

Abstract
Numerous in vitro and in vivo studies have shown that isoflavones exhibit anti-proliferative activity against epidermal growth factor (EGF) receptor-positive malignancies of the breast, colon, skin, and prostate. 7,3′,4′-Trihydroxyisoflavone (7,3′,4′-THIF) is one of the metabolites of daidzein, a well known soy isoflavone, but its chemopreventive activity and the underlying molecular mechanisms are poorly understood. In this study, 7,3′,4′-THIF prevented EGF-induced neoplastic transformation and proliferation of JB6 P+ mouse epidermal cells. It significantly blocked cell cycle progression of EGF-stimulated cells at the G1 phase. As shown by Western blot, 7,3′,4′-THIF suppressed the phosphorylation of retinoblastoma protein at Ser-795 and Ser-807/Ser-811, which are the specific sites of phosphorylation by cyclin-dependent kinase (CDK) 4. It also inhibited the expression of G1 phase-regulatory proteins, including cyclin D1, CDK4, cyclin E, and CDK2. In addition to regulating the expression of cell cycle-regulatory proteins, 7,3′,4′-THIF bound to CDK4 and CDK2 and strongly inhibited their kinase activities. It also bound to phosphatidylinositol 3-kinase (PI3K), strongly inhibiting its kinase activity and thereby suppressing the Akt/GSK-3β/AP-1 pathway and subsequently attenuating the expression of cyclin D1. Collectively, these results suggest that CDKs and PI3K are the primary molecular targets of 7,3′,4′-THIF in the suppression of EGF-induced cell proliferation. These insights into the biological actions of 7,3′,4′-THIF provide a molecular basis for the possible development of new chemoprotective agents.
Keywords: CDK (Cyclin-dependent Kinase), Cell Cycle, Growth Factors, Signal Transduction, Transformation, Cot, MKK4, Skin Cancer, Trihydroxyisoflavone
Introduction
The complex process of carcinogenesis is believed to be comprised of three stages that include initiation, promotion, and progression. The reversible and lengthy stage of tumor promotion is a potential target for prevention strategies. Interventions at this stage appear more likely to be successful than interventions at the tumor initiation stage, which is irreversible and brief (1). Aberrant regulation of growth signaling is implicated in the abnormal biological process of tumorigenesis, and epidermal growth factor (EGF)4 might be one of the tumor promoters needed for the induction of aberrant cell growth (2). Indeed, growth factors, including EGF, are expressed at high levels in a variety of human cancer cells, including those derived from skin, breast, colon, lung, and prostate. These growth factors stimulate cell proliferation, invasiveness, and angiogenesis (3, 4).
The JB6 mouse epidermal cell system of clonal genetic variants, which are promotion-sensitive (P+) or promotion-resistant (P−), provides an ideal model for investigations of the molecular mechanisms involved in neoplastic transformation, promotion, and progression (1). In JB6 P+ cells, EGF induces cell cycle progression and the formation of large, tumorigenic, anchorage-independent colonies in soft agar.
Uncontrolled cell growth is considered a major event in carcinogenesis. Multiple lines of evidence suggest the existence of a strong link between cell cycle deregulation and carcinogenesis (5). In particular, dysregulation of the cyclin-dependent kinases (CDKs) causes increased cell proliferation. CDK activity requires binding of regulatory subunits known as cyclins. The D-type cyclins are initially activated in response to mitogenic signals and preferentially bind to and activate CDK4 during the early G1 stage of the cell cycle (6). Activation of these complexes leads to phosphorylation of retinoblastoma protein (Rb). The phosphorylated Rb (pRb) then binds to transcription factors, including E2Fs, and contributes to the expression of S-phase genes. The association of CDK2 with cyclin E and then with cyclin A is believed to coordinate events as cells progress from G1 through S phase (7). pRb is initially phosphorylated on multiple sites by CDK4 and then additionally phosphorylated by cyclin E-bound CDK2. Therefore, cell cycle progression from G1 to S phase is tightly controlled by interactions among pRb and the cyclin D1-CDK4 and cyclin E-CDK2 complexes.
The phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway is important for cell survival and growth, and PI3K plays a pivotal role in tumorigenesis (8). Elevated expression or excessive activation of PI3K has been observed in many tumor cells (9, 10). Glycogen synthase kinase (GSK)-3β, which acts downstream of the PI3K pathway, induces cyclin D1 degradation in response to mitogenic signals (11, 12). PI3K-dependent phosphorylation of GSK-3β at Ser-9 inhibits activation of GSK-3β and thus stabilizes cyclin D1 (11). Therefore, regulation of the PI3K/Akt/GSK-3β pathway is a promising strategy for cancer chemoprevention.
The isoflavones daidzein and genistein are present at high concentrations in soybean-based foods (13, 14). Numerous in vitro and in vivo studies have shown that isoflavones exhibit anti-proliferative activity against EGF receptor-positive malignancies of breast, colon, skin, and prostate (15–18). Genistein has been the subject of intensive study as a potential anti-carcinogenic compound, whereas daidzein and its metabolites have not received as much attention. Here, we investigated the chemopreventive effects of 7,3′,4′-trihydroxyisoflavone (7,3′,4′-THIF (see Fig. 1)), a daidzein metabolite, on EGF-stimulated cell proliferation and transformation. We report that 7,3′,4′-THIF is a potent inhibitor of CDKs and PI3K and induces cell cycle arrest at G1 phase and suppresses proliferation and transformation of JB6 P+ mouse epidermal cells. The molecular mechanism suggested by this investigation might underlie the chemopreventive activity of 7,3′,4′-THIF and thus could at least partially account for the chemopreventive effect of soy foods.
FIGURE 1.
The chemical structure of 7,3′,4′-trihydroxyisoflavone (7,3′,4′-THIF).
EXPERIMENTAL PROCEDURES
Chemicals
Eagle's minimum essential medium (MEM), basal medium Eagle, gentamicin, and l-glutamine were purchased from Invitrogen. Fetal bovine serum (FBS) was obtained from Gemini Bio-Products (Calabasas, CA), and 7,3′,4′-THIF was obtained from Indofine Chemical Co., Inc. (Hillsborough, NJ). EGF and the β-actin antibody were purchased from Sigma-Aldrich (St. Louis, MO). Antibodies against Ser-795- and Ser-807/Ser-811-phosphorylated Rb, Ser-473-phosphorylated Akt, Ser-9-phosphorylated GSK-3β, total Akt, and total GSK-3β were purchased from Cell Signaling Technology (Danvers, MA), as was the Rb-C fusion protein used as a CDK substrate. Antibodies against cyclin D1, cyclin E, CDK4, and CDK2 were from Santa Cruz Biotechnology (Santa Cruz, CA). Recombinant CDKs were obtained from Millipore (Billerica, MA). CNBr-Sepharose 4B, glutathione-Sepharose 4B, [γ-32P]ATP, and a chemiluminescence detection kit were purchased from Amersham Biosciences. A protein assay kit was obtained from Bio-Rad Laboratories.
