

Abstract
Mammalian thioredoxin reductase (TrxR) is an NADPH-dependent homodimer with three redox-active centers per subunit: a FAD, an N-terminal domain dithiol (Cys59/Cys64), and a C-terminal cysteine/selenocysteine motif (Cys497/Sec498). TrxR has multiple roles in antioxidant defense. Opposing these functions, it may also assume a pro-oxidant role under some conditions. In the absence of its main electron-accepting substrates (e.g. thioredoxin), wild-type TrxR generates superoxide (O ), which was here detected and quantified by ESR spin trapping with 5-diethoxyphosphoryl-5-methyl-1-pyrroline-N-oxide (DEPMPO). The peroxidase activity of wild-type TrxR efficiently converted the O adduct (DEPMPO/HOO•) to the hydroxyl radical adduct (DEPMPO/HO•). This peroxidase activity was Sec-dependent, although multiple mutants lacking Sec could still generate O. Variants of TrxR with C59S and/or C64S mutations displayed markedly reduced inherent NADPH oxidase activity, suggesting that the Cys59/Cys64 dithiol is required for O generation and that O is not derived directly from the FAD. Mutations in the Cys59/Cys64 dithiol also blocked the peroxidase and disulfide reductase activities presumably because of an inability to reduce the Cys497/Sec498 active site. Although the bulk of the DEPMPO/HO• signal generated by wild-type TrxR was due to its combined NADPH oxidase and Sec-dependent peroxidase activities, additional experiments showed that some free HO• could be generated by the enzyme in an H2O2-dependent and Sec-independent manner. The direct NADPH oxidase and peroxidase activities of TrxR characterized here give insights into the full catalytic potential of this enzyme and may have biological consequences beyond those solely related to its reduction of thioredoxin.
), which was here detected and quantified by ESR spin trapping with 5-diethoxyphosphoryl-5-methyl-1-pyrroline-N-oxide (DEPMPO). The peroxidase activity of wild-type TrxR efficiently converted the O adduct (DEPMPO/HOO•) to the hydroxyl radical adduct (DEPMPO/HO•). This peroxidase activity was Sec-dependent, although multiple mutants lacking Sec could still generate O. Variants of TrxR with C59S and/or C64S mutations displayed markedly reduced inherent NADPH oxidase activity, suggesting that the Cys59/Cys64 dithiol is required for O generation and that O is not derived directly from the FAD. Mutations in the Cys59/Cys64 dithiol also blocked the peroxidase and disulfide reductase activities presumably because of an inability to reduce the Cys497/Sec498 active site. Although the bulk of the DEPMPO/HO• signal generated by wild-type TrxR was due to its combined NADPH oxidase and Sec-dependent peroxidase activities, additional experiments showed that some free HO• could be generated by the enzyme in an H2O2-dependent and Sec-independent manner. The direct NADPH oxidase and peroxidase activities of TrxR characterized here give insights into the full catalytic potential of this enzyme and may have biological consequences beyond those solely related to its reduction of thioredoxin.
Keywords: Electron Paramagnetic Resonance (EPR), Oxidase, Peroxidase, Selenium, Superoxide Ion, DEPMPO, NADPH Oxidase, Thioredoxin Reductase
Introduction
Mammalian thioredoxin reductase (TrxR)4 is a ubiquitous NADPH-dependent flavoenzyme that has prominent disulfide reductase activity. The cytosolic (TrxR1) and mitochondrial (TrxR2) isoforms are the only proteins known to reduce their respective thioredoxin substrates (Trx1 and Trx2) (1). The thioredoxins are normally maintained in the reduced form and are responsible for maintaining the thiols of many intracellular proteins in a reduced state (2). As such, the TrxR/Trx system has a critical role in maintaining intracellular thiol redox balance (3). It donates electrons to ribonucleotide reductase and reduces a number of other proteins (e.g. protein-disulfide isomerase, peroxiredoxins, and methionine sulfoxide reductase) (1, 3, 4). Trx also regulates some redox-sensitive transcription factors (1, 3, 4), which enhances their ability to bind DNA. In addition, TrxR can reduce selenite, dehydroascorbate, lipid hydroperoxides, α-lipoic acid, and disulfides in some other proteins (4–6).
The major forms of TrxRs are NADPH-dependent homodimers, and each subunit contains three redox-active sites (1, 3, 4, 7, 8): (a) one FAD that accepts electrons from NADPH, (b) an N-terminal domain dithiol (-CVNVGC- whose Cys residues occupy positions Cys59/Cys64 in TrxR1 studied here) that is reduced by the flavin, and (c) the C-terminal active site, which is believed to accept electrons from the N-terminal dithiol and contains selenocysteine (Sec) within the sequence -Gly-Cys-Sec-Gly (8). The Cys-Sec (Cys497/Sec498) active site is essential for the reduction of oxidized thioredoxins and plays a major role in the reduction of artificial disulfide substrates, such as DTNB (9). However, some substrates (e.g. selenocystine, selenite, and lipoic acid) are perhaps not solely dependent on the Sec and may alternatively be reduced by the N-terminal dithiol, at least for the mitochondrial isoenzyme TrxR2 (10). When the enzyme is reduced by NADPH, the nucleophilic and highly reactive Sec residue becomes surface-exposed and prone to react with either substrates or inhibitors (1, 8). The active site of TrxR can be inhibited by a number of compounds, including some gold-containing organics, cisplatin, nitrosoureas, dinitrohalobenzenes, and others (1, 4, 11–14). Interestingly, different gold compounds may bind to different sites. For example, inhibition by auranofin is strongly associated with the Sec, whereas aurothioglucose is less Sec-dependent and may have some effects on the N-terminal dithiol (10). The N-terminal domains, including the flavin and Cys59/Cys64 dithiol of TrxR, resemble those in glutathione reductase and lipoamide dehydrogenase, but only TrxR has the C-terminal Cys-Sec motif. However, the Cys59/Cys64 dithiol in TrxR may have multiple roles (10) that are not yet well understood.
The TrxR/Trx system is normally considered to have broad antioxidant functions, not only through its reduction of other antioxidant enzyme active sites, lipoic acid, hydroperoxides, and dehydroascorbate (4–6), but also through its attenuation of the effects of ROS on protein thiols by reducing ROS-generated disulfides (3, 4). The inhibition of TrxR compromises Trx reduction and thus the peroxiredoxins that directly reduce H2O2 and alkyl hydroperoxides (3, 4). Recent reports note that peroxiredoxins in cells become oxidized when they are deprived of reducing equivalents from their respective thioredoxins (15, 16). Overall, inhibition of the TrxR/Trx system can promote oxidant susceptibility and cell death (4). Given its important role in promoting cell survival, there is great interest in exploring the inhibition of TrxR as a potential strategy to kill cancer cells. For example, cisplatin irreversibly inhibits TrxR (17), and high levels of TrxR in A549 cells are critical to the effectiveness of cisplatin (18).
In contrast to its antioxidant roles, TrxR may also have pro-oxidant functions under certain conditions. The inhibition of the active site of TrxR may promote pro-oxidant effects in cells, which may directly promote cell death as selenium-compromised thioredoxin reductase-derived apoptotic proteins (SecTRAPs) (1, 19). These cellular effects are not yet well understood, and it is not clear if all inhibitors of TrxR promote these effects. It has been shown that some chemicals promote O generation by TrxR. The electron flow of the natural catalytic cycle of mammalian TrxR (i.e. catalyzing the reduction of substrates, such as oxidized Trx or lipoic acid) may be diverted to O2 under certain conditions, converting the enzyme to a pro-oxidant NADPH oxidase. This was first described for the TrxR-mediated reduction of selenite to selenide, which, when reoxidized to selenite, can mimic a strong NADPH oxidase activity through oxygen-coupled redox cycling (5). The first class of inhibitors described for this enzyme (dinitrohalobenzenes) was also found to facilitate and increase its NADPH oxidase activity, presumably by redox cycling of the covalently attached dinitrophenyl groups (11, 20). Subsequently, a similar induction of NADPH oxidase activity of TrxR was shown for other low molecular weight compounds, including juglone (19, 21), curcumin (22), and motexafen gadolinium (23). However, the catalytic mechanisms of the non-induced inherent NADPH oxidase activity of TrxR have not yet been elucidated.
These studies present an in-depth investigation of the inherent NADPH oxidase activity of TrxR1. A series of site-directed mutation studies and the use of inhibitors provided new insights into the role of the various redox centers of TrxR in its oxidase activity. The studies also provide further insights into its peroxidase activity, including the roles of the Sec and N-terminal dithiol. The ability of TrxR to reduce the O adduct of DEPMPO to the HO• adduct is also described and should reflect its Sec-dependent peroxidase capacity.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents
Tris was from Research Organics (Cleveland, OH). Manganese (III) tetrakis(4-benzoic acid)porphyrin chloride (MnTBAP) and α-phenyl-N-tert-butylnitrone (PBN) were from Alexis Biochemicals (Lausen, Switzerland). 5-Diethoxyphosphoryl-5-methyl-1-pyrroline-N-oxide (DEPMPO) was manufactured by Radical Vision (Marseille, France) and was purchased from Radical Vision or Alexis Biochemicals. Auranofin was from Axxora, LLC (San Diego, CA). Sodium chloride was from VWR Scientific (West Chester, PA), and EDTA was from Fisher. Ferrous ammonium sulfate and hydrogen peroxide were from Mallinckrodt Chemicals (Phillipsburg, NJ). Superoxide dismutase (SOD) was from Sigma (catalog no. S8160, from bovine liver), and catalase was from Calbiochem (catalog no. 219001, from bovine liver). Diethylenetriaminepentaacetic acid (DTPA), deferoxamine mesylate (DFX), 1-chloro-2,4-dintrobenzene (2,4-DNCB), and all other chemicals and reagents were purchased from Sigma.
Recombinant TrxR1 and Site-directed Mutants
Purified wild-type TrxR1 and the site-directed mutants Y116I and Y116T were generated as previously described using a recombinant system that provides for incorporation of the active site Sec (8, 24–27). The truncated variant that is missing Sec498 and Gly499 was generated as described previously (19, 28). Four new TrxR1 mutants were produced for this study, including three single mutants (C59S, C64S, and U498C) and one double mutant (C59S/C64S). The plasmid pTRSTER constructed earlier (29) was used as template for all PCRs. The forward primers used for constructing the C59S, C64S, and C59S/C64S mutants respectively were as follows: C59S-f, 5′-GCGGTCTCGGAACGTCTGTGAACGTGGGCTGCATACC-3′; C64S-f, 5′-GCGGTCTCGGAACGTGTGTGAACGTGGGCTCCATACCTAAAAAACTGATG-3′; C59S/C64S-f, 5′-GCGGTCTCGGAACGTCTGTGAACGTGGGCTCCATACCTAAAAAACTGATG-3′. The same reverse primer was used for constructing these three mutants: C5964S-r, 5′-GCGGTCTCCGTTCCCCCGAGACCCCCATTC-3′. To construct the U498C mutant, the primers were as follows: U498C-f, 5′-GCGGTCTCATGCTGCGGTTAATAATCGGTTGCAGG-3′; U498C-r, 5′-GCGGTCTCCAGCATCCGGACTGGAGGATG-3′. The PCRs were prepared using Phusion® High-Fidelity PCR Master Mix (Finnzyme, Espoo, Finland) with the addition of 20 pmol of each primer and 10 ng of template plasmid in a total volume of 50 μl. Reactions were initiated at 95 °C for 2 min, followed by 30 cycles of amplification (95 °C, 10 s; 60 °C, 10 s; 72 °C, 3 min) and concluded at 72 °C for 10 min. The resulting PCR products were purified using the PCR purification kit (Fermentas), digested with Eco31I and DpnI (Fermentas), repurified with the PCR purification kit, and ligated using T4 DNA ligase (Fermentas). The DNA was then transformed into Escherichia coli BL21(DE3) competent cells, and the mutations were verified by DNA sequencing (GATC Biotech, Konstanz, Germany). For the TrxR variants that contain the C-terminal -Gly-Cys-Sec-Gly motif (wild type, C59S, C64S, C59S/C64S, Y116I, and Y116T), the assistant plasmid pSUABC was co-transformed into the same host bacteria in order to increase selenoprotein yield as described previously (30).
