

Abstract
The pneumococcal epsilon zeta antitoxin toxin (PezAT) system is a chromosomally encoded, class II toxin antitoxin system from the human pathogen Streptococcus pneumnoniae. Neutralization of the bacteriotoxic protein PezT is carried out by complex formation with its cognate antitoxin PezA. Here we study the stability of the inhibitory complex in vivo and in vitro. We found that toxin release is impeded in Escherichia coli and Bacillus subtilis due to the proteolytic resistance of PezA once bound to PezT. These findings are supported by in vitro experiments demonstrating a strong thermodynamic stabilization of both proteins upon binding. A detailed kinetic analysis of PezAT assembly revealed that these particular features of PezAT are based on a strong, electrostatically guided binding mechanism leading to a stable toxin antitoxin complex with femtomolar affinity. Our data show that PezAT complex formation is distinct to all other conventional toxin antitoxin modules and a controlled mode of toxin release is required for activation.
Keywords: Bacterial Toxins, Cell Death, Enzyme Inhibitors, Protein Assembly, Protein-Protein Interactions, Toxin Antitoxin Systems, Assembly Kinetics
Introduction
Toxin-antitoxin (TA)2 systems were initially discovered on prokaryotic low-copy number plasmids, where they ensure stable inheritance in a growing population. By a postsegregational killing mechanism these systems trigger cell death of offspring that have lost the plasmid and were thus also designated as addiction or suicide modules. Most of those TA systems are transcribed from a single bicistronic operon, where the first gene product (antitoxin) inhibits the cytotoxic function upon binding to the product of the succeeding gene (toxin) (1). Addiction to the TA locus is achieved by differential proteolytic stabilities of the two proteins because antitoxic proteins are metabolically less stable and prone to faster degradation by intracellular proteases when compared with their cognate toxins. Hence, loss of the TA-loci results in a faster reduction of the pool of antitoxic proteins than that of toxins due to lack of de novo synthesis, which eventually confronts the host with the freed and corruptive activity of the toxin.
In addition to plasmid-encoded TA systems, similar modules were also discovered to be encoded from chromosomes of free-living bacteria (2). However, the function of these chromosomally encoded TA systems is currently under debate (3, 4) and different functions such as growth control (5), stabilization of mobile genetic elements (6, 7), or programmed cell death (8) have been suggested.
TA systems of the ϵ/ζ family are encoded on various plasmids or chromosomes of Gram-positive pathogens like Streptococcus pyogenes or Streptococcus pneumoniae as well as Gram-negative such as Fusobacterium nucleatum (9–12). The related ζ-toxins are ∼30-kDa monomeric proteins owning a monophosphate kinase-fold and are thought to phosphorylate a still enigmatic substrate (13). In addition to the toxin-inactivating function of the antitoxin ϵ, various homologues harbor a transcriptional repressor domain that regulates the operon (11, 14).
The PezAT system (pneumococcal epsilon zeta) from S. pneumoniae encoded from the chromosomal pneumococcal pathogenicity island 1 (PPI1) (15) is one of the only two ϵ/ζ systems characterized to date. S. pneumoniae strains containing PPI1 encode for various virulence factors that are highly beneficial for infection and these strains show increased pathogenicity. Strains lacking the toxin pezT gene displayed an attenuated infection progress and were out-competed by wild type strains. This finding indicated an important role of PezT for pneumococcal pathogenicity as virulence factor (16).
Expression of PezT leads to growth arrest in Escherichia coli (11). The toxic activity of PezT is inhibited by complex formation with the dimeric three-helix bundle domain of PezA, which is flexibly linked to the amino-terminal helix-turn-helix domain. The crystal structure of the inhibitory complex reveals a dumbbell-shaped heterotetrameric assembly, where the central dimeric three-helix bundle domain of the antitoxin binds two toxins and neutralizes them by blocking the conserved nucleotide binding site. Expression of PezAT is regulated by the transcriptional repressor domain of PezA, which binds to the σ70-like promotor of the bicistronic pezAT operon.
The formation of the inhibitory TA complex is the crucial step in toxin neutralization of bacterial TA systems. We investigated the proteolytic stability of the inhibitory PezAT complex under in vivo-like conditions in bacterial model systems. Furthermore, we performed a detailed characterization of the association/dissociation dynamics of the PezAT system and thereby can provide a general model for heterotetramer formation. Taken together, the results of this study indicate that PezT gets sequestered by PezA in a quasi-irreversible manner indicating that PezAT is a unique TA system.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
PezA, PezT, and all of their variants were expressed from pET28b vectors using the E. coli BL21(DE3)/Codon Plus-RIL strain (Stratagene). Site-directed mutagenesis was performed using the QuikChange protocol (Stratagene). For in vivo studies of protein stability the pezAT open reading frame was cloned into the pASK5+ vector (IBA) using BsaI (NEB). This vector results in an N-terminal Strep II tag fusion with the wild type PezA. All constructs were verified by DNA sequencing. Overexpression of protein constructs from the pET system, as well as purification of PezT variants was carried out as described (11). Note that all PezT constructs bore a non-cleavable C-terminal His6 tag. The initial step of PezA purification using nickel-affinity chromatography was the same as for PezT (11). Subsequently, PezA was dialyzed overnight against buffer A (all buffer compositions are listed under supplemental Table S1) in the presence of thrombin to cleave the N-terminal His tag, except for PezA preparations used in Western blot analysis. The protein was then loaded onto a MonoQ column (GE Healthcare) equilibrated with buffer B and eluted in a gradient to 60% buffer C. Fractions were analyzed by SDS-PAGE, pooled, and further purified by using a Superose 6 (GE Healthcare) size exclusion column equilibrated with buffer D. All protein concentrations were determined by measuring the absorption at 280 nm. PezA, PezT, and all of their variants were stored in buffer D.
Labeling of PezT(D66T) and PezA(M66C) with Fluorescent Dyes
PezT(D66T) was labeled at cysteine 209 with Alexa 488-maleimide (Invitrogen) according to the manufacturer's protocol. Briefly, to a vial containing 1.5 ml of PezT(D66T) (3 mg/ml in buffer E), 140 μl of 10 mm Alexa 488-maleimide was added dropwise under constant stirring and incubated overnight at 4 °C. The reaction was quenched by adding 10 mm dithioerythritol. Precipitated protein was dissolved in buffer D containing 8 m urea and refolded by rapid dilution (1:15) in buffer D. Free fluorescent dye and residual urea were removed by buffer exchange in a 10-kDa cut-off spin column. The label efficiency was estimated spectroscopically to be ∼100%. Proper refolding of the labeled PezT species was verified by circular dichroism and PezA binding experiments. To obtain the fluorescently labeled His-F-PezA variant, dithioerythritol-free His6-PezA(M66C) was labeled with 1,5-IAEDANS (Invitrogen) by diluting 25 μm His6 PezA(M66C) in buffer G.
