where Vn and Vo are the velocities (or rate constant) in a binary solvent or in water, respectively, RS is reactant state, ØR is RS fractionation factor, and ØT is the TS fractionation factor. All data reductions using the simplified forms of this equation for hydrolytic enzymes (Table 1) were performed using the GraFit 3.3 software (Erithacus Software Ltd., Middlesex, UK). The fractionation factors were calculated from the best statistical results, measured by the reduced χ2 and based on the quality of fit and a consistent and reliable mechanism.
TABLE 1.
Models used for fitting the proton inventory data as described by Enyedy and Kovach (18)
| Obtained data | Equation |
|---|---|
| TS1 | Vn = VH (1 − n + nØ1) |
| TS1, solv | Vn = VH (1 − n + nØ1) ØSn |
| 2TS1 | Vn = VH (1 − n + nØ1)2 |
| 2TS1, solv | Vn = VH (1 − n + nØ1)2 ØSn |
| TS1, TS2 | Vn = VH (1 − n + nØ1)(1 − n + n Ø2) |
| TS1, TS2, solv | Vn = VH (1 − n + nØ1)(1 − n + n Ø2) ØSn |
RESULTS
Hydrolysis by SGP of the Peptide Series Abz-KLXSSK-Q-EDDnp and Abz-KLXPSK-Q-EDDnp
Table 2 shows the kinetic parameters for the hydrolysis by SGP of the two series of peptides, Abz-KLXSSK-Q-EDDnp and Abz-KLXPSK-Q-EDDnp, which were cleaved only at the X-S and X-P bonds, respectively. The kcat values for the hydrolysis at the X-P peptide bonds in the series Abz-KLXPSK-Q-EDDnp were significantly lower than the kcat values for the hydrolysis of the X-S bonds in the series Abz-KLXSSK-Q-EDDnp, however, the kcat/Km values for the two series of substrates were quite similar because the Km values are lower in many cases for the X-P cleavage. The peptides with His and Phe at the P1 position were the best substrates for both series. As the kinetic parameters were obtained at pH 4.5, the S1 subsite of SGP accepts both the hydrophobic benzyl group of Phe as well as the positively charged imidazolium group of His. In contrast, the peptides with positively charged amino acids Arg and Lys or aliphatic amino acids Leu, Ile, and Val were all poor substrates. Therefore, the S1 subsite of SGP fits better to the positively charged imidazolium group of His than the guanidinium group of Arg or the ammonium ion of Lys, and also accommodates the aromatic group of Phe better than the aliphatic groups of Leu, Ile, or Val; the peptide with Trp in the Pl site was resistant to hydrolysis. The peptides with Asn and Gln were hydrolyzed with higher efficiency than the peptides with Asp and Glu, indicating that the S1 subsite of SGP did not effectively bind negatively charged side chains.
TABLE 2.
Kinetic parameters for the hydrolysis by SGP of the series of FRET peptides Abz-KLXSSK-Q-EDDnp and Abz-KLXPSK-Q-EDDnp
Hydrolysis conditions were: 100 mm sodium acetate buffer (pH 4.5) at 37 °C. All the peptides were cleaved at the X-S or XP bonds.

pH Profiles of SGP Hydrolytic Activity and Intrinsic Fluorescence
The SGP hydrolytic activity remains after preincubation of the enzyme at different pH values in the range of 2.5 to 7 for 30 min at 37 °C (data not shown). In the same experiments performed in the pH range 8 to 9.5, the enzyme became irreversibly inactivated. The intrinsic fluorescence spectra of SGP at pH 1.2, 4, and 9 are shown in Fig. 1A. The maximum intensity of fluorescence at λmax = 332 nm increased up to pH 3 following a sigmoid curve, from which a pK = 1.4 ± 0.3 can be obtained (Fig. 1B). At pH 8 or higher the maximum fluorescence intensity shifted to λmax = 355 nm. According to the model of discrete states (classes) of Trp residues in proteins (34), at an acidic pH the Trp residue in SGP with λmax = 332 nm is buried and can form the exciplexes (complexes in the excited state) with 2:1 stoichiometry, and at basic pH the Trp residue of SGP having λmax = 355 nm is fully exposed to solvent.