All Sec-containing TrxR1 variants were expressed in E. coli as described previously (25). The truncated and U498C variants of TrxR1, which lack the Sec residue, were expressed in E. coli using standard methods for recombinant protein production (31). All TrxR1 recombinant proteins were purified using 2′,5′-ADP-Sepharose (GE Healthcare), followed by purification of the dimeric forms using gel filtration (ÄKTA-Explorer HPLC system equipped with Superdex 200 10/30 column, GE Healthcare) (32). All enzymes were stored in TE buffer (50 mm Tris-HCl, 2 mm EDTA, pH 7.5) and concentrated by ultrafiltration using a 30-kDa cut-off ultracentrifuge device (PALL). The enzyme concentration was determined spectrophotometrically using the molar extinction coefficient of FAD (13,600 m−1 cm−1 at 463 nm), assuming one FAD per subunit.
General Methods
To remove polyvalent metal ion contaminants, solutions were pretreated with Chelex-100 (4% w/v) for >12 h before use. Experiments under aerobic conditions were conducted in 1.5-ml polypropylene tubes under room air. Experiments under anaerobic conditions were conducted in an anaerobic chamber (Coy Laboratory Products, Grass Lake, MI) (4–5% H2, balance N2) as described previously (33). Buffers and deionized water were preincubated in the anaerobic chamber for ≥24 h before use, and small volumes of more labile reagents (NADPH, DEPMPO, and TrxR) were pre-equilibrated for ≥4 h.
Assay for TrxR Activity
The activity of purified TrxR was measured as the NADPH-dependent reduction of 5,5′-dithiobis(2-nitrobenzoic) acid (DTNB) (20, 34). The system included 0.1 m sodium phosphate, pH 7.4, with 5 mm EDTA, NADPH (0.4 mm), and DTNB (3 mm). Control experiments showed that 4 μm auranofin inhibited essentially all of the DTNB reductase activity in the enzyme preparations, as expected for pure wild-type enzyme (35).
Electron Spin Resonance Spin Trapping
Reactive oxygen species were assessed at fixed time points using ESR spin trapping essentially as described previously (36). Solutions were pretreated with Chelex-100 resin for >12 h prior to use. The buffer (final concentration in assay of 0.15 m KCl, 2.5 mm potassium phosphate, pH 7.35), water, and 0.4 mm NADPH were preincubated for 5 min at 37 °C. TrxR and DEPMPO (14 mm) were added, followed by incubation at 37 °C for the indicated times. In some experiments, 50 mm PBN plus 5% DMSO (v/v) were substituted for DEPMPO. The reactions were stopped by immersion in liquid nitrogen (77 K). Samples were stored in liquid nitrogen, typically for less than 1 week, until analysis by ESR. Freezing and thawing do not change the ESR spectra of DEPMPO adducts (37, 38).
For ESR analysis, samples were quickly thawed and placed in a quartz flat cell, and ESR spectra were obtained without delay at room temperature using a Bruker EMX spectrometer. Instrument settings are indicated under “Results.” ESR spectra were confirmed in replicate experiments. The hyperfine couplings were measured from the ESR data and were confirmed using the simulation software WinSim (version 0.95) (NIEHS, National Institutes of Health) (39). The concentration of each species was obtained by double integration of the simulated spectra using WinSim software. 4-Hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl was used as the standard.
Generation of DEPMPO-Superoxide Adduct
In some experiments, the DEPMPO-superoxide adduct (DEPMPO/HOO•) was preformed, and its decay over time was examined in the presence or absence of various TrxR variants and NADPH. The KO2/18-crown-6 ether system as described by (40, 41) was used to generate O and trap it as DEPMPO/HOO•. Briefly, the system includes 10 mm KO2, 10 mm 18-crown-6, 10% DMSO, and 14 mm DEPMPO in 90 mm phosphate buffer, pH 7.4 (final concentrations). After 1 min at room temperature, SOD was added to a final concentration of 1000 units/ml. After 1 min, TrxR variants (to 1.07 μm) and/or NADPH (to 0.4 mm) were added, the sample was loaded into the flat cell, and 30 consecutive ESR spectra were collected, with an acquisition time of 2.8 min/spectrum (84 min total).
Generation of DEPMPO-Hydroxyl Radical Adduct
In some experiments, DEPMPO/HO• was preformed, and its decay over time was examined in the presence or absence of various TrxR variants and NADPH. In 0.1 m sodium phosphate, pH 7.4, containing 14 mm DEPMPO and 0.2 mm DTPA, HO• was generated by the addition of Fe(NH4)2(SO4)2 to 0.15 mm and H2O2 to 1.7 mm. After 1 min at room temperature, both catalase and SOD were added to 1000 units/ml each. After 1 min, NADPH (to 0.4 mm) and/or TrxR variants (to 1.07 μm) were added, the sample was loaded into the flat cell, and 30 consecutive ESR spectra were collected, with an acquisition time of 2.8 min/spectrum (84 min total).
Miscellaneous Methods
Statistical analyses of three or more groups of data were done using one-way analysis of variance and the Tukey-Kramer post-test (Prism software, GraphPad). Analyses of two groups of data were done using an unpaired Student's t test (Prism software). Significance was assumed at p < 0.05.
RESULTS
Spin Trapping of Oxygen Radicals Formed from the NADPH Oxidase Activity of TrxR; Effect of TrxR Substrates
To characterize the inherent NADPH oxidase activity of TrxR, the spin trap DEPMPO was used to follow the generation of HO• and O under aerobic conditions in the absence of thioredoxin. In time course studies, the DEPMPO/HO• adduct was the predominant signal observed, and its intensity increased dramatically during the first 30 min (Fig. 1A). This signal matches that of DEPMPO/HO• generated by the positive control (Fig. 1B), and its hyperfine coupling constants (aP = 47.2 G, aH = 13.8 G, aN = 13.8 G) are consistent with those of DEPMPO/HO• (42). These signals were dependent on TrxR activity because they were not seen with just TrxR plus DEPMPO (no NADPH) (Fig. 1A) or with DEPMPO plus NADPH without TrxR (see below). There is a very minor component of the TrxR-generated signal (Fig. 1A) that aligns with that of the O-positive control (Fig. 1C) and whose hyperfine coupling constants match those of DEPMPO/HOO•. The DEPMPO/HOO• signal remained at a low level over time (Fig. 1, A, D, and E), whereas the DEPMPO/HO• component increased dramatically over the first 30–60 min (Fig. 1, A and D). In time courses extending from 5 min to 2 h (Fig. 1D) and from 30 min to 6 h (Fig. 1E), the DEPMPO/HO• signal reached a plateau around 1.5–3 h and was still substantial at 6 h. The in vitro half-lives for the spontaneous decay of the DEPMPO/HOO• and DEPMPO/HO• adducts have been reported as less than 30 min (37, 43). If production of O or HO• ceased once the maximal intensity of the adduct signals had been reached, there would have been a noticeable decline in their signals at the later times. Their relatively constant levels at the later times imply a continued rate of production sufficient to offset expected in vitro decay. The signal intensity was directly correlated with the amount of TrxR present, and no signal was seen when TrxR was absent (Fig. 1F).
FIGURE 1.

Representative ESR spectra over time using a constant amount of TrxR and the spin trap DEPMPO. A, a fixed concentration of TrxR (1.07 μm) was incubated at 37 °C under room air for the times indicated with 0.4 mm NADPH and 14 mm DEPMPO. The spectrum at the bottom was also for 30 min but lacked NADPH. B, hydroxyl radical positive control consisting of H2O2 (1.7 mm), ferrous ammonium sulfate (0.15 mm), and DEPMPO (14 mm) (10-min incubation at 37 °C under room air). The components of the signal corresponding to DEPMPO/HO• are indicated by black dots. C, superoxide positive control consisting of xanthine (2 mm), xanthine oxidase (0.2 units), DTPA (0.1 mm), DEPMPO (50 mm), and 50 mm potassium phosphate, pH 7.4 (3-min incubation at 37 °C under room air). The components of the signal corresponding to DEPMPO/HOO• are indicated by open diamonds. The structures for the DEPMPO/HO• and DEPMPO/HOO• adducts are shown beside traces B and C, respectively. D, two different time courses (5–30 min and 10–120 min) showing the amounts of DEPMPO/HO• and DEPMPO/HOO• adducts over time in experiments with 1.07 μm TrxR plus NADPH (0.4 mm). E, a third time course (30–360 min) using the conditions as defined in D. F, relationship between the amount of TrxR and the amount of DEPMPO/HO• adduct (r = 0.969) using a single incubation time of 30 min with different amounts of TrxR plus 0.4 mm NADPH and 14 mm DEPMPO. ESR instrument settings were as follows: modulation amplitude, 1 G; microwave power, 19.92 milliwatts; receiver gain, 6.32 × 104; time constant, 40.96 ms; microwave frequency, 9.76 GHz; sweep width, 200 G, field set, 3480 G; modulation frequency, 100 kHz; scan time, 42 s; number of scans, 9.
Experiments were done to determine if O was required for the generation of the DEPMPO adducts. SOD and the SOD mimetic MnTBAP eliminated both the DEPMPO/HOO• and DEMPO/HO• signals (Fig. 2, a–d). The loss of DEPMPO/HOO• was expected, and the loss of DEPMPO/HO• indicates that O is required for the generation of this signal as well. However, DEPMPO/HOO• does not spontaneously decay to DEPMPO/HO• (37, 44). Although SOD rapidly converts O to H2O2 (rate constant 1.4 × 109 m−1 s−1), the resulting H2O2 does not positively or negatively impact DEPMPO/HO• (see below). H2O2 does not inhibit TrxR activity (e.g. 0.2 mm H2O2 for 2 h did not affect its rate of DTNB reduction (not shown)). The SOD used in these experiments did not affect the DEPMPO/HO• signal generated by Fe(II) + H2O2 (Fig. 2f), so it does not contain contaminants that eliminate H2O2-dependent signals, and it does not convert DEPMPO/HO• to an ESR silent species. The data thereby imply that TrxR generates O and that this O is required for TrxR catalyzing the formation of the DEPMPO/HO• adduct. The mechanisms for the generation of these ESR signals are further explored below.