A 1,5-IAEDANS stock (20 mm in N,N-dimethylformamide) was added dropwise under rigorous stirring to a final concentration of 380 μm. The mixture was further incubated for 1 h at 4 °C before the labeling reaction was quenched with 10 mm dithioerythritol. Free label was removed similarly as described above. The label efficiency was estimated spectroscopically to be ∼130% due to additional labeling of the second, poorly accessible cysteine at position 50.
In Vivo Stability of PezAT
To monitor degradation of PezA and PezT in vivo, E. coli DH5α cells bearing pASK5+(pezAT) or pASK5+(pezA) were grown in 200 ml of LB at 37 °C in presence of 100 μg/ml of ampicillin. At an A600 of 0.5, protein expression was induced for 1 h by addition of 0.2 μg/ml of anhydrotetracycline. After that, chloramphenicol was added to a final concentration of 50 μg/ml to inhibit protein synthesis. Samples of 1 ml were taken in 1-h intervals and cells were harvested at 16,000 × g for 1 min. Subsequently, the cells were resuspended in 100 μl of buffer H and incubated at 95 °C for 5 min. Equivalents of 7.5 μl/A600 were analyzed by SDS-PAGE and subsequent Coomassie staining.
For the experiments probing PezT release, PezAT was expressed as described above. Both cultures expressing either PezA or PezAT were harvested at an A600 of 0.8 by centrifugation (5 min at 6,000 × g, 4 °C). Cells were washed in LB medium after centrifugation and finally resuspended in 100 ml of LB, which was used to inoculate fresh LB medium supplemented with 100 μg/ml of ampicillin to a final A600 of 0.05. The cell growth was monitored at 37 °C by measuring the A600.
PezAT Degradation in Crude Extract
Cellular crude extract was obtained from Bacillus subtilis YB886 overnight cultures grown in LB medium containing 5 μg/ml of kanamycin. Cells were harvested by centrifugation (15 min at 6,000 × g and 4 °C) and 3 g of wet pellet were washed once in buffer I and finally resuspended in 30 ml of buffer I. Cell walls were opened by sonication for 20 min on ice. The resulting crude extract was supplemented with 10 mm MgCl2, 5 mm dithioerythritol and either 1 μm His-F-PezA, 1 μm PezT(D66T) or 1 μm His-F-PezA·PezT(D66T) complex. Individual samples were incubated at room temperature and aliquots were withdrawn at t = 0, 1, 2, and 3 h. Degradation of His-F-PezA and PezT(D66T) was monitored by ECL Western blot analysis using a Penta-His horseradish peroxidase conjugate (Qiagen) following the supplier's instructions. Additionally, degradation of His-F-PezA was monitored by visualizing IAEDANS fluorescence using an Imago compact imaging system (B&L Systems).
Urea Denaturation of PezAT
To monitor unfolding of PezA, 5 μm PezA, or 5 μm PezA·PezT(D66T,W232Y) complex were incubated 15 h at 10 °C and 5 h at 25 °C in buffer D at different urea concentrations ranging from 0 to 7.2 m. The fluorescence spectral center of mass <λ> for each emission spectrum ranging from 335 to 400 nm was calculated by the following equation,
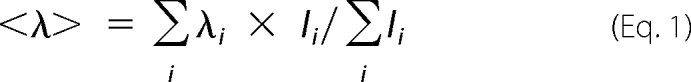 |
where Ii is the fluorescence intensity of the ith recorded point and λi the respective wavelength in nanometer. The change in mean residue ellipticity of the same samples was monitored in a J-810 CD Spectropolarimeter (Jasco) at 222 nm.
Fluorescence Titrations
Toxin-antitoxin binding isotherms were obtained by titrating wild type PezA or PezA(W111F) to a cuvette containing 3 μm PezT(D66T) in buffer D. The change in fluorescence intensity at 347 nm was recorded using a FluoroMax-3 Spectrofluorometer (HORIBA Scientific) with an excitation wavelength at 295 nm.
Stopped-flow Experiments
PezAT association experiments were carried out in a SF61-DX2 stopped-flow instrument from TgK Scientific. The excitation wavelength was set to 295 nm, and the fluorescence was detected using a 320-nm low pass filter. Assembly of PezAT was monitored after rapid mixing of PezT(D66T) with PezA(W111F) in buffer D at 25 °C. The change in fluorescence intensity was measured at 0.25, 0.5, 0.75, 1, 2, 3, and 5 μm toxin antitoxin concentrations for shots with 1:1 stoichiometry and 3/6, 6/3, 2/1, 1.5/1, m and 2/1.5 μm antitoxin/toxin concentrations for asymmetric stoichiometries. 12–15 time traces of each measurement were averaged and analyzed using Dynafit 4.0 (Biokin) (17). In the final fit assuming a two-step model, the apparent rate constants were fitted globally, whereas the trace offsets and amplitudes were fitted locally. Salt dependence of PezAT association was characterized by rapid mixing experiments of PezA and PezT (1 μm and 1:1 stoichiometry) at different NaCl concentrations and by fitting of the two-step model to individual time traces.
Except for measurements at low ionic strength (below 100 mm NaCl), additional time traces for assembly experiments at 3/1 μm antitoxin/toxin concentrations were included in the fit to support convergence into a well defined minimum. To account for any buffer contribution of 50 mm Hepes-NaOH at pH 7.5, 23 mm were added to the overall ionic strength.
The dynamics of the PezA homodimer equilibrium were addressed by rapid dilution experiments where 5 μm PezA was mixed with buffer D to final ratios of 1:15, 1:12, 1:10, 1:8, 1:6, 1:5, and 1:4 in the absence and presence of stoichiometric amounts of tryptophan-free PezT(D66T,W232Y) using a SFM-400 stopped-flow device (BioLogic). Individual time traces from at least 20 shots were averaged per concentration to increase the signal to noise ratio of the intrinsically weak amplitude. These averaged time traces were fitted globally applying a monoexponential assuming perturbation of a one-step dimerization equilibrium (18),
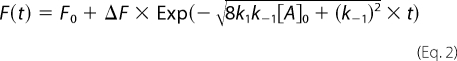 |
where F0 is the fluorescence at time 0, ΔF the increase in fluorescence, k1 the association rate of dimer formation, and k−1 the rate of dissociation.