Cell Culture
JB6 P+ mouse epidermal (JB6 P+) cells were cultured in monolayers in MEM containing 5% FBS, 2 mm l-glutamine, and 25 μg/ml gentamicin at 37 °C under a 5% CO2 atmosphere. The cells were stably transfected with an activator protein-1 (AP-1) luciferase reporter plasmid and maintained in MEM supplemented with 5% FBS and 200 μg/ml G418.
Cell Cycle Analysis
Cell cycle progression was analyzed by a published method (19) with slight modifications. JB6 P+ cells were seeded in 60-mm dishes (1.5 × 105 cells/dish), cultured for 24 h, and then serum-starved in 0.1% FBS-containing MEM (0.1% FBS-MEM) for 36 h to synchronize the cells at G1 phase (19). The cells were then treated with 7,3′,4′-THIF at various concentrations. After 1 h, EGF (final concentration, 10 ng/ml) or FBS (final concentration, 5%) was added to stimulate cell growth. The cells were trypsinized 24 h later, washed with ice-cold Dulbecco's phosphate-buffered saline (36 mg/liter sodium pyruvate, 50 mg/liter streptomycin sulfate, 100 mg/liter kanamycin monosulfate, 1000 mg/liter glucose, calcium chloride, and magnesium chloride), and fixed in ice-cold 70% ethanol at −20 °C overnight. The cells were then washed twice with Dulbecco's phosphate-buffered saline and incubated with 20 μg/ml RNase A and 200 μg/ml propidium iodide in Dulbecco's phosphate-buffered saline at room temperature for 30 min in the dark. The cell cycle phase was determined using a FACSCalibur flow cytometer (BD Biosciences). The data were gathered using ModFit LT software (Verity Software House, Inc., Topsham, ME).
Western Blot Analysis
Cells were cultured in 10-cm dishes (4 × 105 cells/dish) for 24 h and then starved in 0.1% FBS-MEM for an additional 36 h. After treatment with 7,3′,4′-THIF for 1 h, they were treated with EGF for 10 min or 24 h and harvested. The harvested cells were disrupted, and the supernatant fractions were boiled for 5 min. The protein concentration in the lysates was determined using a dye-binding protein assay kit according to the manufacturer's protocol. Lysate protein (30 μg) was subjected to 10% SDS-PAGE and electrophoretically transferred to a polyvinylidene difluoride membrane. After blotting, the membrane was incubated with a primary antibody against cyclin D1, cyclin E, CDK4, CDK2, phosphorylated Akt, GSK-3β, or Rb at 4 °C overnight. Protein bands were visualized using a chemiluminescence detection kit after hybridization with a horseradish peroxidase-conjugated secondary antibody.
Direct CDKs or PI3K Assays
Assays of cyclin D1-CDK4 and cyclin E-CDK2 kinase activity were performed in accordance with instructions provided by Millipore Corp. Briefly, 7,3′,4′-THIF (10, 20, or 40 μm) was incubated with active recombinant cyclin D1-CDK4 or cyclin E-CDK2 protein (80 ng/μl) at 30 °C for 10 min. Then, 10 μl of 1× kinase buffer (5 mm MOPS (pH 7.2), 2.5 mm β-glycerol phosphate, 1 mm EGTA, 0.4 mm EDTA, 5 mm MgCl2, and 0.05 mm dithiothreitol (dithiothreitol)), 2 μl of 200 mm ATP, and 1 μg/μl Rb-C fusion protein were added, and the reaction mixture was incubated at 30 °C for an additional 30 min. Phosphorylation of the Rb-C fusion protein was detected by Western blotting with a pRb (Ser-807/Ser-811) polyclonal antibody.
To determine PI3K activity, an active PI3K protein (100 ng/μl) was incubated with 7,3′,4′-THIF (0, 10, 20, or 40 μm) for 10 min at 30 °C. Then, 20 μl of 0.5 mg/ml phosphatidylinositol (Avanti Polar Lipids, Alabaster, AL) was added, and the mixture was incubated for 5 min at room temperature. Reaction buffer (100 mm HEPES, pH 7.6, 50 mm MgCl2, 250 μm ATP) containing 10 μCi of [γ-32P]ATP was added, and the reaction was incubated for an additional 10 min at 30 °C. The reaction was stopped by the addition of 15 μl of 4 n HCl and 130 μl of chloroform:methanol (1:1) and vortexing. The lower chloroform phase (30 μl) was spotted onto a 1% potassium oxalate-coated silica-gel plate (previously activated for 1 h at 110 °C) and subjected to thin-layer chromatography and autoradiography to visualize the 32P-labeled phosphatidylinositol 3-phosphate product.
Direct Pulldown Assays
The recombinant cyclin D1-CDK4 or cyclin E-CDK2 complex or PI3K (2 μg) was incubated with 7,3′,4′-THIF-conjugated Sepharose 4B (or Sepharose 4B as a negative control) beads (100 μl, 50% slurry) in immunoprecipitation reaction buffer (50 mm Tris-HCl, (pH 7.5), 5 mm EDTA, 150 mm NaCl, 1 mm dithiothreitol, 0.01% Nonidet P-40, 0.02 mm phenylmethylsulfonyl fluoride) containing 2 μg/ml bovine serum albumin and 1× protease inhibitor mixture at 4 °C with gentle rocking overnight. The beads were then washed five times with immunoprecipitation reaction buffer, and the proteins bound to the beads were analyzed by immunoblotting.
Anchorage-independent Transformation Assay
Basal medium Eagle agar (3.5 ml of 0.5%) containing 10% FBS with 7,3′,4′-THIF (0, 10, 20, or 40 μm) was layered onto each well of a 6-well plate. JB6 cells (exposed to 10 ng/ml EGF, 8 × 103 cells/ml) treated with 7,3′,4′-THIF (0, 10, 20, or 40 μm) were mixed with 1 ml of 0.33% basal medium Eagle agar containing 10% FBS and layered on top of the 0.5% agar layer. The separate cultures were maintained at 37 °C under a 5%-CO2 atmosphere for 10 days, and the cell colonies were counted under a microscope with the aid of Image-Pro Plus software (v.4, Media Cybernetics, Silver Spring, MD), as described by Colburn et al. (20).
Luciferase Assay for AP-1 Transactivation
Confluent monolayers of JB6 P+ cells stably transfected with an AP-1 luciferase reporter plasmid were trypsinized, and 8 × 103 viable cells were suspended in 100 μl of 5% FBS-MEM and added to each well of a 96-well plate. The plates were incubated at 37 °C under a humidified 5% CO2 atmosphere. When the cells reached 80–90% confluence, they were serum-starved by culturing in 0.1% FBS-MEM for an additional 24 h. The cells were then treated for 1 h with 7,3′,4′-THIF (0, 10, 20, or 40 μm) and exposed to 10 ng/ml EGF for 24 h. After treatment, cells were disrupted with 100 μl of lysis buffer (0.1 m potassium phosphate buffer (pH 7.8), 1% Triton X-100, 1 mm dithiothreitol, 2 mm EDTA), and luciferase activity was measured using a luminometer (Luminoskan Ascent, Thermo Electron, Helsinki, Finland).