FIGURE 2.

Representative effects of SOD and MnTBAP on the DEPMPO adducts generated by TrxR under aerobic conditions. a and c, a 30-min incubation (37 °C) of 1.07 μm TrxR, 0.4 mm NADPH, and 14 mm DEPMPO. Spectra b and d are the same as a and c with the addition of SOD (333 units/ml) (b) or MnTBAP (0.2 mm) (d). e, ferrous ammonium sulfate plus H2O2 incubated for 10 min. f, the same as e with the addition of SOD. The instrument settings were the same as for Fig. 1.
Experiments were conducted to determine if the spin adduct signals were also generated in the presence of known electron-accepting substrates of TrxR. In the presence of 20 μm selenite, which is directly reduced by TrxR (5), the DEPMPO/HO• signal generated by TrxR plus NADPH was decreased by 55% (supplemental Fig. 1). At this concentration, selenite did not affect the DEPMPO/HO• signal generated by Fe(II) plus H2O2 (supplemental Fig. 1). A concentration of 20 μm selenite is at the estimated Km for calf thymus TrxR (5), so it would not be predicted to fully occupy the enzyme. Although 100 μm selenite similarly decreased the signal by TrxR, it also decreased the signal generated by Fe(II) plus H2O2 (not shown); it may have done so because of its potential to oxidize Fe(II). The data with 20 μm selenite thus imply that TrxR is less prone to generate ROS while it is reducing selenite, probably because electron flow is directed at selenite reduction rather than diverted to O2. It is possible that some of the signal observed in the presence of selenite might result from the redox cycling of reduced selenium species (5). If this is the case, then the ability of selenite to suppress direct generation of ROS by TrxR would be greater than that estimated by supplemental Fig. 1.
Other compounds that are reduced by TrxR were also tested. DTNB markedly suppressed the DEPMPO/HO• signal generated by TrxR, but it also suppressed that generated by Fe(II) plus H2O2 (not shown), suggesting a nonspecific scavenging of ROS. TrxR reduces Trx1, which can then reduce the disulfides in insulin. When Trx1 (5 μm) plus insulin (100 μm) were included, the DEPMPO/HO• signal intensity decreased by 56% (supplemental Fig. 1, spectrum f). In the absence of Trx1, insulin (which is not a direct substrate of TrxR) only decreased the signal by 8% (supplemental Fig. 1, spectrum g). In these experiments (supplemental Fig. 1, spectrum f), where the level of insulin and Trx1 were sufficient to oxidize only about 30% of the NADPH, ROS generation by TrxR could have been supported once insulin reduction was complete. Moreover, a portion of the TrxR analyzed here lacked Sec, which is unable to reduce Trx1 but can still produce O (see below) and may have contributed to the signal generated in the presence of Trx1 (supplemental Fig. 1f). Together, the selenite and insulin data imply that O generation is decreased in the presence of substrates that are reduced by TrxR, probably by diverting electrons from O2 as the alternative electron acceptor.
TrxR-dependent Formation of Hydroxyl Radicals
Experiments were done to further assess the source of the DEPMPO/HO• signal. The secondary radical trapping technique with PBN as the spin trap and DMSO as a HO• scavenger will result in methyl radical (•CH3) production.
 |
PBN forms long lasting spin adducts of carbon-centered radicals, such as •CH3.
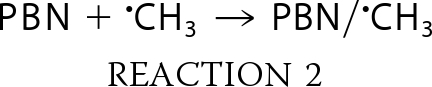 |
In the presence of PBN plus DMSO, TrxR generated a spectrum that is consistent with carbon-centered radical adducts of PBN (Fig. 3A, spectrum a). No signal was seen when TrxR was omitted (Fig. 3A, spectrum b). PBN carbon radical adducts generate six-line signals (three doublets), and a single species will generate a signal in which all peaks are of the same intensity (e.g. computer simulations; Fig. 3B, spectra c and d). However, the TrxR-generated signal (Fig. 3B, spectrum a) had more than six lines, suggesting that it contains more than one type of PBN radical adduct. Simulation of the TrxR spectrum (Fig. 3B, spectrum a) showed that 43% represents the methoxy radical (•OCH3) adduct of PBN, and 55% represents the •CH3 adduct. The generation of PBN/•OCH3 in addition to PBN/•CH3 is not unexpected because •CH3 also reacts with O2 to generate •OCH3, which is also trapped by PBN.
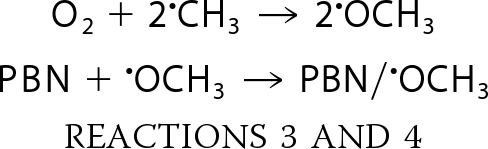 |
The rate constants predict that Reaction 3 is more favorable than Reaction 2, so a mix of both PBN/•CH3 and PBN/•OCH3 is possible, depending on the relative concentrations of the various reactants (45). A mix of both of these PBN adducts has been observed for other methods of HO• generation under room air (36, 46). These PBN/DMSO findings therefore provide additional support for the generation of free HO• by TrxR.
FIGURE 3.

A, representative ESR spectra in which PBN (50 mm) plus DMSO (5%) were substituted for DEPMPO. The samples contained NADPH (0.4 mm) and were incubated for 30 min at 37 °C. Spectrum a included TrxR (1.07 μm), whereas b was without TrxR. B, an expanded view of spectrum a is shown at the center, spanning the region from 3440 to 3500 G, and is compared with computer simulations of the PBN/•OCH3 adduct spectrum (c) (hyperfine splitting constants aN = 15.05 G, aH = 3.32 G) and with a simulated PBN/•CH3 adduct spectrum (d) (hyperfine splitting constants aN = 16.51 G, aH = 3.68 G). In spectrum a, the components of the signal corresponding to PBN/•OCH3 are indicated by open dots, and those for PBN/•CH3 are indicated by black dots. The PBN signals probably underestimate the amount of HO• generated because O can mediate the decay of PBN/•CH3 and PBN/•OCH3 to ESR silent species (71), and the rate constant for the reaction of PBN with •CH3 is about 3–4 orders of magnitude lower than the reaction of HO• with DEPMPO or DMSO (72). The instrument settings were the same as described in the legend to Fig. 1.
Formate was used to confirm HO• generation. Formate is oxidized by HO• to yield a carbon dioxide radical anion, which can be trapped as DEPMPO/CO (42). When formate was included in the reaction mix, a pronounced DEPMPO/CO signal was seen, with lesser amounts of DEPMPO/HO• (supplemental Fig. 2). The fact that the overall DEPMPO/CO signal intensity was greater than the decline in DEPMPO/HO• signal intensity in the presence of formate (supplemental Fig. 2) reflects the increased stability of the DEPMPO/CO adduct relative to that of DEPMPO/HO•.
A typical mechanism for HO• generation is through the one-electron reduction of H2O2 by certain metals, the classical example being iron and the Fenton reaction (47, 48).
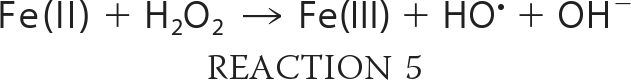 |
These reactions should be stimulated by the addition of H2O2 and markedly inhibited by catalase, which rapidly degrades H2O2. The findings with TrxR, however, were not consistent with this type of mechanism. Catalase did not significantly change the DEPMPO/HO• adduct signal intensity (Fig. 4A), implying that free H2O2 is not required for the DEPMPO/HO• signal generated by TrxR. For in vitro systems with purified enzymes, H2O2 can often be limiting, and its inclusion can markedly enhance metal-mediated HO• generation (36, 49–52). However, the addition of H2O2 did not change the DEPMPO/HO• adduct signal intensity (Fig. 4A). Together, these results imply that H2O2 is not required for the generation of DEPMPO/HO• by TrxR. In Fenton reaction controls (not shown), catalase almost completely eliminated HO• generated by Fe(II) + H2O2, as expected. Neither catalase nor H2O2 affected the small O component of the spectra generated by TrxR (Fig. 4A).
FIGURE 4.

The effects of catalase, H2O2, ANF, metal chelators, and Fe(II) on the DEPMPO adducts generated by wtTrxR under aerobic conditions. A, 30-min incubations (37 °C) of 0.535 μm TrxR, 0.4 mm NADPH, and 14 mm DEPMPO with the following additions as indicated: catalase (833 units/ml), 50 μm H2O2, or 4 μm ANF. B, 30-min incubations (37 °C) of 1.07 μm TrxR, 0.4 mm NADPH, and 14 mm DEPMPO with the following additions as indicated: 0.2 mm DTPA, 0.2 mm DFX, or 15 μm Fe(II) as ferrous ammonium sulfate. The ESR instrument settings were the same as for Fig. 1. Both graphs show the relative molar amounts of DEPMPO/HO• and DEPMPO/HOO• (mean ± S.D. (error bars) for triplicate experiments) for the different treatments. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus the wtTrxR samples.
We then tested the effects of auranofin (ANF), a gold compound that markedly inhibits classical disulfide substrate reduction by TrxR when present at equimolar or higher amounts with respect to TrxR (35, 53). Although 4 μm ANF caused >99% inhibition (not shown) of DTNB reduction by TrxR (a standard assay for the disulfide reductase activity of TrxR), it only decreased the DEPMPO/HO• adduct signal by 28% (Fig. 4A). ANF at the same time increased the DEPMPO/HOO• adduct by 2-fold (Fig. 4A). Overall, ANF thereby influenced the amount of O and HO• adducts generated by TrxR but only to a limited extent compared with its total inhibition of disulfide substrate reduction by the enzyme.
Fenton-type chemistry should be stimulated by the addition of redox-active metals, such as Fe(II), and should be inhibited by chelators that sequester these metal ions (36, 48, 54, 55). The metal chelators DTPA and DFX were thus tested next. Although they each chelate several metals, DFX prevents redox reactions of its sequestered metals, whereas DTPA actually increases HO• generation by the Fenton reaction (56). Consistent with this, DTPA increased the TrxR-generated DEPMPO/HO• adduct by more than 50% (Fig. 4B). Surprisingly, DFX also increased the average TrxR-generated DEPMPO/HO• signal (Fig. 4B), although this increase was not significant. In contrast, DFX completely eliminated the DEPMPO/HO• signal generated by Fe(II) plus H2O2 (not shown), demonstrating that DFX is very effective at stopping iron-mediated reactions under these conditions. If Fenton-type reactions were involved in the generation of HO• by TrxR, then DFX should have significantly reduced the DEPMPO/HO• signals. Because it did not, a role for redox-active metals is not supported. Similarly, DFX blocks the metal-catalyzed decomposition of DEPMPO/HOO• to DEPMPO/HO• (37), so the lack of inhibition by DFX argues against this metal-catalyzed decomposition as a means to generate the DEPMPO/HO• adduct. Although Fe(II) would be predicted to enhance HO• generation from H2O2, the addition of Fe(II) actually decreased the TrxR-generated DEPMPO/HO• signal by 64% (Fig. 4B). This also argues against Fenton-type chemistry and is consistent with the lack of affect by H2O2 and catalase (above). The decreased DEPMPO/HO• signal seen with Fe(II) probably results from inhibition of TrxR by Fe2+. When tested in EDTA-free Tris-HCl buffer (pH 7.5), 25, 50, and 150 μm Fe2+ inhibited DTNB reduction by TrxR by 41, 71, and 96%, respectively. This is consistent with the sensitivity of TrxR to other divalent metals (e.g. Mn2+ and Zn2+) (57). The enhancement of the DEPMPO/HO• signal by DTPA might result from the chelation of trace divalent cations that are causing a partial inhibition of the enzyme. Although the O signal remained a minor component in these experiments (Fig. 4B), it increased 65% in the presence of DTPA and decreased 53% in the presence of 15 μm Fe(II). These trends follow those for DEPMPO/HO• and could reflect increases and decreases in TrxR activity mediated by DTPA and Fe(II), respectively.