Determination of PezAT Dissociation Rate
Displacement of PezT(D66T)Alexa488 from the PezAT complex with unlabeled PezT(D66T) was performed in buffer J at room temperature. 6 μm unlabeled PezT(D66T) was incubated with 0.3 μm of a preformed PezA(W111F)·PezT(D66T)Alexa488 complex. The complex was separated from unbound PezT using a Superdex 200 (GE Healthcare) gel filtration column equilibrated in buffer J, connected to a Waters HPLC system with attached fluorescence detector and autosampler device. Elution volumes for either the complex or PezT alone were determined by calibration runs. 50 μl of sample were injected every 60 min onto the column and fluorescence was recorded at 525 nm with excitation set to 488 nm. Elution profiles of labeled PezAT and PezT were fit to a two-component skew-normal distribution function using CalcCenter 3.0 (Wolfram Research).
RESULTS
PezT Remains Inactive after Inhibition of PezAT Expression in E. coli
In genuine TA systems, toxin activation is caused by selective degradation of the antitoxin by AAA+ chaperone-proteases like Lon, ClpXP, ClpAP, or ClpCP (5, 19–22). To probe liberation of PezT by degradation of PezA in E. coli owning the entire K12 protease background, we expressed PezAT from a tetracycline-inducible promoter in E. coli DH5α. Cells harboring pASK5+(pezAT) were induced at an A600 of 0.5. Subsequently, protein translation was inhibited by adding 50 μg/ml of chloramphenicol after a 1-h expression. Inhibition of cell growth was verified by measuring the optical density in 1-h intervals. In parallel, protein levels of PezA and PezT were monitored using Coomassie-stained PAGE gels loaded with equivalent amounts of lysed cells (Fig. 1A). Intriguingly, we observed only a weak decrease of full-length PezA protein in the presence of PezT during 4 h of growth inhibition. Because PezA is co-expressed with PezT from a single bicistronic operon (11), intracellular PezA concentrations can be expected to be higher than those of PezT due to failure in translation initiation of the second open reading frame. Thus, the observed loss of PezA is probably caused by degradation of free PezA, which is in excess over PezT. This finding was somewhat unexpected, because Lon- and ClpXP-dependent degradation of the PezA homologue ϵ was reported to occur on a time scale of minutes in the presence of ζ toxin when translation was inhibited in B. subtilis (23, 24).
FIGURE 1.

In vivo and semi-in vivo stability of PezAT. A, degradation pattern of PezAT after PezAT expression and subsequent translation inhibition using chloramphenicol. E. coli DH5α cells bearing pASK5+(pezAT) (black) were induced with 0.2 μg/ml of anhydrotetracycline at an A600 of ∼ 0.5 (blue arrow). After 1 h chloramphenicol was added leading to inhibition of protein expression (red arrow). The growth profiles of a control culture bearing pASK5+(pezA) demonstrated that growth inhibition was caused by chloramphenicol only are shown in light gray. Samples with equivalent amounts of cells were analyzed using SDS-gel electrophoresis to monitor PezA and PezT degradation (lower panel). B, growth profiles subsequent to removal of anhydrotetracycline after 1 h of expression of PezAT. E. coli DH5α cells bearing either pASK5+(pezAT) (black) and pASK5+(pezA) (gray) were induced with 0.2 μg/ml at an A600 of ∼0.5. Fresh LB medium was inoculated to a final A600 of ∼0.1 and cell growth was monitored. Cell cultures in which PezAT expression was abolished after removal of inducing agent show similar growth profiles the control. C, degradation of 1 μm of the antitoxin His-F-PezA in B. subtilis raw extract. His-F-PezA was either monitored by ECL Western blot analysis using anti-His5 (left) or imaging of IAEDANS fluorescence (right). D, degradation of 1 μm of the toxin PezT(D66T) in B. subtilis raw extract monitored by Western blot analysis as in C. E, degradation of 1 μm His-F-PezA·PezT(D66T) complex in B. subtilis raw extract similar as in C.
To probe if the observed initial loss of full-length PezA correlates with PezT release or was simply due to terminal truncation and/or degradation of unbound excess PezA, respectively, we selectively switched off expression from the tet promoter to provoke cell poisoning by PezT. Therefore, we induced PezAT expression from pASK5+(pezAT) in DH5α. In contrast to the experiment described above, cells were harvested by centrifugation and washed extensively to remove any tetracycline after 1 h of induction. The bacteria containing the expressed PezAT complex were then used to re-inoculate fresh LB medium and cell growth was monitored (Fig. 1B). Similar to what was shown for the PezT homologue ζ (11, 23, 24) we expected a perceptible growth defect when expression from the tet promoter was impaired indicating a release of PezT upon PezA degradation. Intriguingly, we could not observe a difference between the cell cultures that had either expressed PezAT or, as control, PezA alone, suggesting that no significant release and accumulation of PezT occurred.
The PezAT Core Complex Is Protected from Degradation in B. subtilis Crude Extracts
Because we could not provoke PezT release in E. coli, we speculated about any host-specific degradation and therefore performed a semi-in vivo approach in B. subtilis. In these experiments we could follow degradation of PezA and PezT over time using controlled and stoichiometric concentrations.
As probe for the antitoxin, we used a PezA-mutated variant His6-PezA(M66C) with a 1,5-IAEDANS fluorescence label attached to residue Cys-66 (further simply referred to as His-F-PezA). Cysteine 66 is located in the peptide linker between the N-terminal helix-turn-helix motif and the C-terminal PezT binding domain: the three-helix bundle. This modified variant enabled us to follow degradation of both domains independently. Whereas the Western blot analysis using an anti-His antibody indicated the presence of the N terminus of the polypeptide chain, fluorescence imaging enabled detection of any proteolytic fragments that harbor the peptide linker. Degradation of PezT(D66T) was monitored by Western blot analysis detecting its C-terminal His tag. Both, His-F-PezA and PezT(D66T) were incubated separately and together at a 1:1 molar ratio in highly concentrated, cytosolic extracts from freshly prepared B. subtilis.