Molecular Modeling
Insight II (Accelrys Inc., San Diego, CA) was used for the docking study and structure analysis with the crystal coordinates of PI3K in complex with ATP or myricetin (accession codes 1E8X or 1E90) available in the Protein Data Bank (www.rcsb.org/pdb/).
Statistical Analysis
When applicable, data are expressed as means ± S.D. values, and the Student's t test was used for single statistical comparisons. A probability value of p < 0.05 was used as the criterion for statistical significance.
RESULTS
7,3′,4′-THIF Causes G1 Phase Arrest in JB6 P+ Cells Stimulated with EGF or FBS
To evaluate the possible chemopreventive activity of 7,3′,4′-THIF (Fig. 1), we determined its effect on the proliferation response to EGF stimulation. JB6 P+ cells were synchronized at G0 by serum deprivation for 36 h, pretreated for 1 h with 7,3′,4′-THIF (0, 10, 20, or 40 μm), exposed to EGF for 24 h, and analyzed by flow cytometry. The cell cycle analysis revealed that 7,3′,4′-THIF caused cell cycle arrest at G1 phase. The percentage of cells in G1 phase increased significantly from 48% (EGF-treated group) to 75% (EGF and 10 μm 7,3′,4′-THIF-treated group; Fig. 2A). In contrast, EGF alone increased the percentage of cells in S phase, and pretreatment with 7,3′,4′-THIF before EGF treatment suppressed EGF-stimulated cell cycle progression at S phase. Because the flow cytometry data revealed that the 24-h pretreatment with 7,3′,4′-THIF did not affect the number of quiescent JB6 P+ cells in sub G1 (data not shown), the inhibition caused by 7,3′,4′-THIF was not attributable to cytotoxicity or apoptosis. 7,3′,4′-THIF also induced cell cycle arrest of FBS-stimulated JB6 P+ cells at G1 phase (Fig. 2B).
FIGURE 2.

Effect of 7,3′,4′-THIF on cell cycle phase distribution. In JB6 P+ cells synchronized in G0 and then stimulated with EGF (A) or FBS (B), 7,3′,4′-THIF causes cell cycle arrest at the G1 phase. The cells were serum-starved in 0.1% FBS-MEM for 36 h and then treated or not treated with 7,3′,4′-THIF at the indicated concentrations for 1 h. To stimulate cell growth, 10 ng/ml EGF or 5% FBS was then added. Cell cycle analysis was performed 24 h later, as described under “Experimental Procedures.” a, untreated control; b, 10 ng/ml EGF or 5% FBS alone; c, EGF (or FBS) and 10 μm 7,3′,4′-THIF; d, EGF (or FBS) and 20 μm 7,3′,4′-THIF; e, EGF (or FBS) and 40 μm 7,3′4′-THIF. The results are presented as the percentage of 7,3′,4′-THIF-treated cells in G1, S, or G2/M phase compared with the percentage of non-7,3′,4′-THIF-treated control cells in those phases. Data are shown as means ± S.D. of triplicate samples from three independent experiments. The asterisks indicate a significant (p < 0.01) difference in control versus JB6 cells stimulated with 10 ng/ml EGF or 5% FBS (Student's t test).
7,3′,4′-THIF Inhibits the Expression of G1 Phase-regulatory Proteins
To determine the mechanism responsible for 7,3′,4′-THIF-induced cell cycle arrest at G1 phase, we examined the expression of cyclins and CDKs involved in the G1/S-phase progression. Western blotting results showed that 7,3′,4′-THIF inhibited EGF-induced expression of the early G1 regulators cyclin D1 and CDK4 and of the late G1 regulators cyclin E and CDK2 (Fig. 3A).
FIGURE 3.
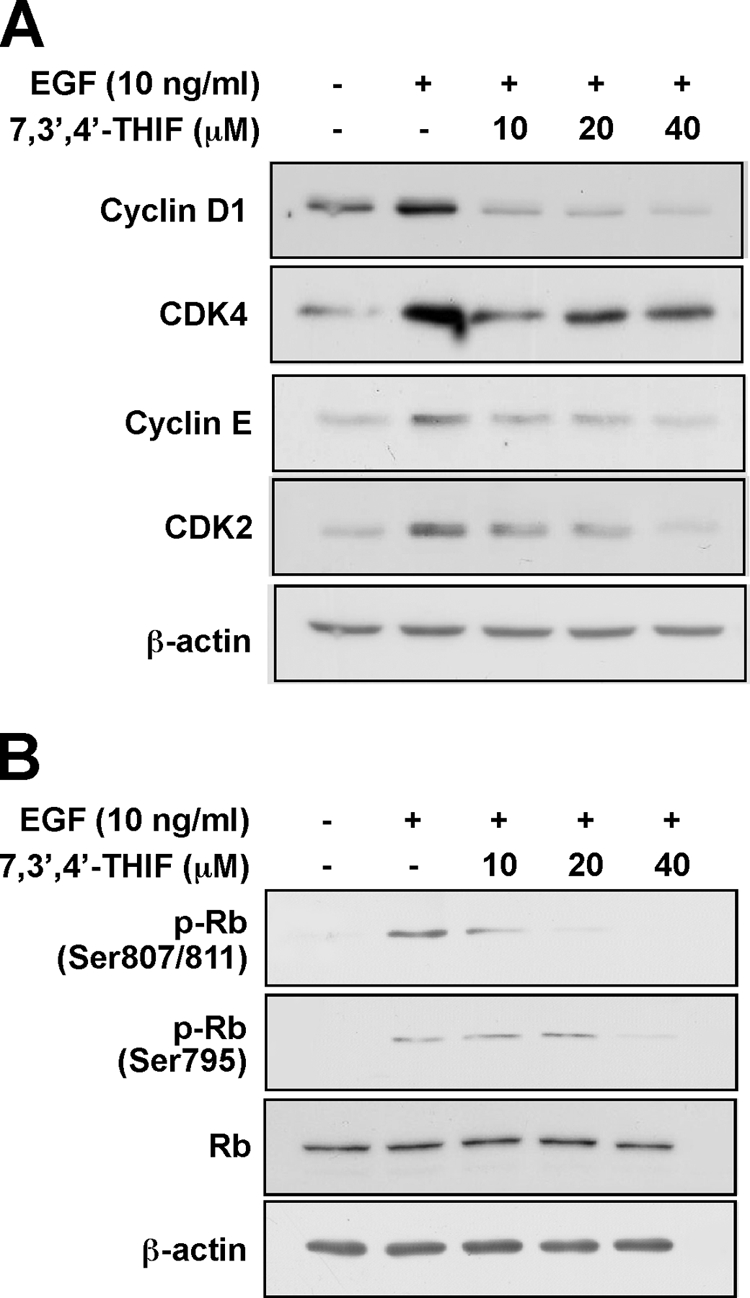
Effect of 7,3′,4′-THIF on G1 phase-related proteins in EGF-stimulated JB6 P+ cells. A, 7,3′,4′-THIF inhibits EGF-induced expression of cyclin D1, CDK4, cyclin E, and CDK2 in JB6 P+ cells. B, 7,3′,4′-THIF suppresses EGF-induced phosphorylation of Rb in JB6 P+ cells. Cells were serum-starved in 0.1% FBS-MEM for 36 h to synchronize cells at G0 and then either treated or not treated with 7,3′,4′-THIF at the indicated concentrations for 1 h before stimulation with 10 ng/ml EGF for 24 h. Protein expression was analyzed by Western blotting as described under “Experimental Procedures” using specific antibodies against the appropriate phosphorylated or total proteins. For A and B, the data shown are representative of three independent experiments.
