

Abstract
The Escherichia coli Hsp40 DnaJ uses its J-domain (Jd) to couple ATP hydrolysis and client protein capture in Hsp70 DnaK. Fusion of the Jd to peptide p5 (as in Jdp5) dramatically increases the apparent affinity of the p5 moiety for DnaK in the presence of ATP, and Jdp5 stimulates ATP hydrolysis in DnaK by several orders of magnitude. NMR experiments with [15N]Jdp5 demonstrated that the peptide tethers the Jd to the ATPase domain. Thus, ATP hydrolysis and client protein binding in DnaK are coupled principally through the association of the client with DnaJ. Overexpression of a recombinant Jd was specifically toxic to cells that simultaneously expressed DnaK. No toxicity was observed when overexpressing Jdp5 or mutant Jd or when co-overexpressing the Jd and the nucleotide exchange factor GrpE. The results suggest that the Jd shifts DnaK to a client-bound form by stimulating the DnaK ATPase but only when the Jd is brought to DnaK by a client-Hsp40 complex.
Keywords: ATPases, Chaperone Chaperonin, Fluorescence, Heat Shock Protein, NMR
Introduction
Hsp70-family molecular chaperones have numerous constitutive and stress-induced roles that involve binding to short hydrophobic stretches within a client protein. Protein folding, protein translocation, and protein disaggregation are a few of the functions that Hsp70s carry out under normal conditions within the cell (1–4).
The two major activities of Hsp70s are located in the two major domains of the protein. The N-terminal 44-kDa domain binds and hydrolyzes ATP, and the 23-kDa peptide binding domain binds client proteins (5). The peptide binding domain is divided into the β-sandwich subdomain where client binding occurs and the α-helical subdomain, which acts as a lid on the β-sandwich subdomain (6). The two major domains are connected by a highly conserved linker region.
Escherichia coli Hsp70/DnaK is the most extensively studied member of the Hsp70s. The behavior of DnaK is like other Hsp70s in that its binding to client proteins is regulated by binding and hydrolysis of ATP. The binding of ATP converts DnaK to the low affinity conformation. This form of the protein binds and releases client proteins quickly, on the time scale of seconds (7, 8). Hydrolysis of ATP slows the off-rate of client proteins by 3 orders of magnitude compared with when ATP is bound (9). Previous studies have shown that a portion of the lid subdomain of DnaK modulates the allosteric behavior. For example, for a lidless form of DnaK (DnaK(1–517)) containing only a portion of helix A, the ATP-stimulated release of peptide is ∼40-fold faster than for wild-type DnaK (10).
The DnaK client binding cycle is regulated by a nucleotide exchange factor, GrpE (11). GrpE interacts with the ATPase domain of DnaK to catalyze the release of ADP (12). Upon release of ADP, DnaK binds ATP, which stimulates the release of bound client.
The structurally and functionally distinct E. coli Hsp40/DnaJ has been described as a co-chaperone for Hsp70/DnaK, and the two proteins are said to operate as a single chaperone machine (13). DnaJ binds to client proteins, and it stimulates the DnaK ATPase activity. The two functions of DnaJ have been assigned to the C terminus and N terminus of the molecular chaperone, respectively. The N-terminal 75-residue J-domain (Jd)2 is responsible for stimulation of DnaK ATPase activity (14, 15). The Jd is also able to couple ATP hydrolysis to peptide capture in DnaK (15). Two distinct models have been suggested for how the Jd couples the DnaK ATPase and peptide binding activities, client protein-recruitment, and triggered capture. In the recruitment model, the Jd brings an associated client protein to DnaK (16–18). In the triggered-capture model, the client protein brings the Jd to DnaK (14, 19, 20). After formation of the three-component complex, the Jd stimulates ATP hydrolysis by DnaK, which firmly stabilizes the client-bound conformation of DnaK. The triggered-capture model more accurately described the role of the Jd in binding of a model client, the peptide p5, because the association rate of the p5 with DnaK was only modestly increased by fusion of p5 with the Jd, whereas the stability of binding to DnaK was dramatically increased (15). Thus, the Jd can act as a specificity factor for DnaK client proteins solely by its ability to stimulate the DnaK ATPase (21).
The mechanism used by the Jd to stimulate the DnaK ATPase is unclear. The Jd could stimulate the Hsp70 ATPase solely through contact with the ATPase domain or by affecting the allosteric coupling between the ATPase and peptide binding domains. Efforts to distinguish between these mechanisms have been confounded by evidence that Hsp40/DnaJ interacts with both the ATPase domain and the peptide binding domain of Hsp70 (19, 22). The binding site for the Jd was localized to the ATPase domain of DnaK by NMR signal perturbations (16), and this interaction was corroborated by the genetic selection of an ATPase domain suppressor of a Jd mutation (23). A probable binding site for the Jd was identified as a positively charged groove at the interface between major domains of the ATPase domain (16, 23, 24). However, recent crystal structures of bovine Hsc70 and yeast Hsp110 (an Hsp70 homolog) suggested that the peptide-binding domain occupies this site in the ADP- and/or ATP-bound conformations (25, 26). Furthermore, mutations that affect interactions with Jd proteins invariably also affected Hsp70 interdomain coupling (25, 27, 28).
The present study employs a fusion protein, Jdp5, in which the peptide p5 was fused to the C terminus of the Jd (15). Jdp5 mimics the binary complex formed between DnaJ and the client peptide. The interaction of Jdp5 with DnaK provides a view on the mechanisms of Jd stimulation of the DnaK ATPase activity and coupling of ATP hydrolysis to peptide capture in DnaK.