We found that when His-F-PezA was incubated in B. subtilis cell extracts, the N-terminal His tag became undetectable within the first hour of incubation (Fig. 1C). In parallel, we observed the occurrence of a faint band with higher electrophoretic mobility, indicating that to some extent N-terminal truncation to a smaller but stable domain occurred. On the other hand, PezT(D66T) turned out to be more stable than PezA and the C-terminal His tag was still detectable after 1 h of incubation (Fig. 1D).
However, when both proteins were incubated together, the His tag of His-F-PezA was still detectable after 2 h of incubation. Apparently, once bound to PezT, full-length His-F-PezA is protected to some extent. In contrast to the previous experiment, almost the entire His-F-PezA was subsequently truncated to the smaller stable domain containing the fluorophore, which was detected by the fluorescence imaging technique (Fig. 1E). Furthermore, PezT remained stable throughout the entire experiment suggesting that the smaller, proteolytically derived C-terminal fragment is sufficient to protect PezT from degradation. We can exclude that neither mutation nor the labeling of PezA have caused the observed stabilizing effect because similar results were obtained with unlabeled wild type PezA (data not shown).
Our experiments show that a PezAT core complex lacking the helix-turn-helix motif of PezA is extremely resistant toward proteolysis when compared with the individual proteins alone. Additional experiments using native gel electrophoresis monitoring IAEDANS fluorescence verified the identity and integrity of this PezAT core complex (supplemental Fig. S1). Thus, PezA undergoes a fast N-terminal truncation, leaving the remaining three-helix bundle associated to PezT. In the core complex the PezA three-helix bundle and PezT are stably associated and both polypeptide chains are resistant toward any further proteolytic cleavage. This result is in good agreement with our stability studies in E. coli (see above) because we observed slight reduction of PezA levels in our PAGE analysis but the cells did not show impaired growth (Fig. 1A).
PezAT Complex Formation Strongly Increases the Thermodynamic Stability of the Two Proteins
Protein susceptibility to degradation depends on their global thermodynamic stability and, if they are substrates for ATP-dependent proteases on the accessibility and local stability of recognition motifs (25–27). To probe if the observed proteolytic stability of the PezAT complex is correlated with an increased thermodynamic stability, we performed unfolding studies of PezAT and both individual proteins using urea-induced denaturation monitored by the red shift in tryptophan fluorescence and circular dichroism. In these experiments a mutated tryptophan-free PezT variant (PezT(D66T,W232Y)) was used, because both, wild type PezA and PezT contain a single tryptophan residue. Whereas Trp-232 of PezT is solvent exposed, Trp-111 of PezA is hidden in the hydrophobic core of the homodimer interface of the C-terminal three-helix bundle (see Fig. 3B). Thus, a change in tryptophan fluorescence upon denaturation was expected to be more pronounced for PezA than for PezT and we could exclusively follow the equilibrium unfolding properties of this domain in the absence and presence of PezT.
FIGURE 3.

Tryptophan fluorescence experiments of PezA and PezT upon complex formation. A, fluorescence titrations of 3 μm PezT(D66T) with wild type PezA (dark gray circles) or tryptophan-free PezA(W111F) (light gray circles) or buffer (white circles). Excitation was set to 295 nm and emission recorded at 347 nm. B, ribbon representation of the PezAT heterotetramer of PezA (red) and PezT (blue). Wild type tryptophan residues Trp-232 of PezT (orange) and Trp-111 (green) are highlighted as stick models. Whereas Trp-232 enables monitoring binding of PezA, Trp-111 of PezA was shown to be sensitive to changes in the homodimer interface of the antitoxin. C, exemplary real time trace of binding of 1 μm PezT(D66T) to 1 μm PezA(W111F) (gray) measured in a stopped-flow device. Tryptophan fluorescence of PezT(D66T) was excited at 296 nm and emission recorded with a 320-nm long-pass filter. The corresponding fit according to the two-step model is shown as black line. D, similar as C but 5 μm PezT(D66T) and 5 μm PezA(W111F). E, similar as C but 3 μm PezT(D66T) and 1.5 μm PezA(W111F). Note the difference in scaling of the time axis.
In the absence of PezT(D66T,W232Y), the unfolding profile of the PezA three-helix bundle shows a broad major transition, with a denaturation mid-point (D½) at 4.1 m urea (Fig. 2A). Interestingly, the presence of PezT(D66T,W232Y) leads to a considerably steeper unfolding transition and the D½ increases to 4.9 m urea, indicating enhanced stability and a more pronounced cooperativity in unfolding of the PezA three-helix bundle (Fig. 2B). We note that we already observed a minor, protein concentration-dependent red shift of PezA tryptophan fluorescence at lower urea concentrations, indicating that the homodimer equilibrium of PezA is disturbed (supplemental Fig. S2).
FIGURE 2.

Urea-induced denaturation experiments. A, unfolding of 5 μm PezA in the absence (white circles) and presence (gray circles) of 5 μm PezT(D66T,W232Y) monitored by the red shift of the spectral center of mass of tryptophan fluorescence. B, PezA denaturation (5 μm) monitored either by tryptophan fluorescence (white circles) or the mean residue ellipticity at 222 nm (white triangles). C, denaturation of PezA (5 μm) in the presence of equimolar concentrations of PezT(D66T,W232Y) monitored by tryptophan fluorescence (gray circles) and denaturation of the complex by mean residue ellipticity at 222 nm (gray triangles). D, denaturation of 5 μm PezT(D66T,W232Y) alone (white diamonds) and in the presence of 5 μm PezA (gray diamonds) monitored by mean residue ellipticity at 222 nm. Note that individual values have been normalized in panel D.
When we monitored the global folding/unfolding equilibrium of PezA and PezAT by circular dichroism at 222 nm, we found a D½ at ∼4.6 m urea for unbound PezA (Fig. 2B). Apparently, the fluorescence and CD response curves of PezA alone have different denaturation midpoints indicating the presence of a folding intermediate. Intriguingly, for the PezA·PezT(D66T,W232Y) complex we observe only a single but steep transition (D½ = 4.9 m) that overlays well with the major transition in tryptophan fluorescence of bound PezA as described above (Fig. 2C). This indicates that PezA shows different unfolding behavior once bound to PezT and an unfolding intermediate occurs only for free PezA. However, both proteins unfold with high cooperativity once they are in complex. Additionally, free PezT(D66T,W232Y) already unfolds at much lower urea concentrations (D½ = 3.5 m) than the complex, indicating a reduced thermodynamic stability of the free toxin compared with the free antitoxin. This rather untypical finding for TA systems seem to be a distinct feature of the family of ϵ/ζ systems because it has also been reported for the ζ toxin from S. pyogenes (23). In summary, our results demonstrate that the proteolytic stability of the PezAT complex correlates with a strong increase in thermodynamic stability of both PezA and PezT once they are bound to each other. Consequently, the heterotetrameric assembly of the complex must provide both proteins with a strong increase in conformational rigidity.