Because phosphorylation of Rb plays a central role in the G1 to S transition, we determined the effect of 7,3′,4′-THIF on Rb phosphorylation at Ser-807/Ser-811 and Ser-795, the sites at which pRb is reportedly phosphorylated by CDK4 (Fig. 3B). Pretreatment of cells with 7,3′,4′-THIF (10, 20, or 40 μm) before EGF stimulation significantly inhibited Rb phosphorylation at these sites (Fig. 3B). These results suggest that 7,3′,4′-THIF-induced inhibition of the expression of G1 regulatory proteins promotes blockage of the G1/S transition of EGF-stimulated JB6 P+ cells.
7,3′,4′-THIF Suppresses the Kinase Activity of Cyclin D1-CDK4 and Cyclin E-CDK2 Complexes
Rb phosphorylation is initiated by the cyclin D1-CDK4 complex and enhances complete pRb phosphorylation by the cyclin E-CDK2 complex, forming a positive feedback loop (11). Therefore, both the cyclin D1-CDK4 and cyclin E-CDK2 complexes are important G1 phase-regulating proteins.
Because CDK4 must form a complex with cyclin D1 for activation, we next examined the effect of 7,3′,4′-THIF on the activity of the cyclin D1-CDK4 complex. Treatment with 7,3′,4′-THIF completely suppressed the kinase activity of the cyclin D1-CDK4 complex at all concentrations tested (Fig. 4A, left panel). Treatment with 7,3′,4′-THIF also partially inhibited the activity of the cyclin E-CDK2 complex (Fig. 4A, right panel), but its inhibitory effect on cyclin E-CDK2 appeared weaker than that on the cyclin D1-CDK4 complex.
FIGURE 4.

Effect of 7,3′,4′-THIF on the kinase activities of the cyclin D1-CDK4 and cyclin E-CDK2 complexes and direct binding to CDK4 and CDK2. A, 7,3′,4′-THIF suppresses the activity of the cyclin D1-CDK4 and cyclin E-CDK2 complexes. A direct kinase assay was performed as described under “Experimental Procedures.” An Rb-C fusion protein was used as the CDK substrate. The level of phosphorylated Rb was determined by Western blot using specific antibodies against Ser-807/Ser-811-phosphorylated Rb-C as described under “Experimental Procedures.” The data shown are representative of three independent experiments. B, 7,3′,4′-THIF specifically binds CDK4 and CDK2. Binding of CDK4 (left panel) or CDK2 (right panel) by 7,3′,4′-THIF was assessed by immunoblotting using specific antibodies against CDK4 or CDK2. Lane 1, input control (cyclin D1-CDK4 or cyclin E-CDK2 protein standard); lanes 2 and 3, pulled down of cyclin D1-CDK4 (left panel) or cyclin E-CDK2 (right panel) with Sepharose 4B beads (control, lane 2) or 7,3′,4′-THIF-conjugated Sepharose 4B beads (lane 3) as described under “Experimental Procedures.” C, 7,3′,4′-THIF does not compete with ATP for binding to cyclin D1-CDK4 or cyclin E-CDK2. The active cyclin D1-CDK4 complex (2 μg) was incubated with ATP (0, 10, or 100 μm) and 100 μl of 7,3′,4′-THIF-Sepharose 4B beads (left panel) or 100 μl of Sepharose 4B beads (negative control) in reaction buffer in a total volume of 500 μl. The mixtures were incubated with shaking at 4 °C overnight. After washing, the pulled down proteins were detected by Western blotting. Lane 1, the cyclin D1-CDK4 (left panel) and cyclin E-CDK2 (right panel) complexes protein standard (input control); lane 2, negative control: the cyclin D1-CDK4 (left panel) and cyclin E-CDK2 (right panel) complexes did not bind Sepharose 4B; lane 3, positive control: cyclin D1-CDK4 complex bound to 7,3′,4′-THIF-Sepharose 4B; lanes 4 and 5, increasing the amount of ATP had no effect on binding of 7,3′,4′-THIF to the cyclin D1-CDK4 or cyclin E-CDK2 complexes.
7,3′,4′-THIF Specifically Binds CDK4 and CDK2 but Does Not Compete with ATP for CDK4 and CDK2 Binding
Our results indicated that the induction of cell cycle arrest by 7,3′,4′-THIF involves the suppression of CDK4 or CDK2 activity, consequently arresting the cells at G1 phase. To determine whether 7,3′,4′-THIF exerts its effects by interacting directly with CDK4 or CDK2, we performed a pulldown assay using 7,3′,4′-THIF-conjugated and non-conjugated Sepharose 4B beads. After an overnight incubation with CDK proteins, 7,3′,4′-THIF-Sepharose 4B or Sepharose 4B-only were subjected to SDS-PAGE. Both CDK4 (Fig. 4B, left panel, lane 3) and CDK2 (Fig. 4B, right panel, lane 3) bound to the 7,3′,4′-THIF-Sepharose 4B beads, but not to the non-conjugated Sepharose-4B beads (Fig. 4B, lane 2), revealing that 7,3′,4′-THIF directly binds CDK4 and CDK2 to suppress their activation. Moreover, ATP did not compete with 7,3′,4′-THIF for binding to CDK4 (Fig. 5C, left panel) or CDK2 (Fig. 5C, right panel). In competition assays, the binding of 7,3′,4′-THIF to CDK4 and CDK2 did not change as the concentration of ATP increased. These results suggest that 7,3′,4′-THIF inhibits CDK4 or CDK2 activity through ATP-independent binding.
FIGURE 5.