Binding experiments found no evidence that the Jd participates in allosteric communication between the ATPase and peptide binding domains of DnaK. Single-turnover ATPase activity assays demonstrated that Jdp5 can dramatically stimulate ATP hydrolysis by DnaK, even when the DnaK peptide-binding domain lacks its lid subdomain. NMR studies on DnaK binding of Jdp5 suggested that the p5 moiety tethers the Jd to its binding site on the ATPase domain. The overexpression of a recombinant Jd caused DnaK-dependent toxicity in an E. coli strain that lacks Jd proteins. The array of factors able to suppress the toxicity was most consistent with the toxicity being due to the Jd stalling the DnaK client binding cycle by non-specifically stimulating DnaK ATPase activity. The results supported the conclusion that the Jd activates the Hsp70 ATPase when it is tethered near its binding site on the ATPase domain through association with a DnaK-bound client protein.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Plasmid Constructs
The Δ3 and Δ4 bacterial strains used for the studies in vivo are derivatives of E. coli K-12. The Δ3 strain is an arabinose-resistant derivative of MC4100 dnaJ::Tn10–42 ΔcbpA::kan ΔdjlA::Ωspc (29). The Δ4 strain was a derivative of MC4100 Δara714 ΔdnaKdnaJ:: kan thr::Tn10(Tet) ΔcbpA::kan ΔdjlA::Ωspc (30). Plasmids pBAD22-dnaJ (pWKG90) (31), pBAD33-dnaJ (30), pWSK29 (32), pSE380ΔNcoI (32), and pSE380ΔNcoI-grpE (pGPE) (30) have been previously described.
The pBAD22 derivatives encoding Jd(1–78), Jd(1–78)D35N, or Jd(1–78)H33Q were constructed by first PCR-amplifying the Jd(1–78) coding sequence from pPLJ(2–78)p5 (15), pPLJD35Np5 (15), or pBAD22-DnaJH33Q (30) using the forward primer 5′-cgagaattcaggaggaaacgaccatggctaagcaagattattacgag-3′ and the reverse primer 5′-gcttgcatgcttagccaccttgctcaaacgcagcat-3′, which introduced NcoI and SphI restriction sites (underlined). The resulting 250-bp fragment was digested with NcoI and SphI and then ligated with pBAD22 (33), which was also digested with the same restriction enzymes. The resulting plasmids were named pEHJ10 (pBAD22-Jd(1–78)), pEHJ11 (pBAD22-Jd(1–78)D35N), and pEHJ13 (pBAD22-Jd(1–78)H33Q). The pBAD33 derivatives of Jd(1–78) and Jd(1–78)D35N were constructed by subcloning the EcoRV-SphI fragment from pEHJ10 or pEHJ11 into the EcoRV-SphI-digested pBAD33 vector (33). The pBAD22 constructs containing Jd(1–78)p5 or Jd(1–78)D35Np5 were constructed in the same manner as pEHJ10 and pEHJ11, except the reverse primer was 5′-gcttgcatgcttaacgacgtggagcgctcaacaa-3′, which allows for amplification of the p5 peptide sequence in pPLJ(2–78)p5 and pPLJD35Np5. The resulting plasmids were named pEHJ3 (pBAD22Jd(1–78)p5) and pEHJ7 (pBAD22Jd(1–78)D35Np5).
To construct DnaK(1–517), the primers 5′-gctgttatcaccgtaccggca-3′ and 5′-gctaagcttttagcgtaccattttctggatttc-3′ were used to PCR-amplify the region of DnaK beginning at base pair 413 to base pair 1551 from pWSK29DnaK+ (30). The reverse primer introduced a HindIII site (underlined) along with a stop codon after base pair 1551 (bold). The resulting 1.1-kbp fragment was digested with EcoRI (cuts at 727 bp in DnaK) and HindIII. The 824-bp fragment from the digest was ligated with pWSK29 DnaK+ that was also digested with EcoRI and HindIII. The resulting plasmid was named pWSKLidless.
Protein Purification and Protein Modification
The plasmids used to express the Jd (pPLJ1–75) and the fusion protein Jdp5 (pPLJ(2–78)p5) have been previously described (15). Jd and Jdp5 were prepared from E. coli NapIV cells (19). [15N]Jd and [15N]Jdp5 were also prepared from E. coli NapIV cells following the protocol described previously (16). DnaK and DnaK (2–388) (Kase) were prepared from NapIV cells containing the plasmids pRLM163 and pRLM157, respectively (16, 34). DnaK(1–517) (“lidless” DnaK(1–517)) was expressed in ΔdnaK52 cells (BB1553) containing the plasmid p517 (10, 35). Purification of lidless DnaK(1–517) was carried out as for DnaK. Both DnaK and lidless DnaK(1–517) were dialyzed in 4 liters of buffer containing 25 mm HEPES-NaOH, pH 7, 50 mm KCl, 5 mm MgCl2, and 5 mm β-mercaptoethanol for 4 days with 3 exchanges per day to remove nucleotide. The plasmid expressing DnaK(387–552)L542Y,L543E (DnaK(387–552)-ye), with an N-terminal histidine tag, has been described previously (36). DnaK(387–552)-ye was expressed in E. coli NapIV cells and purified as previously described (36), except a step gradient up to 200 mm imidazole was used in place of a linear gradient, and protein was not purified from inclusion bodies.
The labeling of Jdp5 with acrylodan (Molecular Probes) was as described previously (15). Mass spectroscopy showed the protein was modified with only one acrylodan group. The protein concentration was determined using the Bio-Rad protein assay. The amount of labeling was determined using the ϵ360 (12900 m−1cm−1) for the acrylodan moiety.
For NMR, [15N]Jdp5 was treated with iodoacetamide as described (37). Briefly, 17 mg of protein in 0.1 m Tris-HCl, pH 8.0, buffer was incubated for 1 h with a 10-fold molar excess of dithiothreitol. A 5-fold molar excess of iodoacetamide over dithiothreitol was added to the solution and allowed to react for an additional hour in the dark. To stop the reaction, an excess of β-mercaptoethanol over iodoacetamide was added. The carbamidomethylated protein ([15N]Jdp5-cam) was dialyzed against 2 liters of 50 mm MOPS-NaOH, pH 6.8, with 3 changes to prepare the sample for NMR.
KD Determinations
Fluorescence-titration experiments were carried out as previously described (15) with the addition of reducing agent 1 mm dithiothreitol to the assay buffer, as described in the legend to Fig. 1. The wavelength of maximum emission for lidless DnaK(1–517) was 510 nm. The average of nine points around 510 nm was used to determine the change in fluorescence.