FIGURE 1.

Intrinsic fluorescence change of SGP as a function of pH. A, representative fluorescence spectrum profiles of SGP in pH 1.2 (dotted line), 4 (solid line), and 9 (dash line). B shows the plot of the intrinsic maximum fluorescence intensity (λem = 332 nm) of SGP as a function of pH. The pH-modified SGP was monitored at λex 280. Measurements were carried out in the standard buffer at 37 °C. Bars are S.D. from triplicate experiments.
The effect of pH on SGP conformation was also evaluated by circular dichroism spectra that were obtained at pH 2, 4, and 9 (Fig. 2). The CD spectra were deconvoluted using CDNN software (Applied Photophysics copyright Gerald Böhm 1997) indicating a high percentage of β-structure (52%) in the SGP at pH 2 and 4, which is consistent with the percentage calculated from the crystal structure (9). Strong alterations were observed at pH 9 when the amount of random structures effectively suppressed the β-structure content of SGP (28% β-structure), indicating that SGP is extensively unfolded in solutions of basic pH.
FIGURE 2.

CD spectra of SGP. Far UV range CD spectrum of SGP in pH 2 (dotted line), 4 (solid line), and 9 (dash line). Assay conditions are described under “Experimental Procedures.”
The pH profiles of the hydrolytic activities of SGP on the substrates MCA-KLF↓SSKQ-EDDnp and MCA-KLH↓SSKQ-EDDnp (arrows indicate cleavage sites) were measured in the pH range from 2 to 7 (Fig. 3). The kcat/Km values conform to “bell-shaped” pH rate profiles, and they fit to Equation 1 with pKe1 = 3.4 ± 0.2 and pKe2 = 5.3 ± 0.1 for the hydrolysis of MCA-KLF↓SSKQ-EDDnp, and pKe1 = 4.4 ± 0.2 and pKe2 = 5.4 ± 0.1 for the hydrolysis of MCA-KLH↓SSKQ-EDDnp. The activity on the acid side of the pH profile for the hydrolysis of MCA-KLF↓SSKQ-EDDnp did not reach zero thereby exhibiting an activity plateau in the extreme acidic region. However, the pH profile curve of MCA-KLH↓SSKQ-EDDnp showed no hydrolysis below pH 2. In the pH 2 to 5 range the imidazole group of His is positively charged because it is fully protonated. Thus, the hydrolysis of MCA-KLH↓SSKQ-EDDnp seems to depend on the ionization of a carboxylic group in the S1 subsite of SGP, whereas this is not necessary for the hydrolysis of MCA-KLF↓SSKQ-EDDnp that fits a benzyl group into the S1 subsite. The structure of SGP containing a fragment of angiotensin II (DRVYIHPF) bound to its catalytic site showed that the imidazole group of His fits into the S1 subsite of SGP. Therefore, the imidazole group is located between the phenyl group of Phe138 and the indole group of Trp67, and the Nϵ2 atom of the substrate imidazole forms a hydrogen bond to the enzyme carboxylate of Asp57 that also interacts with the carboxyl group of Asp65 (1). Thus, the observed differences in the pH profiles for the hydrolysis of MCA-KLF↓SSKQ-EDDnp and MCA-KLH↓SSKQ-EDDnp can be related to how the imidazolium and benzyl groups bind to the SGP S1 subsite. In this view, ionization of a carboxyl group inside of the S1 subsite of SGP (pKe1 = 4.4) is required for the imidazolium group to get into it, and then MCA-KLH↓SSKQ-EDDnp can be hydrolyzed. In contrast, for the hydrolysis of MCA-KLF↓SSKQ-EDDnp the ionization of a carboxylate group inside of the S1 subsite of SGP is not required for the benzyl group to bind into it and then pKe1 = 3.4 could be attributed to the carboxyl group of Glu136 that participates in the catalytic process. The environment of the His and Phe side chains in the S1 pocket are shown in Fig. 4.
FIGURE 3.

pH dependence of kcat/Km for SGP hydrolysis of substrate MCA-KLFSSKQ-EDDnp (○) and MCA-KLHSSKQ-EDDnp (●). The reactions were carried out in the standard buffer at 37 °C under Michaelis-Menten kinetics. The curves drawn through the experimental points were obtained from fitting to Equation 1.