Effects of 7,3′,4′-THIF on the PI3K/Akt/GSK3-β pathway. A, 7,3′,4′-THIF inhibits EGF-induced phosphorylation of Akt and GSK3-β. JB6 P+ cells were serum-starved in 0.1% FBS-MEM for 36 h to synchronize cells at G0 and then treated with 7,3′,4′-THIF at the indicated concentrations for 1 h followed by stimulation with 10 ng/ml EGF for 10 min, and then harvested. The levels of phosphorylated and total Akt and GSK-3β proteins were then determined by Western blot analysis using specific antibodies against the corresponding phosphorylated or total proteins as described under “Experimental Procedures.” The data shown are representative of three independent experiments. B, 7,3′,4′-THIF inhibits PI3K activity. Kinase assays were performed as described under “Experimental Procedures.” The 32P-labeled phosphatidylinositol 3-phosphate product was resolved by TLC and visualized by autoradiography. The data shown are representative of three independent experiments. C, 7,3′,4′-THIF specifically binds to PI3K. Binding of 7,3′,4′-THIF to PI3K was assessed by immunoblotting using a specific p110 antibody. Lane 1, PI3K protein standard (input control); lanes 2 and 3, PI3K pulldown experiments were performed using Sepharose 4B beads (control; lane 2) or 7,3′,4′-THIF-Sepharose 4B beads (lane 3), as described under “Experimental Procedures.” D, 7,3′,4′-THIF competes with ATP for binding to PI3K. Active PI3K (2 μg) was incubated with ATP (0, 10, or 100 μm) and 100 μl of 7,3′,4′-THIF-Sepharose 4B beads or 100 μl of Sepharose 4B (as a negative control) in reaction buffer in a total volume of 500 μl. The mixtures were incubated at 4 °C overnight with shaking. After washing, the pulled down proteins were detected by Western blotting. Lane 1, PI3K protein standard (input control); lane 2, negative control: PI3K did not bind to Sepharose 4B; lane 3, positive control: PI3K bound to 7,3′,4′-THIF-Sepharose 4B; lanes 4 and 5, as the ATP concentration increased, 7,3′,4′-THIF binding to PI3K decreased.
7,3′,4′-THIF Inhibits the Akt/GSK-3β Cell Growth Signal
GSK-3β induces cyclin D1 degradation in response to mitogenic signals (11, 12) and is inhibited by PI3K-dependent phosphorylation of GSK-3β at Ser-9. Thus, PI3K-mediated inhibition of GSK-3β stabilizes cyclin D1 (11). The PI3K/Akt/GSK-3β pathway plays a key regulatory role in cell cycle progression from G1 to S phase. Therefore, we next evaluated the effect of 7,3′,4′-THIF on the Akt/GSK-3β signaling pathway by Western blot. Treatment with 7,3′,4′-THIF inhibited the phosphorylation of both Akt and GSK-3β (Fig. 5A) but had no effect on the phosphorylation of ERKs or p38 (data not shown).
7,3′,4′-THIF Suppresses PI3K Activity by Binding to PI3K
To investigate whether PI3K is a molecular target of 7,3′,4′-THIF in the induction of cell cycle arrest, we next examined the effect of 7,3′,4′-THIF on the kinase activity of PI3K. We found that 7,3′,4′-THIF inhibited PI3K activity (Fig. 5B), indicating that PI3K is an additional important molecular target of 7,3′,4′-THIF in the inhibition of cyclin D1 expression.
We next examined whether 7,3′,4′-THIF exerted this effect by interacting directly with PI3K. In a pulldown assay, PI3K precipitated with 7,3′,4′-THIF-conjugated Sepharose 4B beads (Fig. 5C, lane 3), but not with non-conjugated Sepharose-4B beads (Fig. 5C, lane 2), showing that 7,3′,4′-THIF can bind directly to PI3K to inhibit its activation and downstream signaling. Moreover, the binding of 7,3′,4′-THIF to PI3K decreased as the concentration of ATP increased (Fig. 5D), suggesting that 7,3′,4′-THIF inhibits PI3K activity by competing with ATP for PI3K binding.
7,3′,4′-THIF Inhibits EGF-induced AP-1 Transactivation in and Neoplastic Transformation of JB6 P+ Cells
The promoter region of the cyclin D1 gene contains two binding sites for AP-1, a positive regulator of cyclin transcription (21). Thus, AP-1 activation is thought to play an important role in tumor promoter-induced cell proliferation and neoplastic transformation.
To determine whether the repression of proliferation and cyclin D1 expression by 7,3′,4′-THIF involves inhibition of AP-1, we measured AP-1 transactivation using JB6 cell lines stably transfected with an AP-1 luciferase reporter plasmid. Results showed that 7,3′,4′-THIF inhibited EGF-induced transactivation of AP-1 in a dose-dependent manner (Fig. 6A), suggesting that the inhibition of cell proliferation and cyclin D1 expression by 7,3′,4′-THIF occurs through the suppression of AP-1 activity.
FIGURE 6.
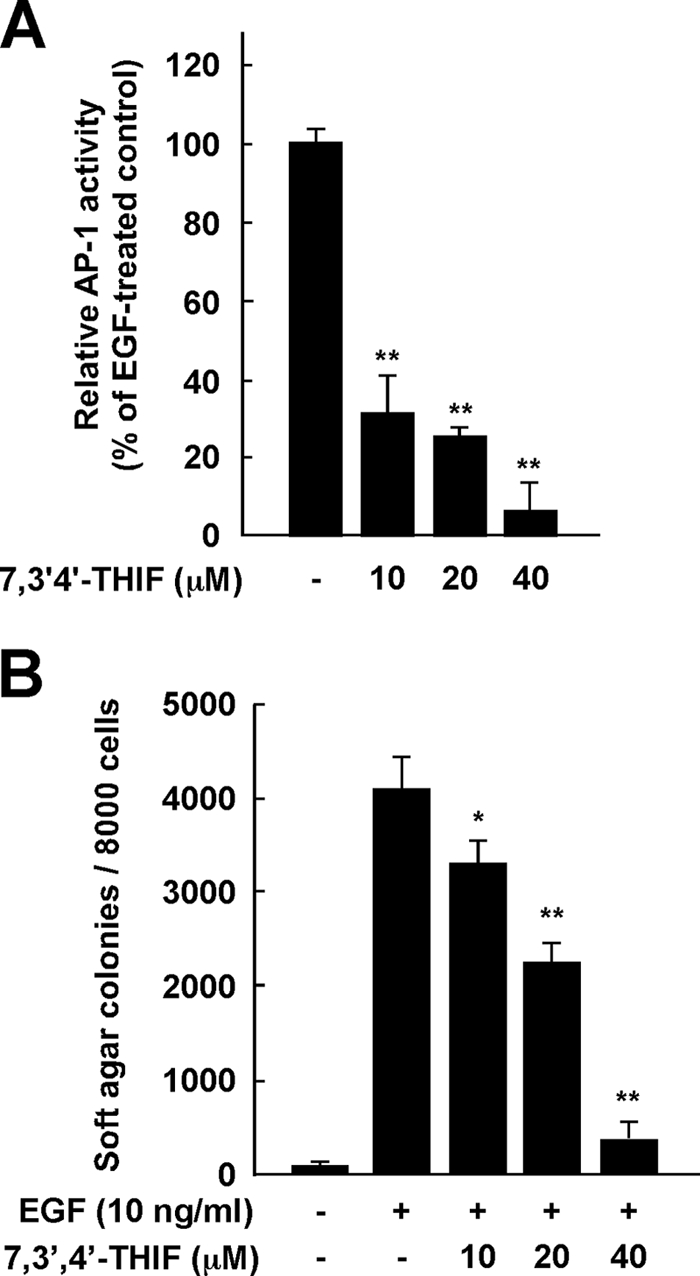
Effects of 7,3′,4′-THIF on EGF-induced neoplastic transformation and AP-1 activation of JB6 P+ cells. A, 7,3′,4′-THIF inhibits EGF-induced AP-1 transactivation. JB6 P+ cells were stably transfected with an AP-1-luciferase reporter plasmid and cultured as described under “Experimental Procedures.” The cells were then serum-starved in 0.1% FBS-MEM, treated or not treated with 7,3′,4′-THIF at the indicated concentrations for 1 h, exposed to 10 ng/ml EGF for 24 h, and assayed for luciferase activity. AP-1 activity is expressed as the percent inhibition relative to cells treated with EGF alone. Data are presented as means ± S.D. of triplicate samples from three independent experiments. The asterisks (**, p < 0.01; *, p < 0.05) indicate a significant difference versus the group treated with EGF alone. B, 7,3′,4′-THIF inhibits EGF-induced transformation of JB6 P+ cells. Cells were treated as described under “Experimental Procedures.” Colonies were counted 10 days later under a microscope with the aid of Image-Pro Plus software (v.4). Lane 1, untreated control; lane 2, 10 ng/ml EGF alone; lane 3, EGF and 10 μm 7,3′,4′-THIF; lane 4, EGF and 20 μm 7,3′,4′-THIF; lane 5, EGF and 40 μm 7,3′,4′-THIF. The results are presented as the percent inhibition of cell transformation relative to EGF-stimulated cells in soft agar. Data are presented as means ± S.D. of triplicate samples from three independent experiments. The asterisks (**, p ≤ 0.01; *, p < 0.05) indicate a significant difference versus the group treated with EGF alone.