FIGURE 1.

Binding of lidless DnaK(1–517) to Jdp5A in the absence and presence of ATP. The lidless DnaK(1–517) was titrated into samples of 10 nm Jdp5A in 50 mm HEPES-NaOH, 10 mm KCl, 1 mm MgCl2, 1 mm dithiothreitol, pH 7.6, and, where indicated, 0.5 mm ATP. Binding to Jdp5A of lidless DnaK(1–517) causes an increase in fluorescence intensity of the acrylodan attached to Jdp5A, and the apparent affinity of Jdp5A is dramatically greater in the presence of ATP. Insets show the similar increases in intensity and shifts in wavelength of maximum emission for Jdp5A and p5A (50 nm) upon binding to lidless DnaK(1–517).
NMR
For analysis of resonance broadening in [15N]Jdp5-cam, a 100 μm stock solution was prepared by combining 166.7 μl of 1.2 mm [15N]Jdp5-cam, 200 μl of D2O, 6 μl of NaN3, 15 μl of (2,2,3,3-D4) sodium-3-trimethylsilylpropionate, 100 μl of 1 m MOPS-NaOH, pH 6.7, and 467 μl of H2O. Aliquots of 600 μl from the 100 μm [15N]Jdp5-cam stock solution were used for each titration experiment. The final concentrations for Kase, DnaK, lidless DnaK(1–517), and DnaK(387–552)-ye were 0, 66.7, 127, and 183 μm, and the final concentration for ATP where it is stated to be present was 1 mm. Two-dimensional 1H,15N HSQC NMR spectra employed the standard Bruker pulse sequence phase sensitive Echo/Antiecho-TPPI gradient selection (38, 39) (invietgp) with 1000 acquisition points for spectral width 2003 Hz in the 1H dimension and 256 increments in t1 for 1800 Hz in the 15N dimension.
Single-turnover ATPase Activity Assays
Single-turnover ATPase assays were performed at room temperature as described before with little modification (15). Briefly, a mixture of 2 nm ATP and 1 nm [α-32P]ATP were preincubated with 2 μm lidless DnaK wild-type or T199A proteins. Then Jd or Jdp5 were added to initiate the hydrolysis reactions. At each specified time point, reactions were quenched by mixing 8 μl of the mixture with 2 μl of 1 n HCl, and the mixture was then put on ice. From each sample, 2 μl was spotted onto a poly(ethylenimine)-cellulose thin-layer chromatography sheet and developed in 1 m formic acid with 0.5 m LiCl. Dried sheets were exposed to a Fuji phosphorimaging plate, and images were recorded by a Fuji BAS 300 and analyzed with Multi Gauge Version 3.0 (Fuji Photo Film Co., Ltd.). The fraction ADP at each time point was corrected from the background level in samples without lidless DnaK, and then data were fit to a first-order rate equation using Origin.
Assays for Jd Function in Vivo
Bacterial viability was determined by first transforming either E. coli Δ3 or E. coli Δ4 with the plasmid encoding the protein of interest or the empty vector. The transformants were grown overnight at 30 °C. The overnight cultures were serially diluted and spotted on to Luria-Bertani agar plates containing the appropriate antibiotics (100 μg/μl ampicillin, 15 μg/μl chloramphenicol) and l-arabinose where indicated. IPTG was used as an inducer for the pGPE plasmid where indicated. Plates were then incubated at 30 °C for 20 h.
RESULTS
The DnaK “Lid” Is Dispensable for Jd Function
To test whether the Jd could function in the context of lidless DnaK(1–517), the binding of a Jd fusion protein was analyzed in the absence and presence of ATP. The fusion protein, Jdp5, consists of an N-terminal Jd and C-terminal peptide p5 (CLLLSAPRR) (15). To monitor the binding of Jdp5 or p5 to DnaK or lidless DnaK(1–517) with fluorescence, the single cysteine in Jdp5 or p5 was modified with acrylodan. Previous studies indicated that the peptide moiety of acrylodan-labeled Jdp5 (Jdp5A) bound to the peptide binding domain of DnaK in a manner similar to that of the acrylodan-labeled peptide (p5A) (15). Moreover, binding experiments showed that Jdp5A competes with p5A for binding to DnaK (data not shown).
The binding affinities of Jdp5A and p5A for lidless DnaK(1–517) were determined by monitoring the change in acrylodan fluorescence as the concentration of lidless DnaK(1–517) was increased (Fig. 1). Acrylodan fluorescence increased and shifted to lower wavelengths with increasing concentrations of lidless DnaK(1–517), and the changes were saturated at the highest concentrations of lidless DnaK(1–517). Lidless DnaK(1–517) caused an ∼1.2-fold increase in fluorescence and a blue shift of 10 nm compared with the 2-fold increase in fluorescence and a blue shift of 15 nm caused by DnaK (15).
In the absence of ATP, fusion of the Jd to the peptide increased peptide affinity for lidless DnaK(1–517) by 10-fold, which is somewhat greater than the 3-fold increase for DnaK (Table 1). In the presence of ATP, fusion of the Jd to the peptide increased apparent affinity for lidless DnaK(1–517) more than 400,000-fold, which is 20 times greater than the increase of 20,000-fold for DnaK (Table 1).
TABLE 1.
Binding of peptide and fusion protein to DnaK and DnaK(1–517)
A DnaK-bound Client-Peptide Fusion Tethers the Jd to the ATPase Domain
To better understand how the Jd interacts with DnaK when the Jd is tethered to a client peptide, NMR perturbations of the carbamidomethylated fusion protein, [15N]Jdp5-cam, were monitored during titration of Kase, DnaK, lidless DnaK(1–517), or DnaK(387–552)-ye, which is a recombinant form of the DnaK peptide binding domain (36). The various DnaK-related proteins are illustrated schematically in Fig. 2. The titration points were ratios of DnaK-related protein to [15N]Jdp5-cam of 0.67:1.00 and 1.27:1.00 and 1.83:1.00. Chemical shift perturbations observed during the titration of Kase into [15N]Jdp5-cam were similar to those for titration of Kase into [15N]Jd (16), and there was overall moderate broadening of resonances (Fig. 3b). During the titration with DnaK, half of the backbone amide resonances of [15N]Jdp5-cam were severely broadened when the concentration of DnaK reached a molar excess (Fig. 3c), and resonances for all backbone amides were severely broadened by the third point in the titration (data not shown). Titration of lidless DnaK(1–517) into [15N]Jdp5-cam caused the most dramatic effect on the spectrum. With the first addition of lidless DnaK(1–517), almost all of the resonances disappeared (Fig. 3d).