Low temperature (10 K) ESR of undiluted TrxR was done to determine if there was significant iron or copper content. A very small signal was observed at g = 4.0 (not shown), which is consistent with a trace of high spin non-heme iron signal typical of most samples. The concentration was estimated to be only ∼0.02 μm, which is >730-fold less than that for the TrxR (14.64 μm) used for this analysis. The 0.2 mm DFX used in Fig. 4B would therefore have been in vast excess to the trace iron contributed by TrxR in spin trapping experiments (1.07 μm TrxR would have contributed 0.00146 μm iron). No signals consistent with Cu2+, another potentially redox-active metal, were observed in undiluted TrxR. Together, the DTPA, DFX, Fe(II), and low temperature EPR data argue against iron- or copper-mediated Fenton-type reactions as a significant contributor to the TrxR-generated DEPMPO/HO• signals.
ESR Spectra, Aerobic versus Anaerobic Conditions
To determine if the TrxR-generated HO• and O adducts were dependent on O2, spin trapping experiments were next conducted under aerobic as well as anaerobic conditions. Both signals were O2-dependent; under anaerobic conditions, the DEPMPO/HO• and DEPMPO/HOO• adducts were decreased by 87 and 65%, respectively (Fig. 5). The small amounts of these adducts under anaerobic conditions probably result from small amounts of residual O2 in the solutions. The anaerobic signal, although small, was more complicated than that under aerobic conditions, with small quantities of one or more species whose hyperfine constants are consistent with a carbon-centered radical (Fig. 5). A similar unidentified radical was previously observed anaerobically with P450 reductase, cytochrome b5, NADPH, and chromate (36). This carbon-centered radical is therefore not unique to TrxR. Altogether, the DEPMPO/HO• and DEPMPO/HOO• adducts are dependent on O2 and do not represent a direct reaction of TrxR with DEPMPO. The O2 dependence is consistent with the ability of SOD and MnTBAP to eliminate these signals (above).
FIGURE 5.

Dependence of the DEPMPO adducts on O2. A, representative ESR spectra of reactions incubated aerobically (top) or anaerobically (bottom). The buffers, water, and NADPH were mixed and incubated for 5 min at 37 °C. Each reaction was started by adding TrxR (1.07 μm final concentration) and DEPMPO (14 mm final concentration). After 12 min at 37 °C, the reaction was loaded into an ESR flat cell and sealed, and spectra were acquired at room temperature. For the anaerobic experiments, the buffers and deionized water were preincubated in the anaerobic chamber for ≥24 h before use, and small volumes of NADPH, DEPMPO, and TrxR were pre-equilibrated for ≥4 h. The aerobic experiments were conducted identically, except all solutions and steps were handled under room air. Instrument settings were as described in the legend to Fig. 1. B, relative quantities of the HO•, O, and carbon radical adducts of DEPMPO (mean ± S.D. for triplicate experiments) observed under aerobic versus anaerobic conditions. **, p < 0.01; ***, p < 0.001 versus the corresponding aerobic samples.
The Role of Redox-active Domains within TrxR
Because ANF only caused a small decrease in the DEPMPO/HO• signal (above), two other inhibitors of the Sec active site were tested. Cisplatin (0.1 mm), which is presumed to covalently bind the Sec (Sec498) (12, 17, 58), inhibited 77–83% of its DTNB reductase activity, consistent with the prior studies. Cisplatin did not significantly decrease the DEPMPO/HO• signal (Fig. 6, a and b) (4.3 ± 0.98 μm with cisplatin versus 4.7 ± 0.48 μm without cisplatin). Similarly, the DEPMPO/HOO• component of the spectrum was also not significantly changed (0.65 ± 0.27 μm with cisplatin versus 0.47 ± 0.12 μm without cisplatin; p > 0.05, n = 3). These data imply that TrxR can still generate DEPMPO/HO• when cisplatin is bound to the enzyme, which agrees well with previous reports of cisplatin-derivatized TrxR in the form of redox-active SecTRAPs (19).
FIGURE 6.

Representative effects of inhibitors of the Sec active site. Each spectrum was recorded after a 30-min aerobic incubation (37 °C) of 1.07 μm TrxR, 0.4 mm NADPH, and 14 mm DEPMPO. The samples for b and d were identical to those for a and c, respectively, except that each contained a TrxR inhibitor, either cisplatin (b) or 2,4-DNCB (d). The sample for e was the same as for d except that it also contained SOD (333 units/ml). The instrument settings were the same as for Fig. 1. In c and d, the components of the spectrum corresponding to DEPMPO/HO• are indicated by black dots above the spectrum, whereas the components corresponding to DEPMPO/HOO• are indicated by open diamonds.
2,4-DNCB irreversibly binds both Cys497 and Sec498 of TrxR, and this dramatically inhibits its ability to reduce DTNB or other disulfide substrates (11). There is no evidence that 2,4-DNCB alkylates Cys59/Cys64. In ESR studies, 2,4-DNCB decreased DEPMPO/HO• by 46%, and it increased DEPMPO/HOO• more than 11-fold (Fig. 6, c and d), making DEPMPO/HOO• the predominant component of the spectrum. This increase in O reflects the previously reported induction of NADPH oxidase activity by 2,4-DNCB, with most of the O generated by redox cycling of the nitro groups of 2,4-DNCB (11, 20). SOD eliminated essentially all of the DEPMPO/HO• and DEPMPO/HOO• signals in the 2,4-DNCB sample (Fig. 6e). There were additional unidentified small signals noted in Fig. 6e, which may result from the nitro anion radicals that would be expected from the one-electron reduction of 2,4-DNCB.
Using additional experiments, we examined the role of the redox-active sites within TrxR in the generation of O and HO• adducts. These sites include the Cys-Sec (Cys497/Sec498), the Cys59/Cys64 dithiol, and the flavin (FAD) (4). The flavin accepts electrons from NADPH and transfers them to Cys59/Cys64, which then presumably reduces Cys497/Sec498 on the other subunit of the homodimer. The crystal structure of catalytically active TrxR1 indicates that the Sec498 is positioned beneath the side chain of Tyr116. Y116I and Y116T mutations result in a 53–57% lower catalytic turnover (kcat) of Trx and 29% lower turnover of DTNB (8). Hence, Tyr116 might influence the generation of O and/or HO• by TrxR.
Various site-directed mutants of TrxR were examined. The kinetic constants for the Tyr116 variants were reported previously (8). The constants for each preparation of TrxR mutants used in this study are shown in Table 1. The mutants lacking Cys59 and/or Cys64 were essentially devoid of DTNB reductase activity (Table 1). Although they retain the active site Cys-Sec that reduces Trx and DTNB, their inability to reduce DTNB and Trx probably represents their inability to reduce the Cys-Sec active site. The kcat for DTNB reduction by U498C was 176-fold lower than that for wild-type TrxR (wtTrxR), and the truncated mutant (lacking Sec498/Gly499) was essentially negative for DTNB reduction (Table 1). The truncated and U498C mutants retain a functional flavin and Cys59/Cys64 dithiol, as was also shown earlier (9, 19).
TABLE 1.
Steady-state kinetic parameters of different TrxR1 variants
This table summarizes the kinetic parameters using DTNB as a model substrate with several different recombinant TrxR1 variants. These kinetic parameters should serve as a comparison with the capacity of these variants to reduce DEPMPO/HOO• and produce O and HO•, as described in the present study.
| Kinetic parametera | Recombinant rat TrxR1 variant |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| wtTrxR1 (Sec-enriched)b | wtTrxR1 (standard preparation)c | C59Sd | C59S/C64Se | C64Sd | U498Cf | Truncatedg | Y116Ib | Y116Tb | |
| kcat (min−1) | 4220 ± 62 | 2040 ± 62 | <2 | <2 | <2 | 11.6 ± 0.1 | <2 | 2990 ± 170 | 2860 ± 140 |
| Km (μm) | 94.0 ± 22.5 | 288 ± 23 | NAh | NA | NA | 44.1 ± 2.6 | NA | 97.1 ± 15.7 | 90.6 ± 13.2 |
| kcat/Km (min−1 μm−1) | 44.9 | 7.1 | NA | NA | NA | 0.26 | NA | 30.8 | 31.6 |
a Kinetic parameters were determined at room temperature (20–22 °C) following the formation of TNB− at 412 nm in TrxR1-catalyzed NADPH-dependent DTNB reduction, using the standard DTNB assay conditions for TrxR1 (20, 34).
b Values obtained with Sec-enriched enzyme preparations using PAO-Sepharose purification as reported earlier (8). In the present study, standard preparations of the enzyme variants were used, typically showing ∼40–60% lower maximal turnover.
c Parameters as determined here for the enzyme preparation used in the present study, which had a specific activity of 17 units/mg.
d These variants were produced in the present study and lacked detectable DTNB reductase activity.
e This variant was produced as described earlier (19).
f Activity as determined here. Similar values for this variant have also been reported earlier (9).
g The truncated TrxR1 variant used in the present study showed very low and close to undetectable DTNB reductase activity, whereas slightly higher values have been reported elsewhere for truncated rat TrxR1 (9).
h NA, not applicable. Given the very low turnover of these variants, the other parameters cannot be reliably determined.
Following a 30-min incubation, the DEPMPO/HO• adducts for Y116I and Y116T were essentially at identical levels to wtTrxR (Fig. 7). Although the DEPMPO/HOO• component of the signal was a very minor part of the spectrum for all three, the Y116I and Y116T did have 2.0- and 2.3-fold more DEPMPO/HOO• than wtTrxR at 30 min (Fig. 7). After only 10 min of incubation, however, the DEPMPO/HO• adducts for Y116I and Y116T were decreased by 37 and 45%, respectively, versus wtTrxR, and the DEPMPO/HOO• adducts were increased by 2.2- and 2.8-fold, respectively (not shown). The 10 min data therefore suggest that Tyr116 mutants are somewhat slower than wtTrxR in generating DEPMPO/HO• but that by 30 min, the Tyr116 mutants are able to match wtTrxR. For DTNB reduction, the kcat values for the Tyr116 variants were previously noted to be ∼30% lower than wtTrxR that was similarly purified using PAO-Sepharose (8).
FIGURE 7.

Comparison of site-directed variants to wild-type TrxR. Representative ESR spectra of reactions incubated aerobically at 37 °C for 30 min. Each reaction contained 1.07 μm TrxR (wild type or the site-directed variants indicated), 14 mm DEPMPO, and 0.4 mm NADPH. The components of the spectra corresponding to DEPMPO/HO• are indicated by black dots, whereas the components corresponding to DEPMPO/HOO• are indicated by open diamonds. DEPMPO adducts were not seen when NADPH was omitted (not shown). Instrument settings were as described in the legend to Fig. 1.