Because anchorage-independent growth, a unique characteristic behavior of cancer cells, is tightly regulated through cell cycle signaling, we next examined the effect of 7,3′,4′-THIF on EGF-induced neoplastic transformation of JB6 P+ cells. As indicated by cell colony numbers, treatment with 7,3′,4′-THIF significantly suppressed EGF-induced neoplastic transformation in a dose-dependent manner (Fig. 6B). At 40 μm, 7,3′,4′-THIF suppressed EGF-induced cell transformation by 17.7% (Fig. 6B).
DISCUSSION
Because chemopreventive agents are likely to be used continually by essentially healthy people, naturally occurring substances are ideal candidates, presuming safety during long term use. Isoflavones are a class of naturally occurring bioactive phytochemicals found in many plants (22). Numerous experimental studies have shown that soy isoflavones such as genistein and daidzein have anti-carcinogenic effects, and soy foods are generally recognized as safe. Several in vitro and in vivo studies have demonstrated that soy extracts and isoflavones have an inhibitory effect on proliferation of EGF-overexpressing cancer cells (17, 23, 24).
A substantial percentage of dietary daidzein is converted to biologically active metabolites by intestinal microflora or liver microsomes (25, 26). We previously showed that equol is one of the most biologically active metabolites of daidzein (27). We also suggested that equol, but not daidzein, is a potent inhibitor of mitogen-activated protein kinase activity and that equol has chemopreventive activity in JB6 P+ cells, suppressing 12-O-tetradecanoylphorbol 13-acetate-induced neoplastic transformation (27).
In rats (28) and humans (25), isoflavones are subject to oxidative biotransformation through hepatic metabolism. One of the major products of the hepatic metabolism of daidzein is 7,3′,4′-THIF (29). However, no reports on the possible chemopreventive effects or molecular mechanism of action of this hepatic metabolite have been published. In the present study, we found that 7,3′,4′-THIF prevented the transition of JB6 P+ cells from G1 to S phase, significantly blocking EGF-stimulated progression of the cell cycle at G1. It also effectively suppressed EGF-induced neoplastic transformation of JB6 P+ cells, whereas we previously found daidzein itself had no effect (27). This study thus demonstrates that 7,3′,4′-THIF is a potent chemopreventive agent against EGF-induced proliferation of JB6 P+ cells. Our findings also provide information about the molecular mechanisms and targets of this activity.
Defective CDK4 inhibition and deregulated CDK4 activity have been observed in many cancers. Phosphorylation of Rb by CDK4 contributes to the release and activation of E2F target genes, including those encoding E- and A-type cyclins, which facilitate progression through G1 phase. In particular, Ser-795 and Ser-807/Ser-811 in Rb are specific sites for phosphorylation by CDK4 and are additionally phosphorylated by cyclin E-bound CDK2 (30, 31). The G1/S-phase transition is tightly regulated by the interactions of the CDK4-cyclin D and CDK2-cyclin E complexes with Rb. Therefore, CDK inhibitors have been considered relevant candidates for anticancer therapeutic agents owing to their potential ability to restore control of the cell cycle (5, 32). The development of a natural inhibitor of CDK4 or CDK2 activity is a promising approach in this chemopreventive strategy. Our results show that 7,3′,4′-THIF completely suppresses CDK4 activity and partially inhibits CDK2 activity by binding directly to these proteins. Thus, 7,3′,4′-THIF exerts its potent chemopreventive effect by suppressing CDK activities.
Overexpression of cyclin D1 has been frequently observed in many human cancers (33–35). Moreover, cyclin D1-deficient mice are resistant to tumors induced by the Ras and Neu oncogenes (8). Growth factor-mediated regulation of cyclin D1 induction is primarily mediated by PI3K/Akt-dependent pathways (8, 12, 36). Akt-catalyzed phosphorylation of GSK-3β at Ser-9 decreases the catalytic activity of GSK-3β and thus contributes to cyclin D1 stabilization. We therefore examined whether suppression of the PI3K/Akt/GSK-3β pathway by 7,3′,4′-THIF would down-regulate EGF-induced expression of cyclin D1 and found that 7,3′,4′-THIF did suppress EGF-induced phosphorylation of Akt and GSK-3β.
We then hypothesized that the molecular target of 7,3′,4′-THIF in the inhibition of EGF-induced cyclin D1 expression might be a kinase upstream of Akt. We found that 7,3′,4′-THIF effectively and directly inhibits PI3K activity, resulting in the down-regulation of Akt and GSK-3β. Furthermore, 7,3′,4′-THIF competes with ATP for PI3K binding, which might explain the reduced kinase activity of 7,3′,4′-THIF-bound PI3K. Collectively, these results suggest that the inhibition of cyclin D1 expression by 7,3′,4′-THIF is primarily the result of direct suppression of PI3K activity.