FIGURE 4.

Different environment around the side chains of P1-His and P1-Phe in SGP crystal structures. Overlay of the S1 subsite in crystal structures of SGP bound to the reaction product of angiotensin II (Protein Data Bank code 1S2K; carbon, wheat) and the inhibitor TA1 (PDB code 2IFW; carbon, green) obtained after a least squares alignment of the two enzyme structures.
Effect of NaCl on SGP Hydrolytic Activity
Fig. 5 shows a substantial decrease of the SGP catalytic efficiency (kcat/Km) as the NaCl concentration is increased up to 1 m. Moreover, in the range up to 200 mm NaCl the effect is mainly in Km because there was no significant decrease in kcat values. A significant decrease in the kcat values was observed as the salt concentration is further increased from 200 to 800 mm NaCl. The SGP structure seems to be unaffected by an increase in salt content, because no change in the protein intrinsic fluorescence was observed at pH 4.5 in the analyzed salt concentration range (data not shown).
FIGURE 5.

Effect of NaCl on kcat/Km (A), kcat (B), and 1/Km (C) for SGP hydrolysis of substrate MCA-KLFSSKQ-EDDnp. The reactions were carried out in the standard buffer at pH 4.5, 37 °C, under Michaelis-Menten kinetics as described under “Experimental Procedures.” Bars are S.D. from triplicate experiments.
Effects of Temperature in the pH Profiles of SGP Activity
Fig. 6A shows the effect of temperature on the pH profiles of the kcat/Km values of SGP hydrolysis of MCA-KLFSSKQ-EDDnp, from which the pKe1 and pKe2 values were obtained at each temperature. The straight line obtained in the plot of the pKe versus 1/T allows one to obtain the enthalpy of ionization (ΔH) of the titratable groups that influence the catalysis (Fig. 6B). The ΔH1 and ΔH2 obtained from the pKe versus 1/T plot were 4 ± 1 and 42 ± 4 kJ/mol, respectively. The ΔH values for carboxyl group ionization is close to the range ± 6 kJ/mol (35, 36), hence it is reasonable to state that the ionization of a carboxyl group occurs in the acid limb of the pH curve. The ΔH2 value is more related to the titration of an ammonium group that can belong to Lys or to the N terminus of the protein. The participation of an imidazole group is ruled out because there are no His residues in the SGP sequence. To explore further the nature of these groups the effect of 20% ethanol on the pH profile of the SGP hydrolytic activity was also investigated. Fig. 7 shows the shift of the acid limb of the pH profile of SGP activity to higher pH values, whereas the basic limb did not change. These results are compatible with the titration of a carboxylic group on the acid limb because the organic solvent impairs hydration of the carboxylate group and the proton that resulted from the carboxyl group ionization (COOH ↔ COO− + H+). In the titration of an amino group (R-NH3+ ↔ R-NH2 + H+) no effect of ethanol would be expected because there are charged and hydrated groups in both sides of the ionization reaction and the dehydration effect of ethanol on R-NH3+ is compensated by its impairment of the hydration of H+.
FIGURE 6.

Temperature and pH dependence of SGP activity. Effect of temperature on the SGP pH profile of hydrolysis of the substrate MCA-KLFSSKQ-EDDnp (A). The kcat/Km values were determined at pseudo first-order reactions measured in 100 mm citric acid-phosphate buffer at 10 (○), 20 (▴), 30 (□), and 40 °C (●). B, the van't Hoff plot for SGP pKe1 (○) and pKe2 (●) temperature dependence.
FIGURE 7.

Effect of ethanol on SGP pH dependence. The pH kcat/Km profile for the hydrolysis of substrate MCA-KLFSSKQ-EDDnp in the absence (○) and presence of 20% ethanol (●). The kcat/Km values were determined at pseudo first-order reactions measured in 100 mm citric acid-phosphate buffer at 30 °C.