FIGURE 2.

Diagrams illustrating the domain structure of DnaJ- and DnaK-related proteins used in this work. The p5 peptide sequence does not correspond to any part of DnaJ or DnaK. DnaJ domains are indicated as follows: J, J-domain; G/F, glycine/phenylalanine-rich domain; Zn, zinc binding domain; CTD, C-terminal domain (13). DnaK domains are indicated as follows: ATPase, ATPase domain; Linker, interdomain linker; PBdβ, β subdomain of the peptide-binding domain; PBdα, α subdomain of the peptide-binding domain.
FIGURE 3.

Binding of [15N]Jdp5 to DnaK-related proteins, as revealed by two-dimensional 1N]H-15N]N HSQC NMR spectra. a, shown is [15N]Jdp5 alone. b, shown are [15N]Jdp5 and Kase at a 1:1.27 molar ratio. c, shown are [15N]Jdp5 and DnaK at a 1:1.27 molar ratio. d, shown are [15N]Jdp5 and lidless DnaK(1–517) at a 1:0.67 molar ratio. In the presence of Kase there is little broadening of [15N]Jdp5 resonances. In the presence of DnaK, approximately half of the resonances are too broad to detect at this contour level. In the presence of substoichiometric lidless DnaK(1–517), backbone resonances are obliterated. Remaining resonances correspond to the side chains of glutamine residues and the C-terminal backbone amide.
The varied broadening of resonances in [15N]Jdp5-cam during binding to the different DnaK molecules was likely controlled by the interaction of the p5 moiety with the peptide binding domain. To show that severe resonance broadening in the Jd moiety depends on binding of the p5 moiety, excess p5 peptide was added to an NMR sample containing [15N]Jdp5-cam and DnaK. As expected, resonance broadening was substantially reversed (Fig. 4, a and b). To determine whether resonance broadening in the Jd moiety depended on the presence of the ATPase domain in DnaK, a recombinant peptide binding domain, DnaK(387–552)-ye (28), was titrated into [15N]Jdp5-cam. At the highest point in the titration, resonances of the Jd moiety showed little change, whereas the resonances corresponding to the p5 moiety were obliterated (Fig. 4c). The addition of excess p5 peptide reversed the broadening (Fig. 4d). These results suggested that the severe resonance broadening in the Jd moiety was caused by its interaction with the ATPase domain, and this interaction depended on binding of the p5 moiety to the peptide binding domain.
FIGURE 4.

The dependence of J-domain broadening on its being tethered to DnaK by the fused peptide. a, shown are [15N]Jdp5 and DnaK at a 1:1.83 molar ratio. b, shown are [15N]Jdp5, DnaK, and p5 peptide at a 1:1.83:10 molar ratio. c, shown are [15N]Jdp5 and DnaK(387–552)-ye at a 1:1.83 molar ratio. d, shown are [15N]Jdp5, DnaK(387–552)-ye, and p5 at a 1:1.83:10 molar ratio. In the presence of DnaK, many more [15N]Jdp5 resonances are evident when excess p5 is added, indicating that the peptide has displaced the peptide moiety of [15N]Jdp5 from the peptide binding domain, resulting in less binding of the J-domain to the ATPase domain. In the presence of DnaK(387–552)-ye, broadening is essentially limited to resonances of the p5 moiety of [15N]Jdp5 (boxes) because the ATPase domain is absent. The addition of excess p5 eliminated broadening of these signals.
The above experiments analyzed [15N]Jdp5-cam binding to DnaK in the absence of ATP, and therefore, they rely on the intrinsic exchange of DnaK between the open and closed states of the peptide-binding domain. Because ATP is present in the biological context, we would like to analyze binding in the presence of ATP. However, the rapid conversion of ATP to ADP in the NMR sample precluded studies with wild-type DnaK or DnaK(1–517), and thus, they were undertaken with the ATPase-defective mutant DnaK(1–517)T199A. DnaKT199A was shown to undergo the ATP-dependent conformational transition, but its rate of ATP hydrolysis is vastly reduced (40). In the absence of ATP, the broadening of [15N]Jdp5-cam resonances by DnaK(1–517)T199A was concentration-dependent and comparable with that caused by DnaK (Fig. 5b). When 1 mm ATP was included, broadening for many resonances increased (Fig. 5c).
FIGURE 5.