It is important to note that even for highly enriched purifications of recombinant wtTrxR and mutants made to contain Sec, the full-length enzyme is predominant (typically ∼65%), but there is also ∼35% truncated enzyme (lacking Sec498/Gly499) present in these preparations (32). This results from inefficient Sec insertion at the UGA codon when trying to express proteins with full Sec content. However, the U498C variant is full-length, and the truncated variant is also homogenous; therefore, these two TrxR variants serve as important controls for determining any activities that are completely Sec-independent. At 10 min, the truncated variant generated 89% less DEPMPO/HO• than wtTrxR, whereas DEPMPO/HOO• was increased by 3.7-fold (not shown). Although the truncated enzyme generated larger signals at 30 min than at 10 min, DEPMPO/HOO• was still the predominant component (4.5-fold larger than wtTrxR), and DEPMPO/HO• was 82% smaller than wtTrxR (Fig. 7). These data imply that the truncated enzyme can still generate some O, but that it is severely compromised in generating DEPMPO/HO•.
The U498C variant, which has a Cys497/C498 dithiol, largely resembled the truncated variant, with DEPMPO/HO• markedly decreased relative to wild-type and DEPMPO/HOO• (the prominent component) increased by 2.9-fold versus wild-type (Fig. 7). These data further imply that the Sec is important for generating DEPMPO/HO• and that having Cys-Cys in the C-terminal active site cannot compensate for a Cys-Sec motif or the absence of the Sec.
C59S and C64S variants were next used to explore the role of the N-terminal dithiol. These variants were made to still contain the C-terminal active site (Cys497/Sec498), although a fraction of these recombinant proteins will also lack Sec498/Gly499, as discussed above. For both C59S and C59S/C64S, the DEPMPO/HO• adduct was <10% of wtTrxR, and the DEPMPO/HOO• adduct signal was also very small and only about 30% of that observed with U498C (Fig. 7). The signals for C64S were also very small relative to wtTrxR, but they were larger than those for the C59S variants (Fig. 7). For C64S, DEPMPO/HOO• was the more predominant component of the spectrum, although both adduct signals were small. For all variants, SOD essentially eliminated all signals (not shown), confirming the previous findings with wild-type TrxR (Fig. 2) and indicating that O is required for the formation of the DEPMPO adducts. Together, these data imply that the N-terminal dithiol (Cys59 in particular) has an important role in O generation by the enzyme. Because O is required for both DEPMPO/HOO• and DEPMPO/HO• adduct formation, the C59S/C64S variants do not generate normal amounts of either species. In contrast, the truncated and U498C variants, which have Cys59/Cys64 but lack the Sec, can still generate some O but are markedly compromised in the generation of DEPMPO/HO•.
Reduction of DEPMPO/HOO• and DEPMPO/HO• Adducts
The formate and PBN data (above) imply that at least a portion of the DEPMPO/HO• signal represents trapping of freely formed HO• by DEPMPO. However, the results with catalase and H2O2 (Fig. 4A) indicate that H2O2 is not required to generate DEPMPO/HO•. The lack of inhibition by DFX (Fig. 4B) and the dependence of the DEPMPO/HO• signal on O (Fig. 2) further suggest that Fenton-like reactions do not generate the bulk of DEPMPO/HO• adducts. Although DEPMPO/HOO• does not spontaneously decay to DEPMPO/HO• in vitro (37, 44), the possibility that TrxR, in addition to generating some free HO•, might reduce DEPMPO/HOO• to DEPMPO/HO• was considered. To test this, DEPMPO was incubated with potassium superoxide (KO2) to generate DEPMPO/HOO•. A large excess of SOD was then added to degrade any remaining O, and then NADPH and/or TrxR were added, and the spin adduct signals were followed over time (30 consecutive scans, 2.8 min each). The resulting time course data are shown in Fig. 8, and examples of the spectra on which they are based are shown in supplemental Fig. 3. The spontaneous decay of DEPMPO/HOO• is shown as none in Fig. 8A, with DEPMPO/HOO• accounting for >94% of the total signal and DEPMPO/HO• only a very minor component (Fig. 8B). The results for NADPH alone and wtTrxR alone were essentially identical to spontaneous decay (Fig. 8A). The half-lives remained constant over the 30 scans, with values of 29.1 ± 1.1, 29.6 ± 2.1, and 29.3 ± 3.3 min for none, NADPH alone, and wtTrxR alone, respectively. These values are consistent with a previous report (59). Over time, these samples showed no increase in DEPMPO/HO• (Fig. 8B and supplemental Fig. 3), indicating that DEPMPO/HOO• was not reduced to DEPMPO/HO• by either NADPH alone or TrxR alone. However, wtTrxR plus NADPH was markedly different, showing a mix of both DEPMPO/HOO• and DEPMPO/HO• throughout the time course (Fig. 8, A and B, and supplemental Fig. 3); DEPMPO/HOO• decay was markedly accelerated (initial half-life of 14.4 min), and DEPMPO/HO• became the predominant component of the spectrum at later times (Fig. 8, A and B). The DEPMPO/HO• signal for wtTrxR plus NADPH declined in intensity over time with an initial half-life of 36.1 min, which progressively lengthened over time (Fig. 8B). Together, these data indicate that wtTrxR can reduce DEPMPO/HOO• to DEPMPO/HO• in an NADPH-dependent manner. Because SOD prevents de novo generation of both DEPMPO/HOO• and DEPMPO/HO• by TrxR (Fig. 2) and because SOD was added before the TrxR in these experiments, the DEPMPO/HO• generated from DEPMPO/HOO• by TrxR plus NADPH (Fig. 8B) must have been generated by reduction of the preformed DEPMPO/HOO•.
FIGURE 8.

Representative data showing the relative amounts of DEPMPO/HOO• (A, C, and E), and DEPMPO/HO• (B, D, and F) for experiments in which preformed DEPMPO/HOO• was the substrate. DEPMPO/HOO• was preformed by incubating KO2 with DEPMPO as described under “Experimental Procedures.” After 1 min at room temperature, SOD (1000 units/ml) was added. One minute later, 0.4 mm NADPH and/or wtTrxR or one of the TrxR variants (1.07 μm) were added. The sample was loaded into the flat cell, and 30 consecutive ESR spectra were collected, with an acquisition time of 2.8 min per spectrum (84 min total). Controls lacking both NADPH and TrxR (none) were also analyzed. The time indicates the start time for the acquisition of each spectrum, with 0 min indicating the start of acquisition of the first spectrum. It takes ∼2 min to load the flat cell and tune the instrument, so 0 min is actually 2 min after adding the NADPH and/or TrxR. ESR instrument settings were as follows: modulation amplitude, 1 G; microwave power, 20.02 milliwatts; receiver gain, 6.32 × 104; time constant, 81.92 ms; microwave frequency, 9.77 GHz; sweep width, 200 G; field set, 3480 G; modulation frequency, 100 kHz; resolution, 2048 points/scan; scan time, 167.8 s; 30 consecutive scans (n = 1 each).
Examples of the complete time course data for DEPMPO/HOO• for the other variants are shown in Fig. 8. Four other variants (U498C, truncated mutant, C59S, and C64S) gave results essentially identical to those for NADPH alone (Fig. 8, C and D), indicating that they cannot detectably reduce DEPMPO/HOO• to DEPMPO/HO•. The truncated mutant and U498C results indicate that the Sec (Sec498) is critical for reduction of DEPMPO/HOO•. Because both U498C and the truncated variation have a functional flavin and N-terminal Cys59/Cys64 dithiol, these N-terminal redox centers cannot substitute for Sec in the reduction of DEPMPO/HOO•. However, because the Cys59/Cys64 dithiol provides electrons to the Cys-Sec active site, the inability of the C59S and C64S variants to reduce DEPMPO/HOO• probably results from their inability to reduce the Cys-Sec. The C59S and C64S variants were also unable to reduce the disulfide substrate DTNB (kcat at least 1000-fold below wtTrxR; Table 1).
Both of the here analyzed Tyr116 variants were similar to wtTrxR in their reduction of DEPMPO/HOO• (Fig. 8E). Because both wtTrxR and the Tyr116 variants contain a fraction of truncated protein that also lacks Sec498/Gly499, the differences between these two can be directly attributed to the Tyr116 substitutions. Both Tyr116 variants also generated DEPMPO/HO• quickly, although the initial levels of DEPMPO/HO• were less than wtTrxR (Fig. 8F). This is consistent with the observation of somewhat slower DEPMPO/HO• generation by the Tyr116 variants after only 10 min, but they were similar to wild type after 30 min (see above). Thus, Tyr116 is not essential for this reaction but may possibly facilitate it, similar to the effects of this residue on overall catalytic capacity, as noted earlier (8). The crystal structure of oxidized TrxR shows that the Sec is packed near the aromatic ring of Tyr116, so it is possible that Tyr116 facilitates the interaction of Cys59 with the Sec (8). In this way, the loss of Tyr116 might impede reduction of the Sec.
Overall, the forms of TrxR that accelerated DEPMPO/HOO• decay in an NADPH-dependent manner (wtTrxR and Tyr116 variants) also resulted in pronounced DEPMPO/HO• generation. In no case did we see accelerated DEPMPO/HOO• decay without DEPMPO/HO• generation (Fig. 8). Therefore, we do not predict that any of the variants directly accelerated conversion of DEPMPO/HOO• to ESR silent species.
In the experiments with preformed DEPMPO/HOO•, the SOD would have rapidly converted any remaining O to H2O2. It should be noted that H2O2 is not an inhibitor of the DTNB reductase activity of TrxR (above), and H2O2 does not significantly accelerate the de novo generation of HO• by TrxR (Fig. 4A). However, because H2O2 is a substrate for wtTrxR, albeit weakly so (9), it is possible that the excess H2O2 could have altered DEPMPO/HO• generation in these particular experiments. In the presence of catalase, the decay of DEPMPO/HOO• by wtTrxR plus NADPH was actually faster, and even more DEPMPO/HO• was generated (Fig. 9A). If a significant amount of the DEPMPO/HO• signal was dependent on SOD-generated H2O2, then catalase should have actually decreased the DEPMPO/HO• signal. Thus, the generation of DEPMPO/HO• in these experiments does not require H2O2. The results in Fig. 9A are not due to reduction of DEPMPO/HOO• by catalase because the decay of DEPMPO/HOO• with NADPH alone was actually slower in the presence of catalase (Fig. 9B). The data therefore suggest that the SOD-generated H2O2 may actually slow the rate of DEPMPO/HOO• reduction by wtTrxR and that catalase alleviates this by degrading H2O2. The catalase data suggest that H2O2 may compete with DEPMPO/HOO• as a substrate of TrxR, although its Km for H2O2 is rather high (∼2.5 mm) (4, 9).
FIGURE 9.

The effects of catalase (1000 units/ml) on the decay of preformed DEPMPO/HOO•. The experiments used NADPH plus wtTrxR (A) or NADPH alone (B). Other experimental details were the same as those described in the legend to Fig. 8.