Solvent Kinetic Isotope Effects
The differences of the pD dependence compared with the pH dependence of kcat/Km and kcat for the SGP hydrolysis of MCA-KLFSSKQ-EDDnp are shown in Fig. 8. The pD profiles for kcat/Km and kcat obtained in D2O were of a doubly sigmoidal character, and similar to those found in the pH profile with water, except that D2O shifted the acid limb of the pD activity curve by 1.1 pKa units toward higher values (pKe1 = 3.5 ± 0.1 in water and 4.4 ± 0.2 in D2O), whereas the pK2 value of the basic limb did not change significantly (pKe2 = 5.3 ± 0.1 in water and 5.6 ± 0.2 in D2O). As summarized in Ref. 15, enzyme pH rate profiles are expected to shift with deuteration of the aqueous environment due to an increase in acid pK values, requiring that the correct analysis of the solvent isotopic effect must be performed with pH and pD independent parameters k(Limit1) (referring to the kcat or kcat/Km obtained in the acid plateau) and k(Limit2) (referring to the kcat or kcat/Km) obtained at pH (or pD)-independent maximum). At low pH values there were significant decreases of kcat/Km and kcat parameters in D2O, with a SKIE value k(limit)1(H2O)/k(limit)1(D2O) of 5.9 ± 1.5 for kcat/km and 3.3 ± 0.8 for kcat. These results indicate that a general acid-base reaction is the rate-limiting step for both parameters. Interestingly, at the maximum limit of activity an inverse kinetic isotopic effect for kcat/Km, with k(limit)2(H2O)/k(limit)2(D2O) = 0.71 ± 0.06, but a normal SKIE for kcat, with k(limit)2(H2O)/k(limit)2(D2O) = 1.42 ± 0.27 are observed.
FIGURE 8.

Effect of pH and pD on SGP activity. The pH kcat/Km (A) and pH kcat (B) profile for hydrolysis of the substrate MCA-KLFSSKQ-EDDnp in water (○) and 99.5% D2O (●).
To clarify further the origins and number of protons transferred in the SKIE for the hydrolysis of MCA-KLFSSKQ-EDDnp by the neutral-pH enzyme form, proton inventories were performed at the pH (or pD)-independent maximum (Limit2). As shown in Fig. 9, the proton inventory for kcat is linear and consistent with a mechanism in which the observed solvent isotope effect originates entirely from deuterium fractionation at a single protonic site in the rate-limiting transition state (ØT = 0.70 ± 0.02). In contrast, for kcat/Km, the inverse and linear isotopic effect suggests that another step, presumably a conformational change or the release of products becomes rate-determining for kcat/Km at pH or pD in the range from 4 to 5.
FIGURE 9.

Proton inventory of the specificity constant kcat/Km (●) and kcat (○) for SGP hydrolysis of MCA-KLFSSKQ-EDDnp. Ratios of kcat/Km and kcat measured in mixed isotopic solvents over the kcat/Km and kcat measured in water were plotted against the atom fraction of deuterium, n. Assay conditions are described under “Experimental Procedures.” Bars are S.D. from triplicate experiments.
Temperature Dependence of SGP Activity
Fig. 10 shows the Eyring plot for the kcat/Km values of the hydrolysis for substrate MCA-KLFSSKQ-EDDnp by SGP in citric acid-phosphate buffer (pH 4.5) over the temperature range from 10 to 40 °C, in which SGP was stable (data not shown). The linear Eyring plot allowed the calculation of entropy (ΔS*), enthalpy (ΔH*), and Gibbs energy of activation (ΔG*) associated with the rate-limiting step of SGP hydrolysis. The reaction is mainly driven by a very positive entropic contribution (ΔS* = 80 J/mol/K) that overcomes the large enthalpic term (ΔH* = 56 kJ/mol). The solvent effects are certainly interfering with the entropy change, although a negative entropic contribution was expected to result from the freezing in translational and rotational motion that occurs when the reactants go from the ground state (EH + S) to the enzyme-substrate complex (EHS).
FIGURE 10.

Eyring plot for the hydrolysis of MCA-KLFSSKQ-EDDnp by SGP over the 10–40 °C temperature range. The obtained parameters for T = 310 (37 °C) were ΔS* = 80 J/mol/K, ΔH* = 56 kJ/mol, and ΔG* = 31 kJ/mol. Assay conditions are described under “Experimental Procedures.”