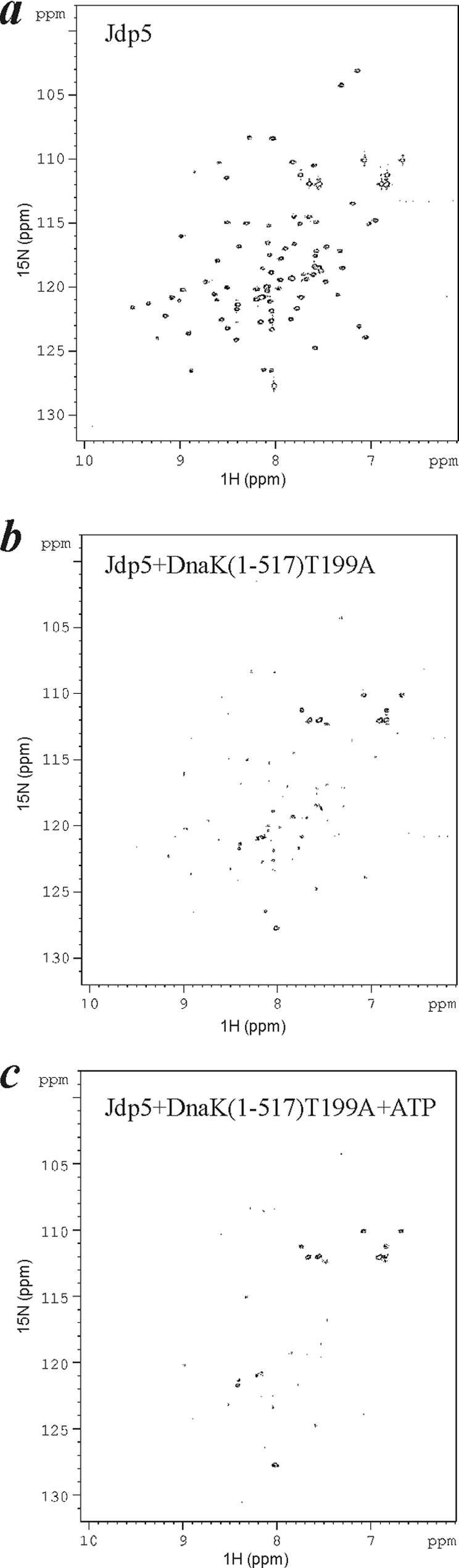
The effect on J-domain broadening by ATP binding to DnaK(1–517)T199A. a, shown is [15N]Jdp5 alone. b, shown are [15N]Jdp5 and DnaK(1–517)T199A at a 1:0.67 molar ratio. c, shown is [15N]Jdp5 with DnaK(1–517)T199A after inclusion of 1 mm ATP. Increased broadening in the presence of ATP suggests that [15N]Jdp5 binding and dissociation with DnaK(1–517)T199A has accelerated toward the intermediate exchange regime.
Jdp5 Stimulates the ATP Hydrolysis by DnaK
When ATP hydrolysis in DnaK is stimulated by DnaJ, ADP/ATP-exchange becomes rate-limiting (11). Therefore, Jdp5 stimulation of ATP hydrolysis by DnaK(1–517) was examined under single-turnover conditions, wherein the enzyme concentration is in large excess over that of ATP. Both Jd and Jdp5 can stimulate the ATPase activity of DnaK(1–517). However, the degree of stimulation by Jdp5 was vastly greater than that by Jd. The rate constant for ATP hydrolysis by DnaK(1–517) in the presence of 20 μm Jd was 0.28 ± 0.047 min−1, which was 4-fold higher than the unstimulated rate (0.07 ± 0.009 min−1). The reaction stimulated by 20 μm Jdp5 went so fast that within 10 s more than 90% of the ATP had been converted to ADP. As a result, the reaction-rate data obtained by manual mixing was inadequate for fitting to a first-order process. We estimated a lower limit of 14 min−1 as the rate of ATP hydrolysis stimulated by Jdp5 under these conditions. The rate constant for unstimulated hydrolysis catalyzed by DnaK(1–517)T199A was 0.007 ± 0.002 min−1, 10-fold lower than that of the DnaK(1–517).
Jd Overexpression Shifts DnaK into Client-bound Complexes
The ability of Jdp5 or the Jd to function in vivo was tested using the E. coli strain Δ3 from which the genes encoding three Jd proteins (DnaJ, CbpA, and DjlA) had been mutated (29). As a consequence of the mutations, the Δ3 strain was unable to grow at temperatures above 30 °C, and neither Jdp5 nor Jd was able to complement the defect under any conditions tested (data not shown).
Overexpression of the Jd (composed of DnaJ residues 1–78) was extremely toxic to E. coli Δ3 (Fig. 6, left panel). To test if the toxicity depended on a functional Jd, the Jd mutants containing the substitution D35N or H33Q were tested, and neither protein was found to be toxic. The lack of toxicity by JdD35N was not due to a lower level of protein expression (Fig. 6, right panel).
FIGURE 6.

The Jd exerts a DnaK-dependent toxic effect on E. coli growth. Left, DnaJ, Jd(1–78), Jd(1–78) mutants and Jd(1–78)p5 were expressed from the l-arabinose-inducible vector pBAD22 in the Δ3 strain at 30 °C in the absence or presence of 1% l-arabinose. In the presence of l-arabinose, the expressed construct Jd(1–78) arrested cell growth. The Jd(1–78) was expressed from the l-arabinose inducible vector pBAD33 in the Δ4 strain at 30 °C in the absence or presence of 1% l-arabinose (ara). The empty vector pWSK29 or the vector pWSK29DnaK+ expressing DnaK under the control of its native promoter was also present. Jd(1–78) toxicity was relieved in the dnaK-null strain Δ4, but Jd(1–78) toxicity returned when DnaK was also co-expressed. Right, 15% SDS-PAGE showing that the lack of toxicity in Jd(1–78) is not due to a lower level of protein expression. Δ3 cells containing the empty vector pBAD22, Jd(1–78), or Jd(1–78)D35N were induced (I) with l-arabinose. After lysis of the cells, the proteins present in the soluble fractions were separated by SDS-PAGE. Uninduced (U) samples were also analyzed.
The toxicity of the Jd could have been due to its effect on DnaK because the Jd is known to stimulate DnaK ATPase activity (15). Indeed Jd toxicity was found to specifically depend on the presence of DnaK, i.e. the Jd toxicity on E. coli growth was relieved when the Jd was expressed in the E. coli strain Δ4 (Fig. 6, left panel), which lacks DnaK in addition to the three Jd proteins (30). To demonstrate that the lack of toxicity is due to the absence of DnaK and not due to accumulation of suppressors that occur in the Δ4 strain, DnaK was co-expressed with the Jd in the Δ4 strain. The toxicity of the Jd was completely restored.
Several mechanisms for Jd toxicity could be proposed, such as the sequestration of DnaK or the sequestration of an important DnaK client protein(s). The existence of the Δ4 strain indicated that DnaK is not necessary for cell viability. Therefore, sequestration of DnaK probably was not the mechanism through which the Jd caused toxicity. It is likely that the Jd stimulation of DnaK ATPase activity trapped an important client(s) in a complex with DnaK. We reasoned that upon overexpressing DnaK nucleotide exchange factor, GrpE, the complex might continue through its reaction cycle and release the client protein(s). To test this hypothesis, GrpE was co-expressed with the Jd in the Δ3 strain. At high levels of GrpE, Jd toxicity was completely relieved (Fig. 7a). This hypothesis is also supported by the fact that overexpression of Jdp5 is not toxic (Fig. 6, left panel). This may be due to the peptide moiety of Jdp5 being trapped in the substrate binding site of DnaK instead of an important client proteins(s).