The reduction of H2O2 by the TrxR variants was tested as described by Zhong and Holmgren (9) by examining the ability of 5 mm H2O2 to stimulate NADPH oxidation. The Tyr116 variants were 15–23% slower than wtTrxR but still showed prominent activity (Fig. 10). In contrast, the U498C and truncated mutants had <10% of wtTrxR activity, and the C59S and C64S mutants were ≤6.5% of wild-type (Fig. 10). This pattern is consistent with the reduction of preformed DEPMPO/HOO• (Fig. 8) (i.e. those that do not reduce DEPMPO/HOO• to DEPMPO/HO• also do not have significant activity with H2O2). Because similar TrxR redox centers are required to reduce both H2O2 and DEPMPO/HOO•, it is possible that the latter may also be handled as a peroxide substrate by TrxR.
FIGURE 10.

The relative rates of NADPH oxidation for the TrxR variants using H2O2 as the substrate. Both cuvettes contained Tris-EDTA buffer (pH 7.4), 0.4 mm NADPH, and 5 mm H2O2. The sample cuvette also contained TrxR (0.1 μm). The rates at 37 °C (determined by the change in absorbance at 340 nm, mean ± S.D. (error bars), n = 3) were taken from the linear portion during the first 50 s of the assay.
It should be noted that KO2 might contain peroxide contaminants (60). However, the H2O2 generated by the SOD was a significant source of peroxide, and it caused only a limited decrease in the rate of DEPMPO/HOO• reduction (Fig. 9). Any peroxide contaminants in KO2 may have therefore, if anything, caused underestimation of the rate of DEPMPO/HOO• reduction by wtTrxR.
Given the ability of wtTrxR to reduce DEPMPO/HOO• to DEPMPO/HO•, we determined if DEPMPO/HO• was also a substrate for wtTrxR or its variants. To test this, DEPMPO was incubated with ferrous iron plus H2O2 plus DTPA to generate DEPMPO/HO•. A large excess of catalase and SOD was then added to degrade any remaining H2O2 and to prevent TrxR from generating new adducts, respectively. NADPH and/or TrxR were then added, and the spin adduct signals were followed over time (30 consecutive scans, 2.8 min each). We noted significant variation in the relative decay rates in the replicate experiments with NADPH and the TrxR variants (not shown). This is consistent with the highly variable decay rates reported in other studies (43, 61). We also noted considerable variability in the initial DEPMPO/HO• signal intensities that was irrespective of the reductant (NADPH alone or with different TrxR variants) (not shown). The results also uncovered a significant relationship between the initial DEPMPO/HO• signal intensity and the initial decay rate (i.e. larger signals showed faster decay (shorter initial half-lives) than smaller signals (supplemental Fig. 4)). Within each time course, the half-lives also got progressively longer as the signal intensity declined. Together, these data imply that the DEPMPO/HO• decay rate is significantly influenced by its concentration. Even when accounting for the influence of the initial signal intensity, there was no convincing evidence that any of the TrxR enzymes accelerated the decay of DEPMPO/HO•. The data therefore suggest that DEPMPO/HO• is a poor substrate (or not a substrate) for TrxR.
Mechanism of Free HO• Generation
Although the bulk of the DEPMPO/HO• signal generated by wtTrxR seemed to result from the two-electron reduction of DEPMPO/HOO•, the PBN/DMSO data (Fig. 3) indicate that free HO• is also generated by TrxR. To further explore this process, additional experiments with PBN were conducted. In the presence of NADPH, PBN, and DMSO, a mix of •CH3 and •OCH3 adducts of PBN was similarly generated by wtTrxR, truncated mutant, and the U498C variant (Fig. 11). The Sec residue of TrxR is therefore not needed to generate free HO•, in contrast to its role in DEPMPO/HOO• reduction. This generation of free HO• was also H2O2-dependent because catalase largely eliminated the PBN adduct signals for all three variants (Fig. 11), which contrasts with the reduction of DEPMPO/HOO• to DEPMPO/HO• that was H2O2-independent (see above).
FIGURE 11.

The generation of free HO• by wtTrxR and the truncated and U498C variants in the presence (right) and absence (left) of catalase (833 units/ml). The samples were incubated aerobically for 30 min at 37 °C and included NADPH (0.4 mm), PBN (50 mm), DMSO (5%, v/v), and the indicated forms of TrxR (1.07 μm). The spectra spanned 3380–3580 G, but only the portion in which signals were observed is shown here. Computer simulations of the spectra at the left are consistent with a mix of PBN/•OCH3 (hyperfine splitting constants aN = 15.05 G, aH = 3.32 G) and PBN/•CH3 (hyperfine splitting constants aN = 16.51 G, aH = 3.68 G) adducts (see Fig. 3). The instrument settings were the same as described in the legend to Fig. 1, except that the microwave frequency was 9.806 GHz.
DISCUSSION
Previous reports have shown that mammalian TrxR can generate large amounts of O as a result of redox cycling with compounds such as 2,4-DNCB, curcumin, and the quinone juglone (11, 20–22). Given the robust generation of O that was stimulated by these agents, O was readily detectable by the adrenochrome or cytochrome c methods. With some of these agents, however, SOD only inhibited a minority of cytochrome c reduction (21). TrxR has some direct cytochrome c reductase activity that must be accounted for when using this method to assess superoxide generation (21). ESR with DEPMPO is ∼20-fold more sensitive than the cytochrome c method for detecting O (38). This additional sensitivity allowed us here to examine the significant inherent NADPH oxidase activity of mammalian TrxR in the absence of redox cycling agents and to determine the role of its redox centers in this activity.
TrxR clearly exhibited enhanced NADPH oxidase activity when it was prevented from donating electrons to substrates, such as oxidized Trx, selenite, or DTNB. The most likely explanation is that electron flow to these substrates decreased electron availability for reaction with O2. In cells, oxidized Trx should in most cases be a major substrate for TrxR, striving to maintain Trx in the reduced state. In untreated human lung epithelial cells (BEAS-2B), >95 and 100% of Trx1 and Trx2 are found in their reduced states, respectively (62). Similarly, in untreated human endothelial cells (HMEC-1), 83 and 100% of Trx1 and Trx2 are in the reduced state (63). Although there is expected to be a continuous flux of electrons through the TrxR/Trx system to maintain intracellular thiol redox balance, there is probably an excess capacity of TrxR under normal conditions. In support of this, Trx1 was maintained in the normal reduced state in HeLa cells in which TrxR activity was inhibited ∼90% by either the TrxR inhibitor aurothioglucose or by small interfering RNA knockdown (64). In A549 cells, TrxR activity was found to be in great excess relative to Trx activity, and small interfering RNA knockdown of TrxR1 by ∼90% did not impede cell growth or promote cell death (18). An excess capacity of TrxR in cells therefore implies the potential for TrxR to divert some electrons to O2 even under normal growth conditions.
Inhibitors that bind the Sec of TrxR1 block its ability to reduce Trx and selenite (9). Such inhibition could promote NADPH oxidase, provided that this particular activity would not be blocked by the inhibitor. Indeed, we noted here that Sec-interacting inhibitors, such as cisplatin and ANF, did not block the NADPH oxidase activity, which implies that an intact Sec residue is not required for the O generation by TrxR. Consistent with this, the Sec-minus mutants still generated significant O. In cells, therefore, TrxR inhibitors, such as cisplatin and ANF, would be predicted to enhance the inherent NADPH oxidase activity of TrxR by preventing electron transfer to Trx. It should be noted that these inhibitors are not redox cycling themselves, so they do not promote O generation by a direct mechanism.
The delivery of Sec-minus mutants or of wtTrxR pretreated with Sec inhibitors, such as cisplatin, into cancer cells was found to promote pro-oxidant effects and rapid cell death (1, 19). The term SecTRAPs describes these gains of function by the otherwise inhibited forms of the enzyme (1, 19). The cells in which SecTRAPs have been introduced still have their normal content of active full-length TrxR, and the SecTRAPs do not block Trx reduction by full-length TrxR (19). However, SecTRAPs induced a strong DCF fluorescence and cell death in these cells (19). The Cys59/Cys64 dithiol is required for SecTRAP activity (19), consistent with our observations that this dithiol is required for inherent NADPH oxidase activity. Overall, conditions that result in SecTRAP activity match those for which we observed NADPH oxidase activity. The inherent NADPH oxidase activity of TrxR could therefore be an important contributor to SecTRAP activity and potentially the cytotoxicity of SecTRAP-forming compounds, such as cisplatin.
Fig. 12 shows a schematic overview of the proposed mechanism for the inherent NADPH oxidase activity of wtTrxR, including the role(s) of its redox centers. Electrons diverted to O2 result in the generation of O by the enzyme. The Cys59/Cys64 dithiol probably represents a major site of O generation. Of these two cysteines, Cys59 proved more critical. Unlike some other flavoproteins, the FAD, which precedes Cys59/Cys64 in the flow of electrons within TrxR, does not directly generate O at any appreciable rate. Although the Sec may contribute to O generation either directly or indirectly by influencing the activity of the Cys59/Cys64 dithiol, the Sec was not required for this O-producing activity. Because the truncated and U498C variants behaved similarly, their results reflect the properties of TrxR upon loss of a normally functional Sec residue. Although there is some truncated enzyme species present in the preparations of wtTrxR and other variants made to contain Sec (as discussed above) (32), the U498C is full-length and homogeneous, so its results cannot be attributed to a truncated form of the enzyme.
FIGURE 12.

Scheme of the inherent NADPH oxidase activity of TrxR and source of the resulting ESR findings. Electrons from NADPH are transferred to the FAD, which reduces the N-terminal dithiol (Cys59/Cys64). This dithiol reduces the C-terminal Cys497/Sec498 active site. During normal catalysis, TrxR cycles between the 2- and 4-electron reduced states shared mainly between Cys59/Cys64 and Cys497/Sec498, with the FAD in the oxidized state or forming a charge-transfer complex with Cys64 (1, 7). In the absence of an electron-accepting substrate (e.g. oxidized thioredoxin), TrxR can reduce O2 to O. Mutants lacking Sec498 (Sec) generate less DEPMPO/HO• but are still capable of generating significant O. However, mutants lacking Cys59 and/or Cys64 generate almost no O or HO• signals, suggesting that this dithiol is a major site of O generation and that the FAD does not directly generate O. O is required for the majority of the DEPMPO/HO• signal because SOD eliminates nearly all of this signal. With wtTrxR, the bulk of the signal is not the O adduct but rather the HO• adduct, which is mostly formed by the 2-electron reduction of DEPMPO/HOO• by TrxR; this process does not require H2O2 but does require the Sec (U498) in the reduced state (*) because the Cys59/Cys64 dithiol is also required to reduce the DEPMPO/HOO• adduct. The 2-electron reduction of DEPMPO/HOO• to DEPMPO/HO• probably reflects its peroxidase activity because the 2-electron reduction of H2O2 similarly requires Sec in the reduced state. H2O2 can slow the rate of reduction of DEPMPO/HOO• to DEPMPO/HO• by acting as a competing substrate for the Sec-dependent peroxidase. A lesser amount of the DEPMPO/HO• adduct can be attributed to the generation of HO• in an H2O2-dependent but Sec-independent mechanism (Minor Pathway), as supported by the PBN/DMSO data (Figs. 3 and 11). The peroxidase activity of wtTrxR would be predicted to diminish the generation of free HO•.