SKIEs for Hydrolysis of Phe-Pro, His-Ser, and His-Pro Peptide Bonds
The pH and pD profile for the kcat/Km values of hydrolysis by SGP of the peptide MCA-KLF↓PSKQ-EDDnp containing Pro in the P1′ position (Fig. 11A) were very similar to the profiles for the hydrolysis of the peptide MCA-KLF↓SSKQ-EDDnp with serine in the P1′ position (Fig. 8A). The two peptides containing histidine at P1, MCA-KLH↓SSKQ-EDDnp and MCA-KLH↓PSKQ-EDDnp (Fig. 11, B and C), were resistant to hydrolysis in extreme acid pD values, similar to that observed in H2O; they exhibited a normal SKIE at the pH or pD independent maximum with k(limit)2(H2O)/k(limit)2(D20) of 1.55 ± 0.35 and 2.6 ± 1.2, respectively. These results indicate that the solvent isotope effects depended on the nature of the P1 amino acid of the scissile peptide bond. The normal SKIEs for the kcat/Km values at the pH or pD independent maximum were observed only with the peptides containing His in the P1 position. These results indicate that proton movement is involved in the formation of the enzyme-substrate complex when His is the P1 residue, and does not occur at the rate-determining step when Phe is at the P1 position. This interpretation is in accordance to the hypothesis suggested above that interaction of the imidazolium group of P1 His-containing substrates with the S1 subsite of SGP depends on the deprotonation of a carboxyl group inside this subsite.
FIGURE 11.

Effect of D2O on SGP pL dependence. The pH kcat/Km profile for hydrolysis of substrate MCA-KLFPSKQ-EDDnp (A), MCA-KLHSSKQ-EDDnp (B), and MCA-KLHPSKQ-EDDnp in water (○) and 99.5% D2O (●).
DISCUSSION
Hydrolysis of peptides with Pro in the P1′ position is a rare event in the activities of peptidases, but SGP hydrolyzes angiotensin II at the His6–Pro7 bond (DRVYIH↓PF) (1, 6, 37). This particular hydrolytic activity of SGP was further examined using the FRET peptides series Abz-KLXPSKQ-EDDnp compared with the series Abz-KLXSSKQ-EDDnp. The preference observed in these two peptide series for Phe and His over Leu, Ile, and Val and Arg and Lys, seems to be related to the particular structure of the S1 subsite of SGP. The crystallographic structure of SGP with angiotensin II bound in the active site showed that the phenyl group of Phe138 and the indole group of Trp67 accommodate the angiotensin II imidazole group of His6 between them (1). However, at pH 4, the imidazole group is fully protonated and the positively charged imidazolium group requires an interaction with carboxylate group inside the S1 subsite. This interaction was reported (1) and both nitrogens of the substrate imidazole group seems to be important for its interactions in the S1 subsite of SGP (Fig. 4). In fact, we synthesized and assayed peptides Abz-KL(ϵ2-methyl)HSSKQ-EDDnp and Abz-KL(δ1-methyl)HSSKQ-EDDnp that were hydrolyzed with kcat/Km values 2.5 and 6.2 μm−1 s−1, respectively, which are significantly lower than the kcat/Km = 363 μm−1 s−1 obtained with Abz-KLHSSKQ-EDDnp (Table 2).
The SGP pH range from 2 to 7 maintains its structure and activity, but at pH 8 or higher it is irreversibly denatured as shown by circular dichroism (Fig. 2) and peptidase inactivation. The intrinsic fluorescence of Trp residues in SGP increased from pH 0.8 up to pH 3 following a sigmoidal-shaped curve from which a pKa = 1.4 ± 0.3 (Fig. 1) could be calculated. This result indicates that the intrinsic fluorescence of the Trp residues were sensitive to titration of carboxyl groups with low pKa values, which could be attributed to the buried Asp57 and/or Asp43 as described in the SGP three-dimensional structure (1).