To investigate the molecular mechanism of the PI3K inhibition by 7,3′,4′-THIF, we carried out a modeling study using the crystal structure of PI3K in complex with ATP or myricetin (37, 38). PI3K consists of four domains: a Ras-binding domain, a C2 domain, a helical domain, and a catalytic domain. The catalytic domain of PI3K consists of an N-lobe and a C-lobe with a fold similar to other protein kinases, and this structural similarity is also conserved in the ATP binding site that is flanked by these two lobes. Consequently, ATP binds between these lobes in a manner similar to ATP binding in many protein kinases. The N- and C-lobes are linked through a loop, which is called the “hinge region.” The backbone of this loop interacts with the adenine moiety of ATP by hydrogen bonding. Considering the experimental result showing that 7,3′,4′-THIF is an ATP-competitive inhibitor of PI3K, we docked the compound to the ATP binding site of PI3K (Fig. 7A). The hydroxyl groups at the 3′ and 4′ positions of 7,3′,4′-THIF could make hydrogen bonds with the backbone atoms of Val-887 in the hinge region of PI3K. The hydroxyl group at the 7 position could also form hydrogen bonds with the side chains of Asp-836. In addition, 7,3′,4′-THIF would be sandwiched by the side chains of the hydrophobic residues in the ATP binding site, including Met-804, Trp-812, Ile-831, Tyr-867, and Ile-879 from the N-lobe and Ala-885, Met-953, Phe-961, and Ile-963 from the C-lobe. The potent inhibitory activity of 7,3′,4′-THIF against PI3K would be due to these hydrogen bonds and hydrophobic interactions. Because 7,3′,4′-THIF was shown to be an ATP-noncompetitive inhibitor of CDKs, a docking study was not possible due to the lack of available structure data for the binding of ATP-noncompetitive inhibitors with CDKs.
FIGURE 7.

A, hypothetical model of PI3K in complex with 7,3′,4′-THIF and an enlarged view. The Ras-binding domain, C2 domain, and helical domain of PI3K are colored in gray. The N-lobe, C-lobe, and hinge region of the catalytic domain are colored in purple, orange, and yellow, respectively. 7,3′,4′-THIF (atomic color) binds to the ATP binding site in the catalytic domain of PI3K. The residues involved in the 7,3′,4′-THIF binding are labeled. The residues in gray ellipses are the hydrophobic residues interacting with 7,3′,4′-THIF. The hydrogen bonds are depicted as white lines. B, hypothetical scheme of the mechanism of the chemopreventive action of 7,3′,4′-THIF.
From the experimental result showing that 40 μm 7,3′,4′-THIF does not affect other protein kinases such as EGF receptor, ERKs, MSK1, Akt, MKK3/6, or p38 kinases (data not shown) and inhibits CDKs in an ATP-noncompetitive manner, we can hypothesize that the selective binding of the compound to the ATP binding site of PI3K is due to a similar, but distinct structure of the ATP binding site of PI3K compared with those of protein kinases such as the CDKs. Binding of 7,3′,4′-THIF to CDKs might induce a structural rearrangement of CDKs into an inactive conformation or weaken the binding affinity of cyclins to CDKs. Further studies with x-ray crystallography to determine the inhibitor complex structures would elucidate the exact binding modes of 7,3′,4′-THIF to PI3K and CDK2/4.
A recently adopted strategy in anticancer therapeutics development has been to develop agents that target a variety of different kinases. The hope is that these agents will be more efficacious than are the highly selective kinase inhibitors administered as single agents. Moreover, a multitargeted kinase inhibitor might have broad-range antitumor efficacy. Two such multitargeted kinase inhibitors, sunitinib and sorafenib, have proven to be effective in clinical testing (39).
Although CDK inhibitors are thought to be relevant drug candidates for cancer therapy, the first-generation CDK inhibitors, such as flavopiridol and UCN-01, have not exhibited significant clinical advantages. A possible explanation for this disappointing result is that different kinds of tumors might have different sensitivities to different types of CDK inhibition, depending on their pathogenic spectrum of mutations (32). Therefore, naturally occurring dietary substances capable of regulating different kinases, rather than or in addition to chemotherapy, might be ideal agents for chemoprevention.
AP-1, which is activated in response to growth factors and tumor promoters, regulates the expression of several genes involved in growth control as well as cell transformation (40, 41). Indeed, many studies have demonstrated the involvement of AP-1 in EGF- or 12-O-tetradecanoylphorbol 13-acetate-induced transformation of JB6 P+ cells (42, 43). The cyclin D1 gene promoter region contains two AP-1 binding sites, suggesting that cyclin D1 is a major target for AP-1 transcriptional activation (44, 45). Therefore, AP-1-mediated cell transformation is implicated as a key event in cell proliferation in carcinogenesis.
In summary, 7,3′,4′-THIF prevents EGF-induced neoplastic transformation and proliferation of JB6 P+ mouse epidermal cells, significantly blocking cell cycle progression at G1 phase after EGF stimulation. 7,3′,4′-THIF suppresses EGF-induced phosphorylation of Rb at Ser-795 and Ser-807/Ser-811 and inhibits the expression of G1 phase-regulatory proteins, including cyclin D1, CDK4, cyclin E, and CDK2. In addition to regulating the expression of cell cycle-regulatory proteins, 7,3′,4′-THIF binds to CDK4 and CDK2 and strongly inhibits their kinase activities. Furthermore, 7,3′,4′-THIF binds to PI3K and suppresses its kinase activity, thereby blocking the Akt/GSK-3β/AP-1 pathway and subsequently suppressing cyclin D1 expression. A simplified depiction of our proposed mechanism for the anti-proliferative effects of 7,3′,4′-THIF is shown in Fig. 7B. Collectively, these results suggest that CDKs and PI3K are the primary molecular targets of 7,3′,4′-THIF in the suppression of EGF-induced cell proliferation and transformation. These insights into the biological actions of 7,3′,4′-THIF might provide a molecular basis for the development of new chemoprotective agents.
This work was supported, in whole or in part, by National Institutes of Health Grants CA027502, CA120388, CA111536, CA077646, and CA081064. This work was also supported by The Hormel Foundation, by grants from the World Class University Program (R31-2008-00-10056-0), Priority Research Centers Program (2009-0093824), and Basic Science Research Program (314-2007-1-F00054, and 2009-0090797) through the National Research Foundation (NRF) of Korea, Funded by the Ministry of Education, Science and Technology, Republic of Korea.
- EGF
- epidermal growth factor
- CDK
- cyclin-dependent kinase
- Rb
- retinoblastoma
- pRb
- phosphorylated Rb
- PI3K
- phosphatidylinositol 3-kinase
- GSK
- glycogen synthase kinase
- 7,3′,4′-THIF
- 7,3′,4′-trihydroxyisoflavone
- MEM
- minimal essential medium
- FBS
- fetal bovine serum
- AP-1
- activator protein-1
- MOPS
- 4-morpholinepropanesulfonic acid
- ERK
- extracellular signal-regulated kinase.