FIGURE 7.

Compensation of Jd toxicity by co-expression of GrpE or lidless DnaK(1–517). a, DnaJ, Jd(1–78), and Jd(1–78)D35N were expressed from the l-arabinose-inducible vector pBAD33 in the Δ3 strain at 30 °C in the absence or presence of 1% l-arabinose (ara). GrpE was simultaneously expressed from the IPTG-inducible vector pSE380 with 0.05 mm IPTG. Overexpression of GrpE also compensated for the toxicity conferred by Jd(1–78) (compare dilutions labeled Jd(1–78)+vector and Jd(1–78)+GrpE for growth in the presence of arabinose and IPTG). b, Jd(1–78) was expressed from the l-arabinose inducible vector pBAD33 in the Δ4 strain at 30 °C in the absence or presence of 1% l-arabinose. The empty vector pWSK29, the vector pWSK29DnaK+ containing DnaK under its native promoter, or the vector pWSKLidless containing lidless DnaK(1–517) also under the DnaK native promoter were present. When lidless DnaK(1–517) was expressed in the place of DnaK, overexpression of Jd(1–78) did not result in any toxicity (compare dilutions labeled Jd(1–78)+DnaK and Jd(1–78)+Lidless for growth in the presence of arabinose). c, 15% SDS-PAGE showing lack of toxicity upon co-overexpression of lidless DnaK(1–517) is not due to a poor expression level of the protein. Δ4 cells containing the empty vector pSE380, DnaK, or lidless DnaK(1–517) were induced (I) with IPTG. After lysis of the cells, the proteins present in the soluble fractions were separated by SDS-PAGE. Uninduced (U) samples were also analyzed.
It had been shown by Slepenkov and Witt (10) that lidless DnaK(1–517) has a shorter lifetime in its complex with peptide, as compared with wild-type DnaK. Due to the ability of lidless DnaK(1–517) to release client faster than full-length DnaK, the presence of excess Jd might not sequester as much of the putative DnaK client and, thus, have reduced toxicity. Consistent with this interpretation, when lidless DnaK(1–517) was co-expressed with the Jd in the Δ4 strain, the Jd did not exhibit toxicity (Fig. 7b). The difference observed for lidless DnaK(1–517) versus full-length DnaK was not due to different levels of protein expression (Fig. 7c).
DISCUSSION
Experiments presented in this study demonstrate that a client peptide, which is bound to the DnaK peptide binding domain, can tether an associated Jd to the DnaK ATPase domain. This is a potential mechanism for DnaJ to couple peptide loading with ATP hydrolysis in DnaK (Fig. 8). We considered an alternative mechanism, namely, that the Jd mediates allosteric communication between the ATPase and peptide binding domains. However, two experiments designed to reveal an interaction of the Jd with the peptide binding domain in the absence of ATP produced negative results. First, the binding to DnaK or lidless DnaK(1–517) of acrylodan-labeled peptide was not affected by the presence of excess free Jd, and second, amide-group NMR perturbations for [15N]Jd binding to DnaK were not affected by the addition of peptide (data not shown). Thus, the Jd and the occupied peptide binding domain did not compete for interactions with the ATPase domain. This conclusion is consistent with a recent NMR study on allosteric behavior in DnaK, suggesting that the two domains of DnaK interact only when ATP is bound (41). Although the Jd could interact with the DnaK peptide binding domain in the presence of ATP, this possibility seems unlikely in view of the fact the Jd binds to DnaK with nearly equal affinity in the presence of ADP and ATP (Ref. 16 and data not shown).
FIGURE 8.

Model for association of Jdp5 with DnaK-ATP. 1, the p5 moiety of Jdp5 binds to the peptide binding domain of DnaK. Tethering of the Jd is illustrated as multiple DnaK-bound conformations of Jdp5, wherein the p5 moiety is fixed, but the Jd explores alternative orientations. 2, the binding of Jd to the DnaK ATPase domain promotes a conformational change in DnaK that accelerates ATP hydrolysis. The activated DnaK is presumed to be transient, and thus, it is illustrated in brackets. 3, ATP hydrolysis stabilizes the closed conformation of the peptide binding domain. 4, nucleotide exchange reopens the peptide binding domain, permitting the dissociation of the p5 moiety. The association of a DnaJ-client complex with DnaK is predicted to be similar to that of Jdp5, except that non-covalently bound DnaJ can dissociate before nucleotide exchange and opening of the peptide binding domain in DnaK.
The results also suggested that Jd function does not depend on the presence of the helical lid portion of the peptide binding domain. Peptide affinity for the DnaK peptide binding domain could be dramatically enhanced by fusion with the Jd even when most of the helical lid was removed from peptide binding domain. Thus, the lid probably does not contact the Jd. This conclusion is important in the context of the recent suggestion on the basis of the structure of the ATP-bound Hsp110/Sse1 from yeast (26) that the lid contacts the ATPase domain in the ATP-bound DnaK.
The artificial protein Jdp5 is intended to mimic an Hsp40-client complex, wherein the client protein would be associated with a Jd through the Hsp40 client-binding domains (15, 42). Upon binding of the client to the peptide-binding domain of Hsp70, the Jd is tethered to the ATPase domain, where the Jd is poised to stimulate ATP hydrolysis.