The pH profiles of the hydrolytic activities of SGP on substrates MCA-KLF↓SSKQ-EDDnp and MCA-KLH↓SSKQ-EDDnp allowed the identification of two carboxylic groups involved in the hydrolytic activity of SGP. The hydrolysis of MCA-KLF↓SSKQ-EDDnp did not reach zero on the acid limb of the pH profile and the hydrolysis of MCA-KLH↓SSKQ-EDDnp is null below pH 2.5. We interpreted these data to correspond to a pKe1 = 3.4 ± 0.2 obtained with MCA-KLF↓SSKQ-EDDnp and implicate the carboxylate of Glu136 of the catalytic center and the pKe1 = 4.4 with MCA-KLH↓SSKQ-EDDnp likely corresponds to a carboxyl group inside the S1 subsite that is necessary for binding the imidazolium group.
It is noteworthy that the decrease of SGP activity with increasing NaCl concentration resulted from the systematic increase of the Km values (Fig. 5C). Equivalent salt effects were observed with the hydrolytic activity of the foot and mouth disease cysteine protease Lbpro (38), whose catalytic center is exposed to the solvent.
Analysis of the SGP pH dependence was performed with the substrate MCA-KLFSSKQ-EDDnp to investigate the catalytic mechanism of the SGP family, especially to address contribution of the catalytic Glu136 in the water activation. The data regarding the enthalpy of ionization as well as the effect of the dielectric constant on pKe1 point to a carboxyl group being deprotonated in the ascendant acid limb of pH profile. If the pKe1 is the result of ionization of Glu136, the activity observed in the extreme acid region suggests that the catalytic activity would occur whether it is protonated or not in the free enzyme form (i.e. prior to the substrate binding). The observation that the maximum activity is achieved when the Glu136 carboxyl group exists as a carboxylate supports the mechanism proposed earlier (1, 9), in which Glu136 acts as a general base in the deprotonation of the catalytic water.
The inverse SKIE obtained with the neutral enzyme form suggests that a physical (conformational change or product release step) rather than a chemical step is rate-determining for MCA-KLFSSKQ-EDDnp hydrolysis, although the viscosity of D2O can also be involved (39). This effect of increased solvent viscosity was accessed with glycerol as viscosogen and no significant effect on SGP activity was observed up to 10% glycerol concentration (data not shown). The occurrence of a conformational change as the rate-determining step is supported by the large entropic contribution observed and by the conformational rearrangements in the SGP structure promoted by inhibitor binding (9). The binding of the transition state mimic inhibitor TA1 to SGP triggers a large conformational change in two opposing loops that tend to bury the inhibitor from solvent exposure (Fig. 12).
FIGURE 12.

Conformational changes in SGP (green) in response to binding of the transition state mimic inhibitor Ac-FKF(3S,4S)-phenylstatinyl-LR-NH2 (designated as TA1, in yellow). Two opposing loops (magenta) of SGP comprising residues Cys141–Cys148 and Tyr71–Gly80 close to the active site to bury the inhibitor from the surrounding solvent.
In the acid plateau, however, the protonated Glu136 could not deprotonate the catalytic water. Based on the Ser-His dyad action in serine peptidases hydrolysis (17), the Glu136 proton could be transferred to an acceptor atom as the substrate bound to enzyme. The decrease in kcat/Km obtained in the presence of D2O, similar to that observed with serine peptidases (18, 33, 40), supports this view. The extreme low pK value expected for such a acceptor residue suggests a carboxylate nature.
The rate-limiting proton-transfer step for kcat observed in both pH regions and the linear normal proton inventory obtained indicates that a single proton is transferred either in the formation or breakdown of the enzyme-bound tetrahedral adduct of substrate and H2O. Because water activation is an obligatory step, and our data supports the carboxylate character of Glu136, it is reasonable to assume that it acts as a general base accepting the proton from the catalytic water and that this step is rate-determining for kcat. This interpretation also supports the assumption that a conformational change and not the release of products is rate-determining for kcat/Km because, otherwise the inverse SKIE should be also observed in kcat.
The resistances to hydrolysis observed with P1-His peptides at extreme acid pH values and the 1 unit increase in pKe1 value obtained are indicative that a carboxylate other than Glu136 is involved in catalysis of these peptides. By structural analysis, it is clear that the P1-His imidazolium group of angiotensin II peptide packs between the phenyl side chain of Phe138 and the indole ring of Trp67, but the Nϵ2 atom of the imidazolium group forms a hydrogen bond to the carboxylate of Asp57 (1) (Fig. 4). By analogy, its negative character could be essential for accommodation of positive P1 residues reflected in the acid limb pH profile for these peptides.