Association of PezT and PezA Leads to a Conformational Change of PezT
Our previous results prompted us to perform a detailed investigation of the dynamics of PezAT complex formation. Recent isothermal calorimetry experiments indicated a strong 1:1 interaction between PezA and PezT with an affinity in the subnanomolar range (11). In an initial tryptophan florescence titration experiment, we found that the tight binding event is accompanied by an overall increase in tryptophan fluorescence of the complex (Fig. 3A). We could confirm that the increased quantum yield can be attributed to Trp-232 of PezT, because titration experiments using the tryptophan-free PezA(W111F) variant showed the same change as observed for wild type proteins. Additionally, our experiments could confirm the previously reported 1:1 stoichiometry, because the fluorescence only increased until equimolar concentrations were reached (Fig. 3A). Thus, we can unambiguously assign the fluorescence increase to a conformational change around Trp-232 of PezT. Trp-232 is located in the loop between the two C-terminal helices of the toxin and, notably, not part of any protein contact interface. Therefore, the enhanced emission intensity observed can only be explained by a local conformational change in the environment of the fluorophore. It is worth mentioning that deletion of Trp-111 in PezA decreased the total tryptophan fluorescence of the complex only marginally, indicating that this fluorophore is highly quenched.
PezT Associates with PezA in a Rapid Two-step Mechanism
Because the absence of any curvature in the binding isotherm did not allow determination of a dissociation constant (Fig. 3A), we decided to measure binding kinetics using rapid mixing techniques. In these experiments, PezT(D66T) was mixed with PezA(W111F) at a set of different protein concentrations in the micromolar range using a stopped-flow device (Fig. 3, C–E). This non-toxic PezT(D66T) variant was previously shown to have impaired toxic activity of PezT in vivo, however, the side chain of Asp-66 is not involved in any PezA/PezT interaction (11).
Intriguingly, these experiments revealed an ultra-rapid formation of the complex, because the kinetic traces level out within milliseconds at low micromolar concentrations. We collected a dataset of 13 time traces at stoichiometric and non-stoichiometric mixing ratios and fitted them globally to different possible association pathways. Such global fitting procedures of different datasets have been shown to be adequate for elucidating sophisticated binding pathways (28, 29). Surprisingly, the minimal association mechanism that describes the data sufficiently is a simplified, quasi-irreversible two-step model (Scheme 1) that involves a conformational change of PezT (T) after binding to an empty PezA binding site (Ai),
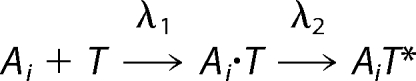 |
where the apparent rate constant λ1 describes formation of a transient complex Ai · T and λ2 describes a rapid conformational change of PezT upon formation of the stereospecific complex, leading to the observed increase in tryptophan fluorescence. The best fit values obtained for the experimental conditions are well defined (supplemental Fig. S3) and converge to 100 μm−1 s−1 for λ1 and 410 s−1 for λ2.
This association pathway is reminiscent to a generalized protein-protein association model for two binders, where formation of an initial transient complex, also often denoted as “encounter complex,” is followed by formation of the stereospecific complex (30, 31). Notably, convergence of the fit to the data were already obtained using this simplified, quasi-irreversible two-step model and including any additional putative rate constant accounting for the oligomeric state of PezA or a transiently occurring trimeric intermediate etc. was not required. This finding together with the fact that the initial binding rate λ1 exceeds the expected value for diffusion-controlled protein-protein association, suggested the presence of long-range electrostatic interactions between PezA and PezT.
The PezAT Complex Forms a Stable Heterotetramer in Solution
Any possible disassembly factor that is capable of separating PezA from PezT would also need to interfere with the homodimer interface of PezA, because dimeric PezA is sandwiched between two PezT molecules in the heterotetrameric complex (see Ref. 11 and Fig. 3B). We therefore investigated the kinetics of PezA homodimerization and asked whether a PezAT heterotetramer or a PezAT heterodimer is the dominating oligomeric state in solution. In relaxation experiments we could monitor the change in tryptophan fluorescence of the highly quenched tryptophan 111 of PezA, which is located in the homodimer interface. Briefly, either 5 μm PezA or PezAT complex (but using the tryptophan free PezT variant) were rapidly diluted to various concentrations using a stopped-flow device (Fig. 4, A and B). Global fit analysis of the resulting relaxation kinetics revealed that dimerization of PezA and PezAT heterodimers can be described by a simple one-step dimerization mechanism. We found that PezA alone forms homodimers with a Kd of 16 nm following moderate association (kon = 6.3 μm−1 s−1) and dissociation (koff = 0.1 s−1) kinetics. In contrast, we observed a slightly decreased affinity of PezA homodimers once bound to PezT resulting in a Kd of 60 nm. Whereas the dissociation rate constant increased to koff = 0.3 s−1, the association rate constant was only reduced modestly to kon = 5.3 μm−1 s−1. Apparently, the homodimer is slightly destabilized in the PezAT complex but assuming that PezA and PezT have similar cytosolic concentrations as reported for the related ϵ/ζ system (23), we speculate that a heterotetramer is the predominating oligomeric state in vivo. Thus, any factor that can separate PezT from PezA needs to interfere with the heterotetrameric PezA/PezT arrangement.
FIGURE 4.

Relaxation kinetics of the PezA homodimerization measured by tryptophan fluorescence. A, exemplary time trace of a rapid 1:15 buffer dilution at 10 μm PezA (gray) and the monoexponential fit describing a one-step dimerization mechanism. B, exemplary relaxation time traces of the PezAT heterodimer/heterotetramer equilibrium measured by rapid 1:15 buffer dilution at 10 μm PezA·PezT(D66T,W232Y) complex (gray) and the corresponding fit (black). Protein fluorescence was excited at 280 nm and emission recorded with a 320-nm long pass filter.
PezAT Association Is Electrostatically Enhanced
Several protein-protein complexes have been described, where complementary electrostatic interactions strongly accelerate association kinetics (32–35). Because we observed association kinetics that are faster than those of a purely diffusion controlled process, we expected PezAT complex formation to be dependent on the ionic strength of the solute. Our assumption was further supported by calculations of electrostatic surface potentials of PezA and PezT using APBS (36–38), which showed that the PezT binding interface of PezA has an overall highly negative surface charge, whereas the cognate interface of PezT is complementarily positively charged (Fig. 5A).