Support for the tethering mechanism was provided by NMR studies on the equilibrium binding of [15N]Jdp5 to the various DnaK constructs. The results suggested that the p5 moiety of [15N]Jdp5 can regulate the frequency of interaction between the Jd moiety and the ATPase domain. The addition of DnaK to a sample of [15N]Jdp5 resulted in the broadening of [15N]Jdp5 amide resonances in proportion to the concentration of DnaK, whereas the addition of even a substoichiometric amount of lidless DnaK(1–517) resulted in the obliteration of the [15N]Jdp5 amide resonances. Because resonance broadening in the Jd moiety was not observed when [15N]Jdp5 was bound to the recombinant peptide-binding domain, broadening depends on Jd interaction with the ATPase domain. A dramatic loss of amide resonances with lidless DnaK(1–517) is expected if the frequency of exchange between free and bound Jd in [15N]Jdp5 is comparable with the chemical-shift differences for resonances of free and bound Jd (intermediate exchange regime). In contrast, the proportional loss of amide resonances with DnaK suggests that exchange between free and bound Jd in this complex is relatively slow (slow exchange regime). Buczynski et al. (9) have shown that peptide dissociation from lidless DnaK(1–517) is ∼100 times faster than from wild-type DnaK, and on-rates were also increased. Thus, the frequency of Jd binding to the ATPase domain could be increased by faster exchange of the p5 moiety of [15N]Jdp5 in and out of the peptide binding domain of lidless DnaK(1–517). Whether Jd and peptide can simultaneously bind to DnaK or DnaK(1–517) and, if so, for what duration remains unclear from our experiments. Further studies using NMR relaxation-dispersion measurements should resolve distinct timescales for exchange of the two moieties of [15N]Jdp5 in their respective binding sites.
The ATP-dependent conversion of DnaK to the open conformation is expected to accelerate the exchange of the p5 moiety of [15N]Jdp5 in and out of the peptide binding domain. However, the effect of ATP on the [15N]Jdp5 HSQC NMR spectrum was not observed due to the rapid conversion of ATP to ADP in the NMR sample, resulting from Jdp5-stimulated ATP hydrolysis in lidless DnaK(1–517) (data not shown). Thus, the effect of ATP was examined for [15N]Jdp5 exchange into the hydrolysis-defective mutant, DnaK(1–517)T199A. The ATP-triggered off-rate of a peptide from full-length DnaKT199A was shown to be the same as that from the wild-type DnaK, although DnaKT199A exhibited a 30-fold slower rate of ATP hydrolysis (40). A similarly reduced rate of hydrolysis in the lidless DnaK(1–517)T199A allowed the recording of an HSQC NMR spectrum before the products ADP and Pi had accumulated. In the absence of ATP, broadening of [15N]Jdp5 NMR signals was less severe with DnaK(1–517)T199A, as compared with DnaK(1–517). This may be due to a slower intrinsic rate of exchange between “open” and “closed” states of the peptide binding domain in the mutant protein. However, as expected, broadening of [15N]Jdp5 resonances increased when ATP was added, indicating that the rate of exchange had increased and was now closer to the intermediate-exchange regime. Thus, the rate of peptide exchange in the ATP-bound DnaK controls access of the Jd to the DnaK ATPase domain by tethering the Jd near its binding site on the ATPase domain.
The data from our experiments in vivo suggested that excessive Jd can shift DnaK into a complex that sequesters a client protein(s) that is crucial for cell growth. In agreement with this model, the overexpression of GrpE relieved Jd toxicity by switching DnaK to the ATP-bound form, which releases the client. The ability of GrpE to release DnaK-sequestered client proteins explains the seemingly paradoxical observation that GrpE is essential even though DnaK is not (43). Replacing DnaK with lidless DnaK(1–517) probably also relieved Jd toxicity because DnaK(1–517) releases client proteins more quickly. The lack of toxicity by Jdp5 overexpression is in agreement with this model, presumably because the peptide portion of the fusion protein displaced the client that became trapped in DnaK upon overexpression of the unfused Jd.
It is interesting that overexpression of DnaJ was not toxic under conditions in which Jd overexpression is extremely toxic. This may be due to a region(s) other than Jd interacting with the peptide binding domain of DnaK and subsequently promoting the release of the important client protein(s) that was transiently captured by DnaK. Indeed, DnaJ has previously been reported to interact with both the ATPase domain and the peptide binding domain of DnaK (22). Karzai and McMacken (19) suggested that the G/F-rich region interacts with the peptide binding domain during DnaJ-mediated stimulation of the DnaK ATPase activity. Although Vázquez-Laslop et al. (44) demonstrated that overexpression of DnaJ is toxic to cells and increases cell survival in the presence of exogenous bacteriocidal antibiotics, the bacterial strain used in their experiments has the full complement of J-domain proteins. Because these other J-domain proteins may compete with DnaJ for binding to clients and/or DnaK, one or more of them may prevent DnaJ from displacing the important client protein(s) from DnaK. Our in vivo toxicity test was conducted in the E. coli strain Δ3, which lacks all three J-domain proteins. Our results are in agreement with previous reports using the same bacterial strain wherein overexpression of full-length DnaJ was not toxic (29, 30).
The potential for DnaK to sequester clients is tightly regulated under normal circumstances. The very low intrinsic ATPase rate of DnaK and the low affinity of the Jd for the ATPase domain ensure that DnaK clients do not bind stably to DnaK unless they simultaneously attract a DnaJ molecule to the complex. Thus, the triggered-capture mechanism requires that the client be recognized by two chaperones to engage the machine. As a result, the potential for toxic DnaK binding to clients is minimized.
This work was supported by grants from the National Science Foundation (to S. J. L.), the Swiss National Science Foundation (to C. G.), the Canton of Geneva (to C. G.), and the CNRS-Action Thématique et Incitative sur Programme (to P. G.).
- Jd
- J-domain
- MOPS
- 4-morpholinepropanesulfonic acid
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- HSQC
- heteronuclear single quantum correlation.