In conclusion our data supports a glutamic peptidase mechanism that involves a nucleophilic attack of a general base (Glu136)-activated water and establish a fundamental role of S1 subsite interactions in promoting the catalytic mechanism. Further studies must be performed to clarify the identity of the essential ionizable group related to pKe2. Initially our results point to an amine group either from a lysine residue or from the enzyme N terminus. Because usually the pK of lysine groups in enzymes are higher (∼9) then observed here and there is no lysine in the catalytic cleft, the latter hypothesis seems more reasonable.
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), Brazil, and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
- SGP
- scytalidoglutamic peptidase
- SKIEs
- solvent kinetic isotope effects
- FRET
- fluorescence resonance energy transfer
- MCA
- 4-methyl coumarin-7-amide
- Q-EDDnp
- glutaminyl-[N-(2,4-dinitrophenyl)-ethylenediamine]
- Abz
- ortho-aminobenzoic acid
- TA1
- SGP transition state mimic inhibitor Ac-FKF(3S,4S)-phenylstatinyl-LR-NH2
- HPLC
- high pressure liquid chromatography
- MES
- 4-morpholineethanesulfonic acid.
REFERENCES
- 1.Fujinaga M., Cherney M. M., Oyama H., Oda K., James M. N. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 3364–3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rawlings N. D., Morton F. R., Kok C. Y., Kong J., Barrett A. J. (2008) Nucleic Acids Res. 36, D320–D325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sims A. H., Dunn-Coleman N. S., Robson G. D., Oliver S. G. (2004) FEMS Microbiol. Lett. 239, 95–101 [DOI] [PubMed] [Google Scholar]
- 4.O'Donoghue A. J., Mahon C. S., Goetz D. H., O'Malley J. M., Gallagher D. M., Zhou M., Murray P. G., Craik C. S., Tuohy M. G. (2008) J. Biol. Chem. 283, 29186–29195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murao S., Oda K. (1985) in Aspartic Proteinases and Their Inhibitors (Kostka V. ed) pp. 379–399, Walter de Gruyter, Berlin [Google Scholar]
- 6.Oda K. (2004) in Handbook of Proteolytic Enzymes (Barrett A. J., Rawlings N. D., Woessner J. F. eds) pp. 219–221, Elsevier Academic Press, London [Google Scholar]
- 7.Kataoka Y., Takada K., Oyama H., Tsunemi M., James M. N., Oda K. (2005) FEBS Lett. 579, 2991–2994 [DOI] [PubMed] [Google Scholar]
- 8.Yabuki Y., Kubota K., Kojima M., Inoue H., Takahashi K. (2004) FEBS Lett. 569, 161–164 [DOI] [PubMed] [Google Scholar]
- 9.Pillai B., Cherney M. M., Hiraga K., Takada K., Oda K., James M. N. (2007) J. Mol. Biol. 365, 343–361 [DOI] [PubMed] [Google Scholar]
- 10.James M. N. (2006) Biol. Chem. 387, 1023–1029 [DOI] [PubMed] [Google Scholar]
- 11.Sasaki H., Kojima M., Sawano Y., Kubota K., Suganuma M., Muramatsu T., Takahashi K., Tanokura M. (2005) Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 81, 441–446 [Google Scholar]
- 12.Schechter I., Berger A. (1967) Biochem. Biophys. Res. Commun. 27, 157–162 [DOI] [PubMed] [Google Scholar]
- 13.Fersht A. (ed) (1999) Structure and Mechanism in Protein Science, pp. 54–102, W.H. Freeman and Co., New York [Google Scholar]