FIGURE 5.

PezA and PezT binding is accelerated by electrostatic interactions. A, open book representation of the PezA and PezT binding interface showing complementary charged surface potentials at moderate ionic strength. Charge distributions have been calculated using the PyMOL APBPS, PDB code 2PQR at 0.2 m NaCl. B, dependence of the apparent rate constants λ1 (dark gray) and λ2 (light gray) on the ionic strength. The black line corresponds to the fit of λ1 using Equation 1 with a fixed value of 6 Å for A. Best fit values were −9.5 for U/kT and 1 × 106 m−1 s−1 for k(∞).
The existence of intermolecular electrostatic forces can be verified by measuring the rate dependences of interactions on the ionic strength of the solute. To probe the PezAT toxin antitoxin association for such an electrostatic enhancement, we measured formation of 1 μm PezAT complex at increasing concentrations of NaCl. Plotting λ1 versus the ionic strength of the solute verified the electrostatic interaction experimentally (Fig. 5B). Indeed, λ1 is ∼230 μm−1 s−1 at 0.05 m NaCl and decreases by more than an order of magnitude to less than 15 μm−1 s−1 at 1 m NaCl. On the other hand, the second observed rate constant λ2 seems to be affected only mildly by varying the salt concentration. It retained close to a value of 400 s−1 at 0.1 to 0.4 m NaCl and remained fast but indeterminable at higher salt concentrations due to the increasing rate limitation by λ1. This is reminiscent to the Colicin E9-Im9 complex, where the rate of the second association step is not grossly affected by the ionic strength of the solute indicating that λ2 is mostly dependent on short-range interactions (35).
The salt dependence of the rate describing the formation of transient protein-protein complexes can be well explained on the basis of Debye's (39–41) treatment of the association rates of charged spheres,
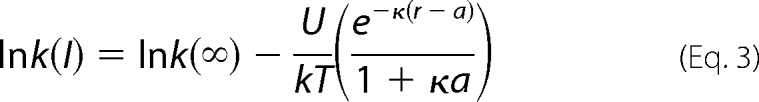 |
where k(∞) is the rate at infinity ionic strength, U the interaction energy between the two proteins at zero ionic strength, and κ the Debye-Hückel screening parameter directly dependent on the ionic strength of the solute. According to Gabdoulline and Wade (42), a represents the double distance between the protein charges relevant for association and the ion centers in the solvent (∼6 Å for NaCl) and r the relative separation of the protein charges in the transient complex. When fitting the salt dependence of λ1 using Equation 1, we obtain a value of ∼7 Å for r indicating that PezA and PezT are already in very close proximity prior to the second binding step. Thus, in the transient complex both proteins are most probably in close proximity and in a conformation, which is similar to the final stereospecific complex. This is corroborated by the quasi-irreversibility of the first reaction step of the global fit.
The PezAT Complex Has Femtomolar Affinity at Physiological Ionic Strength
Anticipated from the fast association kinetics, we were interested in the dissociation kinetics of the PezAT complex. Initial experiments hinted at a very slow toxin exchange rate from the complex. Because such kinetics are difficult to investigate by conventional stopped-flow measurements, we established a long-time displacement assay using Alexa 488-labeled PezT(D66T). Briefly, PezT(D66T)Alexa488 in complex with PezA(W111F) was incubated with a 20-fold excess of unlabeled PezT(D66T) over a time period of 2 days. Samples taken at individual time points were separated by size exclusion chromatography. The integrity of the complex was verified by SDS-PAGE analysis after long time storage in the buffer system used in the experiment (data not shown). Exchange kinetics were monitored by the observed decrease of the fluorescence signal of the PezAT complex due to displacement of labeled PezT(D66T) by unlabeled species and an increase of the fluorescence signal of labeled monomeric PezT (Fig. 6A). Both, occurrence of detectable free PezT(D66T) and disappearance of the complex could be fitted with a monoexponential yielding an overall dissociation rate constant of PezT replaced from the PezAT heterotetramer with λoff = 6.5 × 10−6 s−1 resulting in an average half-life of t½ of ∼30 h for the PezAT interaction (Fig. 6B).
FIGURE 6.

Dissociation kinetics of the PezAT complex measured by exchange kinetics. 0.3 μm preassembled PezA(W111F)/PezT(D66T)Alexa488 was incubated with 3 μm unlabeled PezT(D66T). A, the decrease of fluorescent PezAT species and increase of free, labeled PezT was monitored by analytical gel filtration. B, data after peak integration and normalization were fit in parallel with a monoexponential, yielding an apparent dissociation constant of 6.5 × 10−6 s−1.
Even though our kinetic data do not provide all information necessary to describe every forward and backward rate constant of a classical two-state model in detail, the quasi-irreversibility of the system allowed for calculation of an apparent Kd from the ratio of the observed constants λoff/λ1 as described for other high affinity complexes (35, 43). Thus, the PezAT interaction displays an intriguingly tight binding with an apparent Kd of 65 × 10−15 m (65 femtomolar) at physiological ionic strength.
DISCUSSION
In this study we investigated the overall stability of the chromosomally encoded PezAT TA system from the human pathogen S. pneumoniae. Our data suggest an intriguing resistance of the inhibitory complex toward proteolysis in E. coli and B. subtilis preventing release of toxin under the conditions tested. Results from in vitro unfolding experiments further indicated that there is a tight interaction between toxin and antitoxin that has a great impact on the thermodynamic stability of both proteins. Fig. 7 summarizes the detailed kinetic analysis of the PezAT interaction determined using rapid mixing methods and time-resolved size exclusion chromatography. According to our findings, PezA forms homodimers with nanomolar affinity that are rapidly attracted to unbound PezT due to complementary electrostatic charges. After formation of an initial, transient complex, a fast rearrangement locks the PezAT assembly in a predominantly heterotetrameric state.
FIGURE 7.

Model of the PezAT assembly dynamics. The onset of PezAT heterotetramer formation is driven by rapid association of PezT with either PezA monomers or dimers. This initial association step is based on long range electrostatic interactions resulting in a transient PezAT complex in which both proteins are still separated by ∼7 Å. A subsequent rapid rearrangement eventually leads to the final stereospecific inhibitory PezAT complex, which is predominantly a heterotetramer due to the nanomolar affinity of the central PezA homodimer interface.