Although O is easily generated by wtTrxR, the HO• adduct of DEPMPO was the predominant species in the resulting ESR spectra. Because this was the result of the 2-electron reduction of DEPMPO/HOO• to DEPMPO/HO•, this means that the sum of the HOO• and HO• adducts largely reflects the initial generation of O. We suggest that the reduction of DEPMPO/HOO• represents the peroxidase activity of wtTrxR, and the ESR studies performed here helped us to further define this activity. The inability of the U498C and truncated variants to catalyze either DEPMPO/HOO• or H2O2 reduction shows that this activity is Sec-dependent. The results with the U498C variant showed that a Cys at position 498 cannot substitute for the Sec, in agreement with earlier studies of the H2O2 reduction by TrxR (9). The mere presence of Sec is not sufficient, however, because the C59S and C64S variants constructed here maintain their Sec residue but could still not reduce DEPMPO/HOO• or H2O2. Because Cys59/Cys64 is believed to donate electrons to the Cys-Sec active site, the Sec in C59S and C64S variants should be oxidized in the form of an essentially inert selenenylsulfide motif (8). This is consistent with a notion that the peroxidase activity requires electron flow through the Sec, with Cys59/Cys64 serving as the immediate Sec-reducing entity of the enzyme. The Cys59/Cys64 dithiol in itself clearly does not have peroxidase activity because the U498C and truncated variants have functional Cys59/Cys64 yet cannot reduce DEPMPO/HOO• or H2O2. The stepwise mechanism by which the reduced Sec of wtTrxR may reduce DEPMPO/HOO• is shown in supplemental Fig. 5. In this mechanism, the Sec directly reduces the hydroperoxyl moiety of the spin adduct, and Cys497 and Cys59/Cys64 resolve the oxidized Sec and return the Cys-Sec active site to the reduced state, respectively.
The complexity of the effects of inhibitors on TrxR function was further revealed herein. Both cisplatin and ANF presumably bind to the Sec residue of the enzyme and markedly inhibit its disulfide reductase activity (10, 12, 17, 35, 53, 58). However, these inhibitors had little to no effect on the DEPMPO/HO• signal intensity (i.e. little effect on the NADPH oxidase and peroxidase activities that underlie these signals). As noted above, there is some (∼35%) truncated enzyme present in recombinant wtTrxR. One might thus have speculated that the ANF and cisplatin blocked all Sec-dependent activity of wtTrxR and that the remaining ESR signals were due to the truncated contaminant that is not inhibited by ANF or cisplatin. However, this cannot account for the results. Although the truncated enzyme indeed has NADPH oxidase activity, it lacks the peroxidase activity necessary to generate the vast majority of the DEPMPO/HO• signal that was still predominant in the experiments with ANF and cisplatin. The results do suggest, however, that DEPMPO/HOO• is not handled identically to H2O2. Although ANF only caused a 28% decrease in the DEPMPO/HO• signal generated by wtTrxR, it inhibited the reduction of 5 mm H2O2 by 94% (not shown). If both substrates were reduced as peroxides, it is clear that ANF better inhibits the reduction of some peroxides, such as H2O2, although the reasons for these differences are not yet clear. Because TrxR reduces peroxynitrite at relatively slow rates (65), an alternative possibility is that DEPMPO/HOO• is recognized as a peroxynitrite-like substrate. This seems less likely, however, because the conversion of the HOO• adduct to the HO• adduct is consistent with a peroxidase activity. In addition, the structure of DEPMPO/HOO• (Fig. 1C) is different from peroxynitrite in that the former has a carbon atom between the nitroxyl and peroxyl groups.
Glutathione peroxidases are GSH-dependent enzymes that reduce H2O2 and various organic peroxides to their corresponding alcohols (66). Most forms of glutathione peroxidase contain a catalytic Sec residue. Glutathione peroxidase can reduce the O adducts of the spin traps DMPO, DEPMPO, mito-DEPMPO, and 5-tert-butoxycarbonyl 5-methyl-1-pyrroline N-oxide to their corresponding HO• adducts (37, 67–69). We are not aware of studies that have explored the requirement for the Sec residue in these glutathione peroxidase-catalyzed reactions, but the results with TrxR suggest that the glutathione peroxidase Sec may be involved in reducing the hydroperoxyl group of these O adducts. The Sec is also required for H2O2 reduction by wtTrxR (9) (Fig. 10) and may also be necessary for the reduction of other peroxides, such as hydroperoxyeicosatetraenoic acid (6). Hydroperoxyeicosatetraenoic acid reduction is stimulated severalfold by the addition of selenocystine (6), and the selenium-containing low molecular weight compound ebselen strongly stimulates H2O2 reduction by the thioredoxin system (70). The physiological advantage or role of a peroxidase activity as an additional function of TrxR is not known, but it has been suggested that it may serve as a means to decrease the proximal generation of HO•, which could theoretically damage the enzyme (9). Our findings of a direct production of a small amount of free HO• by the enzyme are notable in this context.
The formate and PBN/DMSO data demonstrated the potential for TrxR to generate some free HO•. This mechanism is H2O2-dependent but Sec-independent (Fig. 12, Minor Pathway) and is therefore distinct from that of its Sec-dependent peroxidase activity. Furthermore, its peroxidase activity would be expected to limit the generation of free HO•, as depicted in Fig. 12 (Minor Pathway). This free HO• probably does not result from a one-step 2-electron reduction of O because such a mechanism should be H2O2-independent.
There has been considerable interest in the conversion of spin-trapped adducts of O to their HO• counterparts (60). With DMPO adducts, this conversion can be spontaneous with an expected half-life of <1 min (37). However, DEPMPO/HOO• does not spontaneously decay to DEPMPO/HO• (37, 44). The present studies are the first to show that the Sec-dependent peroxidase activity of TrxR can catalyze this 2-electron conversion. It is possible that TrxR may similarly catalyze reduction of other nitroxide spin adducts of O. It is known that the half-life of spin adducts is significantly shorter in cells than in aqueous solution (43) and that cells can reduce the HOO• adducts of 5-tert-butoxycarbonyl 5-methyl-1-pyrroline N-oxide, DMPO, and DEPMPO to the corresponding HO• adducts (59). Cellular TrxR may thus contribute to this reduction, at least with DEPMPO. However, it would be difficult in cells to determine the portion of this reduction that is due to TrxR without using knock-out or knockdown approaches because DEPMPO/HO• generation is clearly not blocked by the TrxR inhibitors, such as cisplatin and ANF. The potent Sec-dependent conversion of DEPMPO/HOO• to DEPMPO/HO• by TrxR1 that was found here probably reflects the significant capacity of this enzyme to also act as a direct peroxidase. Future studies are warranted to better understand the cellular conditions or tissue types in which this peroxidase activity of TrxR is of biological importance in addition to its roles in maintaining cellular Trx in a reduced state.
Although our studies were conducted with TrxR1 (cytosolic), mitochondrial TrxR2 has analogous redox centers. TrxR2 may therefore also have inherent NADPH oxidase activity when the reduction of its normal substrates is impeded. However, the N-terminal dithiol of TrxR2 has additional measurable activities in the reduction of selenite or some other substrates (10), so it may not behave identically to TrxR1 in regard to its peroxidase activities.
To conclude, we have here presented an analysis of the inherent NADPH oxidase activity of TrxR1, finding that the enzyme can catalyze a significant production of O, some of which may subsequently be converted to free HO•. This apparently contradictory property of TrxR, considered to be one of the more important antioxidant enzymes in cells, is a vivid reflection of the “yin and yang” character of many redox active proteins in general and perhaps especially so for TrxR (1). This type of inherent prooxidant capacity becomes pronounced in the absence of reduction of its natural substrates. It is probably of less importance under non-stressed and non-perturbed cellular growth conditions. However, when TrxR becomes inhibited by certain compounds, including cisplatin or auranofin studied here, or when the cellular need for reduction of Trx is significantly less than the overall capacity of cellular TrxR1, the pro-oxidant properties of TrxR1 may become of larger importance. This NADPH oxidase capacity may then contribute to a promotion of cytotoxicity related to an increased oxidative stress and to the therapeutic potential of TrxR-targeting inhibitors.
Supplementary Material
Acknowledgment
We are grateful to Dr. Jeannette Vásquez-Vivar (Medical College of Wisconsin) for discussions and insights on the spin trapping experiments.
This work was supported, in whole or in part, by National Institutes of Health Grant R01ES012707 (to C. R. M.). This work was also supported by grants from the Swedish Research Council (Medicine), the Swedish Cancer Society, the Knut and Alice Wallenberg Foundation, and Karolinska Institutet (to E. S. J. A.). The EPR facilities of the Department of Biophysics of the Medical College of Wisconsin are supported by National Institutes of Health National Biomedical ESR Center Grant EB001980.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–5.
- TrxR
- thioredoxin reductase
- Trx
- thioredoxin
- wtTrxR
- wild type TrxR
- 2,4-DNCB
- 2,4-dinitrochlorobenzene
- ANF
- auranofin
- DEPMPO
- 5-diethoxyphosphoryl-5-methyl-1-pyrroline-N-oxide
- DFX
- deferoxamine
- DMPO
- 5′,5′-dimethylpyrroline N-oxide
- DTNB
- 5,5′-dithiobis(2-nitrobenzoic) acid
- DTPA
- diethylenetriaminepentaacetic acid
- ESR
- electron spin resonance
- HO•
- hydroxyl radical
- MnTBAP
- manganese (III) tetrakis(4-benzoic acid)porphyrin chloride
- •CH3
- methyl radical
- •OCH3
- methoxy radical
- O
- superoxide
- PBN
- α-phenyl-N-tert-butylnitrone
- Sec or U
- selenocysteine
- SecTRAP
- selenium-compromised thioredoxin reductase-derived apoptotic protein
- SOD
- superoxide dismutase
- ROS
- reactive oxygen species.