REFERENCES
- 1.Chai Y., Koppenhafer S. L., Bonini N. M., Paulson H. L. (1999) J. Neurosci. 19, 10338–10347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frydman J. (2001) Annu. Rev. Biochem. 70, 603–647 [DOI] [PubMed] [Google Scholar]
- 3.Young B. P., Craven R. A., Reid P. J., Willer M., Stirling C. J. (2001) EMBO J. 20, 262–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greener T., Grant B., Zhang Y., Wu X., Greene L. E., Hirsh D., Eisenberg E. (2001) Nat. Cell Biol. 3, 215–219 [DOI] [PubMed] [Google Scholar]
- 5.Flaherty K. M., DeLuca-Flaherty C., McKay D. B. (1990) Nature 346, 623–628 [DOI] [PubMed] [Google Scholar]
- 6.Zhu X., Zhao X., Burkholder W. F., Gragerov A., Ogata C. M., Gottesman M. E., Hendrickson W. A. (1996) Science 272, 1606–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Slepenkov S. V., Witt S. N. (1998) Biochemistry 37, 16749–16756 [DOI] [PubMed] [Google Scholar]
- 8.Gisler S. M., Pierpaoli E. V., Christen P. (1998) J. Mol. Biol. 279, 833–840 [DOI] [PubMed] [Google Scholar]
- 9.Buczynski G., Slepenkov S. V., Sehorn M. G., Witt S. N. (2001) J. Biol. Chem. 276, 27231–27236 [DOI] [PubMed] [Google Scholar]
- 10.Slepenkov S. V., Witt S. N. (2002) Biochemistry 41, 12224–12235 [DOI] [PubMed] [Google Scholar]
- 11.Liberek K., Marszalek J., Ang D., Georgopoulos C., Zylicz M. (1991) Proc. Natl. Acad. Sci. U.S.A. 88, 2874–2878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Packschies L., Theyssen H., Buchberger A., Bukau B., Goody R. S., Reinstein J. (1997) Biochemistry 36, 3417–3422 [DOI] [PubMed] [Google Scholar]
- 13.Genevaux P., Georgopoulos C., Kelley W. L. (2007) Mol. Microbiol. 66, 840–857 [DOI] [PubMed] [Google Scholar]
- 14.Misselwitz B., Staeck O., Rapoport T. A. (1998) Mol. Cell 2, 593–603 [DOI] [PubMed] [Google Scholar]
- 15.Wittung-Stafshede P., Guidry J., Horne B. E., Landry S. J. (2003) Biochemistry 42, 4937–4944 [DOI] [PubMed] [Google Scholar]
- 16.Greene M. K., Maskos K., Landry S. J. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 6108–6113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheetham M. E., Caplan A. J. (1998) Cell Stress Chaperones 3, 28–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laufen T., Mayer M. P., Beisel C., Klostermeier D., Mogk A., Reinstein J., Bukau B. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 5452–5457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karzai A. W., McMacken R. (1996) J. Biol. Chem. 271, 11236–11246 [DOI] [PubMed] [Google Scholar]
- 20.Mayer M. P., Schröder H., Rüdiger S., Paal K., Laufen T., Bukau B. (2000) Nat. Struct. Biol. 7, 586–593 [DOI] [PubMed] [Google Scholar]
- 21.Russell R., Wali, Karzai A., Mehl A. F., McMacken R. (1999) Biochemistry 38, 4165–4176 [DOI] [PubMed] [Google Scholar]
- 22.Mayer M. P., Laufen T., Paal K., McCarty J. S., Bukau B. (1999) J. Mol. Biol. 289, 1131–1144 [DOI] [PubMed] [Google Scholar]
- 23.Suh W. C., Burkholder W. F., Lu C. Z., Zhao X., Gottesman M. E., Gross C. A. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 15223–15228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gässler C. S., Buchberger A., Laufen T., Mayer M. P., Schröder H., Valencia A., Bukau B. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 15229–15234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang J., Prasad K., Lafer E. M., Sousa R. (2005) Mol. Cell 20, 513–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Q., Hendrickson W. A. (2007) Cell 131, 106–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davis J. E., Voisine C., Craig E. A. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 9269–9276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Awad W., Estrada I., Shen Y., Hendershot L. M. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 1164–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Genevaux P., Schwager F., Georgopoulos C., Kelley W. L. (2002) Genetics 162, 1045–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cajo G. C., Horne B. E., Kelley W. L., Schwager F., Georgopoulos C., Genevaux P. (2006) J. Biol. Chem. 281, 12436–12444 [DOI] [PubMed] [Google Scholar]
- 31.Kelley W. L., Georgopoulos C. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 3679–3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Genevaux P., Keppel F., Schwager F., Langendijk-Genevaux P. S., Hartl F. U., Georgopoulos C. (2004) EMBO Rep. 5, 195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guzman L. M., Belin D., Carson M. J., Beckwith J. (1995) J. Bacteriol. 177, 4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Russell R., Jordan R., McMacken R. (1998) Biochemistry 37, 596–607 [DOI] [PubMed] [Google Scholar]
- 35.Bukau B., Walker G. C. (1989) J. Bacteriol. 171, 6030–6038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Swain J. F., Schulz E. G., Gierasch L. M. (2006) J. Biol. Chem. 281, 1605–1611 [DOI] [PubMed] [Google Scholar]
- 37.Coligan J. E., Dunn B. M., Ploegh H. L., Speicher D. W., Wingfield P. T. (eds) (1998) Current Protocols in Protein Science, pp. 15.1.3–15.1.5, Vol. 2, John Wiley and Sons, Inc., New York [Google Scholar]
- 38.Palmer A. G., Cavanagh J., Wright P. E., Rance M. (1991) J. Magn. Reson. 93, 151–170 [Google Scholar]
- 39.Kay L. E., Keifer P., Saarinen T. (1992) J. Am. Chem. Soc. 114, 10663–10665 [Google Scholar]
- 40.Barthel T. K., Zhang J., Walker G. C. (2001) J. Bacteriol. 183, 5482–5490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swain J. F., Dinler G., Sivendran R., Montgomery D. L., Stotz M., Gierasch L. M. (2007) Mol. Cell 26, 27–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han W., Christen P. (2003) J. Biol. Chem. 278, 19038–19043 [DOI] [PubMed] [Google Scholar]
- 43.Ang D., Georgopoulos C. (1989) J. Bacteriol. 171, 2748–2755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vázquez-Laslop N., Lee H., Neyfakh A. A. (2006) J. Bacteriol. 188, 3494–3497 [DOI] [PMC free article] [PubMed] [Google Scholar]