This outstanding femtomolar affinity of PezA and PezT is the strongest reported among all TA systems, which have been characterized so far. Intriguingly, other TA systems like PhD/Doc (44), RelB/RelE (45), and CcdB/CcdA (46) have been described to have affinities 3 to 8 orders of magnitudes lower than PezAT. Even for the PezAT homologue ϵ/ζ, only just a micromolar affinity has been reported (23). In summary, PezAT is rather reminiscent to endotoxin-inhibitor complexes like barnase/bastar (34) or members of the colicin/immunoprotein family (33, 35), which are not meant to dissociate in the host cell but kill susceptible target cells after secretion.
The difference in affinity within the family of ϵ/ζ TA systems is somewhat unexpected, because given the structural similarity of PezT and ζ (11, 13), one can expect that both proteins have a similar toxic function. Furthermore, site-directed mutagenesis studies have shown that conserved amino acids are involved in toxicity, although the target of PezT and ζ is still enigmatic. A probable explanation for these contradicting findings might be different cellular missions of PezAT and ϵ/ζ. Whereas disruption of the PezT open reading frame in S. pneumoniae resulted in impaired infectiousness of the pathogen, which implies that PezAT acts as a virulence factor (16), ϵ/ζ was reported to be a classical TA system ensuring stable inheritance of the plasmid pSM19035 (14). It seems plausible that toxin release rates have been fine tuned and adapted to perform different cellular tasks in their specific host.
Activation of classical TA systems usually involves accessibility of the antitoxin toward degradation by cellular proteases leading to an accumulation of freed toxin when protein expression from the TA operon is impaired. However, the high affinity of PezAT and the resulting stabilization of PezA upon complex formation with PezT seem to impair toxin release by simple dissociation. Additionally, it is unlikely that PezT release upon simple dissociation is caused by proteolytic removal of the transcriptional repressor domain of PezA, because the C-terminal domains binds to PezT with comparable affinity as full-length protein (supplemental Fig. S4). Our results rather suggest that specific degradation of PezA from the complex either requires a host-specific protease, which might be absent in the organisms used in this study, or a triggered activation of an adaptor molecule by an external queue required for proper recognition of PezA by a constitutive AAA+ protease.
It is tempting to speculate that the absence of “leakiness” in PezT inhibition renders the host-cell capable to actively control toxin release. This could be in contrast to other TA systems like ϵ/ζ, CcdA/CcdB, mazE/mazF, RelE/RelB, or PemI/PemK, which have been shown to respond directly to loss of either the TA open reading frame or general stress conditions that impair protein translation (14, 19, 47–50). On the other hand, examples of TA systems have been reported to require bacterial signaling pathways to carry out sophisticated functions like programmed cell death upon fruit body formation in Myxococcus xanthus or growth regulation by quorum sensing in E. coli (5, 8). Moreover, TA systems are key players during persister cell formation and the development of bacterial biofilms (51–54). Given the observed lack of proteolytic degradation and the reported thermodynamic properties of the PezAT system, PezAT most likely is similar to the latter described type of TA systems.
In general, activation of any TA system will enable new therapeutic approaches in our antimicrobial defense. Thus, understanding the assembly and disassembly pathways and kinetics is of crucial importance for such challenging goals. However, in the case of PezAT activation of toxin by antitoxin displacement using peptidic mimics as recently suggested for other TA systems (55, 56) might not be feasible.
Supplementary Material
Acknowledgments
We thank H. Salmen, C. Nürnberger, and M. Gebhardt for experimental assistance. We are grateful to C. C. Yeo, B. Loll, B. Lunde, and R. L. Shoeman for helpful discussions and I. Schlichting for continuous encouragement and support.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and Figs. S1–S4.
- TA
- toxin-antitoxin
- Pez
- pneumococcal epsilon zeta
- 1,5-IAEDANS
- 5-({2-[(iodoacetyl)amino]ethyl}amino)naphthalene-1-sulfonic acid
- AAA
- ATPases associated with various cellular activities.
REFERENCES
- 1.Gerdes K., Christensen S. K., Løbner-Olesen A. (2005) Nat. Rev. Microbiol. 3, 371–382 [DOI] [PubMed] [Google Scholar]
- 2.Pandey D. P., Gerdes K. (2005) Nucleic Acids Res. 33, 966–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsilibaris V., Maenhaut-Michel G., Mine N., Van Melderen L. (2007) J. Bacteriol. 189, 6101–6108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Magnuson R. D. (2007) J. Bacteriol. 189, 6089–6092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kolodkin-Gal I., Hazan R., Gaathon A., Carmeli S., Engelberg-Kulka H. (2007) Science 318, 652–655 [DOI] [PubMed] [Google Scholar]
- 6.Wozniak R. A., Waldor M. K. (2009) PLoS Genet. 5, e1000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szekeres S., Dauti M., Wilde C., Mazel D., Rowe-Magnus D. A. (2007) Mol. Microbiol. 63, 1588–1605 [DOI] [PubMed] [Google Scholar]
- 8.Nariya H., Inouye M. (2008) Cell 132, 55–66 [DOI] [PubMed] [Google Scholar]
- 9.Ceglowski P., Boitsov A., Karamyan N., Chai S., Alonso J. C. (1993) Mol. Gen. Genet. 241, 579–585 [DOI] [PubMed] [Google Scholar]
- 10.O'Connor E. B., O'Sullivan O., Stanton C., Danielsen M., Simpson P. J., Callanan M. J., Ross R. P., Hill C. (2007) Plasmid 58, 115–126 [DOI] [PubMed] [Google Scholar]