REFERENCES
- 1.Arnér E. S. (2009) Biochim. Biophys. Acta 1790, 495–526 [DOI] [PubMed] [Google Scholar]
- 2.Arnér E. S., Holmgren A. (2000) Eur. J. Biochem. 267, 6102–6109 [DOI] [PubMed] [Google Scholar]
- 3.Powis G., Montfort W. R. (2001) Annu. Rev. Biophys. Biomol. Struct. 30, 421–455 [DOI] [PubMed] [Google Scholar]
- 4.Nordberg J., Arnér E. S. (2001) Free Radic. Biol. Med. 31, 1287–1312 [DOI] [PubMed] [Google Scholar]
- 5.Kumar S., Björnstedt M., Holmgren A. (1992) Eur. J. Biochem. 207, 435–439 [DOI] [PubMed] [Google Scholar]
- 6.Björnstedt M., Hamberg M., Kumar S., Xue J., Holmgren A. (1995) J. Biol. Chem. 270, 11761–11764 [DOI] [PubMed] [Google Scholar]
- 7.Zhong L., Arnér E. S., Holmgren A. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 5854–5859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng Q., Sandalova T., Lindqvist Y., Arnér E. S. (2009) J. Biol. Chem. 284, 3998–4008 [DOI] [PubMed] [Google Scholar]
- 9.Zhong L., Holmgren A. (2000) J. Biol. Chem. 275, 18121–18128 [DOI] [PubMed] [Google Scholar]
- 10.Lothrop A. P., Ruggles E. L., Hondal R. J. (2009) Biochemistry 48, 6213–6223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nordberg J., Zhong L., Holmgren A., Arnér E. S. (1998) J. Biol. Chem. 273, 10835–10842 [DOI] [PubMed] [Google Scholar]
- 12.Witte A. B., Anestål K., Jerremalm E., Ehrsson H., Arnér E. S. (2005) Free Radic. Biol. Med. 39, 696–703 [DOI] [PubMed] [Google Scholar]
- 13.Gromer S., Urig S., Becker K. (2004) Med. Res. Rev. 24, 40–89 [DOI] [PubMed] [Google Scholar]
- 14.Urig S., Becker K. (2006) Semin. Cancer Biol. 16, 452–465 [DOI] [PubMed] [Google Scholar]
- 15.Myers C. R., Myers J. M. (2009) Toxicology 257, 95–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Myers J. M., Myers C. R. (2009) Free Radic. Biol. Med. 47, 1477–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arnér E. S., Nakamura H., Sasada T., Yodoi J., Holmgren A., Spyrou G. (2001) Free Radic. Biol. Med. 31, 1170–1178 [DOI] [PubMed] [Google Scholar]
- 18.Eriksson S. E., Prast-Nielsen S., Flaberg E., Szekely L., Arnér E. S. (2009) Free Radic. Biol. Med. 47, 1661–1671 [DOI] [PubMed] [Google Scholar]
- 19.Anestål K., Prast-Nielsen S., Cenas N., Arnér E. S. (2008) PLoS ONE 3, e1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arnér E. S., Björnstedt M., Holmgren A. (1995) J. Biol. Chem. 270, 3479–3482 [DOI] [PubMed] [Google Scholar]
- 21.Cenas N., Nivinskas H., Anusevicius Z., Sarlauskas J., Lederer F., Arnér E. S. (2004) J. Biol. Chem. 279, 2583–2592 [DOI] [PubMed] [Google Scholar]
- 22.Fang J., Lu J., Holmgren A. (2005) J. Biol. Chem. 280, 25284–25290 [DOI] [PubMed] [Google Scholar]
- 23.Hashemy S. I., Ungerstedt J. S., Zahedi Avval F., Holmgren A. (2006) J. Biol. Chem. 281, 10691–10697 [DOI] [PubMed] [Google Scholar]
- 24.Arnér E. S. (2002) Methods Enzymol. 347, 226–235 [DOI] [PubMed] [Google Scholar]
- 25.Cheng Q., Stone-Elander S., Arnér E. S. (2006) Nat. Protoc. 1, 604–613 [DOI] [PubMed] [Google Scholar]
- 26.Johansson L., Chen C., Thorell J. O., Fredriksson A., Stone-Elander S., Gafvelin G., Arnér E. S. (2004) Nat. Methods 1, 61–66 [DOI] [PubMed] [Google Scholar]
- 27.Rengby O., Johansson L., Carlson L. A., Serini E., Vlamis-Gardikas A., Kårsnäs P., Arnér E. S. (2004) Appl. Environ. Microbiol. 70, 5159–5167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anestål K., Arnér E. S. (2003) J. Biol. Chem. 278, 15966–15972 [DOI] [PubMed] [Google Scholar]
- 29.Arnér E. S., Sarioglu H., Lottspeich F., Holmgren A., Böck A. (1999) J. Mol. Biol. 292, 1003–1016 [DOI] [PubMed] [Google Scholar]
- 30.Rengby O., Arnér E. S. (2007) Appl. Environ. Microbiol. 73, 432–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sambrook J., Fritsch E. F., Maniatis T. (eds) (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., Vol. 3, pp. 17.1–17.44, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 32.Rengby O., Cheng Q., Vahter M., Jörnvall H., Arnér E. S. (2009) Free Radic. Biol. Med. 46, 893–904 [DOI] [PubMed] [Google Scholar]
- 33.Myers C. R., Myers J. M. (1998) Carcinogenesis 19, 1029–1038 [DOI] [PubMed] [Google Scholar]
- 34.Holmgren A. (1977) J. Biol. Chem. 252, 4600–4606 [PubMed] [Google Scholar]
- 35.Gromer S., Arscott L. D., Williams C. H., Jr., Schirmer R. H., Becker K. (1998) J. Biol. Chem. 273, 20096–20101 [DOI] [PubMed] [Google Scholar]
- 36.Borthiry G. R., Antholine W. E., Kalyanaraman B., Myers J. M., Myers C. R. (2007) Free Radic. Biol. Med. 42, 738–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frejaville C., Karoui H., Tuccio B., Le Moigne F., Culcasi M., Pietri S., Lauricella R., Tordo P. (1995) J. Med. Chem. 38, 258–265 [DOI] [PubMed] [Google Scholar]
- 38.Tordo P. (1998) in Electron Paramagnetic Resonance: Specialist Periodical Reports (Gilbert B. C., Atherton N. M., Davies M. J. eds) Vol. 16, pp. 116–144, The Royal Society of Chemistry, Cambridge [Google Scholar]
- 39.Duling D. R. (1994) J. Magn. Reson. B 104, 105–110 [DOI] [PubMed] [Google Scholar]
- 40.Hardy M., Chalier F., Finet J. P., Rockenbauer A., Tordo P. (2005) J. Org. Chem. 70, 2135–2142 [DOI] [PubMed] [Google Scholar]
- 41.Chalier F., Hardy M., Ouari O., Rockenbauer A., Tordo P. (2007) J. Org. Chem. 72, 7886–7892 [DOI] [PubMed] [Google Scholar]
- 42.Karoui H., Hogg N., Fréjaville C., Tordo P., Kalyanaraman B. (1996) J. Biol. Chem. 271, 6000–6009 [DOI] [PubMed] [Google Scholar]
- 43.Khan N., Wilmot C. M., Rosen G. M., Demidenko E., Sun J., Joseph J., O'Hara J., Kalyanaraman B., Swartz H. M. (2003) Free Radic. Biol. Med. 34, 1473–1481 [DOI] [PubMed] [Google Scholar]
- 44.Vásquez-Vivar J., Hogg N., Martásek P., Karoui H., Tordo P., Pritchard K. A., Jr., Kalyanaraman B. (1999) Free Radic. Res. 31, 607–617 [DOI] [PubMed] [Google Scholar]
- 45.Perkins M. J. (1980) in Advances in Physical Organic Chemistry (Gold V., Bethel D. eds) Vol. 17, pp. 1–64, Academic Press, Inc., New York [Google Scholar]
- 46.Britigan B. E., Coffman T. J., Buettner G. R. (1990) J. Biol. Chem. 265, 2650–2656 [PubMed] [Google Scholar]
- 47.Halliwell B., Gutteridge J. M. (1984) Biochem. J. 219, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liochev S. I., Fridovich I. (1994) Free Radic. Biol. Med. 16, 29–33 [DOI] [PubMed] [Google Scholar]
- 49.Shi X. G., Dalal N. S. (1990) Arch. Biochem. Biophys. 277, 342–350 [DOI] [PubMed] [Google Scholar]
- 50.Shi X., Dong Z., Dalal N. S., Gannett P. M. (1994) Biochim. Biophys. Acta 1226, 65–72 [DOI] [PubMed] [Google Scholar]
- 51.Shi X., Mao Y., Knapton A. D., Ding M., Rojanasakul Y., Gannett P. M., Dalal N., Liu K. (1994) Carcinogenesis 15, 2475–2478 [DOI] [PubMed] [Google Scholar]
- 52.Shi X. L., Dalal N. S. (1990) Free Radic. Res. Commun. 10, 17–26 [DOI] [PubMed] [Google Scholar]
- 53.Pia Rigobello M., Messori L., Marcon G., Agostina Cinellu M., Bragadin M., Folda A., Scutari G., Bindoli A. (2004) J. Inorg. Biochem. 98, 1634–1641 [DOI] [PubMed] [Google Scholar]
- 54.Shi X. G., Sun X. L., Gannett P. M., Dalal N. S. (1992) Arch. Biochem. Biophys. 293, 281–286 [DOI] [PubMed] [Google Scholar]
- 55.Halliwell B. (1989) Free Radic. Biol. Med. 7, 645–651 [DOI] [PubMed] [Google Scholar]
- 56.Yamazaki I., Piette L. H. (1990) J. Biol. Chem. 265, 13589–13594 [PubMed] [Google Scholar]
- 57.Rigobello M. P., Callegaro M. T., Barzon E., Benetti M., Bindoli A. (1998) Free Radic. Biol. Med. 24, 370–376 [DOI] [PubMed] [Google Scholar]
- 58.Millet R., Urig S., Jacob J., Amtmann E., Moulinoux J. P., Gromer S., Becker K., Davioud-Charvet E. (2005) J. Med. Chem. 48, 7024–7039 [DOI] [PubMed] [Google Scholar]
- 59.Shi H., Timmins G., Monske M., Burdick A., Kalyanaraman B., Liu Y., Clément J. L., Burchiel S., Liu K. J. (2005) Arch. Biochem. Biophys. 437, 59–68 [DOI] [PubMed] [Google Scholar]
- 60.Finkelstein E., Rosen G. M., Rauckman E. J. (1982) Mol. Pharmacol. 21, 262–265 [PubMed] [Google Scholar]
- 61.Villamena F. A., Zweier J. L. (2004) Antioxid. Redox Signal. 6, 619–629 [DOI] [PubMed] [Google Scholar]
- 62.Myers J. M., Antholine W. E., Myers C. R. (2008) Toxicology 246, 222–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Szadkowski A., Myers C. R. (2008) Toxicology 243, 164–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Watson W. H., Heilman J. M., Hughes L. L., Spielberger J. C. (2008) Biochem. Biophys. Res. Commun. 368, 832–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Arteel G. E., Briviba K., Sies H. (1999) Chem. Res. Toxicol. 12, 264–269 [DOI] [PubMed] [Google Scholar]
- 66.Lu J., Holmgren A. (2009) J. Biol. Chem. 284, 723–727 [DOI] [PubMed] [Google Scholar]
- 67.Rosen G. M., Freeman B. A. (1984) Proc. Natl. Acad. Sci. U.S.A. 81, 7269–7273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hardy M., Rockenbauer A., Vásquez-Vivar J., Felix C., Lopez M., Srinivasan S., Avadhani N., Tordo P., Kalyanaraman B. (2007) Chem. Res. Toxicol. 20, 1053–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhao H., Joseph J., Zhang H., Karoui H., Kalyanaraman B. (2001) Free Radic. Biol. Med. 31, 599–606 [DOI] [PubMed] [Google Scholar]
- 70.Zhao R., Masayasu H., Holmgren A. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 8579–8584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kosaka H., Katsuki Y., Shiga T. (1992) Arch. Biochem. Biophys. 293, 401–408 [DOI] [PubMed] [Google Scholar]
- 72.Rhodes C. J., Tran T. T., Morris H. (2004) Spectrochim. Acta A Mol. Biomol. Spectrosc. 60, 1401–1410 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.