REFERENCES
- 1.Bode A. M., Dong Z. (2006) Mol. Carcinog. 45, 422–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Furuse J. (2008) Crit. Rev. Oncol. Hematol. 67, 8–15 [DOI] [PubMed] [Google Scholar]
- 3.Khalil M. Y., Grandis J. R., Shin D. M. (2003) Expert. Rev. Anticancer Ther. 3, 367–380 [DOI] [PubMed] [Google Scholar]
- 4.Mimeault M., Pommery N., Hénichart J. P. (2003) Growth Factors 21, 1–14 [DOI] [PubMed] [Google Scholar]
- 5.Vermeulen K., Van Bockstaele D. R., Berneman Z. N. (2003) Cell Prolif 36, 131–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shapiro G. I. (2006) J. Clin. Oncol 24, 1770–1783 [DOI] [PubMed] [Google Scholar]
- 7.Lee M. H., Yang H. Y. (2003) Cancer Metastasis Rev. 22, 435–449 [DOI] [PubMed] [Google Scholar]
- 8.Shukla S., Gupta S. (2007) Cell Cycle 6, 1102–1114 [DOI] [PubMed] [Google Scholar]
- 9.Steelman L. S., Stadelman K. M., Chappell W. H., Horn S., Bäsecke J., Cervello M., Nicoletti F., Libra M., Stivala F., Martelli A. M., McCubrey J. A. (2008) Expert Opin Ther Targets 12, 1139–1165 [DOI] [PubMed] [Google Scholar]
- 10.Yuan T. L., Cantley L. C. (2008) Oncogene 27, 5497–5510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liang J., Slingerland J. M. (2003) Cell Cycle 2, 339–345 [PubMed] [Google Scholar]
- 12.Takahashi-Yanaga F., Sasaguri T. (2008) Cell Signal 20, 581–589 [DOI] [PubMed] [Google Scholar]
- 13.Messina M. J., Persky V., Setchell K. D., Barnes S. (1994) Nutr. Cancer 21, 113–131 [DOI] [PubMed] [Google Scholar]
- 14.Wu A. H., Ziegler R. G., Nomura A. M., West D. W., Kolonel L. N., Horn-Ross P. L., Hoover R. N., Pike M. C. (1998) Am. J. Clin. Nutr. 68, 1437S–1443S [DOI] [PubMed] [Google Scholar]
- 15.Bennink M. R. (2001) Adv. Exp. Med. Biol. 492, 11–17 [DOI] [PubMed] [Google Scholar]
- 16.Farina H. G., Pomies M., Alonso D. F., Gomez D. E. (2006) Oncol. Rep. 16, 885–891 [PubMed] [Google Scholar]
- 17.Hewitt A. L., Singletary K. W. (2003) Cancer Lett. 192, 133–143 [DOI] [PubMed] [Google Scholar]
- 18.Jian L. (2009) Mol. Nutr. Food Res. 53, 217–226 [DOI] [PubMed] [Google Scholar]
- 19.Ahmad N., Feyes D. K., Nieminen A. L., Agarwal R., Mukhtar H. (1997) J. Natl. Cancer Inst. 89, 1881–1886 [DOI] [PubMed] [Google Scholar]
- 20.Colburn N. H., Former B. F., Nelson K. A., Yuspa S. H. (1979) Nature 281, 589–591 [DOI] [PubMed] [Google Scholar]
- 21.Shaulian E., Karin M. (2001) Oncogene 20, 2390–2400 [DOI] [PubMed] [Google Scholar]
- 22.Song W. O., Chun O. K., Hwang I., Shin H. S., Kim B. G., Kim K. S., Lee S. Y., Shin D., Lee S. G. (2007) J. Med. Food. 10, 571–580 [DOI] [PubMed] [Google Scholar]
- 23.Wang J., Eltoum I.-E., Lamartiniere C. A. (2004) Mol. Cell. Endocrinol. 219, 171–180 [DOI] [PubMed] [Google Scholar]
- 24.Sarkar F. H., Adsule S., Padhye S., Kulkarni S., Li Y. (2006) Mini. Rev. Med. Chem. 6, 401–407 [DOI] [PubMed] [Google Scholar]
- 25.Kulling S. E., Honig D. M., Metzler M. (2001) J. Agric. Food Chem. 49, 3024–3033 [DOI] [PubMed] [Google Scholar]
- 26.Rüfer C. E., Glatt H., Kulling S. E. (2006) Drug. Metab. Dispos 34, 51–60 [DOI] [PubMed] [Google Scholar]
- 27.Kang N. J., Lee K. W., Rogozin E. A., Cho Y. Y., Heo Y. S., Bode A. M., Lee H. J., Dong Z. (2007) J. Biol. Chem. 282, 32856–32866 [DOI] [PubMed] [Google Scholar]
- 28.Kulling S. E., Honig D. M., Simat T. J., Metzler M. (2000) J. Agric. Food Chem. 48, 4963–4972 [DOI] [PubMed] [Google Scholar]
- 29.Kulling S. E., Lehmann L., Metzler M. (2002) J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 777, 211–218 [DOI] [PubMed] [Google Scholar]
- 30.Day P. J., Cleasby A., Tickle I. J., O'Reilly M., Coyle J. E., Holding F. P., McMenamin R. L., Yon J., Chopra R., Lengauer C., Jhoti H. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 4166–4170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elangovan S., Hsieh T. C., Wu J. M. (2008) Anticancer Res. 28, 2641–2647 [PubMed] [Google Scholar]
- 32.Malumbres M., Barbacid M. (2009) Nat. Rev. Cancer 9, 153–166 [DOI] [PubMed] [Google Scholar]
- 33.Diehl J. A. (2002) Cancer Biol. Ther. 1, 226–231 [DOI] [PubMed] [Google Scholar]
- 34.Petty W. J., Dragnev K. H., Dmitrovsky E. (2003) Lung Cancer 41, Suppl. 1, S155–S161 [DOI] [PubMed] [Google Scholar]
- 35.Yokota T., Matsuzaki Y., Koyama M., Hitomi T., Kawanaka M., Enoki-Konishi M., Okuyama Y., Takayasu J., Nishino H., Nishikawa A., Osawa T., Sakai T. (2007) Cancer Sci. 98, 1447–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diehl J. A., Cheng M., Roussel M. F., Sherr C. J. (1998) Genes Dev. 12, 3499–3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walker E. H., Perisic O., Ried C., Stephens L., Williams R. L. (1999) Nature 402, 313–320 [DOI] [PubMed] [Google Scholar]
- 38.Walker E. H., Pacold M. E., Perisic O., Stephens L., Hawkins P. T., Wymann M. P., Williams R. L. (2000) Mol. Cell 6, 909–919 [DOI] [PubMed] [Google Scholar]
- 39.Sebolt-Leopold J. S., English J. M. (2006) Nature 441, 457–462 [DOI] [PubMed] [Google Scholar]
- 40.Brown P. H., Alani R., Preis L. H., Szabo E., Birrer M. J. (1993) Oncogene 8, 877–886 [PubMed] [Google Scholar]
- 41.Dong Z., Birrer M. J., Watts R. G., Matrisian L. M., Colburn N. H. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 609–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma C., Wang J., Gao Y., Gao T. W., Chen G., Bower K. A., Odetallah M., Ding M., Ke Z., Luo J. (2007) Cancer Res. 67, 7756–7764 [DOI] [PubMed] [Google Scholar]
- 43.Rogozin E. A., Lee K. W., Kang N. J., Yu H., Nomura M., Miyamoto K., Conney A. H., Bode A. M., Dong Z. (2008) Carcinogenesis 29, 1228–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ouyang W., Li J., Ma Q., Huang C. (2006) Carcinogenesis 27, 864–873 [DOI] [PubMed] [Google Scholar]
- 45.Zhang D., Li J., Gao J., Huang C. (2009) Toxicol. Appl. Pharmacol. 235, 18–24 [DOI] [PMC free article] [PubMed] [Google Scholar]