- 11.Khoo S. K., Loll B., Chan W. T., Shoeman R. L., Ngoo L., Yeo C. C., Meinhart A. (2007) J. Biol. Chem. 282, 19606–19618 [DOI] [PubMed] [Google Scholar]
- 12.Sletvold H., Johnsen P. J., Hamre I., Simonsen G. S., Sundsfjord A., Nielsen K. M. (2008) Plasmid 60, 75–85 [DOI] [PubMed] [Google Scholar]
- 13.Meinhart A., Alonso J. C., Sträter N., Saenger W. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 1661–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zielenkiewicz U., Ceglowski P. (2005) J. Bacteriol. 187, 6094–6105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown J. S., Gilliland S. M., Holden D. W. (2001) Mol. Microbiol. 40, 572–585 [DOI] [PubMed] [Google Scholar]
- 16.Brown J. S., Gilliland S. M., Spratt B. G., Holden D. W. (2004) Infect. Immun. 72, 1587–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuzmic P. (1996) Anal. Biochem. 237, 260–273 [DOI] [PubMed] [Google Scholar]
- 18.Bernasconi C. F. (1976) Relaxation Kinetics, Academic Press, New York [Google Scholar]
- 19.Christensen S. K., Maenhaut-Michel G., Mine N., Gottesman S., Gerdes K., Van Melderen L. (2004) Mol. Microbiol. 51, 1705–1717 [DOI] [PubMed] [Google Scholar]
- 20.Donegan N. P., Thompson E. T., Fu Z., Cheung A. L. (2010) J. Bacteriol. 192, 1416–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim Y., Wang X., Zhang X. S., Grigoriu S., Page R., Peti W., Wood T. K. (2010) Environ. Microbiol. 12, 1105–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lehnherr H., Yarmolinsky M. B. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 3274–3277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Camacho A. G., Misselwitz R., Behlke J., Ayora S., Welfle K., Meinhart A., Lara B., Saenger W., Welfle H., Alonso J. C. (2002) Biol. Chem. 383, 1701–1713 [DOI] [PubMed] [Google Scholar]
- 24.Lioy V. S., Martín M. T., Camacho A. G., Lurz R., Antelmann H., Hecker M., Hitchin E., Ridge Y., Wells J. M., Alonso J. C. (2006) Microbiology 152, 2365–2379 [DOI] [PubMed] [Google Scholar]
- 25.Kenniston J. A., Burton R. E., Siddiqui S. M., Baker T. A., Sauer R. T. (2004) J. Struct. Biol. 146, 130–140 [DOI] [PubMed] [Google Scholar]
- 26.McLendon G., Radany E. (1978) J. Biol. Chem. 253, 6335–6337 [PubMed] [Google Scholar]
- 27.Parsell D. A., Sauer R. T. (1989) J. Biol. Chem. 264, 7590–7595 [PubMed] [Google Scholar]
- 28.Kress W., Mutschler H., Weber-Ban E. (2007) Biochemistry 46, 6183–6193 [DOI] [PubMed] [Google Scholar]
- 29.Werbeck N. D., Kellner J. N., Barends T. R., Reinstein J. (2009) Biochemistry 48, 7240–7250 [DOI] [PubMed] [Google Scholar]
- 30.Alsallaq R., Zhou H. X. (2007) Structure 15, 215–224 [DOI] [PubMed] [Google Scholar]
- 31.Schreiber G., Haran G., Zhou H. X. (2009) Chem. Rev. 109, 839–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee F. S., Shapiro R., Vallee B. L. (1989) Biochemistry 28, 225–230 [DOI] [PubMed] [Google Scholar]
- 33.Li W., Keeble A. H., Giffard C., James R., Moore G. R., Kleanthous C. (2004) J. Mol. Biol. 337, 743–759 [DOI] [PubMed] [Google Scholar]
- 34.Schreiber G., Fersht A. R. (1996) Nat. Struct. Mol. Biol. 3, 427–431 [DOI] [PubMed] [Google Scholar]
- 35.Wallis R., Moore G. R., James R., Kleanthous C. (1995) Biochemistry 34, 13743–13750 [DOI] [PubMed] [Google Scholar]
- 36.Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dolinsky T. J., Czodrowski P., Li H., Nielsen J. E., Jensen J. H., Klebe G., Baker N. A. (2007) Nucleic Acids Res. 35, W522–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dolinsky T. J., Nielsen J. E., McCammon J. A., Baker N. A. (2004) Nucleic Acids Res. 32, W665–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Debye P. (1942) J. Electrochem. Soc. 82, 265–272 [Google Scholar]
- 40.Selzer T., Schreiber G. (1999) J. Mol. Biol. 287, 409–419 [DOI] [PubMed] [Google Scholar]
- 41.Vijayakumar M., Wong K. Y., Schreiber G., Fersht A. R., Szabo A., Zhou H. X. (1998) J. Mol. Biol. 278, 1015–1024 [DOI] [PubMed] [Google Scholar]
- 42.Gabdoulline R. R., Wade R. C. (1999) J. Mol. Recognit. 12, 226–234 [DOI] [PubMed] [Google Scholar]
- 43.Schreiber G., Fersht A. R. (1993) Biochemistry 32, 5145–5150 [DOI] [PubMed] [Google Scholar]
- 44.Gazit E., Sauer R. T. (1999) J. Biol. Chem. 274, 16813–16818 [DOI] [PubMed] [Google Scholar]
- 45.Overgaard M., Borch J., Gerdes K. (2009) J. Mol. Biol. 394, 183–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dao-Thi M. H., Van Melderen L., De Genst E., Afif H., Buts L., Wyns L., Loris R. (2005) J. Mol. Biol. 348, 1091–1102 [DOI] [PubMed] [Google Scholar]
- 47.Van Melderen L., Bernard P., Couturier M. (1994) Mol. Microbiol. 11, 1151–1157 [DOI] [PubMed] [Google Scholar]
- 48.Tsuchimoto S., Nishimura Y., Ohtsubo E. (1992) J. Bacteriol. 174, 4205–4211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sat B., Reches M., Engelberg-Kulka H. (2003) J. Bacteriol. 185, 1803–1807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hazan R., Sat B., Engelberg-Kulka H. (2004) J. Bacteriol. 186, 3663–3669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kolodkin-Gal I., Verdiger R., Shlosberg-Fedida A., Engelberg-Kulka H. (2009) PLoS ONE 4, e6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harrison J. J., Wade W. D., Akierman S., Vacchi-Suzzi C., Stremick C. A., Turner R. J., Ceri H. (2009) Antimicrob. Agents Chemother. 53, 2253–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lewis K. (2008) Curr. Top. Microbiol. Immunol. 322, 107–131 [DOI] [PubMed] [Google Scholar]
- 54.Lewis K. (2005) Biochemistry 70, 267–274 [DOI] [PubMed] [Google Scholar]
- 55.Lioy V. S., Rey O., Balsa D., Pellicer T., Alonso J. C. (2010) Plasmid 63, 31–39 [DOI] [PubMed] [Google Scholar]
- 56.Nieto C., Pellicer T., Balsa D., Christensen S. K., Gerdes K., Espinosa M. (2006) Mol. Microbiol. 59, 1280–1296 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.