- 14.Schowen K. B., Limbach H. H., Denisov G. S., Schowen R. L. (2000) Biochim. Biophys. Acta 1458, 43–62 [DOI] [PubMed] [Google Scholar]
- 15.Schowen K. B., Schowen R. L. (1982) Methods Enzymol. 87, 551–606 [PubMed] [Google Scholar]
- 16.Bender M. L. (1962) J. Am. Chem. Soc. 84, 2582–2590 [Google Scholar]
- 17.Polgár L. (2005) Cell Mol. Life Sci. 62, 2161–2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Enyedy E. J., Kovach I. M. (2004) J. Am. Chem. Soc. 126, 6017–6024 [DOI] [PubMed] [Google Scholar]
- 19.Polgár L. (1979) Eur. J. Biochem. 98, 369–374 [DOI] [PubMed] [Google Scholar]
- 20.Szawelski R. J., Wharton C. W. (1981) Biochem. J. 199, 681–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Theodorou L. G., Lymperopoulos K., Bieth J. G., Papamichael E. M. (2001) Biochemistry 40, 3996–4004 [DOI] [PubMed] [Google Scholar]
- 22.Hyland L. J., Tomaszek T. A., Jr., Meek T. D. (1991) Biochemistry 30, 8454–8463 [DOI] [PubMed] [Google Scholar]
- 23.Polgár L. (1987) FEBS Lett. 219, 1–4 [DOI] [PubMed] [Google Scholar]
- 24.Northrop D. B. (2001) Acc. Chem. Res. 34, 790–797 [DOI] [PubMed] [Google Scholar]
- 25.Izquierdomartin M., Stein R. L. (1992) J. Am. Chem. Soc. 114, 325–331 [Google Scholar]
- 26.Izquierdo M. C., Stein R. L. (1990) J. Am. Chem. Soc. 112, 6054–6062 [Google Scholar]
- 27.Hershcovitz Y. F., Gilboa R., Reiland V., Shoham G., Shoham Y. (2007) FEBS J. 274, 3864–3876 [DOI] [PubMed] [Google Scholar]
- 28.Korkmaz B., Attucci S., Juliano M. A., Kalupov T., Jourdan M. L., Juliano L., Gauthier F. (2008) Nat. Protocols 3, 991–1000 [DOI] [PubMed] [Google Scholar]
- 29.Hirata I. Y., Cezari M. H. S., Nakaie C., Boschcov P., Ito A. S., Juliano M. A., Juliano L. (1994) Lett. Pept. Sci. 1, 299–308 [Google Scholar]
- 30.Glasoe P. K., Long F. A. (1960) J. Phys. Chem. 64, 188–190 [Google Scholar]
- 31.Maita T., Nagata S., Matsuda G., Maruta S., Oda K., Murao S., Tsuru D. (1984) J. Biochem. 95, 465–475 [DOI] [PubMed] [Google Scholar]
- 32.Ellis K. J., Morrison J. F. (1982) Methods Enzymol. 87, 405–426 [DOI] [PubMed] [Google Scholar]
- 33.Okamoto D. N., Kondo M. Y., Santos J. A., Nakajima S., Hiraga K., Oda K., Juliano M. A., Juliano L., Gouvea I. E. (2009) Biochim. Biophys. Acta 1794, 367–373 [DOI] [PubMed] [Google Scholar]
- 34.Reshetnyak Y. K., Burstein E. A. (2001) Biophys. J. 81, 1710–1734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cohn E. J., Edsall J. T. (eds) (1943) Proteins, Amino Acids and Peptides as Ions and Dipolar Ions, pp. 444–505, Reinhold, New York [Google Scholar]
- 36.Leskovac V. (ed) (2003) Comprehensive Enzyme Kinetics, pp. 317–327, Kluwer Academic, New York [Google Scholar]
- 37.Majima E., Oda K., Murao S., Ichishima E. (1988) Agric. Biol. Chem. 52, 787–793 [Google Scholar]
- 38.Santos J. A., Gouvea I. E., Júdice W. A., Izidoro M. A., Alves F. M., Melo R. L., Juliano M. A., Skern T., Juliano L. (2009) Biochemistry 48, 7948–7958 [DOI] [PubMed] [Google Scholar]
- 39.Karsten W. E., Lai C. J., Cook P. F. (1995) J. Am. Chem. Soc. 117, 5914–5918 [Google Scholar]
- 40.Polgar L. (ed) (1989) Mechanisms of Protease Action, pp. 16–18, CRC Press, Boca Raton, FL [Google Scholar]